
Methylmercury Induced Neurotoxicity and the Influence of Selenium in the Brains of Adult Zebrafish (Danio rerio)

Abstract
:1. Introduction
2. Results
2.1. General Observations and Accumulation of Mercury and Selenium
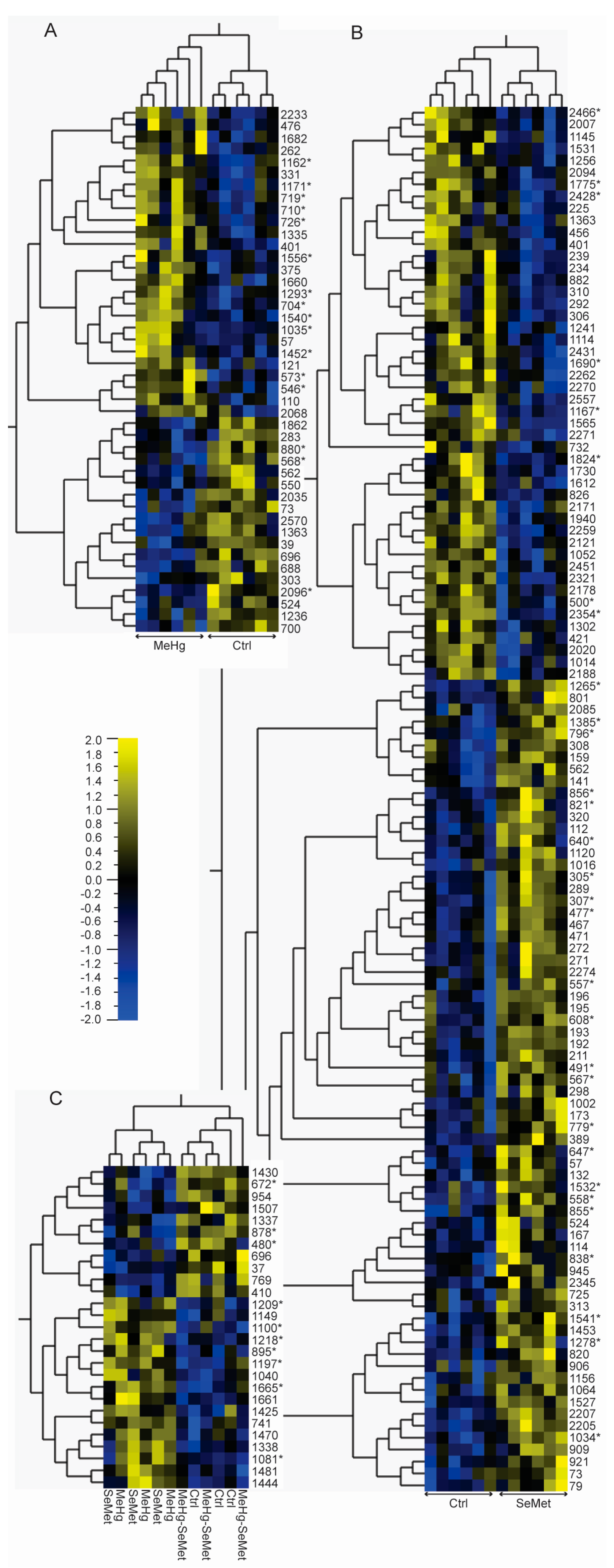
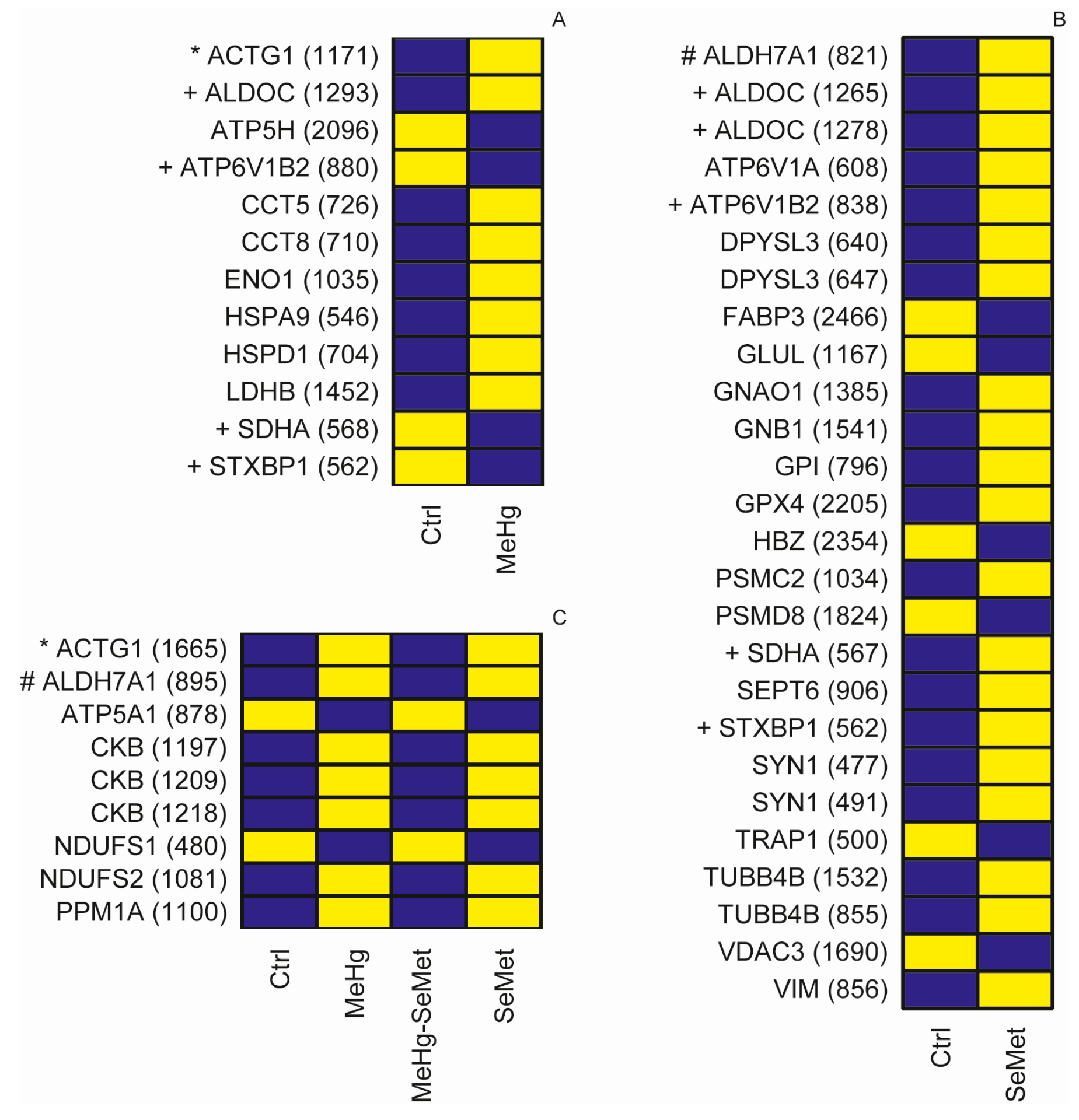
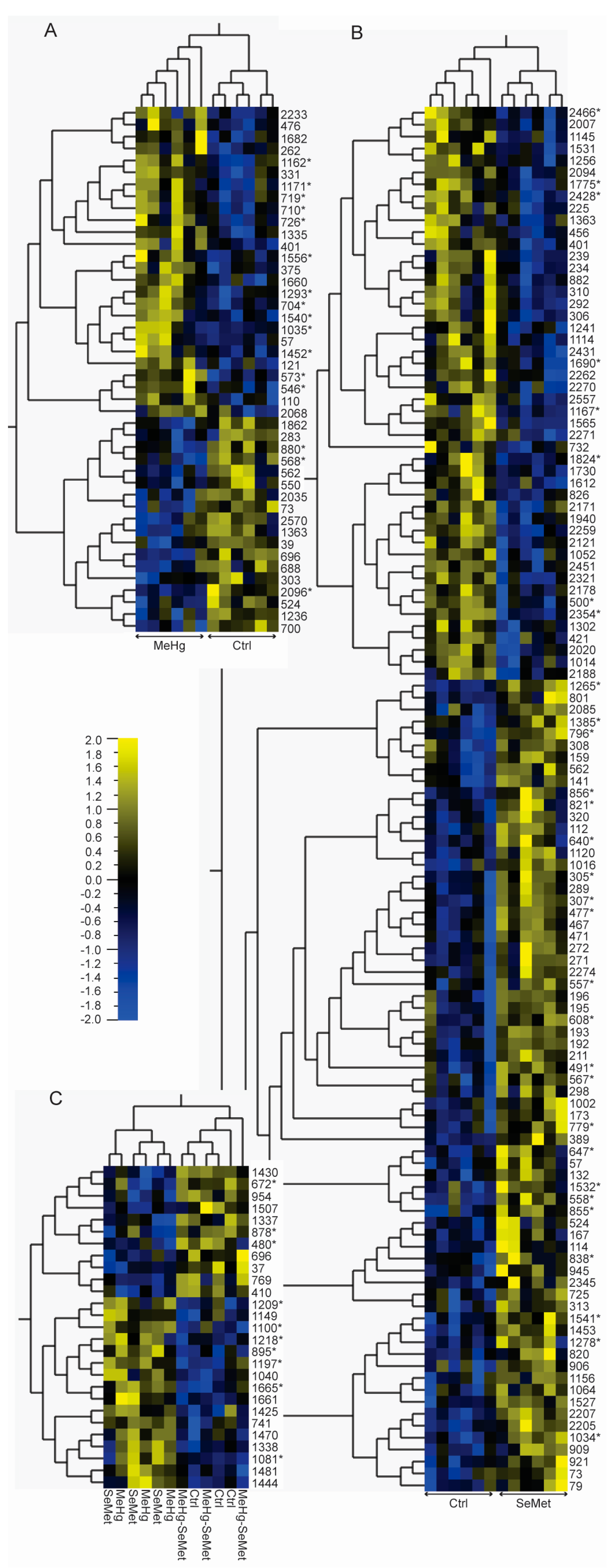
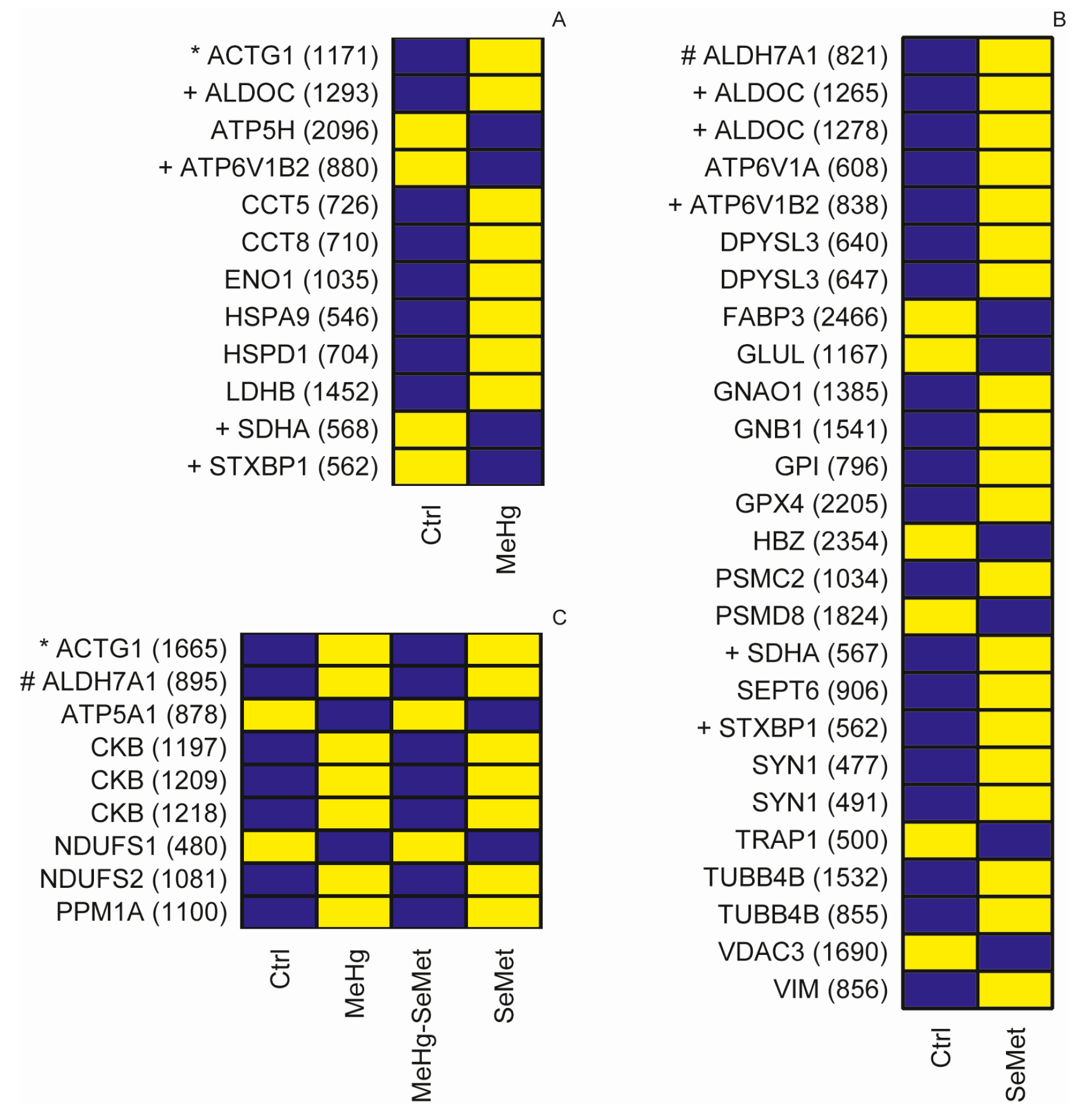
2.2. Protein Expression Analysis and Protein Identification
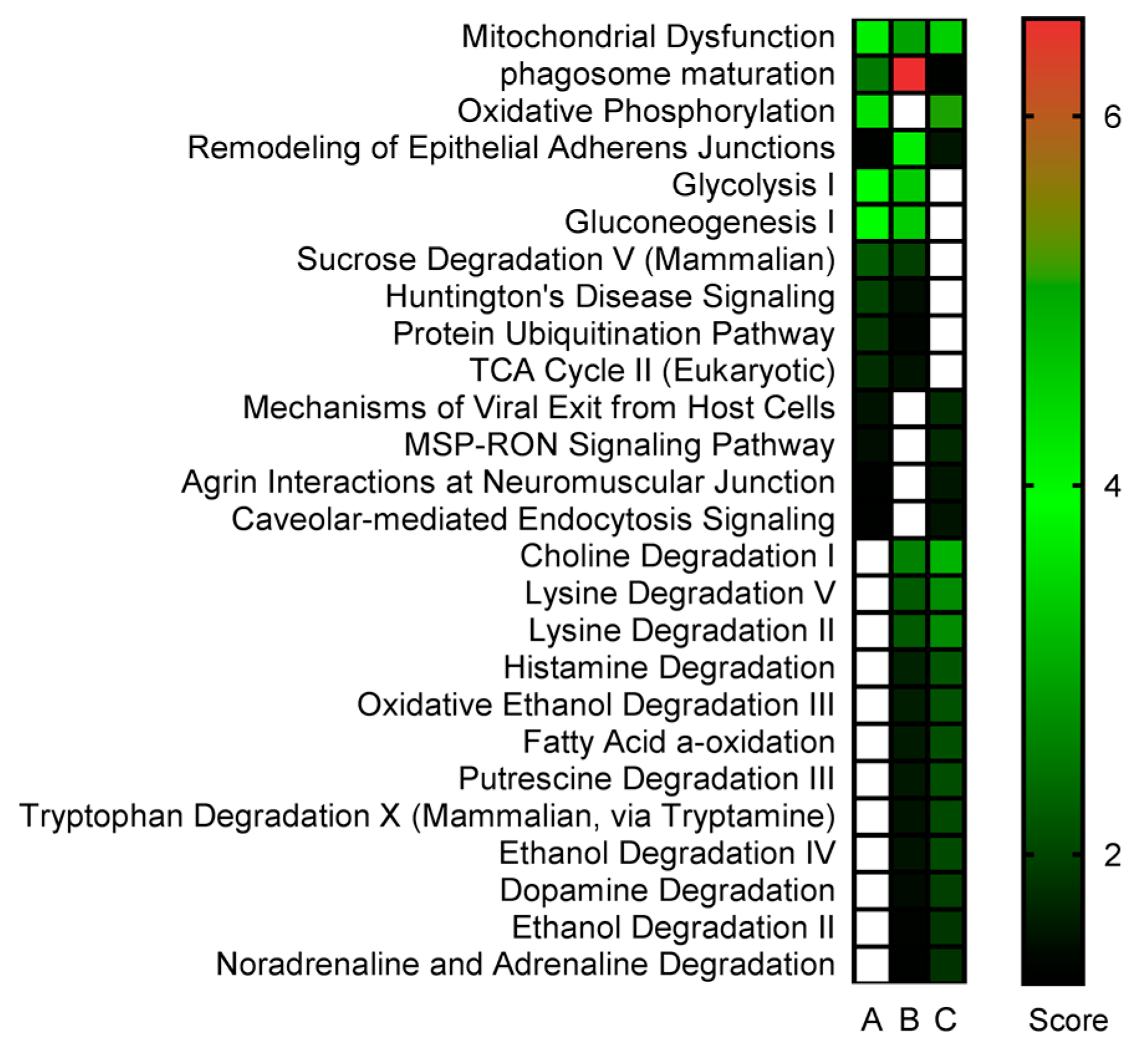
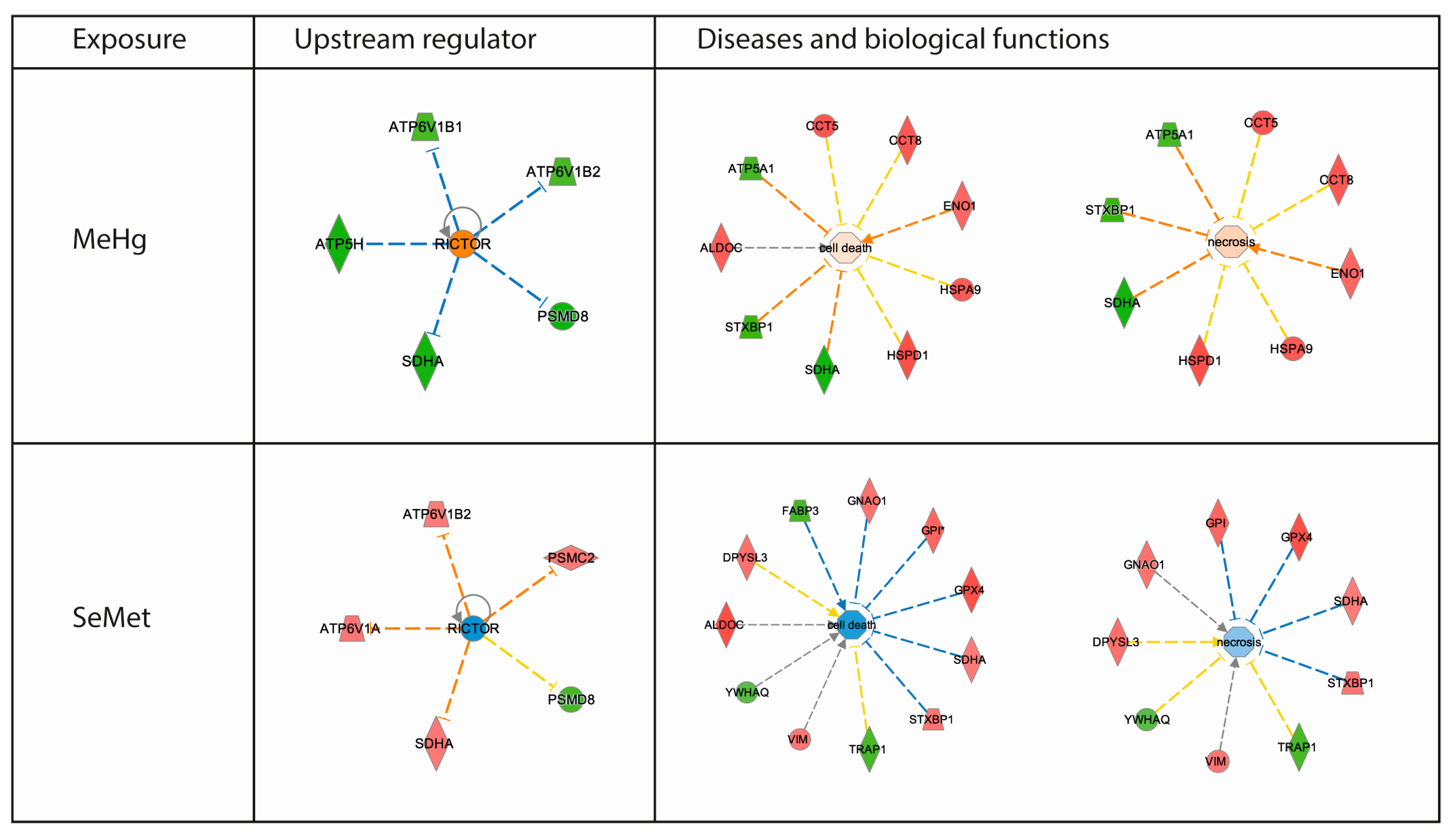
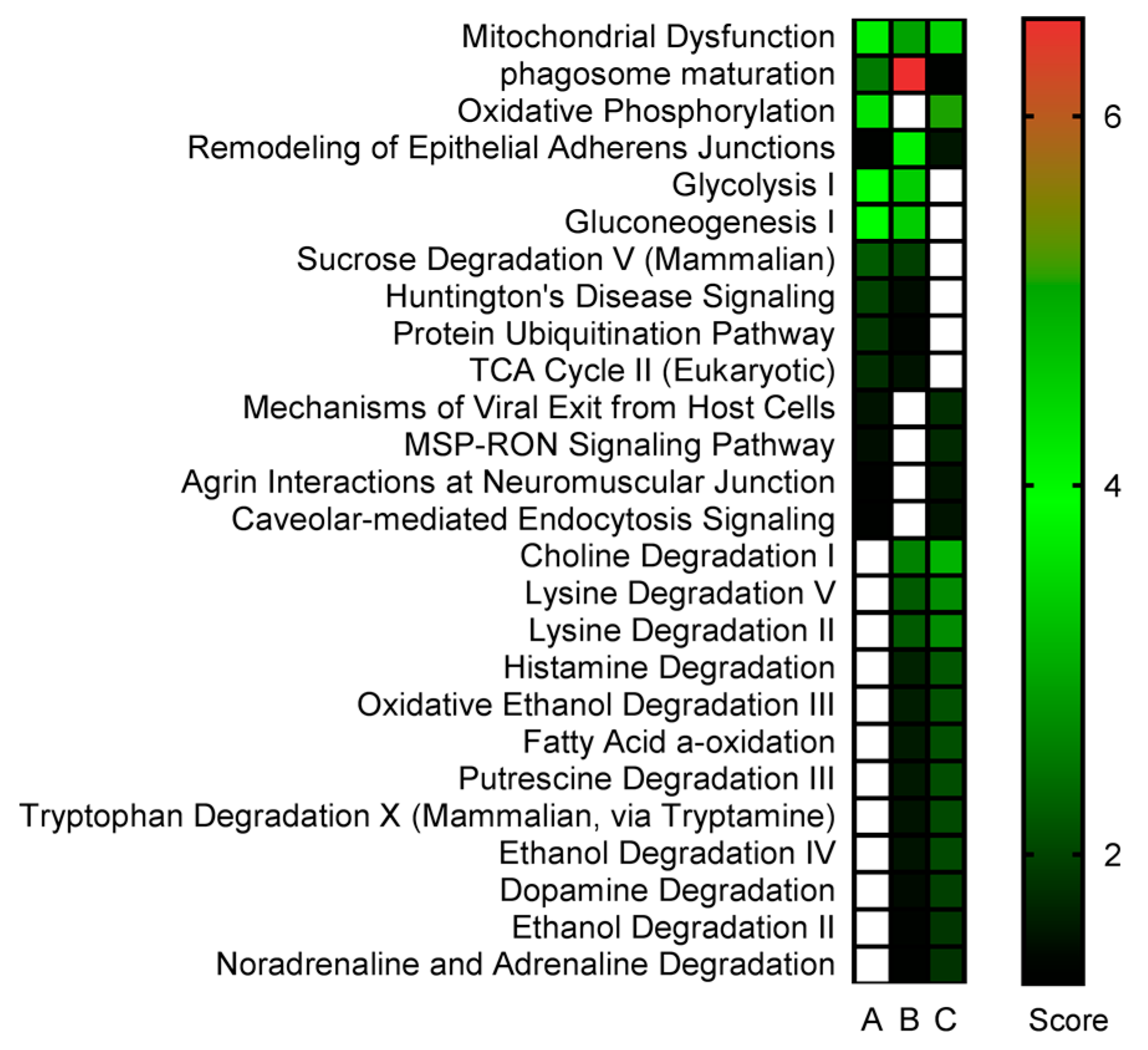
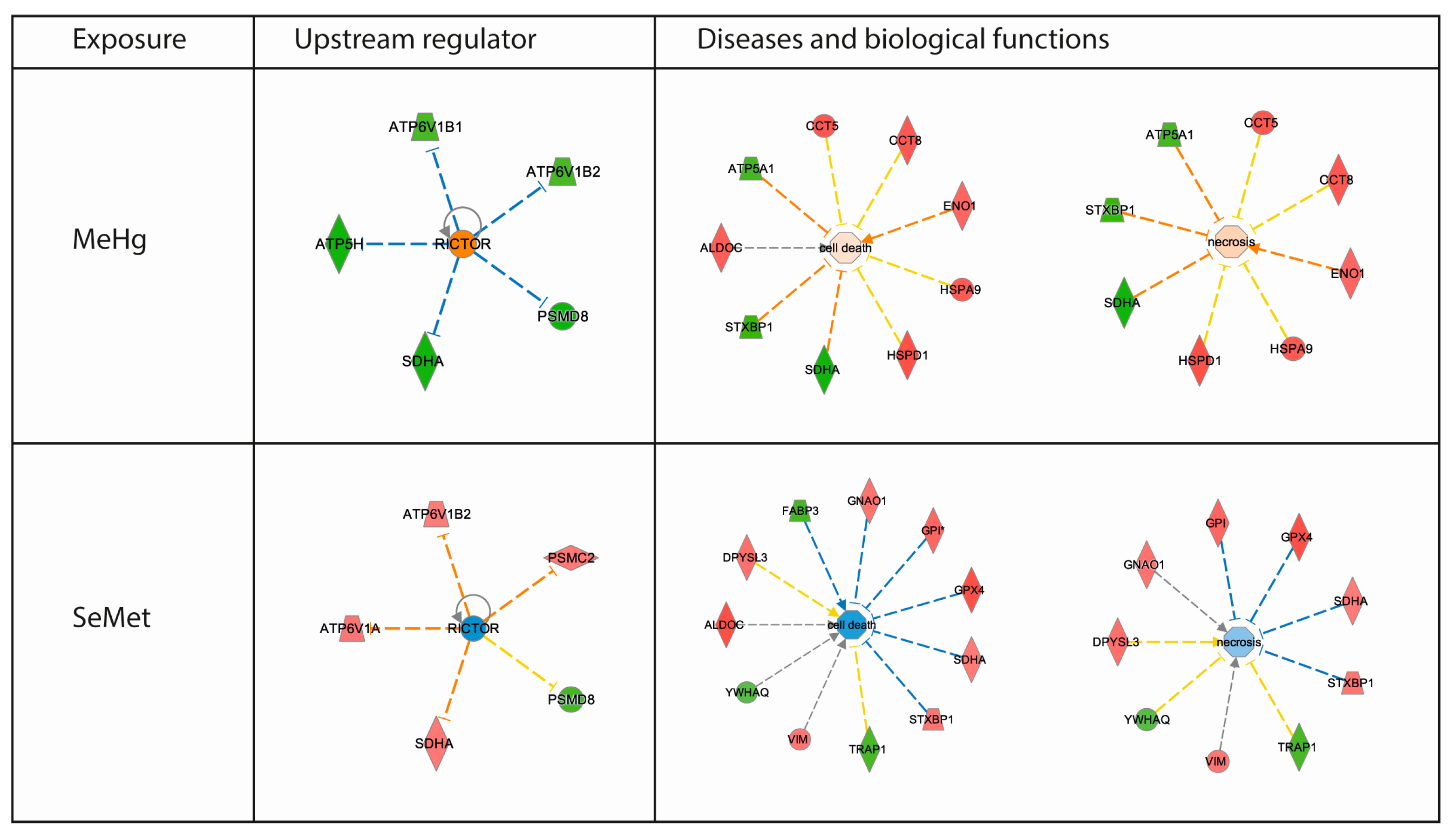
2.3. Proteomics Bioinformatics and Pathway Analysis
3. Discussion
4. Materials and Methods
4.1. Experimental Design and Sampling
4.2. Mercury and Selenium Determination by Inductively Coupled Plasma Mass Spectrometry
4.3. Proteomics Sample Preparation and Cydye Labeling
4.4. Difference in Gel Electrophoresis
4.5. Image Acquisition and Analysis
4.6. Mass Spectrometry
4.7. Bioinformatics
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| IPA | Ingenuity pathway analysis |
| LOQ | Limit of quantification |
| MeHg | Methylmercury |
| mTOR | Mammalian target of rapamycin |
| RICTOR | Rapamycin-insensitive binding partner of mTOR |
| ROS | Reactive oxygen species |
| SeMet | Selenomethionine |
References
- Dale, K.; Rasinger, J.D.; Thorstensen, K.L.; Penglase, S.; Ellingsen, S. Vitamin E reduces endosulfan-induced toxic effects on morphology and behavior in early development of zebrafish (Danio rerio). Food Chem. Toxicol. 2017, 101, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Petriello, M.C.; Newsome, B.J.; Dziubla, T.D.; Hilt, J.Z.; Bhattacharyya, D.; Hennig, B. Modulation of persistent organic pollutant toxicity through nutritional intervention: Emerging opportunities in biomedicine and environmental remediation. Sci. Total Environ. 2014, 491, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, A.; Berntssen, M.H.G.; Lundebye, A.-K.; Røyneberg Alvheim, A.; Secher Myrmel, L.; Fjære, E.; Torstensen, B.E.; Kristiansen, K.; Madsen, L.; Brattelid, T.; et al. Marine fatty acids aggravate hepatotoxicity of α-HBCD in juvenile female BALB/c mice. Food Chem. Toxicol. 2016, 97, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Bergés-Tiznado, M.E.; Fernando Márquez-Farías, J.; Torres-Rojas, Y.; Galván-Magaña, F.; Páez-Osuna, F. Mercury and selenium in tissues and stomach contents of the migratory sailfish, Istiophorus platypterus, from the Eastern Pacific: Concentration, biomagnification, and dietary intake. Mar. Pollut. Bull. 2015, 101, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Karagas, M.R.; Choi, A.L.; Oken, E.; Horvat, M.; Schoeny, R.; Kamai, E.; Cowell, W.; Grandjean, P.; Korrick, S. Evidence on the human health effects of low-level methylmercury exposure. Environ. Health Perspect. 2012, 120, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, W.F.; Mason, R.P. Biochemical cycling of mercury in the marine environment. In Mercury and Its Effects on Environment and Biology; Sigel, A., Sigel, H., Eds.; Mercel Decker, Inc.: New York, NY, USA, 1997; Volume 34, pp. 53–111. [Google Scholar]
- Morel, F.M.M.; Kraepiel, A.M.L.; Amyot, M. The chemical cycle and bioaccumulation of mercury. Annu. Rev. Ecol. Syst. 1998, 29, 543–566. [Google Scholar] [CrossRef]
- Simmons-Willis, T.A.; Koh, A.S.; Clarkson, T.W.; Ballatori, N. Transport of a neurotoxicant by moleclar mimicry: The methylmercury-l-cysteine complex is a substrate for human l-type large neutral amino acid transporter (LAT) 1 and LAT2. Biochem. J. 2002, 367, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Jiang, H.; Syversen, T.; Rocha, J.B.T.; Farina, M.; Aschner, M. The methylmercury-l-cysteine conjugate is a substrate for the l-type large neutral amino acid transporter. J. Neurochem. 2008, 107, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Antunes dos Santos, A.; Appel Hort, M.; Culbreth, M.; López-Granero, C.; Farina, M.; Rocha, J.B.T.; Aschner, M. Methylmercury and brain development: A review of recent literature. J. Trace Elem. Med. Biol. 2016, 38, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.; Puntervoll, P.; Valdersnes, S.; Goksøyr, A. Responses in the brain proteome of Atlantic cod (Gadus morhua) exposed to methylmercury. Aquat. Toxicol. 2010, 100, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Berntssen, M.H.G.; Aatland, A.; Handy, R.D. Chronic dietary mercury exposure causes oxidative stress, brain lesions, and altered behaviour in Atlantic salmon (Salmo salar) parr. Aquat. Toxicol. 2003, 65, 55–72. [Google Scholar] [CrossRef]
- Webber, H.M.; Haines, T.A. Mercury effects on predator avoidance behavior of a forage fish, golden shiner (Notemigonus crysoleucas). Environ. Toxicol. Chem. 2003, 22, 1556–1561. [Google Scholar] [CrossRef] [PubMed]
- Sanfeliu, C.; Sebastià, J.; Cristòfol, R.; Rodríguez-Farré, E. Neurotoxicity of organomercurial compounds. Neurotox. Res. 2003, 5, 283–305. [Google Scholar] [PubMed]
- Vinceti, M.; Mandrioli, J.; Borella, P.; Michalke, B.; Tsatsakis, A.; Finkelstein, Y. Selenium neurotoxicity in humans: Bridging laboratory and epidemiologic studies. Toxicol. Lett. 2014, 230, 295–303. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Dietetic Products; Nutrition and Allergies. Scientific opinion on dietary reference values for selenium. EFSA J. 2014, 12. [Google Scholar] [CrossRef]
- Farina, M.; Aschner, M.; Rocha, J.B.T. Oxidative stress in MeHg-induced neurotoxicity. Toxicol. Appl. Pharmacol. 2011, 256, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M.A.K.; Wang, F. Mercury-selenium compounds and their toxicological significance: Towards a molecular understanding of the mercury-selenium antagonism. Environ. Toxicol. Chem. 2009, 28, 1567–1577. [Google Scholar] [CrossRef] [PubMed]
- Farina, M.; Rocha, J.B.T.; Aschner, M. Mechanisms of methylmercury-induced neurotoxicity: Evidence from experimental studies. Life Sci. 2011, 89, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Yamamoto, M.; Figeys, D.; Ning, Z.; Chan, H.M. Proteomic analysis of cerebellum in common marmoset exposed to methylmercury. Toxicol. Sci. 2015, 146, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Rabilloud, T.; Lescuyer, P. Proteomics in mechanistic toxicology: History, concepts, achievements, caveats, and potential. Proteomics 2015, 15, 1051–1074. [Google Scholar] [CrossRef] [PubMed]
- Aleström, P.; Holter, J.L.; Nourizadeh-Lillabadi, R. Zebrafish in functional genomics and aquatic biomedicine. Trends Biotechnol. 2006, 24, 15–21. [Google Scholar] [PubMed]
- Amlund, H.; Lundebye, A.-K.; Boyle, D.; Ellingsen, S. Dietary selenomethionine influences the accumulation and depuration of dietary methylmercury in zebrafish (Danio rerio). Aquat. Toxicol. 2015, 158, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Rasinger, J.D.; Carroll, T.S.; Lundebye, A.K.; Hogstrand, C. Cross-omics gene and protein expression profiling in juvenile female mice highlights disruption of calcium and zinc signalling in the brain following dietary exposure to CB-153, BDE-47, HBCD or TCDD. Toxicology 2014, 321, 1–12. [Google Scholar] [PubMed]
- Penglase, S.; Hamre, K.; Ellingsen, S. Selenium and mercury have a synergistic negative effect on fish reproduction. Aquat. Toxicol. 2014, 149, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Yamamoto, M.; Figeys, D.; Ning, Z.; Chan, H.M. Proteome profiling reveals regional protein alteration in cerebrum of common marmoset (Callithrix jacchus) exposed to methylmercury. Toxicology 2016, 347, 29–39. [Google Scholar] [PubMed]
- De Oliveira Souza, V.C.; de Marco, K.C.; Laure, H.J.; Rosa, J.C.; Barbosa, F., Jr. A brain proteome profile in rats exposed to methylmercury or thimerosal (ethylmercury). J. Toxicol. Environ. Health A 2016, 79, 502–512. [Google Scholar] [PubMed]
- Sarbassov, D.D.; Sabatini, D.M. Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. J. Biol. Chem. 2005, 280, 39505–39509. [Google Scholar] [CrossRef] [PubMed]
- Switon, K.; Kotulska, K.; Janusz-Kaminska, A.; Zmorzynska, J.; Jaworski, J. Molecular neurobiology of mTOR. Neuroscience 2017, 341, 112–153. [Google Scholar] [CrossRef] [PubMed]
- Takei, N.; Nawa, H. mTOR signaling and its roles in normal and abnormal brain development. Front. Mol. Neurosci. 2014, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Liu, C.; Liu, L.; Zhang, R.; Zhang, H.; Chen, S.; Luo, Y.; Chen, L.; Huang, S. Rapamycin prevents cadmium-induced neuronal cell death via targeting both mTORC1 and mTORC2 pathways. Neuropharmacology 2015, 97, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xu, B.; Liu, L.; Luo, Y.; Zhou, H.; Chen, W.; Shen, T.; Han, X.; Kontos, C.D.; Huang, S. Cadmium induction of reactive oxygen species activates the mTOR pathway, leading to neuronal cell death. Free Radic. Biol. Med. 2011, 50, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Al-Waeli, A.; Pappas, A.C.; Zoidis, E.; Georgiou, C.A.; Fegeros, K.; Zervas, G. The role of selenium in cadmium toxicity: Interactions with essential and toxic elements. Br. Poult. Sci. 2012, 53, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, L.; Zhao, J.; Wang, C.; Bao, J.; Li, J. Effects of selenium and cadmium on ion profiles in the brains of chickens. Biol. Trace Elem. Res. 2016, 174, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Penglase, S.; Hamre, K.; Ellingsen, S. Selenium prevents downregulation of antioxidant selenoprotein genes by methylmercury. Free Radic. Biol. Med. 2014, 75, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Branco, V.; Canário, J.; Lu, J.; Holmgren, A.; Carvalho, C. Mercury and selenium interaction in vivo: Effects on thioredoxin reductase and gluthathione peroxidase. Free Radic. Biol. Med. 2012, 52, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Ralston, N.V.C.; Ralston, C.R.; Blackwell, J.L., 3rd; Raymond, L.J. Dietary and tissue selenium in relation to methylmercury toxicity. Neurotoxicology 2008, 29, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Julshamn, K.; Maage, A.; Norli, H.S.; Grobecker, K.H.; Jorhem, L.; Fecher, P. Determination of arsenic, cadmium, mercury, and lead by inductively coupled plasma/mass spectrometry in foods after pressure digestion: NMKL1 interlaboratory study. J. AOAC Int. 2007, 90, 844–856. [Google Scholar] [PubMed]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Wilm, M.; Shevchenko, A.; Houthaeve, T.; Breit, S.; Schweigerer, L.; Fotsis, T.; Mann, M. Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature 1996, 379, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Vaudel, M.; Barsnes, H.; Berven, F.S.; Sickmann, A.; Martens, L. SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics 2011, 11, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Vaudel, M.; Burkhart, J.M.; Zahedi, R.P.; Oveland, E.; Berven, F.S.; Sickmann, A.; Martens, L.; Barsnes, H. PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat. Biotechnol. 2015, 33, 22–24. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diseases and Biofunctions | Score | Activation | ||
|---|---|---|---|---|
| MeHg | SeMet | MeHg | SeMet | |
| Cell death and survival | ||||
| Cell death | 3.0 | 1.8 | ||
| Cell death of neuroblastoma cell lines | 6.6 | |||
| Cell death of tumor cell lines | 2.8 | |||
| Necrosis | 3.0 | 1.7 | ||
| Cell death and survival, neurological disease | ||||
| Neuronal cell death | 2.4 | |||
| Cellular assembly and organization | ||||
| Formation of cytoskeleton | 2.6 | |||
| Cellular assembly and organization, cellular function and maintenance | ||||
| Microtubule dynamics | 1.9 | |||
| Organization of cytoskeleton | 2.2 | |||
| Cellular assembly and organization, tissue development | ||||
| Formation of filaments | 3.5 | |||
| Cellular compromise | ||||
| Degeneration of cells | 3.9 | |||
| Cellular growth and proliferation | ||||
| Proliferation of cells | 3.0 | |||
| Infectious diseases | ||||
| Viral infection | 2.4 | |||
| Molecular transport | ||||
| Transport of molecule | 2.4 | |||
| Neurological disease | ||||
| Degeneration of nervous system | 3.1 | |||
| Neurodegeneration | 3.0 | |||
| Organismal survival | ||||
| Organismal death | 1.9 | |||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasinger, J.D.; Lundebye, A.-K.; Penglase, S.J.; Ellingsen, S.; Amlund, H. Methylmercury Induced Neurotoxicity and the Influence of Selenium in the Brains of Adult Zebrafish (Danio rerio). Int. J. Mol. Sci. 2017, 18, 725. https://doi.org/10.3390/ijms18040725
Rasinger JD, Lundebye A-K, Penglase SJ, Ellingsen S, Amlund H. Methylmercury Induced Neurotoxicity and the Influence of Selenium in the Brains of Adult Zebrafish (Danio rerio). International Journal of Molecular Sciences. 2017; 18(4):725. https://doi.org/10.3390/ijms18040725
Chicago/Turabian StyleRasinger, Josef Daniel, Anne-Katrine Lundebye, Samuel James Penglase, Ståle Ellingsen, and Heidi Amlund. 2017. "Methylmercury Induced Neurotoxicity and the Influence of Selenium in the Brains of Adult Zebrafish (Danio rerio)" International Journal of Molecular Sciences 18, no. 4: 725. https://doi.org/10.3390/ijms18040725
APA StyleRasinger, J. D., Lundebye, A.-K., Penglase, S. J., Ellingsen, S., & Amlund, H. (2017). Methylmercury Induced Neurotoxicity and the Influence of Selenium in the Brains of Adult Zebrafish (Danio rerio). International Journal of Molecular Sciences, 18(4), 725. https://doi.org/10.3390/ijms18040725