
Pro-Resolving Molecules—New Approaches to Treat Sepsis?


Abstract
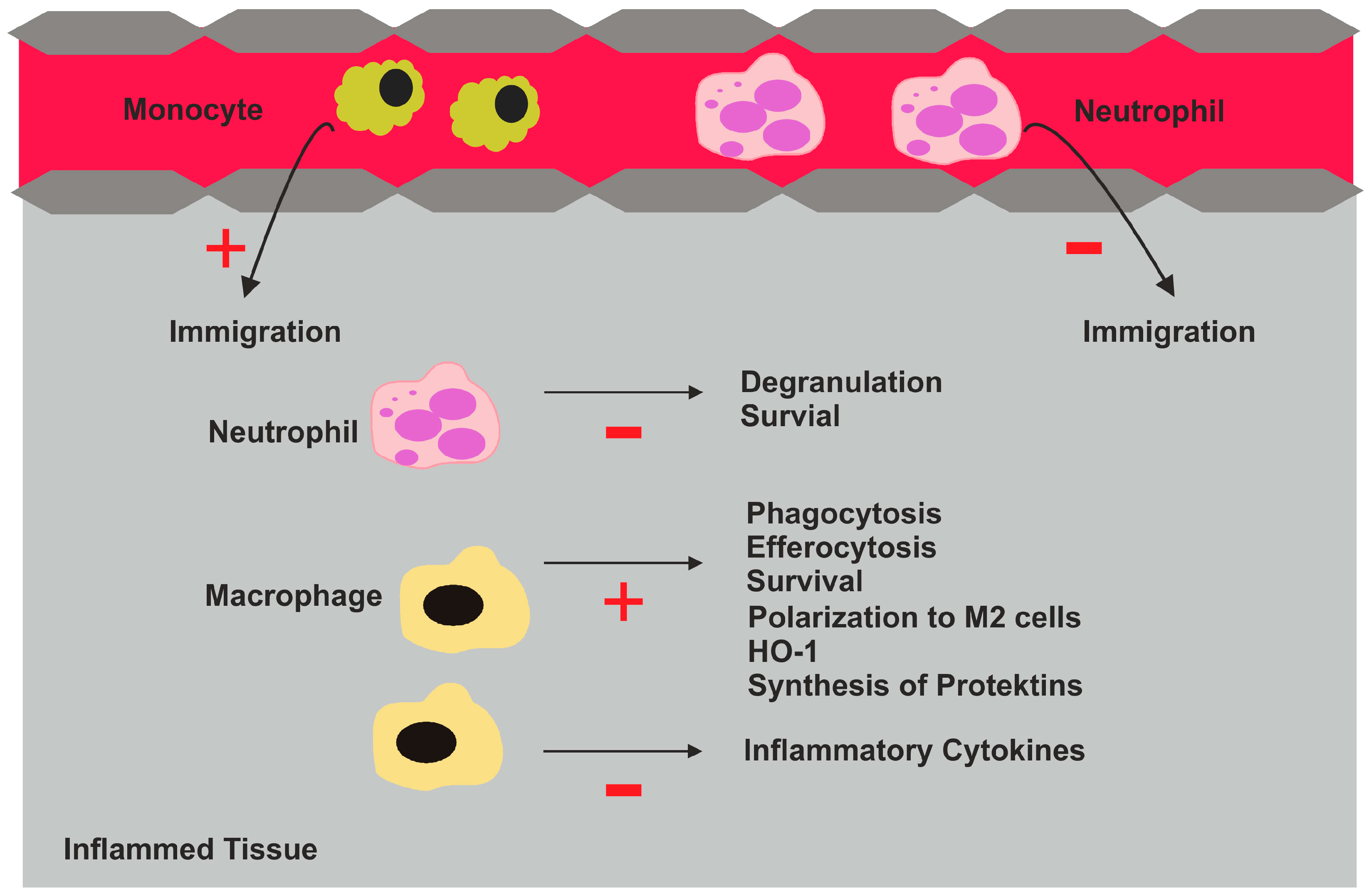
:1. Introduction
2. Molecules Involved in Resolution of Inflammation
2.1. The Antiinflammatory Cytokine Interleukin (IL)-10
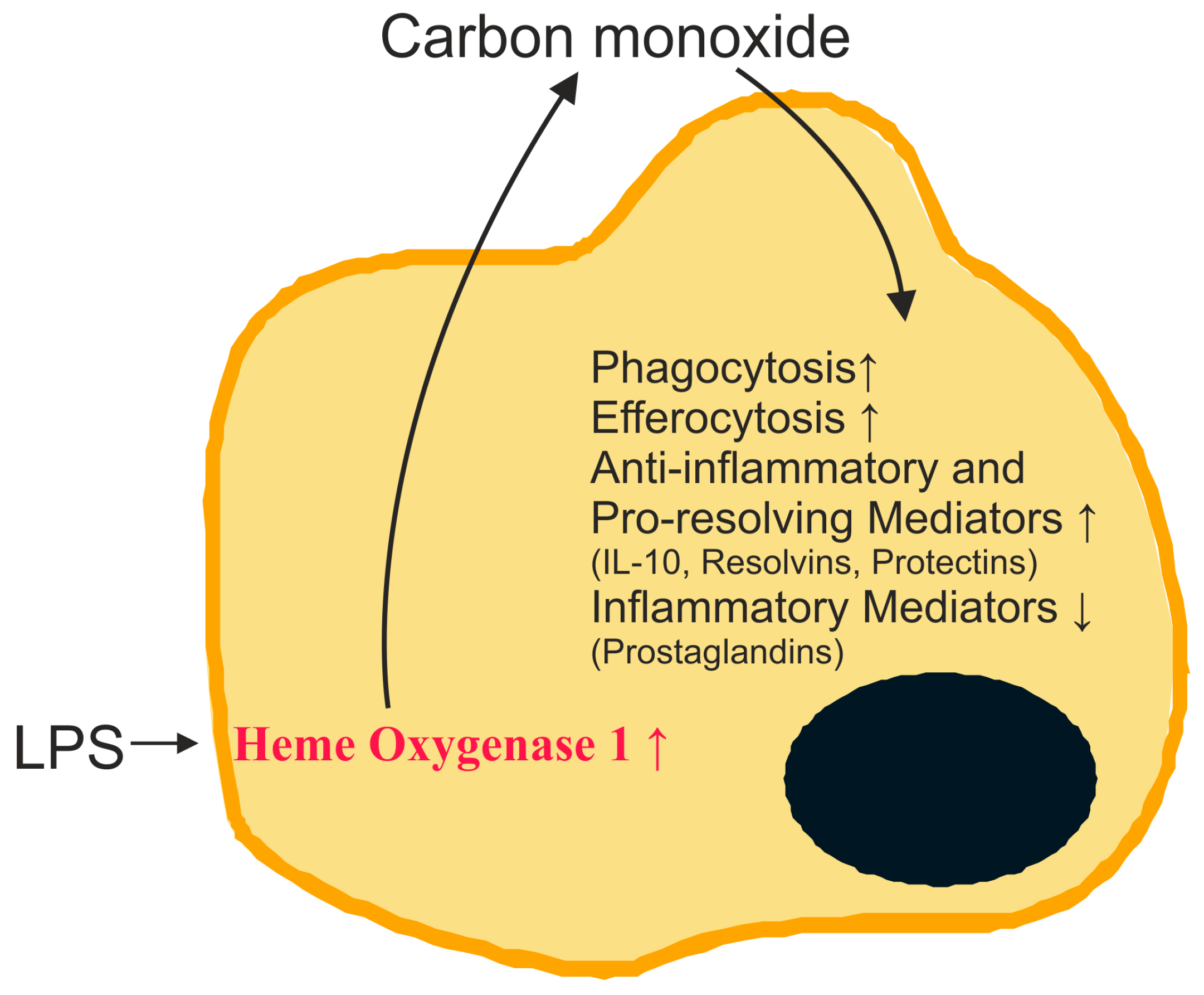
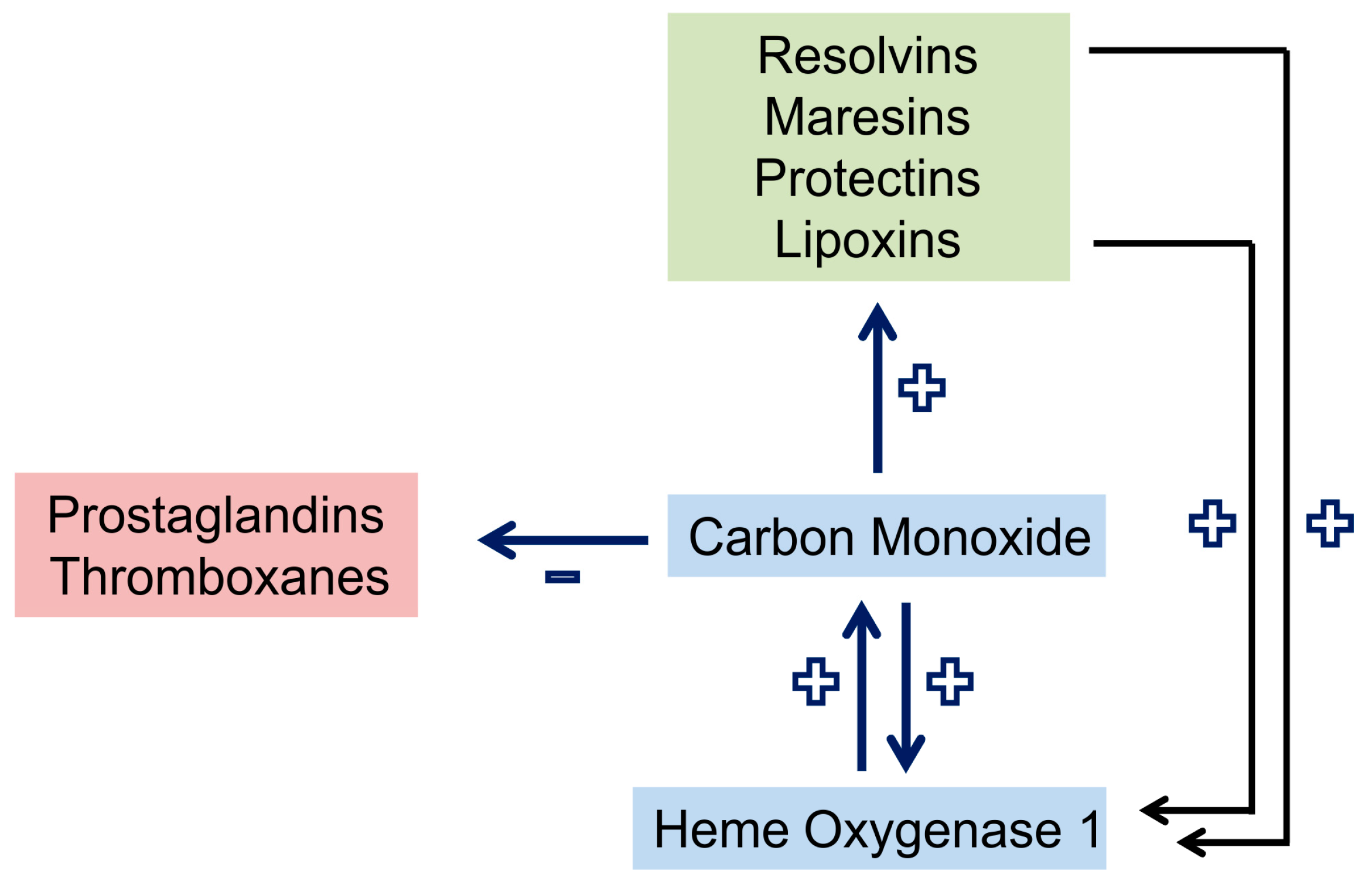
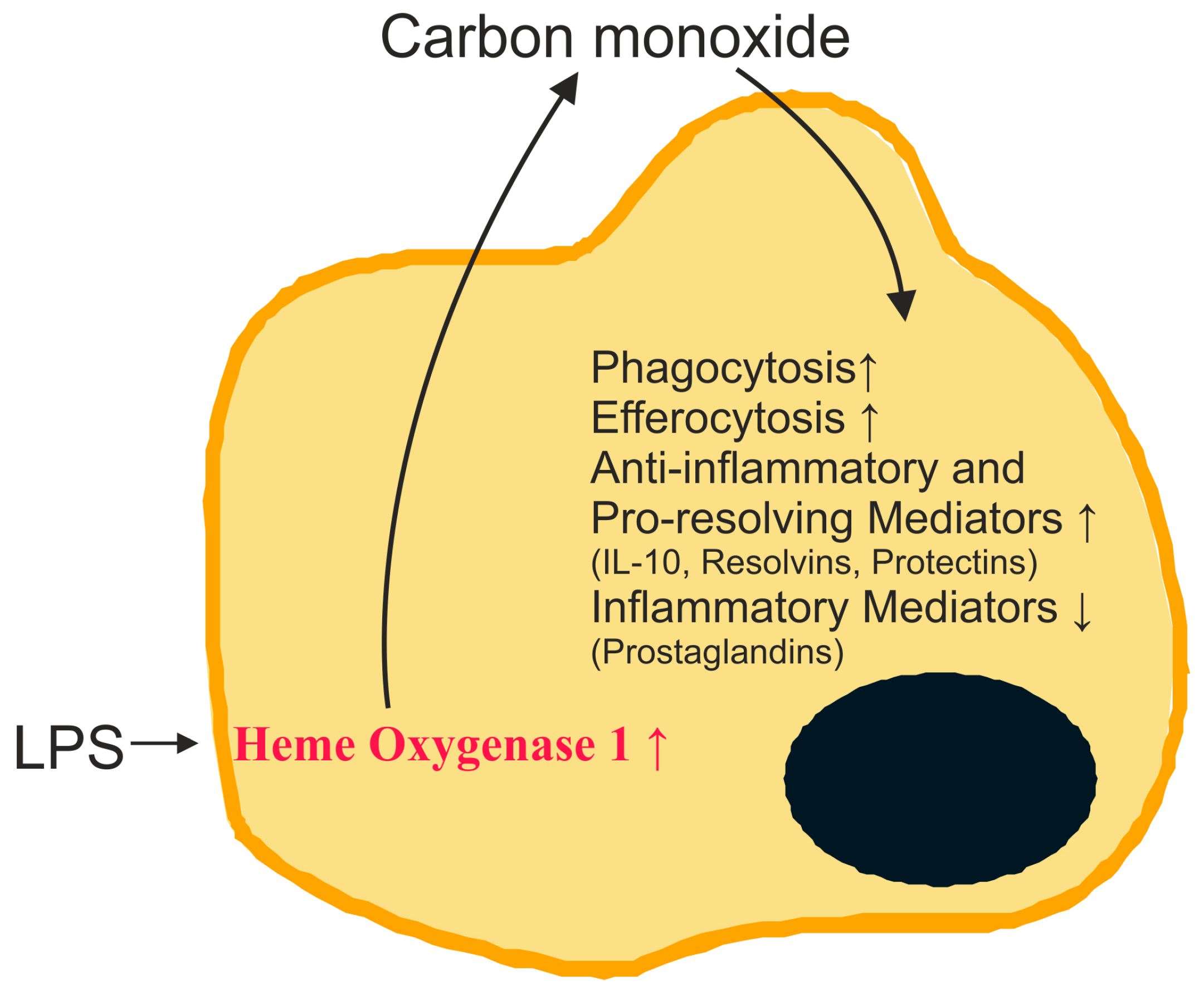
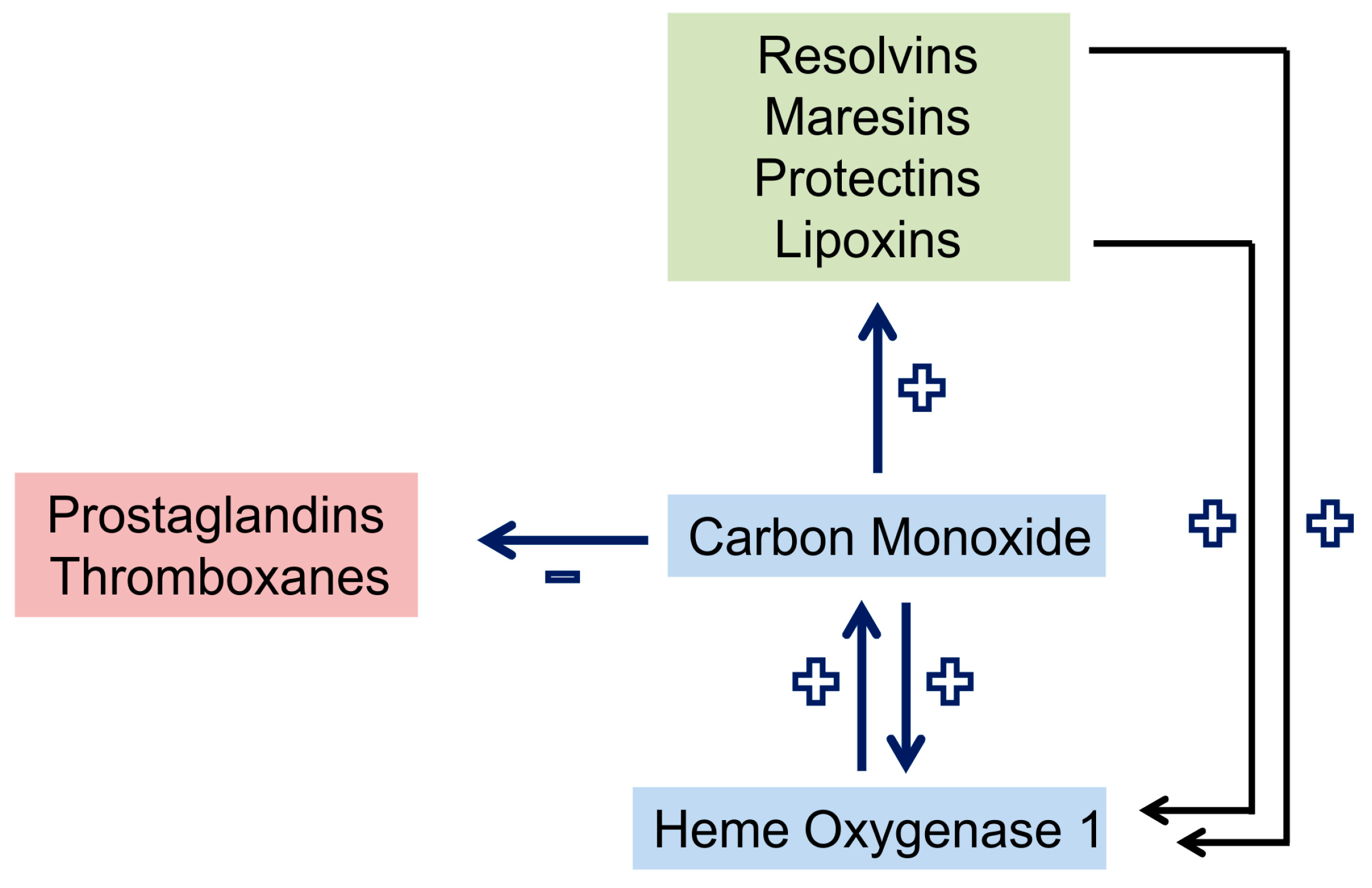
2.2. Carbon Monoxide and Heme Oxygenase
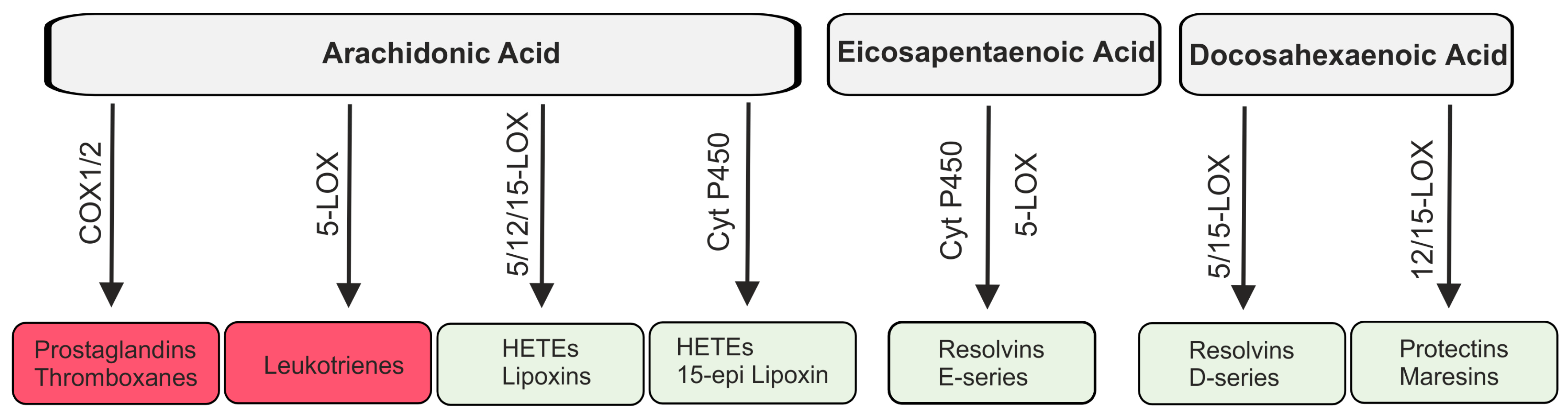
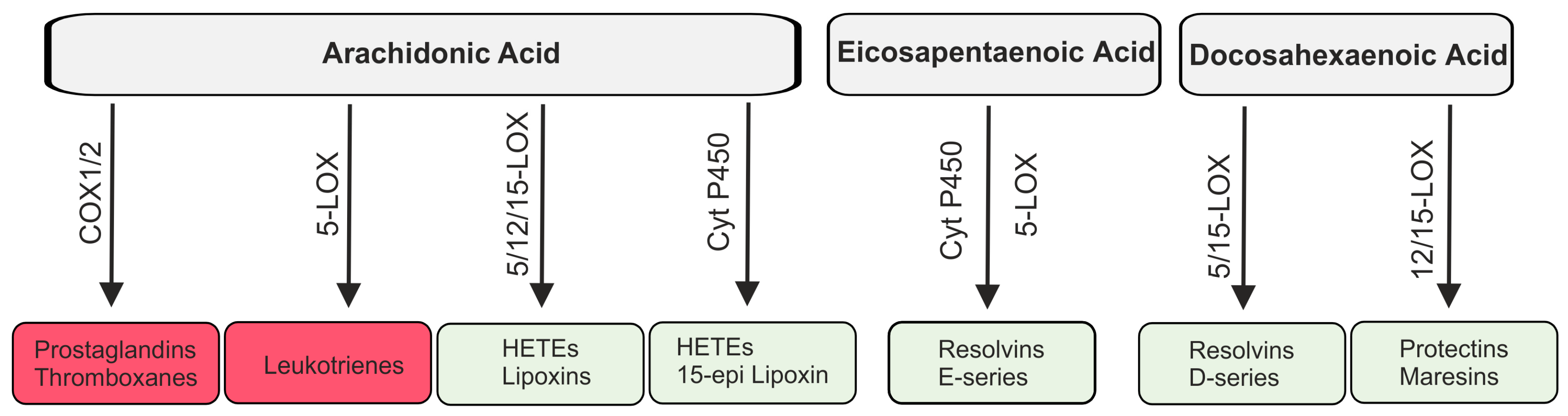
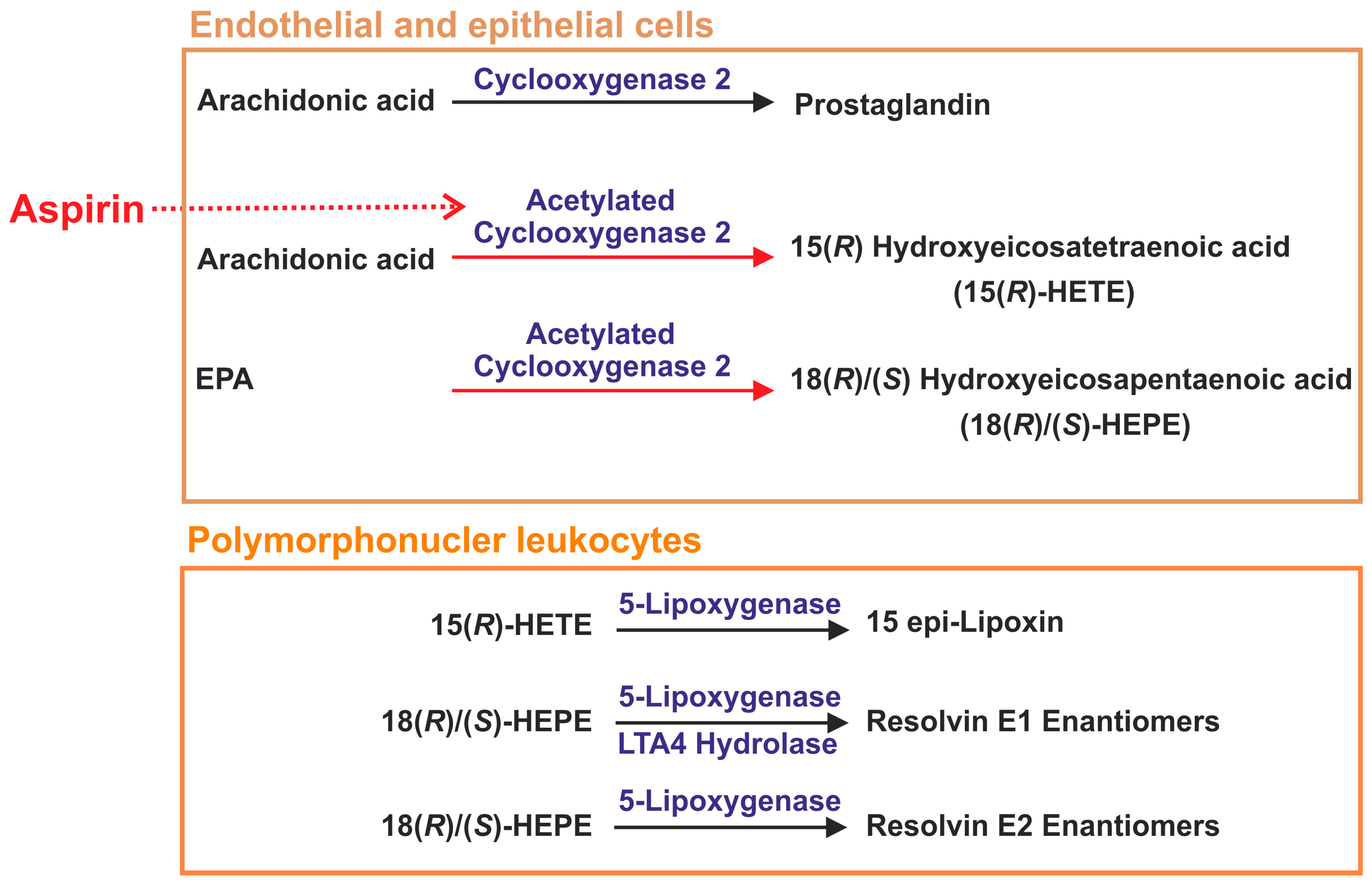
2.3. Lipid Mediators Derived from Arachidonic Acid
2.4. Lipid Mediators Derived from Omega-3 Fatty Acids
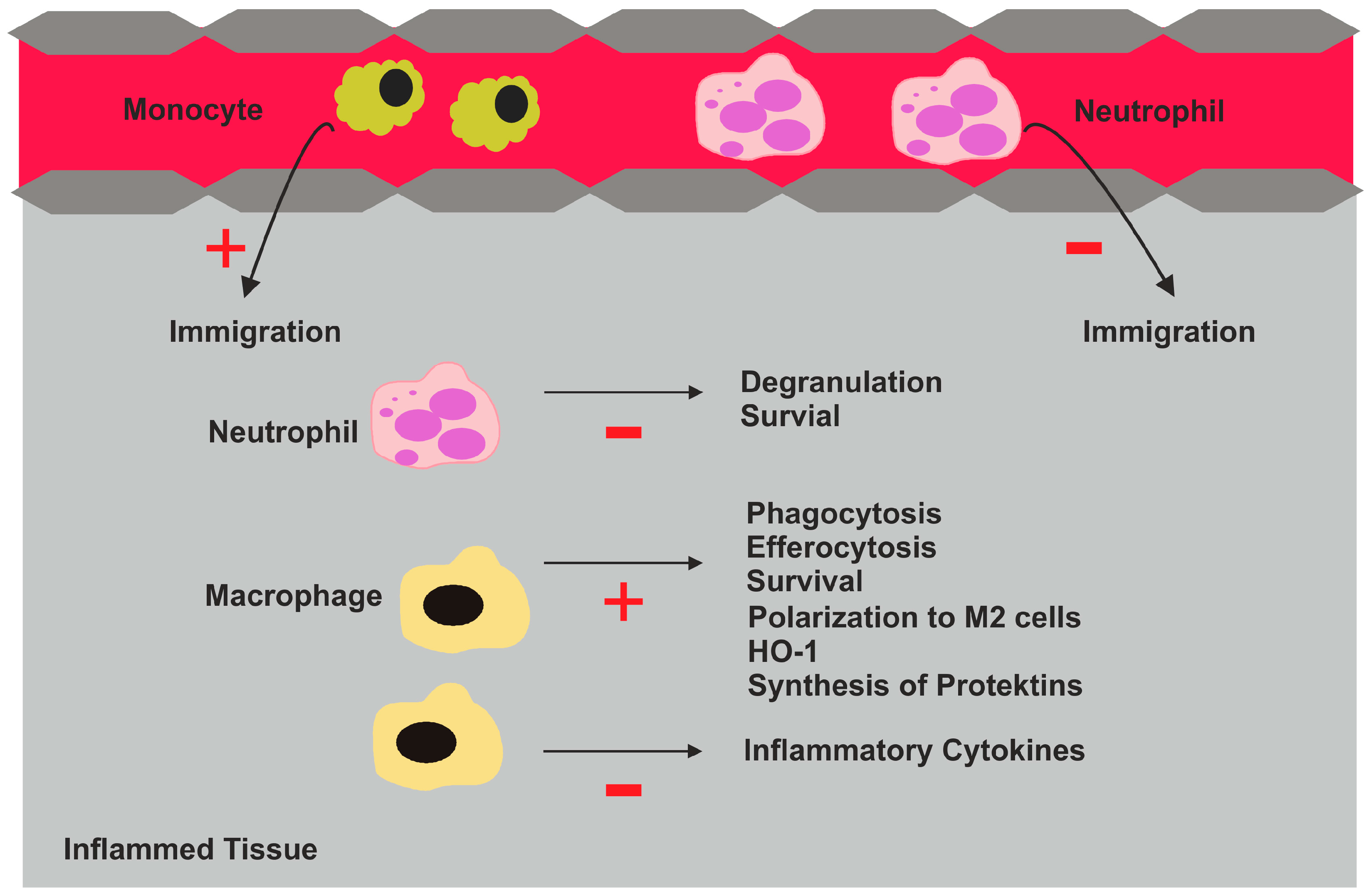
2.5. Biologic Activities of Pro-Resolving Lipids
2.6. Receptors for Pro-Resolving Lipids
2.7. Specialized Pro-Resolving Mediators (SPM) in Sepsis Models
2.8. Systemic Levels of Pro-Resolving Lipid Species in Healthy Volunteers and Non-Septic Patients
2.9. Pro-Resolving Lipid Species in Sepsis Patients
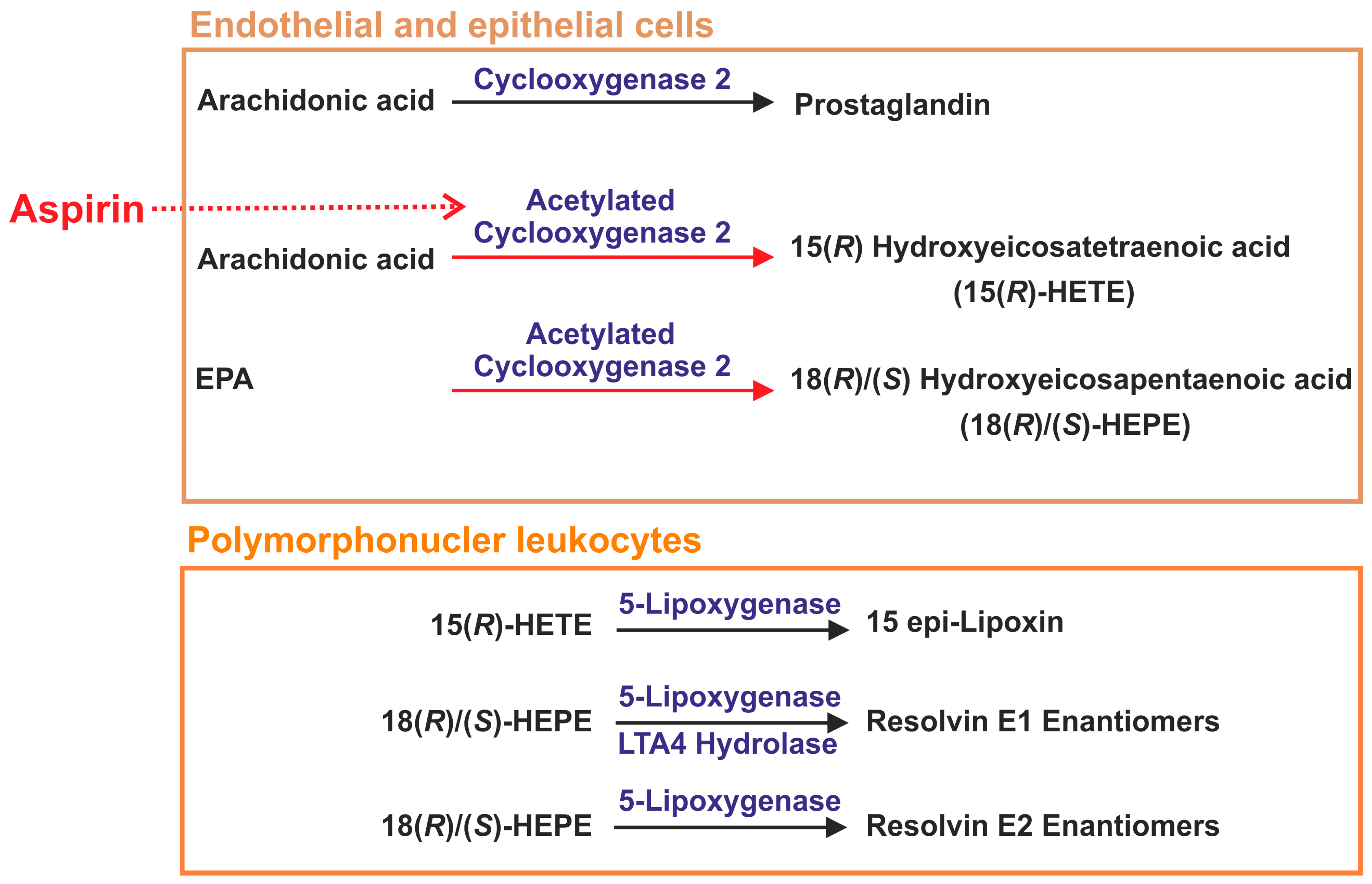
2.10. Aspirin in Sepsis
2.11. Carbon Monoxide in Sepsis Models
2.12. Carbon Monoxide in Clinical Studies
3. Conclusions
Author Contributions
Conflicts of Interest
References
- Leliefeld, P.H.; Wessels, C.M.; Leenen, L.P.; Koenderman, L.; Pillay, J. The role of neutrophils in immune dysfunction during severe inflammation. Crit. Care 2016, 20, 73. [Google Scholar] [CrossRef] [PubMed]
- Mizgerd, J.P. Acute lower respiratory tract infection. N. Engl. J. Med. 2008, 358, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Perretti, M. The resolution of inflammation: New mechanisms in patho-physiology open opportunities for pharmacology. Semin. Immunol. 2015, 27, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.A.; Sousa, L.P.; Pinho, V.; Perretti, M.; Teixeira, M.M. Resolution of inflammation: What Controls Its Onset? Front. Immunol. 2016, 7, 160. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.A. TLRs and innate immunity. Blood 2009, 113, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Mohammad Hosseini, A.; Majidi, J.; Baradaran, B.; Yousefi, M. Toll-like receptors in the pathogenesis of autoimmune diseases. Adv. Pharm. Bull. 2015, 5, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Molteni, M.; Gemma, S.; Rossetti, C. The Role of toll-like receptor 4 in infectious and noninfectious inflammation. Mediat. Inflamm. 2016, 2016, 6978936. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.C. Why have clinical trials in sepsis failed? Trends Mol. Med. 2014, 20, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Opal, S.M.; Dellinger, R.P.; Vincent, J.L.; Masur, H.; Angus, D.C. The next generation of sepsis clinical trial designs: What is next after the demise of recombinant human activated protein C? Crit. Care Med. 2014, 42, 1714–1721. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, N.A.; Unsinger, J.; Hotchkiss, R.S.; Ayala, A. The new normal: Immunomodulatory agents against sepsis immune suppression. Trends Mol. Med. 2014, 20, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Basil, M.C.; Levy, B.D. Specialized pro-resolving mediators: Endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 2016, 16, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Hong, S.; Gronert, K.; Colgan, S.P.; Devchand, P.R.; Mirick, G.; Moussignac, R.L. Resolvins: A family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 2002, 196, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Shinohara, M.; Dalli, J.; Mirakaj, V.; Kibi, M.; Choi, A.M.; Serhan, C.N. Inhaled carbon monoxide accelerates resolution of inflammation via unique proresolving mediator-heme oxygenase-1 circuits. J. Immunol. 2013, 190, 6378–6388. [Google Scholar] [CrossRef] [PubMed]
- Kyokane, T.; Norimizu, S.; Taniai, H.; Yamaguchi, T.; Takeoka, S.; Tsuchida, E.; Naito, M.; Nimura, Y.; Ishimura, Y.; Suematsu, M. Carbon monoxide from heme catabolism protects against hepatobiliary dysfunction in endotoxin-treated rat liver. Gastroenterology 2001, 120, 1227–1240. [Google Scholar] [CrossRef] [PubMed]
- Gobbetti, T.; Cooray, S.N. Annexin A1 and resolution of inflammation: Tissue repairing properties and signalling signature. Biol. Chem. 2016, 397, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Rostoker, R.; Yaseen, H.; Schif-Zuck, S.; Lichtenstein, R.G.; Rabinovich, G.A.; Ariel, A. Galectin-1 induces 12/15-lipoxygenase expression in murine macrophages and favors their conversion toward a pro-resolving phenotype. Prostaglandins Other Lipid Mediat. 2013, 107, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, F.; Tamassia, N.; Rossato, M.; Cassatella, M.A. Understanding the molecular mechanisms of the multifaceted IL-10-mediated anti-inflammatory response: Lessons from neutrophils. Eur. J. Immunol. 2010, 40, 2360–2368. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.K.; Takeda, K.; Akira, S.; Schreiber, R.D. Interleukin-10 receptor signaling through the JAK-STAT pathway: Requirement for two distinct receptor-derived signals for anti-inflammatory action. J. Biol. Chem. 1999, 274, 16513–16521. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, H.W. Molecular mechanism in tolerance to lipopolysaccharide. J. Inflamm. 1995, 45, 13–26. [Google Scholar] [PubMed]
- Adib-Conquy, M.; Adrie, C.; Moine, P.; Asehnoune, K.; Fitting, C.; Pinsky, M.R.; Dhainaut, J.F.; Cavaillon, J.M. NF-kappa B expression in mononuclear cells of patients with sepsis resembles that observed in lipopolysaccharide tolerance. Am. J. Respir. Crit. Care Med. 2000, 162, 1877–1883. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L. The p50-homodimer mechanism in tolerance to LPS. J. Endotoxin Res. 2001, 7, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Pena, O.M.; Pistolic, J.; Raj, D.; Fjell, C.D.; Hancock, R.E. Endotoxin tolerance represents a distinctive state of alternative polarization (M2) in human mononuclear cells. J. Immunol. 2011, 186, 7243–7254. [Google Scholar] [CrossRef] [PubMed]
- Rackov, G.; Hernandez-Jimenez, E.; Shokri, R.; Carmona-Rodriguez, L.; Manes, S.; Alvarez-Mon, M.; Lopez-Collazo, E.; Martinez, A.C.; Balomenos, D. p21 mediates macrophage reprogramming through regulation of p50-p50 NF-kappa B and IFN-beta. J. Clin. Investig. 2016, 126, 3089–3103. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, H.; Zhou, J.; Zhong, Y.; Ali, M.M.; McGuire, F.; Nagarkatti, P.S.; Nagarkatti, M. Role of cytokines as a double-edged sword in sepsis. In Vivo 2013, 27, 669–684. [Google Scholar] [PubMed]
- Serhan, C.N. Treating inflammation and infection in the 21st century: New hints from decoding resolution mediators and mechanisms. FASEB J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sacerdoti, D.; Pesce, P.; di Pascoli, M.; Bolognesi, M. EETs and HO-1 cross-talk. Prostaglandins Other Lipid Mediat. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, M.; Graversen, J.H.; Jacobsen, C.; Sonne, O.; Hoffman, H.J.; Law, S.K.; Moestrup, S.K. Identification of the haemoglobin scavenger receptor. Nature 2001, 409, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Philippidis, P.; Mason, J.C.; Evans, B.J.; Nadra, I.; Taylor, K.M.; Haskard, D.O.; Landis, R.C. Hemoglobin scavenger receptor CD163 mediates interleukin-10 release and heme oxygenase-1 synthesis: Antiinflammatory monocyte-macrophage responses in vitro, in resolving skin blisters in vivo, and after cardiopulmonary bypass surgery. Circ. Res. 2004, 94, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Choi, A.M. Carbon monoxide in the treatment of sepsis. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, B.; Larsen, R.; Gallo, D.; Chin, B.Y.; Harris, C.; Mannam, P.; Kaczmarek, E.; Lee, P.J.; Zuckerbraun, B.S.; Flavell, R.; et al. Macrophages sense and kill bacteria through carbon monoxide-dependent inflammasome activation. J. Clin. Investig. 2014, 124, 4926–4940. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Kim, H.P.; Geng, X.H.; Nakao, A.; Wang, X.; Murase, N.; Drain, P.F.; Sasidhar, M.; Nabel, E.G.; Takahashi, T.; et al. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J. Exp. Med. 2006, 203, 2377–2389. [Google Scholar] [CrossRef] [PubMed]
- Ricciotti, E.; Fitzgerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Tam, V.C. Lipidomic profiling of bioactive lipids by mass spectrometry during microbial infections. Semin. Immunol. 2013, 25, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.D.; Clish, C.B.; Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Bandeira-Melo, C.; Serra, M.F.; Diaz, B.L.; Cordeiro, R.S.; Silva, P.M.; Lenzi, H.L.; Bakhle, Y.S.; Serhan, C.N.; Martins, M.A. Cyclooxygenase-2-derived prostaglandin E2 and lipoxin A4 accelerate resolution of allergic edema in Angiostrongylus costaricensis-infected rats: Relationship with concurrent eosinophilia. J. Immunol. 2000, 164, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Morris, T.; Stables, M.; Hobbs, A.; de Souza, P.; Colville-Nash, P.; Warner, T.; Newson, J.; Bellingan, G.; Gilroy, D.W. Effects of low-dose aspirin on acute inflammatory responses in humans. J. Immunol. 2009, 183, 2089–2096. [Google Scholar] [CrossRef] [PubMed]
- Claria, J.; Serhan, C.N. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc. Natl. Acad. Sci. USA 1995, 92, 9475–9479. [Google Scholar] [CrossRef] [PubMed]
- Vonakis, B.M.; Vanderhoek, J.Y. 15-Hydroxyeicosatetraenoic acid (15-HETE) receptors. Involvement in the 15-HETE-induced stimulation of the cryptic 5-lipoxygenase in PT-18 mast/basophil cells. J. Biol. Chem. 1992, 267, 23625–23631. [Google Scholar] [PubMed]
- Norris, P.C.; Gosselin, D.; Reichart, D.; Glass, C.K.; Dennis, E.A. Phospholipase A2 regulates eicosanoid class switching during inflammasome activation. Proc. Natl. Acad. Sci. USA 2014, 111, 12746–12751. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Hamberg, M.; Samuelsson, B. Lipoxins: Novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc. Natl. Acad. Sci. USA 1984, 81, 5335–5339. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Sheppard, K.A. Lipoxin formation during human neutrophil-platelet interactions: Evidence for the transformation of leukotriene A4 by platelet 12-lipoxygenase in vitro. J. Clin. Investig. 1990, 85, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Le Bel, M.; Brunet, A.; Gosselin, J. Leukotriene B4, an endogenous stimulator of the innate immune response against pathogens. J. Innate Immun. 2014, 6, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Edin, M.L.; Theken, K.N.; Schuck, R.N.; Flake, G.P.; Kannon, M.A.; DeGraff, L.M.; Lih, F.B.; Foley, J.; Bradbury, J.A.; et al. Endothelial CYP epoxygenase overexpression and soluble epoxide hydrolase disruption attenuate acute vascular inflammatory responses in mice. FASEB J. 2011, 25, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Petasis, N.A. Resolvins and protectins in inflammation resolution. Chem. Rev. 2011, 111, 5922–5943. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.F.; Pillai, P.S.; Recchiuti, A.; Yang, R.; Serhan, C.N. Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J. Clin. Investig. 2011, 121, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Clish, C.B.; Brannon, J.; Colgan, S.P.; Chiang, N.; Gronert, K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 2000, 192, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Fredman, G.; Yang, R.; Karamnov, S.; Belayev, L.S.; Bazan, N.G.; Zhu, M.; Winkler, J.W.; Petasis, N.A. Novel proresolving aspirin-triggered DHA pathway. Chem. Biol. 2011, 18, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Xuan, W.; Fan, G.H. Roles of resolvins in the resolution of acute inflammation. Cell Biol. Int. 2015, 39, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, A.T.; Fokin, V.V.; Petasis, N.A.; Serhan, C.N.; Madara, J.L. LXA4, aspirin-triggered 15-epi-LXA4, and their analogs selectively downregulate PMN azurophilic degranulation. Am. J. Physiol. 1999, 276, 988–994. [Google Scholar]
- Serhan, C.N.; Maddox, J.F.; Petasis, N.A.; Akritopoulou-Zanze, I.; Papayianni, A.; Brady, H.R.; Colgan, S.P.; Madara, J.L. Design of lipoxin A4 stable analogs that block transmigration and adhesion of human neutrophils. Biochemistry 1995, 34, 14609–14615. [Google Scholar] [CrossRef] [PubMed]
- El Kebir, D.; Jozsef, L.; Pan, W.; Wang, L.; Petasis, N.A.; Serhan, C.N.; Filep, J.G. 15-epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am. J. Respir. Crit. Care Med. 2009, 180, 311–319. [Google Scholar] [CrossRef] [PubMed]
- El Kebir, D.; Gjorstrup, P.; Filep, J.G. Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc. Natl. Acad. Sci. USA 2012, 109, 14983–14988. [Google Scholar] [CrossRef]
- Ariel, A.; Fredman, G.; Sun, Y.P.; Kantarci, A.; van Dyke, T.E.; Luster, A.D.; Serhan, C.N. Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat. Immunol. 2006, 7, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Eickmeier, O.; Hilberath, J.N.; Zielen, S.; Haworth, O. Pro-resolving lipid mediators in inflammatory lung diseases. Pneumologie 2011, 65, 149–158. [Google Scholar] [CrossRef]
- Kang, J.W.; Lee, S.M. Resolvin D1 protects the liver from ischemia/reperfusion injury by enhancing M2 macrophage polarization and efferocytosis. Biochim. Biophys. Acta 2016, 1861, 1025–1035. [Google Scholar] [CrossRef]
- Ramon, S.; Dalli, J.; Sanger, J.M.; Winkler, J.W.; Aursnes, M.; Tungen, J.E.; Hansen, T.V.; Serhan, C.N. The Protectin PCTR1 is produced by human M2 macrophages and enhances resolution of infectious inflammation. Am. J. Pathol. 2016, 186, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.N.; Surh, Y.J. Resolvin D1-mediated NOX2 inactivation rescues macrophages undertaking efferocytosis from oxidative stress-induced apoptosis. Biochem. Pharmacol. 2013, 86, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Prieto, P.; Rosales-Mendoza, C.E.; Terron, V.; Toledano, V.; Cuadrado, A.; Lopez-Collazo, E.; Bannenberg, G.; Martin-Sanz, P.; Fernandez-Velasco, M.; Bosca, L. Activation of autophagy in macrophages by pro-resolving lipid mediators. Autophagy 2015, 11, 1729–1744. [Google Scholar] [CrossRef] [PubMed]
- Aliberti, J.; Hieny, S.; Reis e Sousa, C.; Serhan, C.N.; Sher, A. Lipoxin-mediated inhibition of IL-12 production by DCs: A mechanism for regulation of microbial immunity. Nat. Immunol. 2002, 3, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Arita, M.; Bianchini, F.; Aliberti, J.; Sher, A.; Chiang, N.; Hong, S.; Yang, R.; Petasis, N.A.; Serhan, C.N. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 2005, 201, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Haworth, O.; Cernadas, M.; Yang, R.; Serhan, C.N.; Levy, B.D. Resolvin E1 regulates interleukin 23, interferon-gamma and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat. Immunol. 2008, 9, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Ariel, A.; Chiang, N.; Arita, M.; Petasis, N.A.; Serhan, C.N. Aspirin-triggered lipoxin A4 and B4 analogs block extracellular signal-regulated kinase-dependent TNF-alpha secretion from human T cells. J. Immunol. 2003, 170, 6266–6272. [Google Scholar] [CrossRef] [PubMed]
- Ariel, A.; Li, P.L.; Wang, W.; Tang, W.X.; Fredman, G.; Hong, S.; Gotlinger, K.H.; Serhan, C.N. The docosatriene protectin D1 is produced by TH2 skewing and promotes human T cell apoptosis via lipid raft clustering. J. Biol. Chem. 2005, 280, 43079–43086. [Google Scholar] [CrossRef] [PubMed]
- Maddox, J.F.; Hachicha, M.; Takano, T.; Petasis, N.A.; Fokin, V.V.; Serhan, C.N. Lipoxin A4 stable analogs are potent mimetics that stimulate human monocytes and THP-1 cells via a G-protein-linked lipoxin A4 receptor. J. Biol. Chem. 1997, 272, 6972–6978. [Google Scholar] [CrossRef] [PubMed]
- Godson, C.; Mitchell, S.; Harvey, K.; Petasis, N.A.; Hogg, N.; Brady, H.R. Cutting edge: Lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J. Immunol. 2000, 164, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.D.; Fokin, V.V.; Clark, J.M.; Wakelam, M.J.; Petasis, N.A.; Serhan, C.N. Polyisoprenyl phosphate (PIPP) signaling regulates phospholipase D activity: A 'stop' signaling switch for aspirin-triggered lipoxin A4. FASEB J. 1999, 13, 903–911. [Google Scholar] [PubMed]
- Wang, C.W.; Colas, R.A.; Dalli, J.; Arnardottir, H.H.; Nguyen, D.; Hasturk, H.; Chiang, N.; van Dyke, T.E.; Serhan, C.N. Maresin 1 biosynthesis and proresolving anti-infective functions with human-localized aggressive periodontitis leukocytes. Infect. Immun. 2016, 84, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Liu, H.; Wu, J.; Qi, H.; Wu, Z.Y.; Shu, H.Q.; Li, H.B.; Chen, L.; Wang, Y.X.; Li, B.; et al. Maresin 1 prevents lipopolysaccharide-induced neutrophil survival and accelerates resolution of acute lung injury. Shock 2015, 44, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Schwab, J.M.; Chiang, N.; Arita, M.; Serhan, C.N. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 2007, 447, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Herova, M.; Schmid, M.; Gemperle, C.; Hersberger, M. ChemR23, the receptor for chemerin and resolvin E1, is expressed and functional on M1 but not on M2 macrophages. J. Immunol. 2015, 194, 2330–2337. [Google Scholar] [CrossRef] [PubMed]
- Arita, M.; Yoshida, M.; Hong, S.; Tjonahen, E.; Glickman, J.N.; Petasis, N.A.; Blumberg, R.S.; Serhan, C.N. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc. Natl. Acad. Sci. USA 2005, 102, 7671–7676. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.F.; Dona, M.; Fredman, G.; Krishnamoorthy, S.; Irimia, D.; Serhan, C.N. Resolvin E2 formation and impact in inflammation resolution. J. Immunol. 2012, 188, 4527–4534. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Urabe, D.; Yokokura, Y.; Arai, H.; Arita, M.; Inoue, M. Total synthesis and bioactivity of resolvin E2. Org. Lett. 2009, 11, 3602–3605. [Google Scholar] [CrossRef] [PubMed]
- Pope, N.H.; Salmon, M.; Davis, J.P.; Chatterjee, A.; Su, G.; Conte, M.S.; Ailawadi, G.; Upchurch, G.R., Jr. D-series resolvins inhibit murine abdominal aortic aneurysm formation and increase M2 macrophage polarization. FASEB J. 2016, 30, 4192–4201. [Google Scholar] [CrossRef] [PubMed]
- Tjonahen, E.; Oh, S.F.; Siegelman, J.; Elangovan, S.; Percarpio, K.B.; Hong, S.; Arita, M.; Serhan, C.N. Resolvin E2: Identification and anti-inflammatory actions: Pivotal role of human 5-lipoxygenase in resolvin E series biosynthesis. Chem. Biol. 2006, 13, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Norris, P.C.; Arnardottir, H.; Sanger, J.M.; Fichtner, D.; Keyes, G.S.; Serhan, C.N. Resolvin D3 multi-level proresolving actions are host protective during infection. Prostaglandins Leukot. Essent. Fatty Acids 2016. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Winkler, J.W.; Colas, R.A.; Arnardottir, H.; Cheng, C.Y.; Chiang, N.; Petasis, N.A.; Serhan, C.N. Resolvin D3 and aspirin-triggered resolvin D3 are potent immunoresolvents. Chem. Biol. 2013, 20, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.W.; Orr, S.K.; Dalli, J.; Cheng, C.Y.; Sanger, J.M.; Chiang, N.; Petasis, N.A.; Serhan, C.N. Resolvin D4 stereoassignment and its novel actions in host protection and bacterial clearance. Sci. Rep. 2016, 6, 18972. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, J.; Hasegawa, S.; Kasubuchi, M.; Ichimura, A.; Nakajima, A.; Kimura, I. Nutritional Signaling via free fatty acid receptors. Int. J. Mol. Sci. 2016, 17, 450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.; Kim, Y.J.; Yang, S.J.; Lee, Y.; Lee, M. Nutrigenomic Functions of PPARs in Obesogenic Environments. PPAR Res. 2016, 2016, 4794576. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Dong, J.; Wu, W.; Yang, T.; Wang, T.; Guo, L.; Chen, L.; Xu, D.; Wen, F. Resolvin D1 attenuates inflammation in lipopolysaccharide-induced acute lung injury through a process involving the PPARgamma/NF-kappa B pathway. Respir Res. 2012, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Fiore, S.; Maddox, J.F.; Perez, H.D.; Serhan, C.N. Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J. Exp. Med. 1994, 180, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Krishnamoorthy, S.; Recchiuti, A.; Chiang, N. Novel anti-inflammatory: Pro-resolving mediators and their receptors. Curr. Top. Med. Chem. 2011, 11, 629–647. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Godson, C.; Guiry, P.J.; Agerberth, B.; Haeggstrom, J.Z. Leukotriene B4/antimicrobial peptide LL-37 proinflammatory circuits are mediated by BLT1 and FPR2/ALX and are counterregulated by lipoxin A4 and resolvin E1. FASEB J. 2011, 25, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Arita, M.; Ohira, T.; Sun, Y.P.; Elangovan, S.; Chiang, N.; Serhan, C.N. Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J. Immunol. 2007, 178, 3912–3917. [Google Scholar] [CrossRef] [PubMed]
- Buechler, C. Chemerin in liver diseases. Endocrinol. Metab. Syndr. 2014, 3, 1–6. [Google Scholar]
- Wittamer, V.; Franssen, J.D.; Vulcano, M.; Mirjolet, J.F.; le Poul, E.; Migeotte, I.; Brezillon, S.; Tyldesley, R.; Blanpain, C.; Detheux, M.; et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J. Exp. Med. 2003, 198, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.C.; Sinal, C.J. Chemerin: At the crossroads of inflammation and obesity. Trends Endocrinol. Metab. 2010, 21, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Horn, P.; Metzing, U.B.; Steidl, R.; Romeike, B.; Rauchfuss, F.; Sponholz, C.; Thomas-Ruddel, D.; Ludewig, K.; Birkenfeld, A.L.; Settmacher, U.; et al. Chemerin in peritoneal sepsis and its associations with glucose metabolism and prognosis: A translational cross-sectional study. Crit. Care 2016, 20, 39. [Google Scholar] [CrossRef] [PubMed]
- Ohira, T.; Arita, M.; Omori, K.; Recchiuti, A.; van Dyke, T.E.; Serhan, C.N. Resolvin E1 receptor activation signals phosphorylation and phagocytosis. J. Biol. Chem. 2010, 285, 3451–3461. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Yacoubian, S.; Lee, C.H.; Yang, R.; Petasis, N.A.; Serhan, C.N. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl. Acad. Sci. USA 2010, 107, 1660–1665. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Gemperle, C.; Rimann, N.; Hersberger, M. Resolvin D1 polarizes primary human macrophages toward a proresolution phenotype through GPR32. J. Immunol. 2016, 196, 3429–3437. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Dalli, J.; Colas, R.A.; Serhan, C.N. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J. Exp. Med. 2015, 212, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; de la Rosa, X.; Libreros, S.; Serhan, C.N. Novel Resolvin D2 Receptor Axis in Infectious Inflammation. J. Immunol. 2017, 198, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Park, C.K.; Xu, Z.Z.; Liu, T.; Lu, N.; Serhan, C.N.; Ji, R.R. Resolvin D2 is a potent endogenous inhibitor for transient receptor potential subtype V1/A1, inflammatory pain, and spinal cord synaptic plasticity in mice: Distinct roles of resolvin D1, D2, and E1. J. Neurosci. 2011, 31, 18433–18438. [Google Scholar] [CrossRef] [PubMed]
- Buras, J.A.; Holzmann, B.; Sitkovsky, M. Animal models of sepsis: Setting the stage. Nat. Rev. Drug. Discov. 2005, 4, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.; Dichter, E.; Lacorte, G.; Kerner, D.; Spur, B.; Rodriguez, A.; Yin, K. Lipoxin A4 increases survival by decreasing systemic inflammation and bacterial load in sepsis. Shock 2011, 36, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Capilato, J.; Pham, M.P.; Walker, J.; Spur, B.; Rodriguez, A.; Perez, L.J.; Yin, K. Lipoxin A4 augments host defense in sepsis and reduces Pseudomonas aeruginosa virulence through quorum sensing inhibition. FASEB J. 2016, 30, 2400–2410. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, Z.; Zhao, S.; Sun, C.; Yang, M. Effect of BML-111 on the intestinal mucosal barrier in sepsis and its mechanism of action. Mol. Med. Rep. 2015, 12, 3101–3106. [Google Scholar] [CrossRef] [PubMed]
- Gobbetti, T.; Coldewey, S.M.; Chen, J.; McArthur, S.; le Faouder, P.; Cenac, N.; Flower, R.J.; Thiemermann, C.; Perretti, M. Nonredundant protective properties of FPR2/ALX in polymicrobial murine sepsis. Proc. Natl. Acad. Sci. USA 2014, 111, 18685–18690. [Google Scholar] [CrossRef] [PubMed]
- Khadaroo, R.G.; Marshall, J.C. ARDS and the multiple organ dysfunction syndrome: Common mechanisms of a common systemic process. Crit. Care Clin. 2002, 18, 127–141. [Google Scholar] [CrossRef]
- Sordi, R.; Menezes-de-Lima, O., Jr.; Horewicz, V.; Scheschowitsch, K.; Santos, L.F.; Assreuy, J. Dual role of lipoxin A4 in pneumosepsis pathogenesis. Int. Immunopharmacol. 2013, 17, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Spite, M.; Norling, L.V.; Summers, L.; Yang, R.; Cooper, D.; Petasis, N.A.; Flower, R.J.; Perretti, M.; Serhan, C.N. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 2009, 461, 1287–1291. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Shetty, S.; Zhang, P.; Gao, R.; Hu, Y.; Wang, S.; Li, Z.; Fu, J. Aspirin-triggered resolvin D1 down-regulates inflammatory responses and protects against endotoxin-induced acute kidney injury. Toxicol. Appl. Pharmacol. 2014, 277, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Fan, X.H.; Wu, Y.P.; Zhu, J.L.; Wang, F.; Bo, L.L.; Li, J.B.; Bao, R.; Deng, X.M. Resolvin D1 improves survival in experimental sepsis through reducing bacterial load and preventing excessive activation of inflammatory response. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Suzuki, K.; Tamura, H.; Nagaoka, I. Suppressive action of resolvin D1 on the production and release of septic mediators in D-galactosamine-sensitized endotoxin shock mice. Exp. Ther. Med. 2011, 2, 57–61. [Google Scholar] [PubMed]
- Seki, H.; Fukunaga, K.; Arita, M.; Arai, H.; Nakanishi, H.; Taguchi, R.; Miyasho, T.; Takamiya, R.; Asano, K.; Ishizaka, A.; et al. The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. J. Immunol. 2010, 184, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Grenon, S.M.; Owens, C.D.; Nosova, E.V.; Hughes-Fulford, M.; Alley, H.F.; Chong, K.; Perez, S.; Yen, P.K.; Boscardin, J.; Hellmann, J.; et al. Short-term, high-dose fish oil supplementation increases the production of omega-3 fatty acid-derived mediators in patients with peripheral artery disease (the OMEGA-PAD I Trial). J. Am. Heart Assoc. 2015, 4, e002034. [Google Scholar] [PubMed]
- Rangel-Huerta, O.D.; Aguilera, C.M.; Mesa, M.D.; Gil, A. Omega-3 long-chain polyunsaturated fatty acids supplementation on inflammatory biomakers: A systematic review of randomised clinical trials. Br. J. Nutr. 2012, 107, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Chiang, N.; Dalli, J. The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Semin. Immunol. 2015, 27, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Domenichiello, A.F.; Kitson, A.P.; Bazinet, R.P. Is docosahexaenoic acid synthesis from alpha-linolenic acid sufficient to supply the adult brain? Prog. Lipid Res. 2015, 59, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, Y.; Brooks, J.; Reider, C.; Fulgoni, V.L., 3rd. US adults are not meeting recommended levels for fish and omega-3 fatty acid intake: Results of an analysis using observational data from NHANES 2003–2008. Nutr. J. 2014, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Skarke, C.; Alamuddin, N.; Lawson, J.A.; Li, X.; Ferguson, J.F.; Reilly, M.P.; Fitzgerald, G.A. Bioactive products formed in humans from fish oils. J. Lipid Res. 2015, 56, 1808–1820. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Konkel, A.; Mehling, H.; Blossey, K.; Gapelyuk, A.; Wessel, N.; von Schacky, C.; Dechend, R.; Muller, D.N.; Rothe, M.; et al. Dietary omega-3 fatty acids modulate the eicosanoid profile in man primarily via the CYP-epoxygenase pathway. J. Lipid Res. 2014, 55, 1150–1164. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.C. Specialized pro-resolving mediators: Do they circulate in plasma? J. Lipid Res. 2015, 56, 1641–1642. [Google Scholar] [CrossRef] [PubMed]
- Markworth, J.F.; Kaur, G.; Miller, E.G.; Larsen, A.E.; Sinclair, A.J.; Maddipati, K.R.; Cameron-Smith, D. Divergent shifts in lipid mediator profile following supplementation with n-3 docosapentaenoic acid and eicosapentaenoic acid. FASEB J. 2016, 30, 3714–3725. [Google Scholar] [CrossRef] [PubMed]
- Polus, A.; Zapala, B.; Razny, U.; Gielicz, A.; Kiec-Wilk, B.; Malczewska-Malec, M.; Sanak, M.; Childs, C.E.; Calder, P.C.; Dembinska-Kiec, A. Omega-3 fatty acid supplementation influences the whole blood transcriptome in women with obesity, associated with pro-resolving lipid mediator production. Biochim. Biophys. Acta 2016, 1861, 1746–1755. [Google Scholar] [CrossRef] [PubMed]
- Barden, A.; Mas, E.; Croft, K.D.; Phillips, M.; Mori, T.A. Short-term n-3 fatty acid supplementation but not aspirin increases plasma proresolving mediators of inflammation. J. Lipid Res. 2014, 55, 2401–2407. [Google Scholar] [CrossRef] [PubMed]
- Colas, R.A.; Shinohara, M.; Dalli, J.; Chiang, N.; Serhan, C.N. Identification and signature profiles for pro-resolving and inflammatory lipid mediators in human tissue. Am. J. Physiol. Cell. Physiol. 2014, 307, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Bays, H. Clinical overview of Omacor: A concentrated formulation of omega-3 polyunsaturated fatty acids. Am. J. Cardiol. 2006, 98, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Yin, X.; Jiang, Q. Natural forms of vitamin E and 13'-carboxychromanol, a long-chain vitamin E metabolite, inhibit leukotriene generation from stimulated neutrophils by blocking calcium influx and suppressing 5-lipoxygenase activity, respectively. J. Immunol. 2011, 186, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Meloni, C.; Manca-di-Villahermosa, S.; Cococcetta, N.; Casciani, C.U.; Finazzi-Agro, A.; Taccone-Gallucci, M. Vitamin E suppresses 5-lipoxygenase-mediated oxidative stress in peripheral blood mononuclear cells of hemodialysis patients regardless of administration route. Am. J. Kidney Dis. 2001, 37, 964–969. [Google Scholar] [CrossRef]
- Dalli, J.; Colas, R.A.; Quintana, C.; Barragan-Bradford, D.; Hurwitz, S.; Levy, B.D.; Choi, A.M.; Serhan, C.N.; Baron, R.M. Human sepsis eicosanoid and proresolving lipid mediator temporal profiles: Correlations with survival and clinical outcomes. Crit. Care Med. 2016, 45, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Uno, H.; Furukawa, K.; Suzuki, D.; Shimizu, H.; Ohtsuka, M.; Kato, A.; Yoshitomi, H.; Miyazaki, M. Immunonutrition suppresses acute inflammatory responses through modulation of resolvin E1 in patients undergoing major hepatobiliary resection. Surgery 2016, 160, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Seymour, C.W.; Liu, V.X.; Iwashyna, T.J.; Brunkhorst, F.M.; Rea, T.D.; Scherag, A.; Rubenfeld, G.; Kahn, J.M.; Shankar-Hari, M.; Singer, M.; et al. Assessment of clinical criteria for sepsis: For the third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 2016, 315, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Mebazaa, A.; Laterre, P.F.; Russell, J.A.; Bergmann, A.; Gattinoni, L.; Gayat, E.; Harhay, M.O.; Hartmann, O.; Hein, F.; Kjolbye, A.L.; et al. Designing phase 3 sepsis trials: Application of learned experiences from critical care trials in acute heart failure. J. Intensive Care 2016, 4, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toner, P.; McAuley, D.F.; Shyamsundar, M. Aspirin as a potential treatment in sepsis or acute respiratory distress syndrome. Crit. Care 2015, 19, 374. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.H.; Shih, C.H.; Yu, Y.B.; Hsu, H.C. Plasma levels in sepsis patients of annexin A1, lipoxin A4, macrophage inflammatory protein-3a, and neutrophil gelatinase-associated lipocalin. J. Chin. Med. Assoc. 2013, 76, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Manzanares, W.; Dhaliwal, R.; Jurewitsch, B.; Stapleton, R.D.; Jeejeebhoy, K.N.; Heyland, D.K. Parenteral fish oil lipid emulsions in the critically ill: A systematic review and meta-analysis. J. Parenter. Enteral. Nutr. 2014, 38, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.J.; Ho, C.K.; Ajibola, O.; Avenell, A. The role of omega-3 fatty acid supplemented parenteral nutrition in critical illness in adults: A systematic review and meta-analysis. Crit. Care Med. 2013, 41, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Spite, M.; Serhan, C.N. Novel lipid mediators promote resolution of acute inflammation: Impact of aspirin and statins. Circ. Res. 2010, 107, 1170–1184. [Google Scholar] [CrossRef] [PubMed]
- Halushka, P.V.; Wise, W.C.; Cook, J.A. Studies on the beneficial effects of aspirin in endotoxic shock: Relationship to inhibition of arachidonic acid metabolism. Am. J. Med. 1983, 74, 91–96. [Google Scholar] [CrossRef]
- Chen, W.; Janz, D.R.; Bastarache, J.A.; May, A.K.; O'Neal, H.R., Jr.; Bernard, G.R.; Ware, L.B. Prehospital aspirin use is associated with reduced risk of acute respiratory distress syndrome in critically ill patients: A propensity-adjusted analysis. Crit. Care Med. 2015, 43, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Eisen, D.P.; Reid, D.; McBryde, E.S. Acetyl salicylic acid usage and mortality in critically ill patients with the systemic inflammatory response syndrome and sepsis. Crit. Care Med. 2012, 40, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Falcone, M.; Russo, A.; Cangemi, R.; Farcomeni, A.; Calvieri, C.; Barilla, F.; Scarpellini, M.G.; Bertazzoni, G.; Palange, P.; Taliani, G.; et al. Lower mortality rate in elderly patients with community-onset pneumonia on treatment with aspirin. J. Am. Heart Assoc. 2015, 4, e001595. [Google Scholar] [CrossRef] [PubMed]
- Kor, D.J.; Erlich, J.; Gong, M.N.; Malinchoc, M.; Carter, R.E.; Gajic, O.; Talmor, D.S. Association of prehospitalization aspirin therapy and acute lung injury: Results of a multicenter international observational study of at-risk patients. Crit. Care Med. 2011, 39, 2393–2400. [Google Scholar] [CrossRef] [PubMed]
- Wiewel, M.A.; de Stoppelaar, S.F.; van Vught, L.A.; Frencken, J.F.; Hoogendijk, A.J.; Klein Klouwenberg, P.M.; Horn, J.; Bonten, M.J.; Zwinderman, A.H.; Cremer, O.L.; et al. Chronic antiplatelet therapy is not associated with alterations in the presentation, outcome, or host response biomarkers during sepsis: A propensity-matched analysis. Intensive Care Med. 2016, 42, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Hetzel, S.; DeMets, D.; Schneider, R.; Borzak, S.; Schneider, W.; Serebruany, V.; Schroder, H.; Hennekens, C.H. Aspirin increases nitric oxide formation in chronic stable coronary disease. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Poss, K.D.; Tonegawa, S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10925–10930. [Google Scholar] [CrossRef] [PubMed]
- Cepinskas, G.; Katada, K.; Bihari, A.; Potter, R.F. Carbon monoxide liberated from carbon monoxide-releasing molecule CORM-2 attenuates inflammation in the liver of septic mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, S.J.; Coronata, A.A.; Fredenburgh, L.E.; Chung, S.W.; Perrella, M.A.; Nakahira, K.; Ryter, S.W.; Choi, A.M. Carbon monoxide confers protection in sepsis by enhancing beclin 1-dependent autophagy and phagocytosis. Antioxid. Redox Signal. 2014, 20, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Lancel, S.; Hassoun, S.M.; Favory, R.; Decoster, B.; Motterlini, R.; Neviere, R. Carbon monoxide rescues mice from lethal sepsis by supporting mitochondrial energetic metabolism and activating mitochondrial biogenesis. J. Pharmacol. Exp. Ther. 2009, 329, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qin, W.; Song, M.; Zhang, Y.; Sun, B. Exogenous carbon monoxide inhibits neutrophil infiltration in LPS-induced sepsis by interfering with FPR1 via p38 MAPK but not GRK2. Oncotarget 2016, 7, 34250–34265. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Kibi, M.; Riley, I.R.; Chiang, N.; Dalli, J.; Kraft, B.D.; Piantadosi, C.A.; Choi, A.M.; Serhan, C.N. Cell-cell interactions and bronchoconstrictor eicosanoid reduction with inhaled carbon monoxide and resolvin D1. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Tsoyi, K.; Hall, S.R.; Dalli, J.; Colas, R.A.; Ghanta, S.; Ith, B.; Coronata, A.; Fredenburgh, L.E.; Baron, R.M.; Choi, A.M.; et al. Carbon monoxide improves efficacy of mesenchymal stromal cells during sepsis by production of specialized proresolving lipid mediators. Crit. Care Med. 2016, 44, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Choi, A.M. Carbon monoxide in exhaled breath testing and therapeutics. J. Breath Res. 2013, 7, 017111. [Google Scholar] [CrossRef] [PubMed]
- Biernacki, W.A.; Kharitonov, S.A.; Barnes, P.J. Exhaled carbon monoxide in patients with lower respiratory tract infection. Respir. Med. 2001, 95, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Pan, F.; Li, H.; Pan, J.; Qin, S.; Jiang, D.; Shen, C. Plasma carbon monoxide levels in term newborn infants with sepsis. Biol. Neonate 2000, 78, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Takaki, S.; Takeyama, N.; Kajita, Y.; Yabuki, T.; Noguchi, H.; Miki, Y.; Inoue, Y.; Nakagawa, T. Beneficial effects of the heme oxygenase-1/carbon monoxide system in patients with severe sepsis/septic shock. Intensive Care Med. 2010, 36, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Morimatsu, H.; Takahashi, T.; Maeshima, K.; Inoue, K.; Kawakami, T.; Shimizu, H.; Takeuchi, M.; Yokoyama, M.; Katayama, H.; Morita, K. Increased heme catabolism in critically ill patients: Correlation among exhaled carbon monoxide, arterial carboxyhemoglobin, and serum bilirubin IX alpha concentrations. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Zegdi, R.; Perrin, D.; Burdin, M.; Boiteau, R.; Tenaillon, A. Increased endogenous carbon monoxide production in severe sepsis. Intensive Care Med. 2002, 28, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Mayr, F.B.; Spiel, A.; Leitner, J.; Marsik, C.; Germann, P.; Ullrich, R.; Wagner, O.; Jilma, B. Effects of carbon monoxide inhalation during experimental endotoxemia in humans. Am. J. Respir. Crit. Care Med. 2005, 171, 354–360. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SPM | Target Cell | Effect | Reference |
|---|---|---|---|
| Lipoxin A4 | Dendritic cell | IL-12 ↓ | [60] |
| Monocyte | Chemotaxis ↑ | [65] | |
| Monocyte | Adhesion to laminin ↑ | [65] | |
| Macrophage | Efferocytosis ↑ | [66] | |
| Macrophage | Heme oxygenase 1 ↑ | [14] | |
| PMN | Chemotaxis ↓ | [51] | |
| PMN | Transendothelial/–epithelial migration ↓ | [51] | |
| PMN | Degranulation ↓ | [50] | |
| PMN | Superoxide anion ↓ | [67] | |
| PMN | CCR5 ↑ | [54] | |
| T cell | TNF secretion ↓ | [63] | |
| Maresin 1 | Macrophage | Phagocytosis ↑ | [68] |
| Macrophage | Killing of Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans ↑ | [68] | |
| Macrophage | Heme oxygenase 1 ↑ | [14] | |
| PMN | Phagocytosis ↑ | [68] | |
| PMN | LPS-induced resistance to apoptosis ↓ | [69] | |
| Protectin D1 | Macrophage | Efferocytosis ↑ | [70] |
| Macrophage | Heme oxygenase 1 ↑ | [14] | |
| PMN | Transendothelial/–epithelial migration ↓ | [13] | |
| PMN | CCR5 ↑ | [54] | |
| T cell | TNF, IFNγ secretion ↓ | [64] | |
| T-cell | Apoptosis ↑ | [64] | |
| RvE1 | Dendritic cell | LPS induced IL-6 ↓ | [62] |
| Dendritic cell | IL-12 ↓ | [61] | |
| Dendritic cell | LPS induced IL-23 ↓ | [62] | |
| Dendritic cell | LPS induced TNF ↓ | [62] | |
| Dendritic cell | Migration ↓ | [61] | |
| Macrophage | Efferocytosis ↑ | [70] | |
| Macrophage | Heme oxygenase 1 ↑ | [14] | |
| Macrophage | IL-10 ↑ | [71] | |
| Neutrophil | Apoptosis ↑ | [53] | |
| PMN | Transendothelial/–epithelial migration ↓ | [47,72] | |
| PMN | CCR5 ↑ | [54] | |
| RvE2 | Macrophage | Phagocytosis ↑ | [73] |
| Macrophage | LPS-induced IL-10 ↑ | [73] | |
| PMN | Chemotaxis ↓ | [73] | |
| PMN | Infiltration ↓ | [74] | |
| RvD1 | Macrophage | M2 polarization ↑ | [56] |
| Macrophage | Efferocytosis ↑ | [56] | |
| Macrophage | Autophagy ↑ | [59] | |
| Macrophage | Heme oxygenase 1 ↑ | [14] | |
| Macrophage | Apoptosis ↓ | [58,59] | |
| RvD2 | Macrophage | M2 polarization ↑ | [75] |
| Macrophage | Heme oxygenase 1 ↑ | [14] | |
| PMN | Infiltration ↓ | [74,76] | |
| RvD3 | Leukocyte | Phagocytosis of E. coli ↑ | [77] |
| Macrophage | Efferocytosis ↑ | [77,78] | |
| Macrophage | Phagocytosis ↑ | [77,78] | |
| PMN | Transmigration ↓ | [78] | |
| PMN | Bacterial phagocytosis ↑ | [77] | |
| PMN | Intracellular reactive oxygen species ↑ | [77] | |
| PMN | Platelet-PMN aggregation ↓ | [77] | |
| RvD4 | PMN | Infiltration ↓ | [79] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buechler, C.; Pohl, R.; Aslanidis, C. Pro-Resolving Molecules—New Approaches to Treat Sepsis? Int. J. Mol. Sci. 2017, 18, 476. https://doi.org/10.3390/ijms18030476
Buechler C, Pohl R, Aslanidis C. Pro-Resolving Molecules—New Approaches to Treat Sepsis? International Journal of Molecular Sciences. 2017; 18(3):476. https://doi.org/10.3390/ijms18030476
Chicago/Turabian StyleBuechler, Christa, Rebekka Pohl, and Charalampos Aslanidis. 2017. "Pro-Resolving Molecules—New Approaches to Treat Sepsis?" International Journal of Molecular Sciences 18, no. 3: 476. https://doi.org/10.3390/ijms18030476