
Evaluation of the Potential Risk of Drugs to Induce Hepatotoxicity in Human—Relationships between Hepatic Steatosis Observed in Non-Clinical Toxicity Study and Hepatotoxicity in Humans-

Abstract
:1. Introduction
2. Results
2.1. One-Month Repeated Oral Dose Study of Compound X in Rats
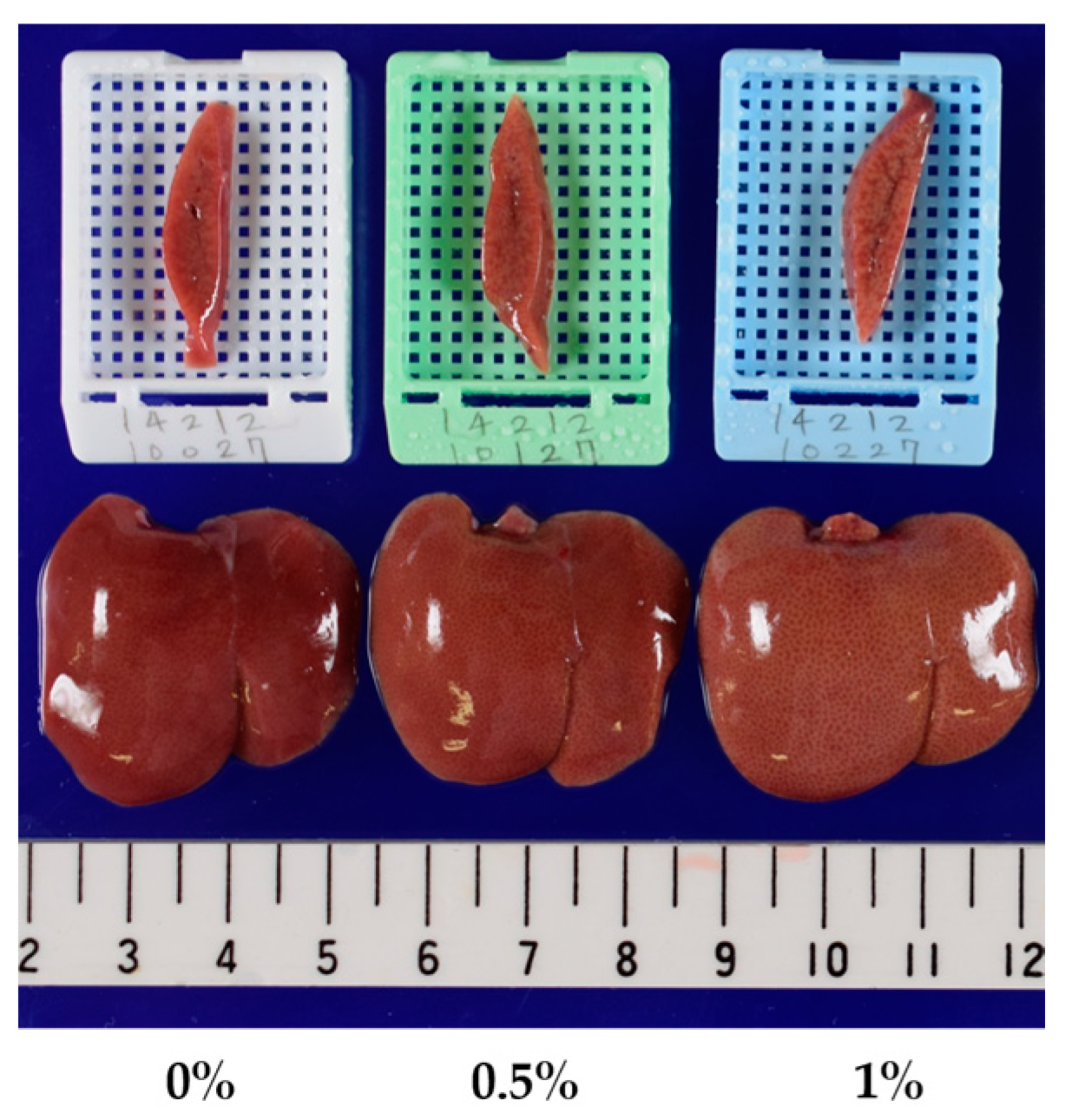
2.2. Three-Month Repeated Oral Dose Study of Compound X in Rats
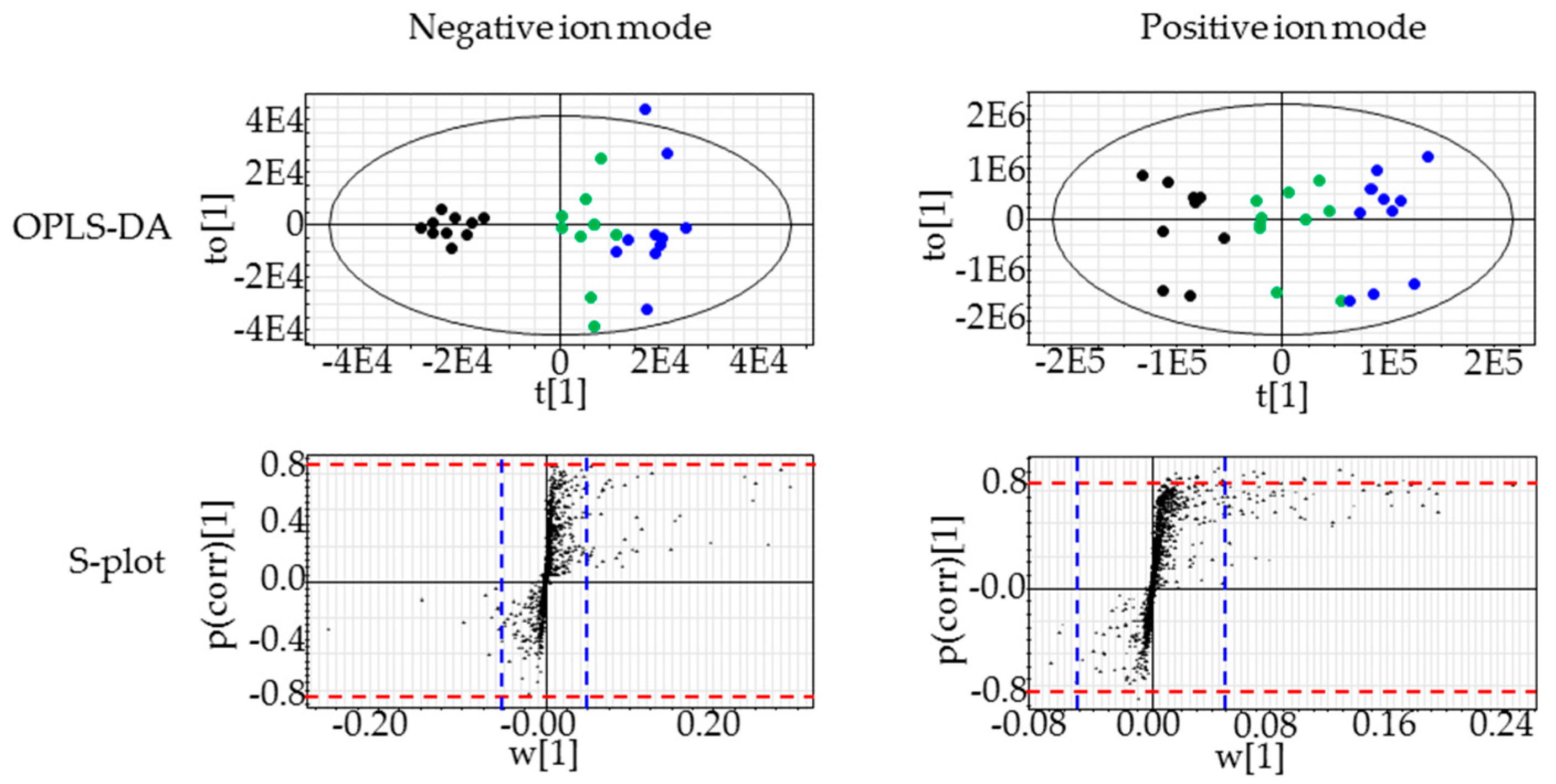
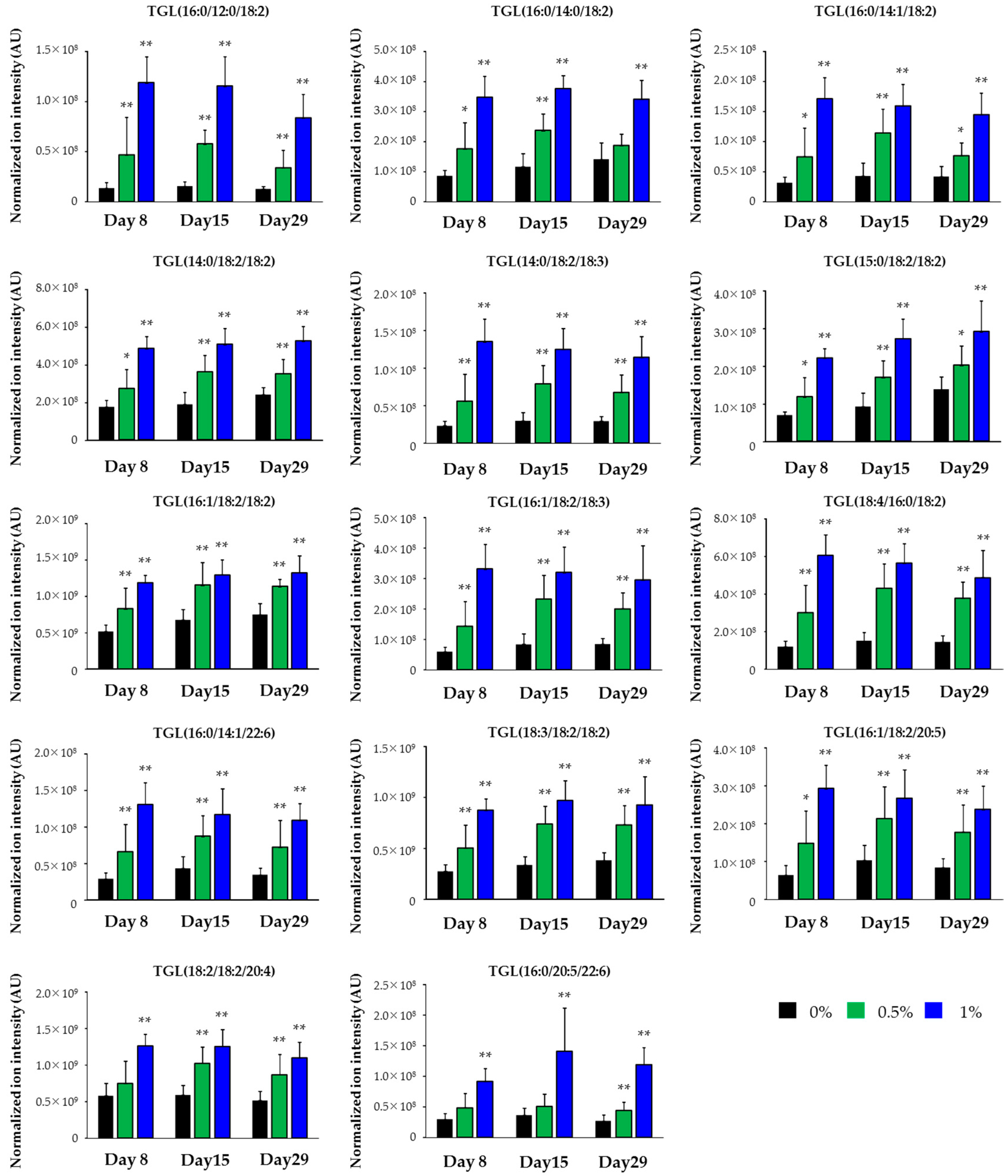
2.3. Lipidomics for the Liver of Rats Treated with Compound X for One Month
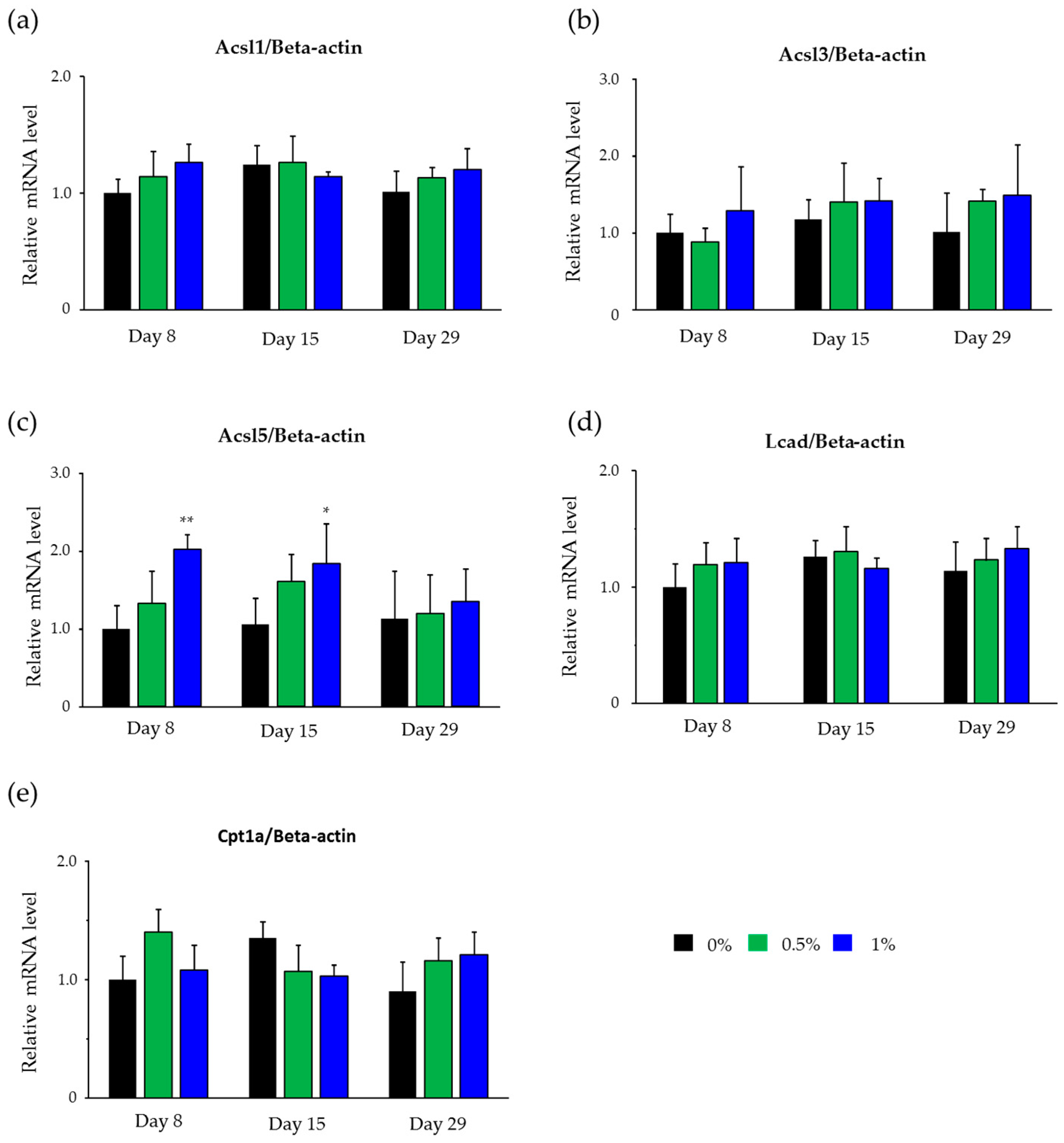
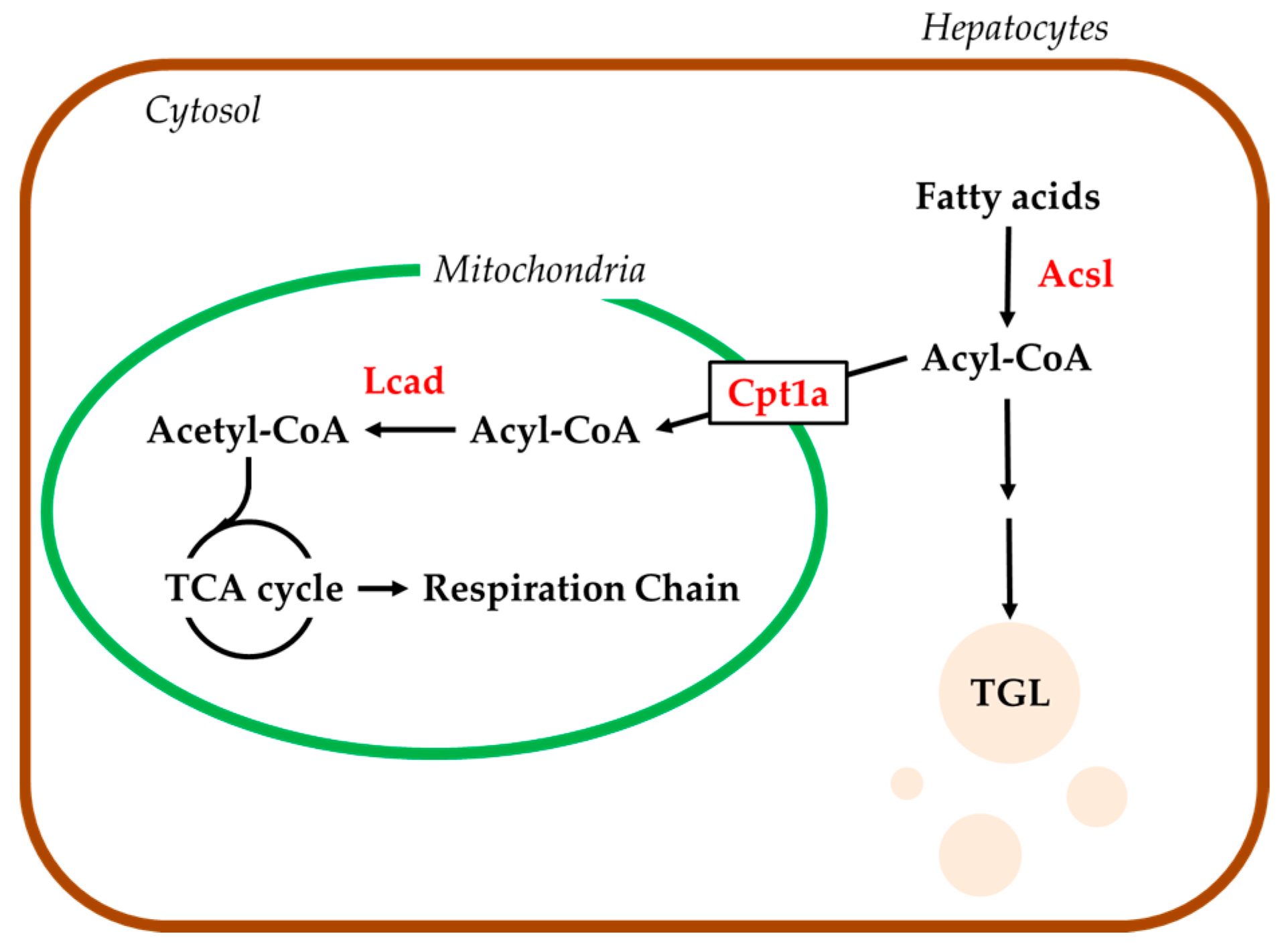
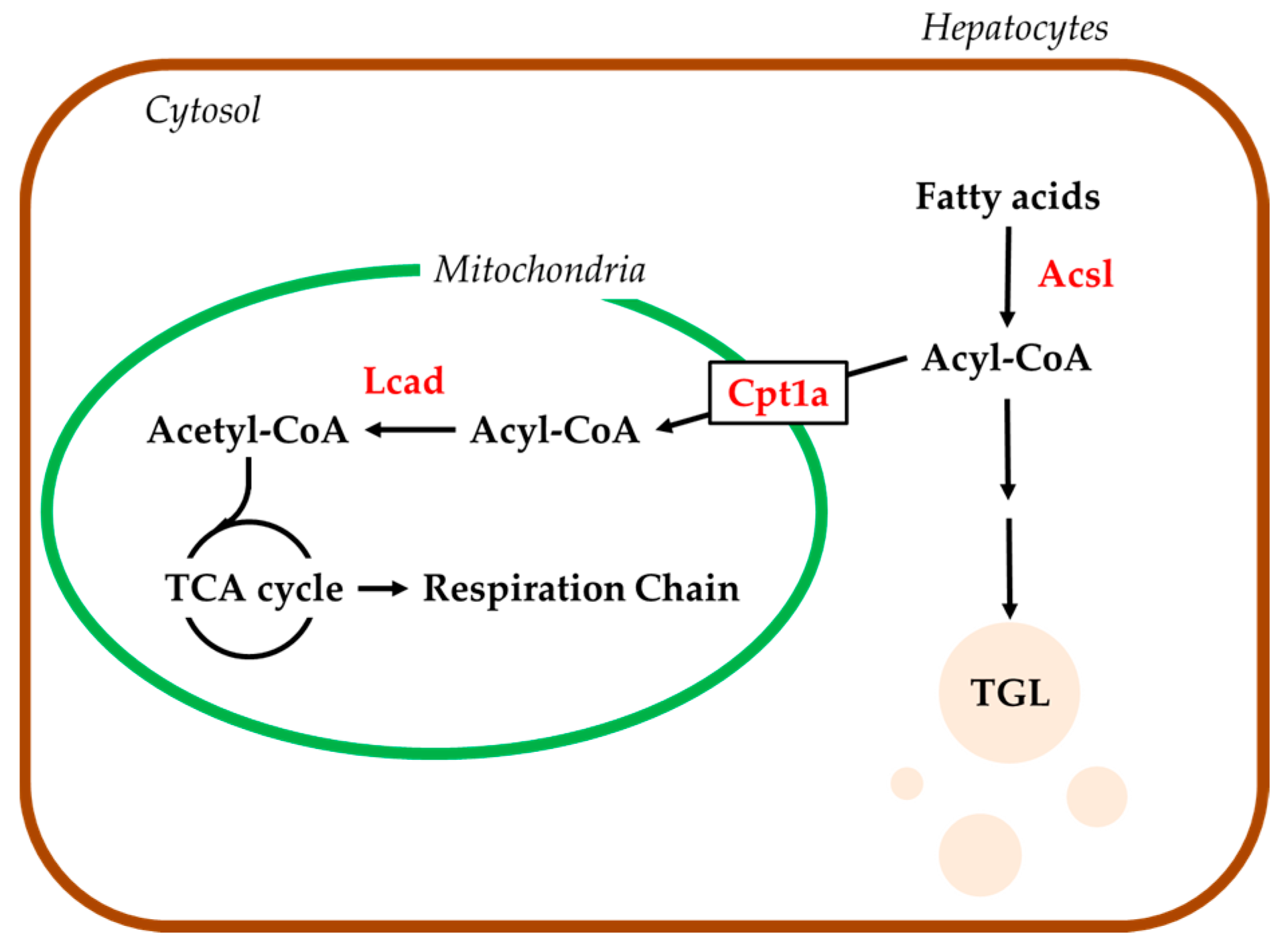
2.4. Hepatic mRNA Levels of Enzymes Related to Beta-Oxidation in Rats Treated with Compound X
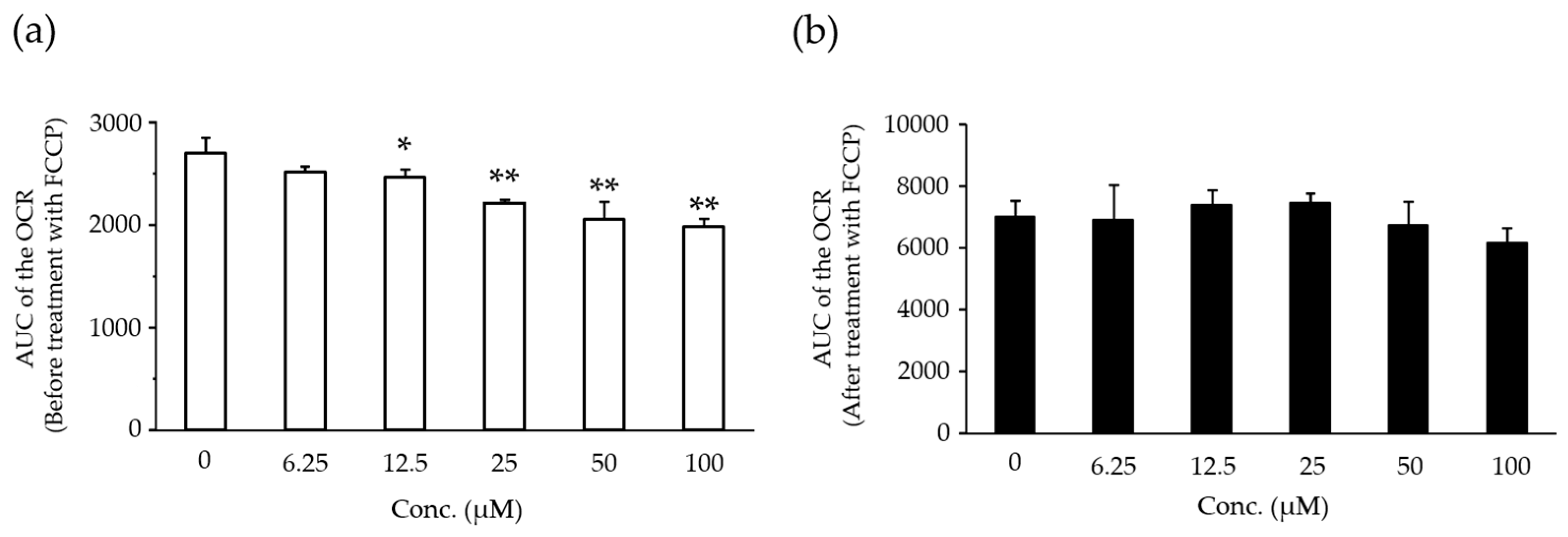
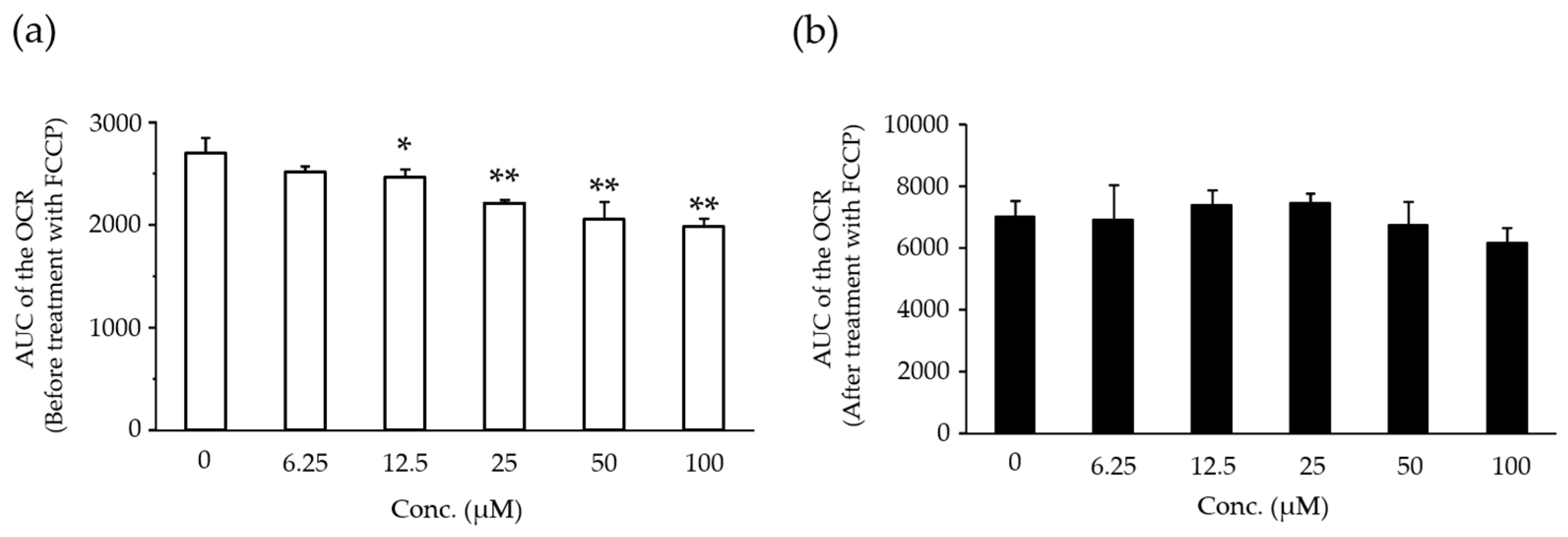
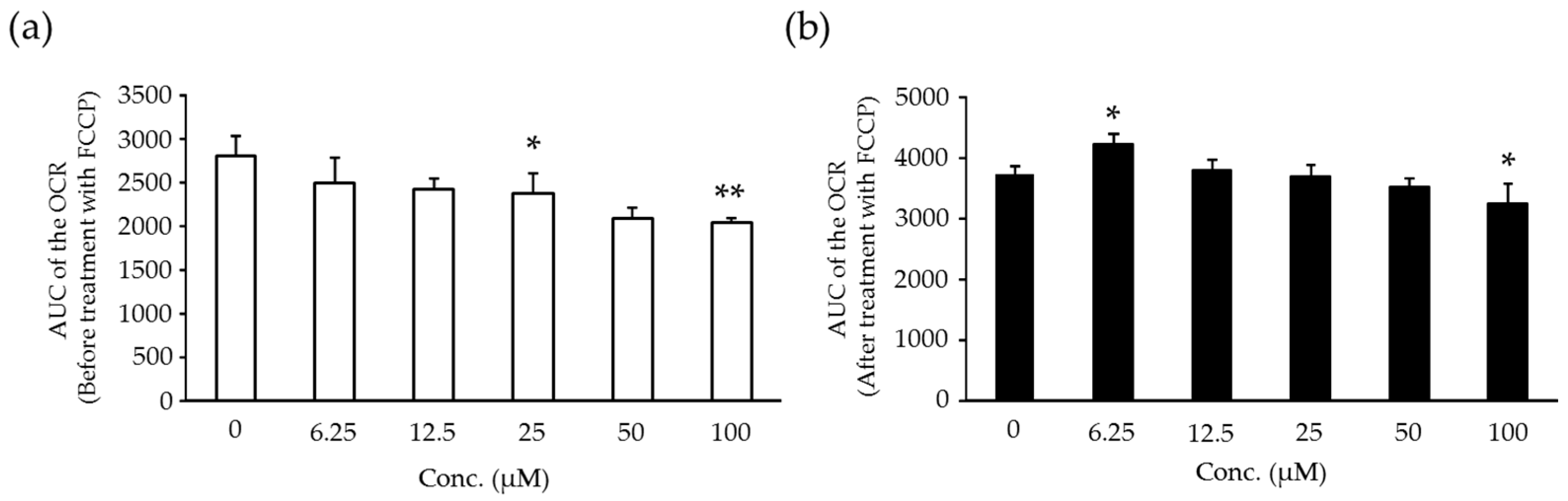
2.5. Effects of Compound X on Mitochondrial Oxygen Consumption in Rat Hepatocyte
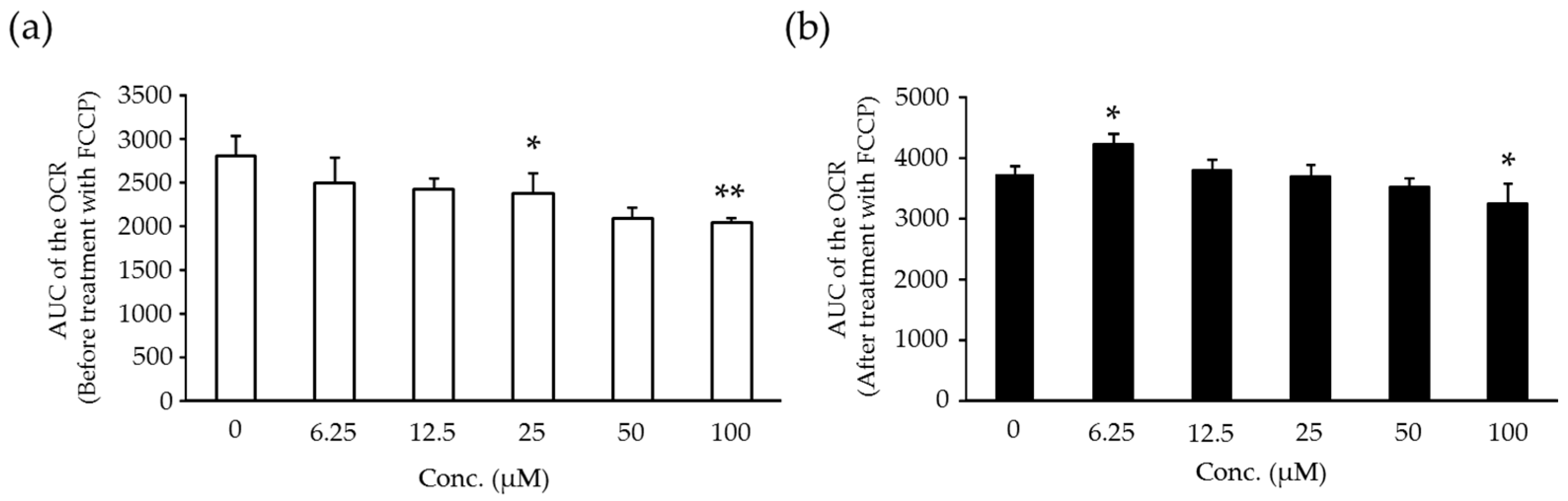
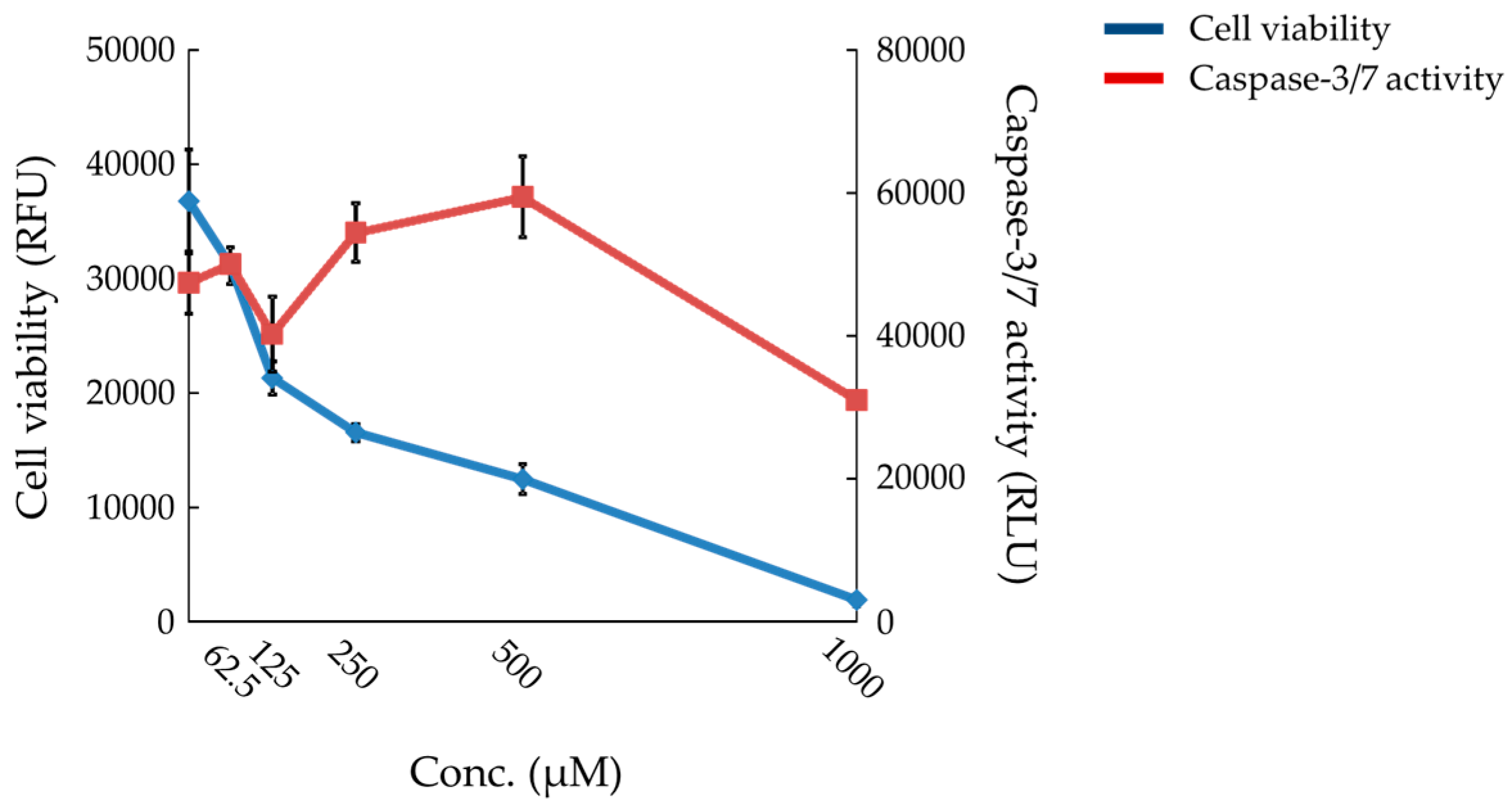
2.6. Effects of Compound X on Mitochondrial Oxygen Consumption and Apoptotic Action of Compound X in Human Hepatocytes
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals
4.3. One and Three-Month Repeated Oral Dose Studies
4.3.1. Dosing
4.3.2. Sampling of Blood Samples
4.3.3. Measurements of Plasma Liver Function Parameters and TGL Levels
4.3.4. Histopathology
4.3.5. Toxicokinetic Analyses
4.4. Lipidomics Study
4.4.1. Dosing
4.4.2. Sampling of Blood and Liver
4.4.3. Lipidomics
4.4.4. OPLS-DA and Lipid Identification
4.4.5. Real-Time PCR
4.5. In Vitro Studies
4.5.1. Rat Hepatocytes
4.5.2. Human Hepatocytes
4.5.3. Measurements of Cell Viability and Caspase-3/7 Activity
4.5.4. Measurements of OCR
4.6. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lee, W.M. Drug-induced hepatotoxicity. N. Eng. J. Med. 2003, 349, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.A.; Schmid, E.F. Drug withdrawals and the lessons within. Curr. Opin. Drug Discov. Devel. 2006, 9, 38–46. [Google Scholar] [PubMed]
- Geisler, C.E.; Hepler, C.; Higgins, M.R.; Renquist, B.J. Hepatic adaptations to maintain metabolic homeostasis in response to fasting and refeeding in mice. Nutr. Metab. 2016, 13, 62. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, T.C.; Pollak, P.T.; Hanniman, E.A.; Sinal, C.J. Disruption of hepatic lipid homeostasis in mice after amiodarone treatment is associated with peroxisome proliferator-activated receptor-α target gene activation. J. Pharmacol. Exp. Ther. 2004, 311, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Gudbrandsen, O.A.; Rost, T.H.; Berge, R.K. Causes and prevention of tamoxifen-induced accumulation of triacylglycerol in rat liver. J. Lipid. Res. 2006, 47, 2223–2232. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, R.G.; Bacon, J.A.; Brass, E.P.; Cramer, C.T.; Petrella, D.K.; Sun, E.L. Metabolic, idiosyncratic toxicity of drugs: Overview of the hepatic toxicity induced by the anxiolytic, panadiplon. Chem. Biol. Interact. 2001, 134, 251–270. [Google Scholar] [CrossRef]
- Silva, M.F.; Aires, C.C.; Luis, P.B.; Ruiter, J.P.; IJlst, L.; Duran, M.; Wanders, R.J.; Tavares de Almeida, I. Valproic acid metabolism and its effects on mitochondrial fatty acid oxidation: A review. J. Inherit. Metab. 2008, 31, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Le Dinh, T.; Freneaux, E.; Labbe, G.; Letteron, P.; Degott, C.; Geneve, J.; Berson, A.; Larrey, D.; Pessayre, D. Amineptine, a tricyclic antidepressant, inhibits the mitochondrial oxidation of fatty acids and produces macrovesicular steatosis of the liver in mice. J. Pharmacol. Exp. Ther. 1988, 247, 745–750. [Google Scholar] [PubMed]
- Luiken, J.J.; Niessen, H.E.; Coort, S.L.; Hoebers, N.; Coumans, W.A.; Schwenk, R.W.; Bonen, A.; Glatz, J.F. Etomoxir-induced partial carnitine palmitoyltransferase-I (CPT-I) inhibition in vivo does not alter cardiac long-chain fatty acid uptake and oxidation rates. Biochem. J. 2009, 419, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Lee, C.H.; Lee, K.Y.; Jung, S.H.; Lee, B.H. Increased hepatic Fatty Acid uptake and esterification contribute to tetracycline-induced steatosis in mice. Toxicol. Sci. 2015, 145, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Sahini, N.; Selvaraj, S.; Borlak, J. Whole genome transcript profiling of drug induced steatosis in rats reveals a gene signature predictive of outcome. PLoS ONE 2014, 9, e114085. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Kim, M.; Lee, B.H.; Kim, J.H.; Kang, K.S.; Kim, H.L.; Yoon, B.I.; Chung, H.; Kong, G.; Lee, M.O. Subchronic effects of valproic acid on gene expression profiles for lipid metabolism in mouse liver. Toxicol. Appl. Pharmacol. 2008, 226, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Knapp, A.C.; Todesco, L.; Beier, K.; Terracciano, L.; Sägesser, H.; Reichen, J.; Krähenbühl, S. Toxicity of valproic acid in mice with decreased plasma and tissue carnitine stores. J. Pharmacol. Exp. Ther. 2008, 324, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Sobaniec-Lotowska, M.E. Effects of long-term administration of the antiepileptic drug—Sodium valproate upon the ultrastructure of hepatocytes in rats. Exp. Toxicol. Pathol. 1997, 49, 225–232. [Google Scholar] [CrossRef]
- Chopra, I.; Roberts, M. Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.Q.; Kim, M.; Kim, J.H.; Kong, G.; Lee, M.O.; Kang, K.S.; Yoon, B.I.; Kim, H.L.; Lee, B.H. Hepatic gene expression profiling and lipid homeostasis in mice exposed to steatogenic drug, tetracycline. Toxicol. Sci. 2006, 94, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.Y.; Wang, B.L.; Zhao, J.; Yao, X.M.; Gu, Y.; Li, Y. Protective effect of bicyclol on tetracycline-induced fatty liver in mice. Toxicology 2009, 261, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, J.D.; Guo, G.L. Mechanistic review of drug-induced steatohepatitis. Toxicol. Appl. Pharmacol. 2015, 289, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Liu, Z.X.; Choi, C.S.; Tian, L.; Kibbey, R.; Dong, J.; Cline, G.W.; Wood, P.A.; Shulman, G.I. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 17075–17080. [Google Scholar] [CrossRef] [PubMed]
- Boelsterli, U.A.; Lim, P.L. Mitochondrial abnormalities—A link to idiosyncratic drug hepatotoxicity? Toxicol. Appl. Pharmacol. 2007, 220, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Kerner, J.; Hoppel, C. Fatty acid import into mitochondria. Biochim. Biophys. Acta 2000, 1486, 1–17. [Google Scholar] [CrossRef]
- Kobayashi, A.; Suzuki, Y.; Kuno, H.; Sugai, S.; Sakakibara, H.; Shimoi, K. Effects of fenofibrate on plasma and hepatic transaminase activities and hepatic transaminase gene expression in rats. J. Toxicol. Sci. 2009, 34, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T. Toxicology Research Laboratories, Central Pharmaceutical Research Institute; Internal Report; Japan Tobacco Inc.: Hadano, Japan, 2008. [Google Scholar]
- Kemmochi, Y. Toxicology Research Laboratories, Central Pharmaceutical Research Institute; Internal Report; Japan Tobacco Inc.: Hadano, Japan, 2009. [Google Scholar]
- Mashek, D.G.; Li, L.O.; Coleman, R.A. Long-chain acyl-CoA synthetases and fatty acid channeling. Future Lipidol. 2007, 2, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Mashek, D.G.; Li, L.O.; Coleman, R.A. Rat long-chain acyl-CoA synthetase mRNA, protein, and activity vary in tissue distribution and in response to diet. J. Lipid Res. 2006, 47, 2004–2010. [Google Scholar] [CrossRef] [PubMed]
- Watkins, P.A. Very-long-chain acyl-CoA synthetases. J. Biol. Chem. 2008, 283, 1773–1777. [Google Scholar] [CrossRef] [PubMed]
- Bu, S.Y.; Mashek, D.G. Hepatic long-chain acyl-CoA synthetase 5 mediates fatty acid channeling between anabolic and catabolic pathways. J. Lipid. Res. 2010, 51, 3270–3280. [Google Scholar] [CrossRef] [PubMed]
- Mashek, D.G.; McKenzie, M.A.; van Horn, C.G.; Coleman, R.A. Rat long chain acyl-CoA synthetase 5 increases fatty acid uptake and partitioning to cellular triacylglycerol in McArdle-RH7777 cells. J. Biol. Chem. 2006, 281, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Goda, K.; Takahashi, T.; Kobayashi, A.; Shoda, T.; Kuno, H.; Sugai, S. Usefulness of in vitro combination assays of mitochondrial dysfunction and apoptosis for the estimation of potential risk of idiosyncratic drug induced liver injury. J. Toxicol. Sci. 2016, 41, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Goda, K.; Kobayashi, A.; Yamada, N.; Maekawa, K.; Saito, Y.; Sugai, S. Arachidonic acid-containing phosphatidylcholine characterized by consolidated plasma and liver lipidomics as an early onset marker for tamoxifen-induced hepatic phospholipidosis. J. Appl. Toxicol. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Saito, K.; Yamada, H.; Nakatsu, N.; Maekawa, K.; Saito, Y. Plasma lipid profiling of different types of hepatic fibrosis induced by carbon tetrachloride and lomustine in rats. Lipids Health Dis. 2016, 15, 74. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Maekawa, K.; Saito, K.; Senoo, Y.; Urata, M.; Murayama, M.; Tajima, Y.; Kumagai, Y.; Saito, Y. Plasma and serum lipidomics of healthy white adults shows characteristic profiles by subjects’ gender and age. PLoS ONE 2014, 9, e91806. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Risk of DILI in Humans | Effects on Mitochondria | Animal Species with Steatosis in Non-Clinical Studies |
|---|---|---|---|
| Amiodarone | + | Inhibition of β-oxidation (BO) and respiratory chain (RC) | Mouse and rat |
| Tamoxifen | + | Inhibition of BO and RC | Rat |
| Panadiplon * | + | Inhibition of BO | Monkey and rabbit |
| Valproic acid | + | Inhibition of BO and RC | Mouse and rat |
| Amineptin | + | Inhibition of BO | Mouse |
| Etomoxir | + | Inhibition of BO | Rat |
| Tetracycline | + | Inhibition of BO and tricarboxylic acid cycle (TC) | Mouse and rat |
| Dose (mg/kg/day) | 0 | 30 | 100 | 300 |
| Number of Animals | 12 | 12 | 12 | 12 |
| Systemic exposure; AUC0–24 (μmol·h/L) (Day 27) | NA | 1.5 | 9.2 | 54.1 |
| Clinical Chemistry | ||||
| AST (U/L) | 66.3 ± 6.5 | 63.1 ± 6.6 | 66.5 ± 8.9 | 64.0 ± 5.4 |
| ALT (U/L) | 28.9 ± 5.7 | 27.3 ± 5.5 | 29.8 ± 6.3 | 30.5 ± 4.8 |
| TGL (mg/dL) | 67.9 ± 25.2 | 72.6 ± 26.4 | 60.8 ± 17.6 | 58.3 ± 16.7 |
| Liver Weights | ||||
| Absolute (g) | 10.328 ± 1.329 | 10.915 ± 0.741 | 10.121 ± 1.056 | 11.213 ± 1.233 |
| Relative (g/100 g BW) | 2.868 ± 0.193 | 2.857 ± 0.128 | 2.792 ± 0.137 | 3.018 ± 0.126 * |
| Macroscopic findings | ||||
| Liver | - | - | - | - |
| Microscopic findings | ||||
| Liver | - | - | - | - |
| Dose (mg/kg/day) | 0 | 100 | 300 | 600 |
| Number of Animals | 15 | 15 | 15 | 15 |
| Systemic exposure; AUC0–24 (μmol·h/L) (Day 90) | NA | 9.2 | 50.5 | 101.1 |
| Clinical Chemistry | ||||
| AST (U/L) | 69.1 ± 11.5 | 65.3 ± 8.0 | 61.3 ± 8.0 | 92.9 ± 18.5 ** |
| ALT (U/L) | 31.9 ± 6.8 | 30.5 ± 6.0 | 29.8 ± 4.9 | 71.3 ±19.4 ** |
| LDH (U/L) | 55.9 ± 10.8 | 59.9 ± 23.3 | 54.1 ± 12.0 | 69.3 ± 19.2 |
| ALP (U/L) | 194.2 ± 36.0 | 196.9 ± 21.2 | 238.5 ± 23.0 ** | 249.6 ± 38.9 ** |
| BIL (mg/dL) | 0.051 ± 0.007 | 0.051 ± 0.007 | 0.050 ± 0.007 | 0.049 ± 0.010 |
| TGL (mg/dL) | 83.5 ± 34.3 | 80.3 ± 26.8 | 70.3 ± 21.5 | 46.1 ± 12.2 ** |
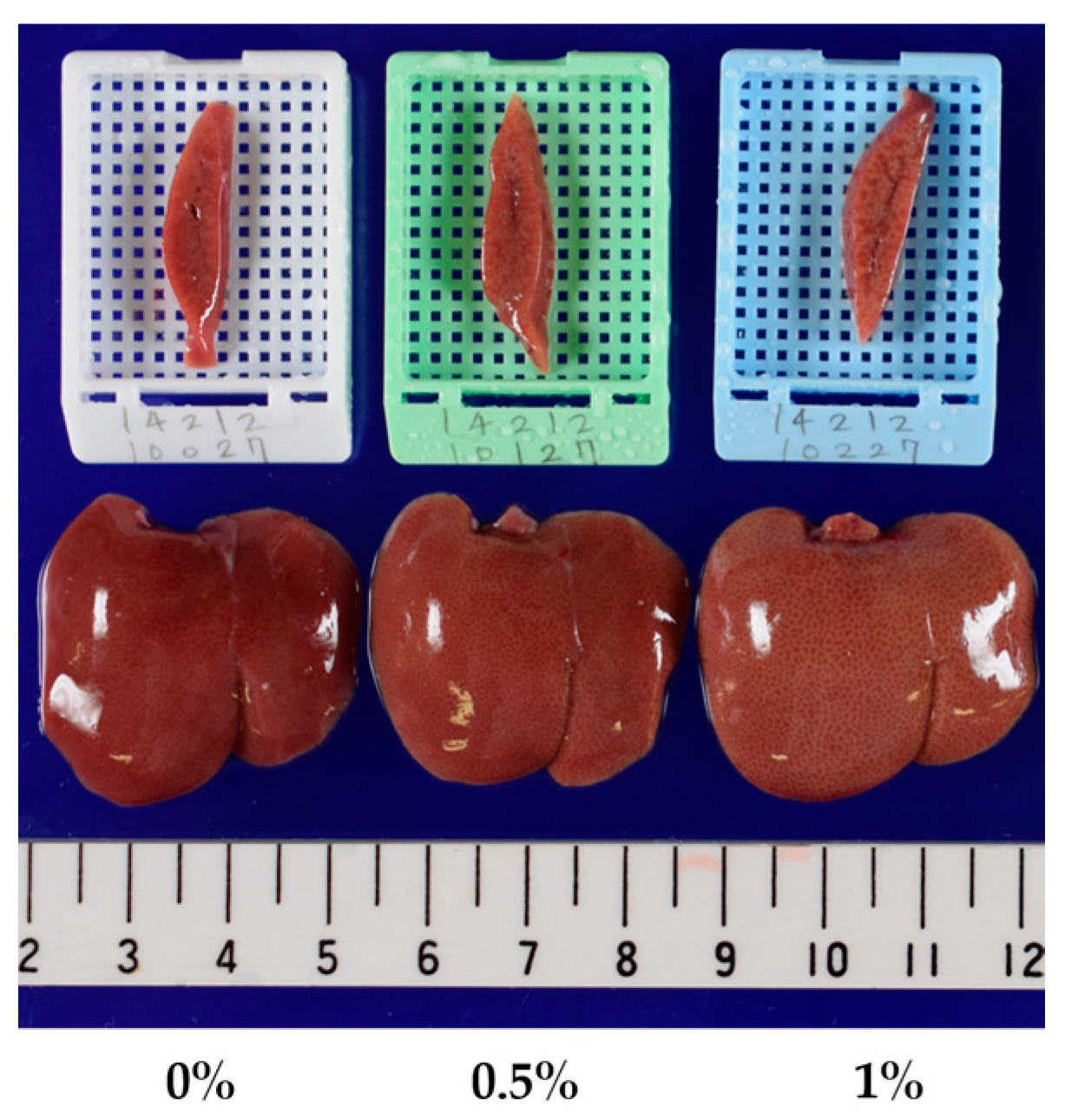
| Macroscopic Findings of the liver | ||||
| Yellowish discoloration | - : 15 | - : 15 | - : 14 | - : 4 |
| P : 1 | P : 11 | |||
| Liver Weights | ||||
| Absolute (g) | 14.327 ± 1.497 | 15.560 ± 2.024 | 16.021 ± 2.106 * | 17.622 ± 1.555 ** |
| Relative (g/100 g BW) | 2.569 ± 0.169 | 2.572 ± 0.173 | 2.673 ± 0.128 | 2.988 ± 0.161 ** |
| Microscopic findings of the liver | ||||
| Fatty change, hepatocyte, periportal | ± : 6 | ± : 5 | ± : 4 | 2+ : 7 |
| + : 4 | + : 8 | + : 11 | 3+ : 8 | |
| 2+ : 5 | 2+ : 2 | |||
| Dose (mg/kg/day) | 0% | 0.5% | 1% | |
| Number of animals/sampling point | 10 | 10 | 10 | |
| Chemical Intake (mg/kg/day) | NA | 380 | 793 | |
| Clinical Chemistry | ||||
| AST (U/L) | Day 8 | 64.0 ± 6.1 | 63.5 ± 5.6 | 67.1 ± 8.7 |
| Day 15 | 54.8 ± 6.0 | 53.8 ± 7.4 | 67.2 ± 12.4 ** | |
| Day 29 | 47.9 ± 7.8 | 53.3 ± 3.3 | 75.3 ± 15.0 ** | |
| ALT (U/L) | Day 8 | 15.4 ± 2.1 | 16.4 ± 2.8 | 17.1 ± 2.4 |
| Day 15 | 14.9 ± 2.2 | 15.0 ± 3.5 | 22.3 ± 7.9 ** | |
| Day 29 | 15.2 ± 3.1 | 17.5 ± 4.0 | 33.6 ± 15.3 ** | |
| LDH (U/L) | Day 8 | 58.2 ± 15.8 | 58.9 ± 23.9 | 65.4 ± 21.5 |
| Day 15 | 38.6 ± 10.1 | 43.8 ± 5.6 | 53.4 ± 10.1 ** | |
| Day 29 | 40.7 ± 15.9 | 41.8 ± 8.0 | 48.8 ± 11.4 | |
| ALP (U/L) | Day 8 | 696.2 ± 151.7 | 742.3 ± 106.9 | 611.1 ± 97.3 |
| Day 15 | 583.7 ± 108.2 | 583.1 ± 93.4 | 579.2 ± 68.2 | |
| Day 29 | 373.0 ± 51.9 | 466.0 ± 79.9 * | 452.3 ± 79.4 * | |
| BIL (mg/dL) | Day 8 | 0.023 ± 0.005 | 0.020 ± 0.005 | 0.018 ± 0.006 |
| Day 15 | 0.026 ± 0.007 | 0.025 ± 0.005 | 0.021 ± 0.006 | |
| Day 29 | 0.024 ± 0.007 | 0.029 ± 0.006 | 0.027 ± 0.009 | |
| TGL (mg/dL) | Day 8 | 33.5 ± 17.4 | 28.1 ± 11.0 | 24.9 ± 10.5 |
| Day 15 | 30.4 ± 10.9 | 27.0 ± 19.4 | 19.0 ± 5.3 | |
| Day 29 | 42.3 ± 18.0 | 34.9 ± 15.8 | 18.3 ± 8.0 ** | |
| Macroscopic Findings of the liver | ||||
| Yellowish discoloration | Day 8 | - : 10 | - : 10 | - : 10 |
| Day 15 | - : 10 | - : 10 | P : 10 | |
| Day 29 | - : 10 | - : 9 | P : 10 | |
| P : 1 | ||||
| Liver Weights | ||||
| Day 8 | Absolute (g) | 7.642 ± 0.645 | 7.678 ± 0.463 | 8.367 ± 0.528 * |
| Relative (g/100 g BW) | 3.176 ± 0.237 | 3.260 ± 0.144 | 3.531 ± 0.177 ** | |
| Day 15 | Absolute (g) | 8.710 ± 0.484 | 8.730 ± 0.644 | 9.113 ± 0.641 |
| Relative (g/100 g BW) | 2.967 ± 0.119 | 2.999 ± 0.141 | 3.269 ± 0.159 ** | |
| Day 29 | Absolute (g) | 10.215 ± 1.449 | 10.555 ± 1.243 | 10.875 ± 0.940 |
| Relative (g/100 g BW) | 2.812 ± 0.194 | 2.760 ± 0.149 | 2.999 ± 0.100 * | |
| Microscopic findings of the liver | ||||
| Fatty change, hepatocyte, periportal | Day 8 | - : 6 | - : 1 | ± : 5 |
| ± : 4 | ± : 9 | + : 5 | ||
| Day 15 | - : 1 | - : 2 | + : 2 | |
| ± : 9 | ± : 8 | 2+ : 8 | ||
| Day 29 | ± : 4 | - : 2 | + : 1 | |
| + : 6 | ± : 5 | 2+ : 8 | ||
| + : 3 | 3+ : 1 | |||
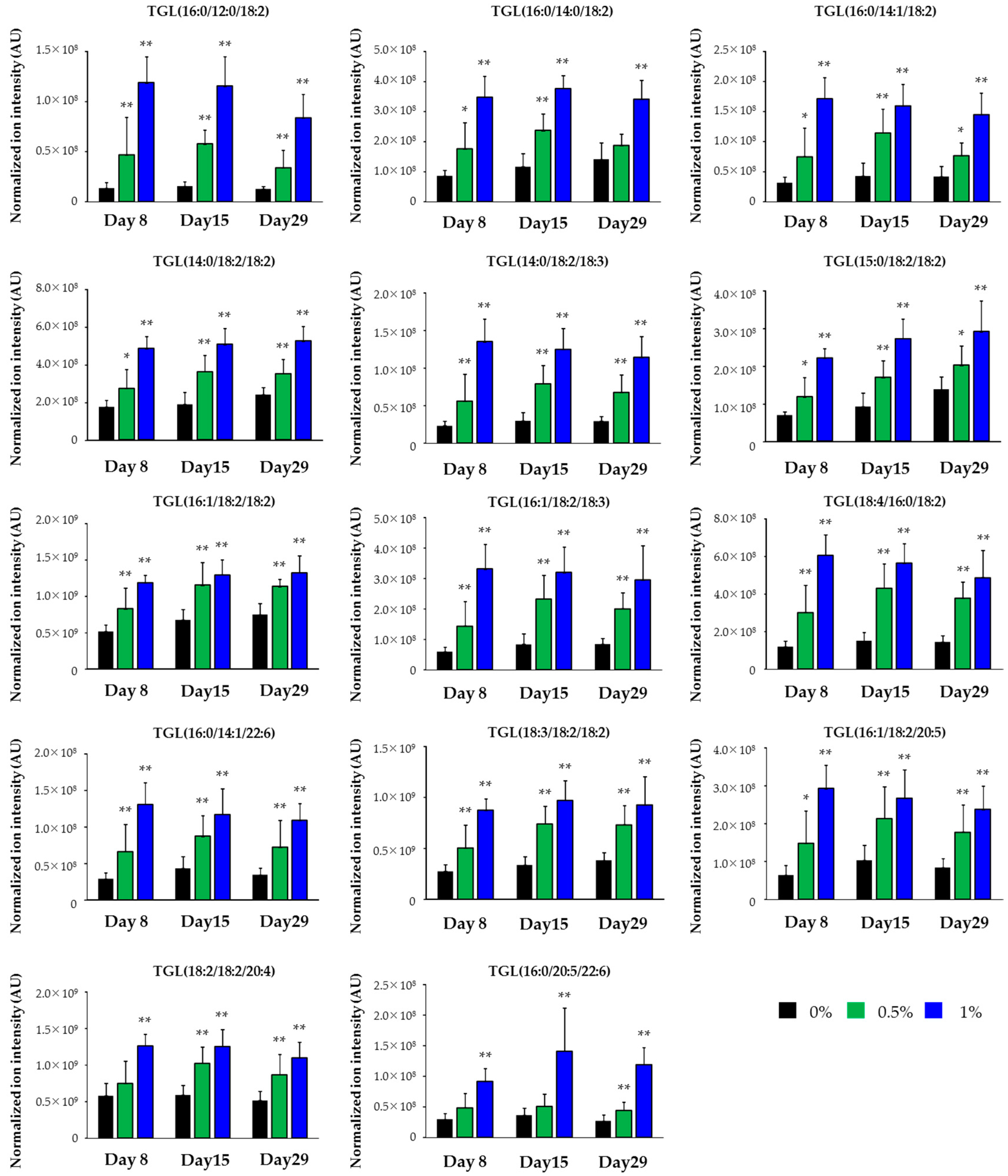
| Class | Lipid | Polarity | Response |
|---|---|---|---|
| TGL | TGL(16:0/12:0/18:2) | Posi | Increase |
| TGL | TGL(16:0/14:0/18:2) | Posi | Increase |
| TGL | TGL(16:0/14:1/18:2) | Posi | Increase |
| TGL | TGL(14:0/18:2/18:2) | Posi | Increase |
| TGL | TGL(14:0/18:2/18:3) | Posi | Increase |
| TGL | TGL(15:0/18:2/18:2) | Posi | Increase |
| TGL | TGL(16:1/18:2/18:2) | Posi | Increase |
| TGL | TGL(16:1/18:2/18:3) | Posi | Increase |
| TGL | TGL(18:4/16:0/18:2) | Posi | Increase |
| TGL | TGL(16:0/14:1/22:6) | Posi | Increase |
| TGL | TGL(18:3/18:2/18:2) | Posi | Increase |
| TGL | TGL(16:1/18:2/20:5) | Posi | Increase |
| TGL | TGL(18:2/18:2/20:4) | Posi | Increase |
| TGL | TGL(16:0/20:5/22:6) | Posi | Increase |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goda, K.; Kobayashi, A.; Takahashi, A.; Takahashi, T.; Saito, K.; Maekawa, K.; Saito, Y.; Sugai, S. Evaluation of the Potential Risk of Drugs to Induce Hepatotoxicity in Human—Relationships between Hepatic Steatosis Observed in Non-Clinical Toxicity Study and Hepatotoxicity in Humans-. Int. J. Mol. Sci. 2017, 18, 810. https://doi.org/10.3390/ijms18040810
Goda K, Kobayashi A, Takahashi A, Takahashi T, Saito K, Maekawa K, Saito Y, Sugai S. Evaluation of the Potential Risk of Drugs to Induce Hepatotoxicity in Human—Relationships between Hepatic Steatosis Observed in Non-Clinical Toxicity Study and Hepatotoxicity in Humans-. International Journal of Molecular Sciences. 2017; 18(4):810. https://doi.org/10.3390/ijms18040810
Chicago/Turabian StyleGoda, Keisuke, Akio Kobayashi, Akemi Takahashi, Tadakazu Takahashi, Kosuke Saito, Keiko Maekawa, Yoshiro Saito, and Shoichiro Sugai. 2017. "Evaluation of the Potential Risk of Drugs to Induce Hepatotoxicity in Human—Relationships between Hepatic Steatosis Observed in Non-Clinical Toxicity Study and Hepatotoxicity in Humans-" International Journal of Molecular Sciences 18, no. 4: 810. https://doi.org/10.3390/ijms18040810