
A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Definition of Gaucher Disease
3. Epidemiology
4. Pathophysiology
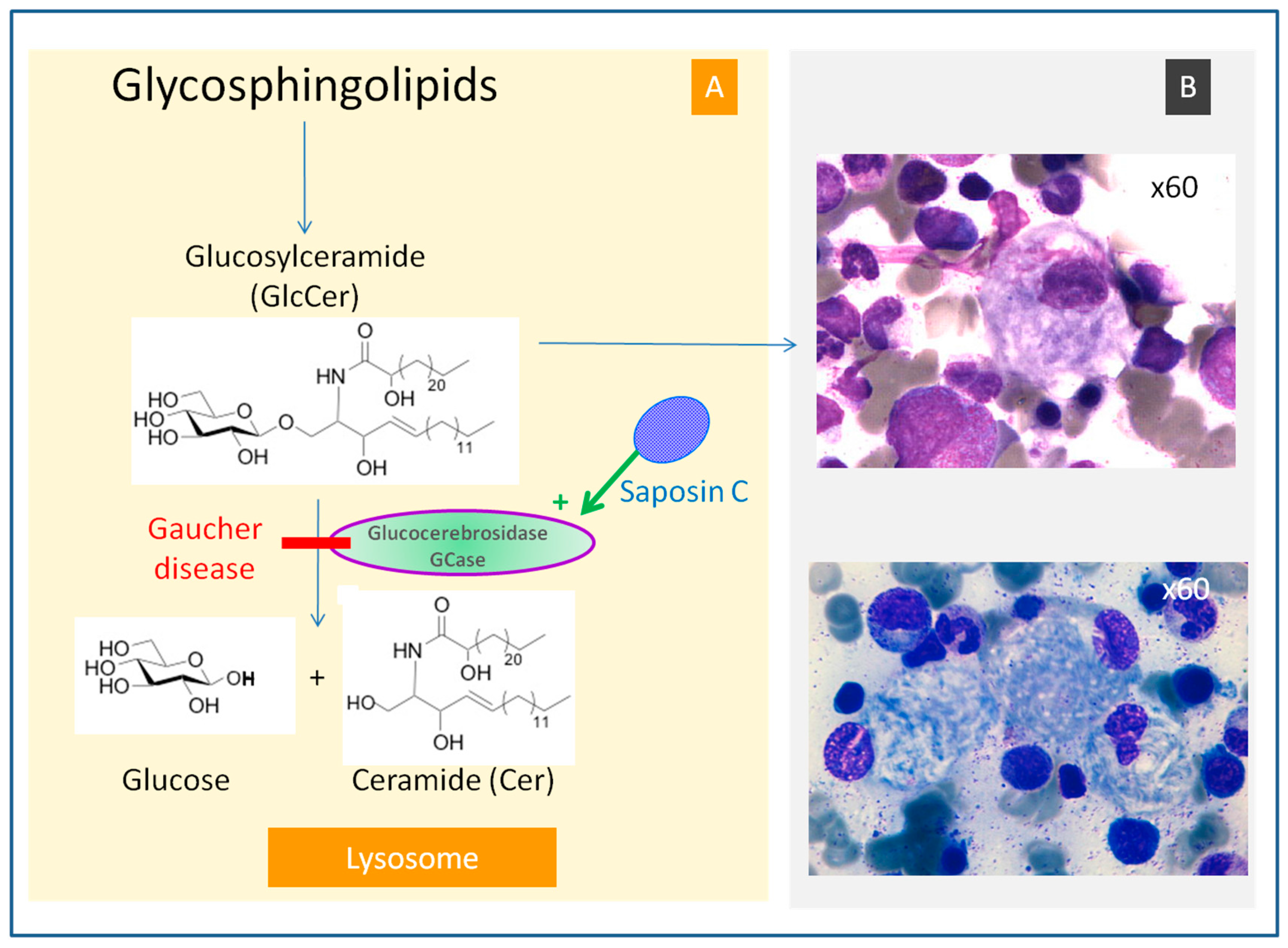
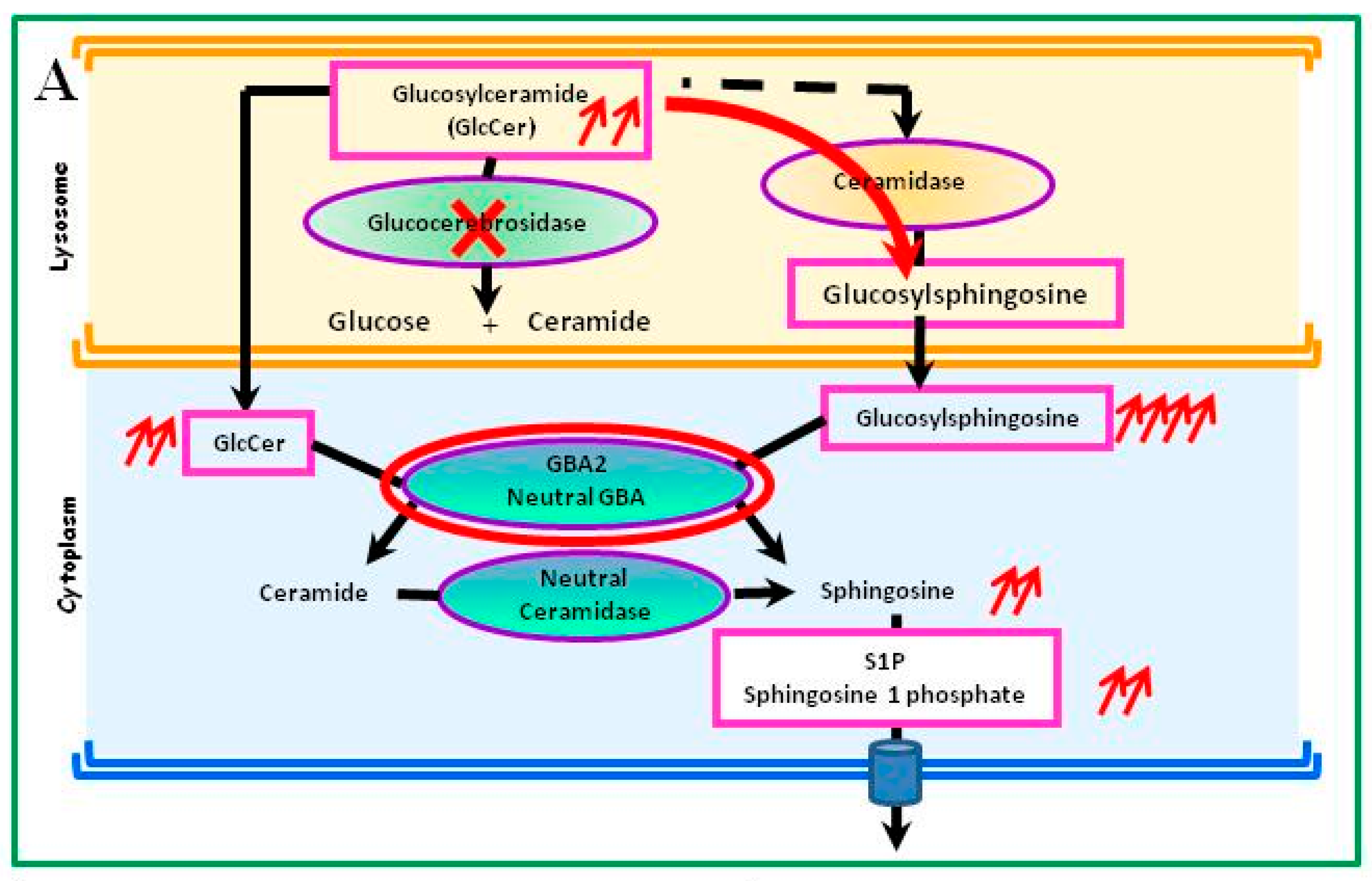
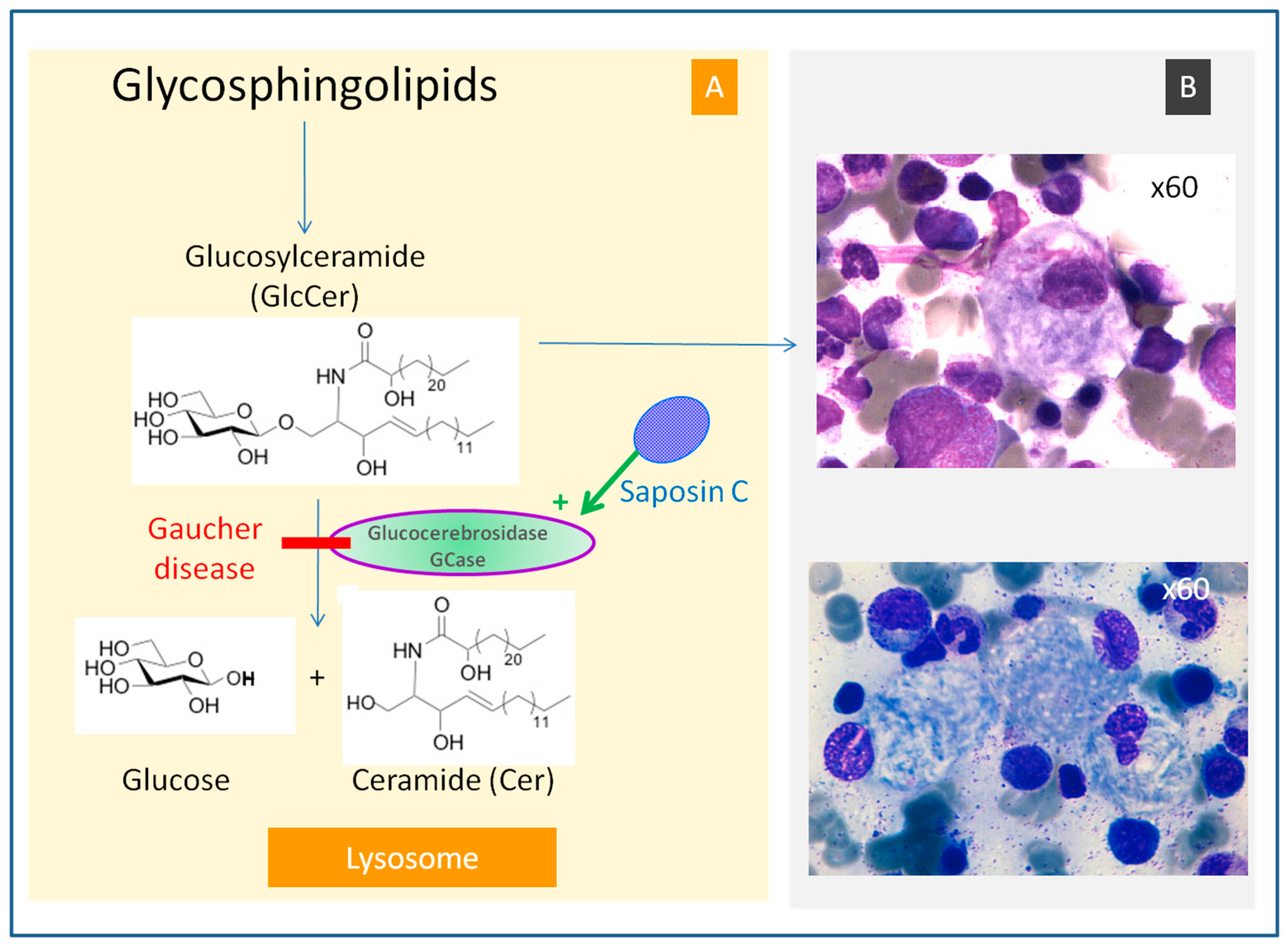
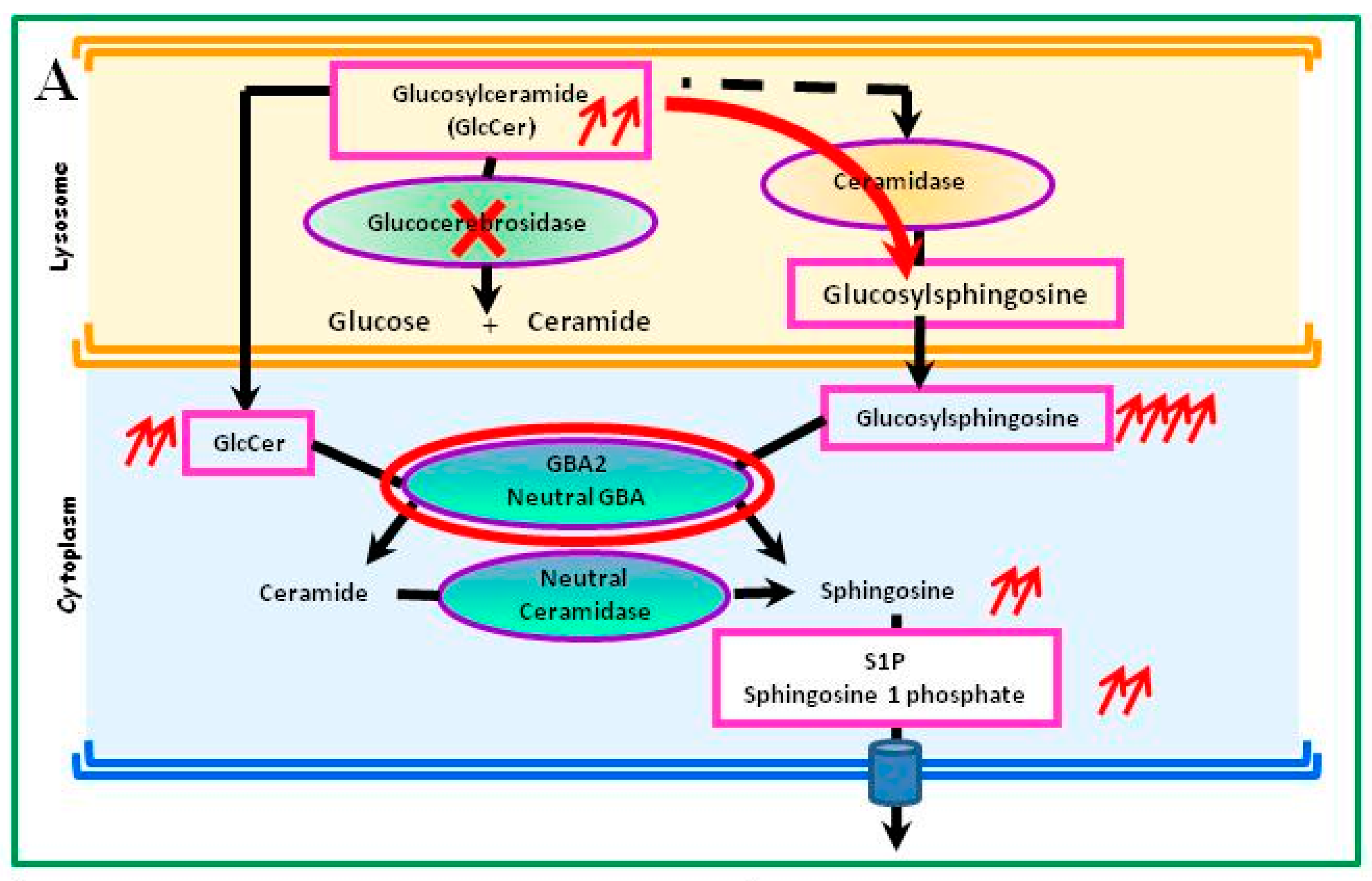
4.1. Glucosylceramide Accumulation
4.2. Subpopulation of Gaucher Cells, a Specific Cell Subpopulation
4.3. Metabolic Consequences Other Than Accumulation of Glucosylceramide in Gaucher Cells
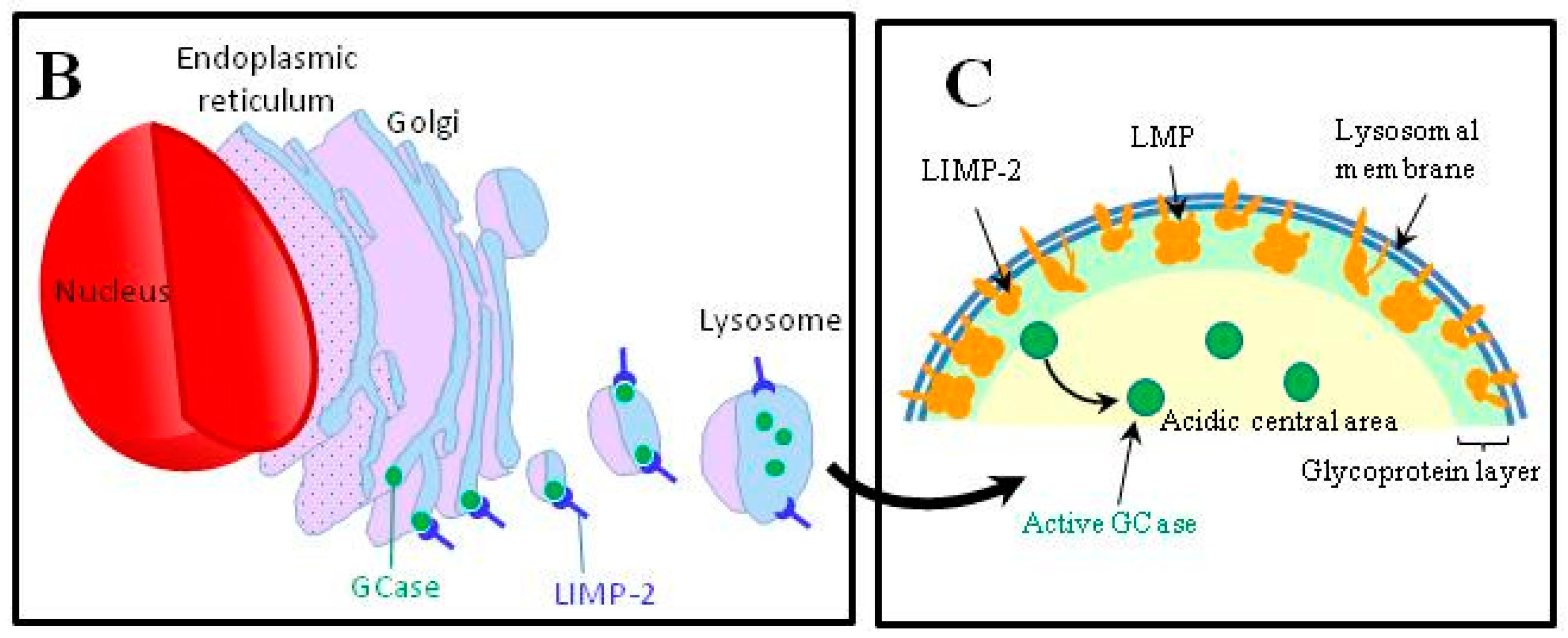
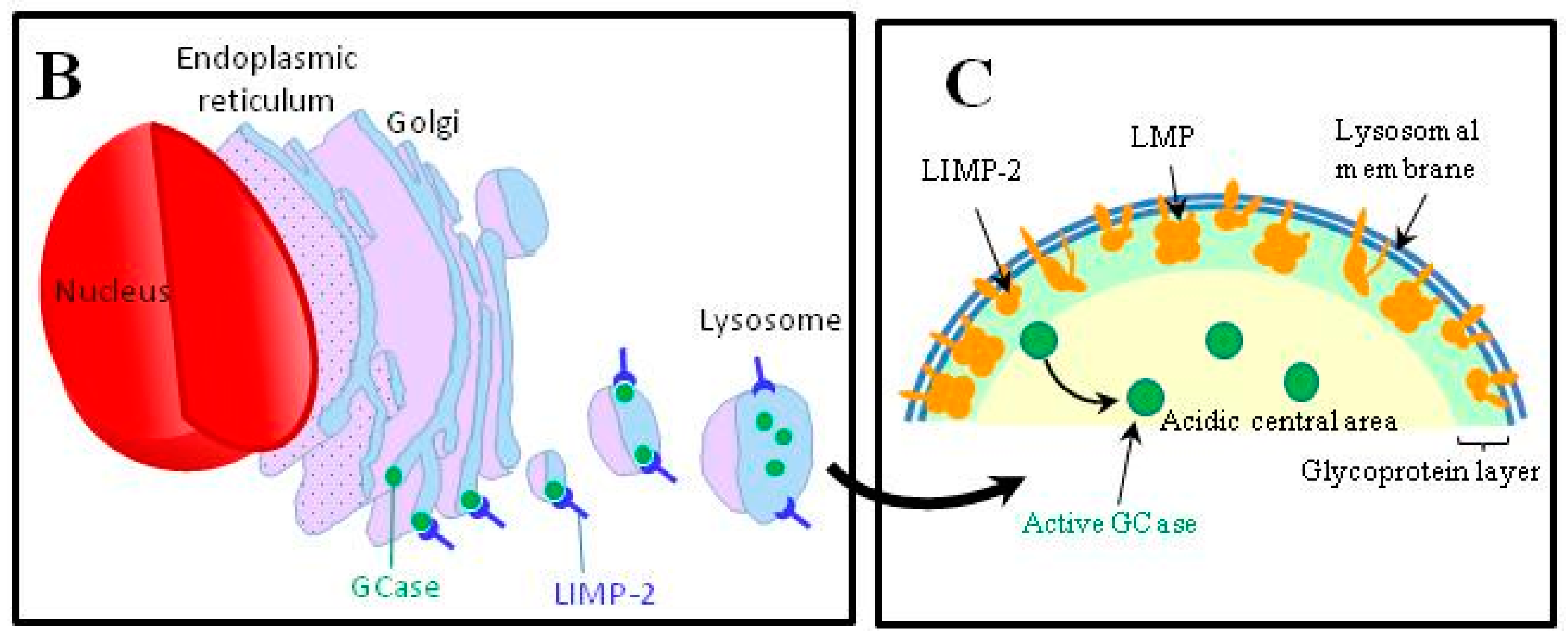
4.4. Abnormalities in the Intracellular Trafficking of Glucocerebrosidase
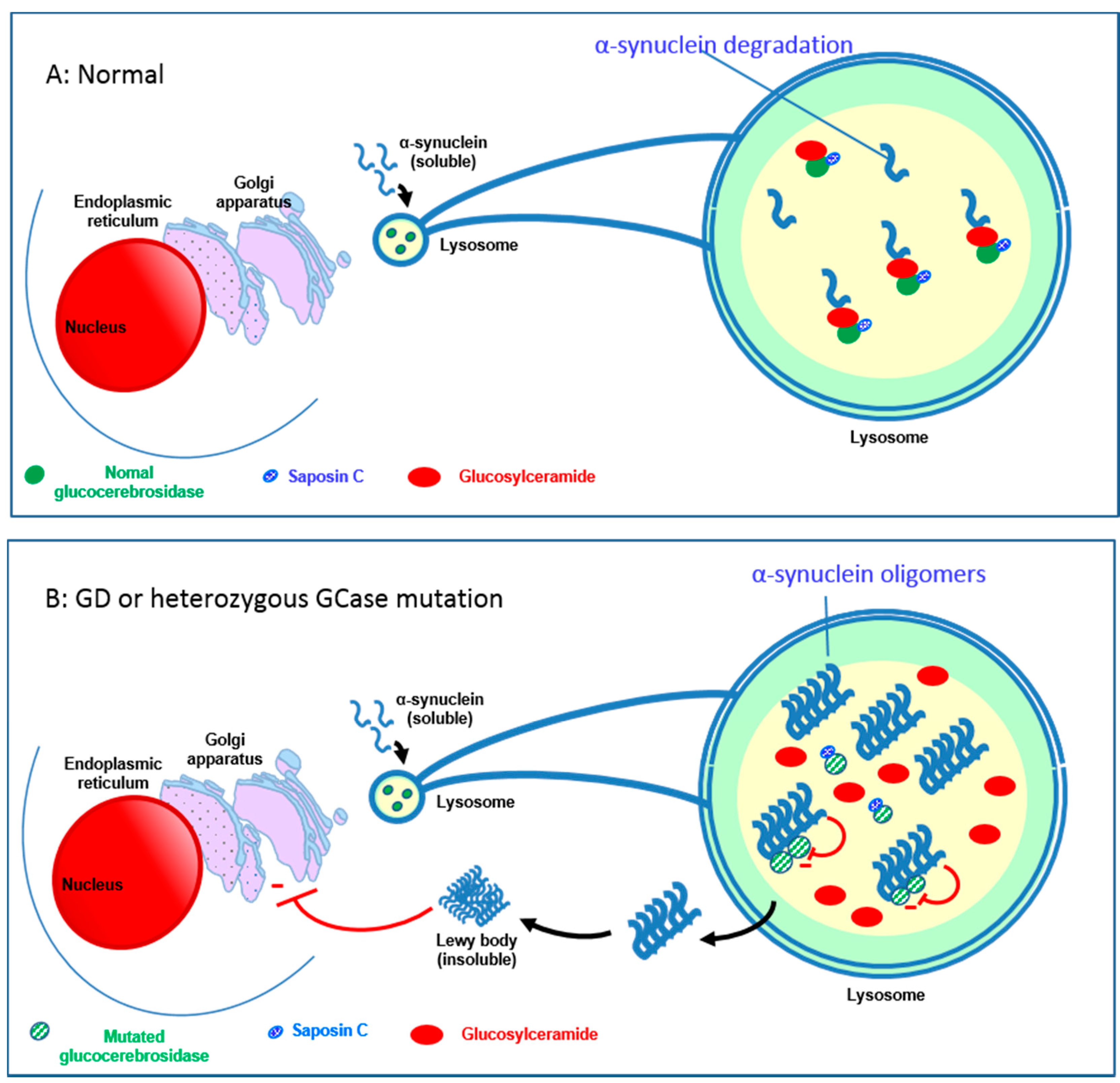
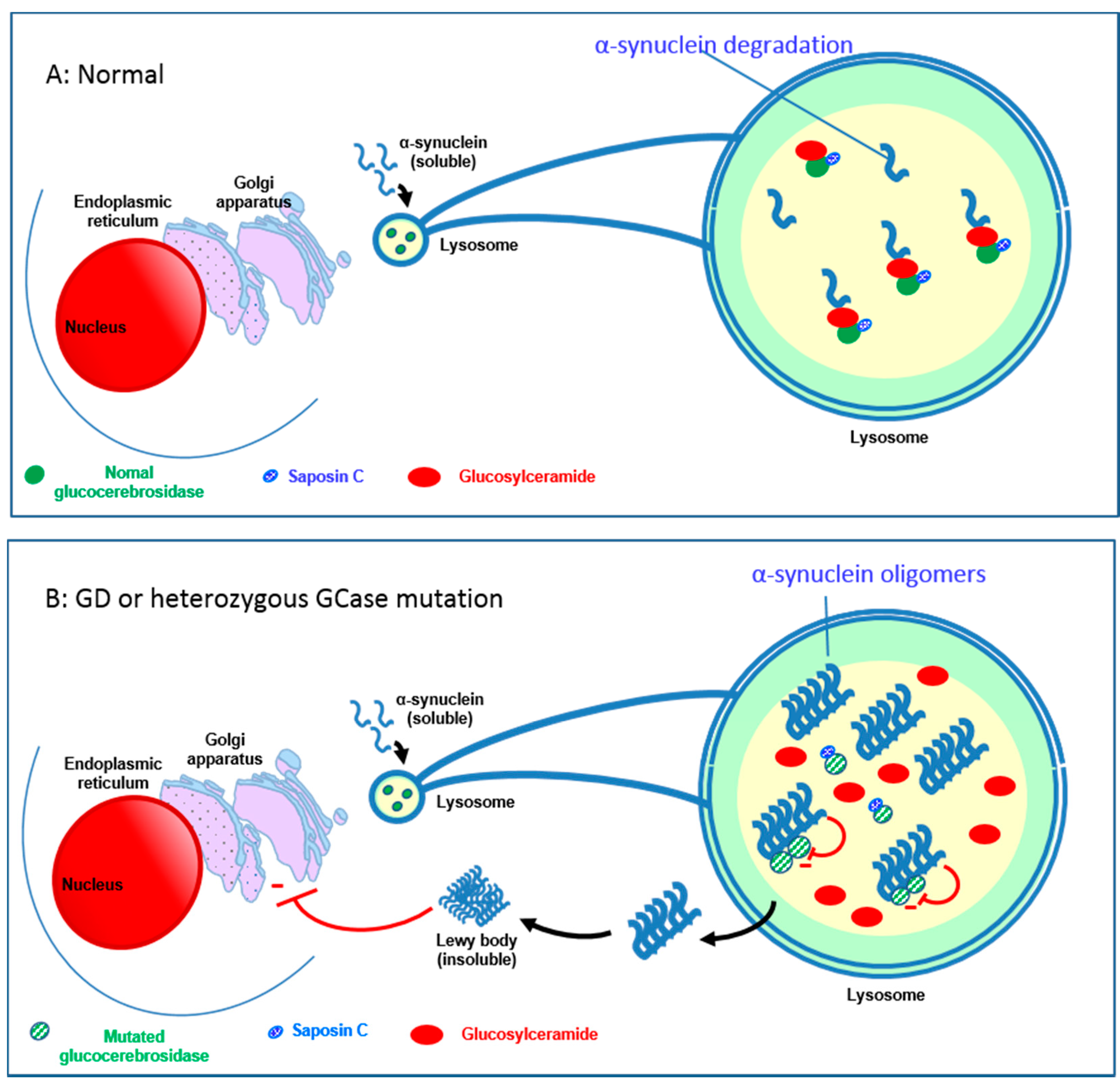
4.5. Relationship between the GBA1 Gene and Parkinson’s Disease
4.6. Relationship between GCase Deficiency and Neoplasia
4.7. Altered Iron Metabolism
5. Clinical Presentations
5.1. Type-1 Gaucher Disease (ORPHA77259)
5.2. Type-3 Gaucher Disease (ORPHA77261)
5.3. Type-2 Gaucher Disease (ORPHA77260)
6. Diagnosis of Gaucher Disease
6.1. GCase Activity
6.2. Bone Marrow Aspiration
6.3. GBA1 Mutations
6.4. Prenatal Diagnosis
7. Laboratory Abnormalities
7.1. Hemogram
7.2. Hemostasis
7.3. Proteinemia, Serum Immunofixation and Electrophoresis
7.4. Disease Biomarkers
7.5. Others Biological Tests
8. Radiological Investigations
9. Management
9.1. Usual Specific Treatments
9.1.1. Enzyme Repacement Therapy
9.1.2. Substrate Reduction Therapy
9.2. Other Specific Treatments
9.2.1. Gene Therapy
9.2.2. Molecular Chaperones
9.3. Symptomatic Treatments
10. Monitoring
11. Prognosis
12. Conclusions
Acknowledgments
Authors Contributions
Conflicts of Interest
Abbreviations
| AVN | Avascular Necrosis |
| ERT | Enzyme Replacement Therapy |
| GCase | glucocerebrosidase |
| GlcCer | glucosylceramide |
| GD | Gaucher Disease |
| SRT | Substrate Reduction Therapy |
References
- Ginzburg, L.; Kacher, Y.; Futerman, A.H. The pathogenesis of glycosphingolipid storage disorders. Semin. Cell Dev. Biol. 2004, 15, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, A.M.; Motta, M.; Tatti, M.; Scarpa, S.; Masuelli, L.; Bhat, M.; Vanier, M.T.; Tylki-Szymanska, A.; Salvioli, R. Saposin C mutations in Gaucher disease patients resulting in lysosomal lipid accumulation, saposin C deficiency, but normal prosaposin processing and sorting. Hum. Mol. Genet. 2010, 19, 2987–2997. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E. Gaucher disease: Insights from a rare Mendelian disorder. Discov. Med. 2012, 14, 273–281. [Google Scholar] [PubMed]
- Grabowski, G.A. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet 2008, 372, 1263–1271. [Google Scholar] [CrossRef]
- Stirnemann, J.; Vigan, M.; Hamroun, D.; Heraoui, D.; Rossi-Semerano, L.; Berger, M.G.; Rose, C.; Camou, F.; de Roux-Serratrice, C.; Grosbois, B.; et al. The French Gaucher’s disease registry: Clinical characteristics, complications and treatment of 562 patients. Orphanet J. Rare Dis. 2012, 7, 77. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.E. The fine structure of the cerebroside occurring in Gaucher’s disease. Proc. Natl. Acad. Sci. USA 1968, 61, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Mikosch, P.; Hughes, D. An overview on bone manifestations in Gaucher disease. Wiener Med. Wochenschr. 2010, 160, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Orvisky, E.; Park, J.K.; LaMarca, M.E.; Ginns, E.I.; Martin, B.M.; Tayebi, N.; Sidransky, E. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: Correlation with phenotype and genotype. Mol. Genet. Metab. 2002, 76, 262–270. [Google Scholar] [CrossRef]
- Kinghorn, K.J.; Gronke, S.; Castillo-Quan, J.I.; Woodling, N.S.; Li, L.; Sirka, E.; Gegg, M.; Mills, K.; Hardy, J.; Bjedov, I.; et al. A Drosophila model of neuronopathic Gaucher disease demonstrates lysosomal-autophagic defects and altered mTOR signalling and is functionally rescued by rapamycin. J. Neurosci. 2016, 36, 11654–11670. [Google Scholar] [CrossRef] [PubMed]
- Boven, L.A.; van Meurs, M.; Boot, R.G.; Mehta, A.; Boon, L.; Aerts, J.M.; Laman, J.D. Gaucher cells demonstrate a distinct macrophage phenotype and resemble alternatively activated macrophages. Am. J. Clin. Pathol. 2004, 122, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Mucci, J.M.; Cuello, M.F.; Kisinovsky, I.; Larroude, M.; Delpino, M.V.; Rozenfeld, P.A. Proinflammatory and proosteoclastogenic potential of peripheral blood mononuclear cells from Gaucher patients: Implication for bone pathology. Blood Cells Mol. Dis. 2015, 55, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Aflaki, E.; Moaven, N.; Borger, D.K.; Lopez, G.; Westbroek, W.; Chae, J.J.; Marugan, J.; Patnaik, S.; Maniwang, E.; Gonzalez, A.N.; et al. Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell 2016, 15, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.J.; Myer, B.J.; Khokher, A.M.; Rushton, N.; Cox, T.M. Pro-inflammatory cytokines and the pathogenesis of Gaucher’s disease: Increased release of interleukin-6 and interleukin-10. Mon. J. Assoc. Phys. 1997, 90, 19–25. [Google Scholar] [CrossRef]
- Barak, V.; Acker, M.; Nisman, B.; Kalickman, I.; Abrahamov, A.; Zimran, A.; Yatziv, S. Cytokines in Gaucher’s disease. Eur. Cytokine Netw. 1999, 10, 205–210. [Google Scholar]
- Hollak, C.E.; Evers, L.; Aerts, J.M.; van Oers, M.H. Elevated levels of M-CSF, sCD14 and IL8 in type 1 Gaucher disease. Blood Cells Mol. Dis. 1997, 23, 201–212. [Google Scholar] [CrossRef] [PubMed]
- van Breemen, M.J.; de Fost, M.; Voerman, J.S.; Laman, J.D.; Boot, R.G.; Maas, M.; Hollak, C.E.; Aerts, J.M.; Rezaee, F. Increased plasma macrophage inflammatory protein (MIP)-1α and MIP-1β levels in type 1 Gaucher disease. Biochim. Biophys. Acta 2007, 1772, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Liu, J.; Yang, M.; Nottoli, T.; McGrath, J.; Jain, D.; Zhang, K.; Keutzer, J.; Chuang, W.L.; Mehal, W.Z.; et al. Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc. Natl. Acad. Sci. USA 2010, 107, 19473–19478. [Google Scholar] [CrossRef] [PubMed]
- Dekker, N.; van Dussen, L.; Hollak, C.E.; Overkleeft, H.; Scheij, S.; Ghauharali, K.; van Breemen, M.J.; Ferraz, M.J.; Groener, J.E.; Maas, M.; et al. Elevated plasma glucosylsphingosine in Gaucher disease: Relation to phenotype, storage cell markers, and therapeutic response. Blood 2011, 118, e118–e127. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Liu, J.; Sun, L.; Chuang, W.L.; Yuen, T.; Yang, R.; Lu, P.; Zhang, K.; Li, J.; Keutzer, J.; et al. Glucocerebrosidase 2 gene deletion rescues type 1 Gaucher disease. Proc. Natl. Acad. Sci. USA 2014, 111, 4934–4939. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.B.; Kim, E.Y.; Jung, S.C. Down-regulation of Bcl-2 in the fetal brain of the Gaucher disease mouse model: A possible role in the neuronal loss. J. Hum. Genet. 2004, 49, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Rolfs, A.; Giese, A.K.; Grittner, U.; Mascher, D.; Elstein, D.; Zimran, A.; Bottcher, T.; Lukas, J.; Hubner, R.; Golnitz, U.; et al. Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS ONE 2013, 8, e79732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef]
- Lecourt, S.; Vanneaux, V.; Cras, A.; Freida, D.; Heraoui, D.; Herbi, L.; Caillaud, C.; Chomienne, C.; Marolleau, J.P.; Belmatoug, N.; et al. Bone marrow microenvironment in an in vitro model of Gaucher disease: Consequences of glucocerebrosidase deficiency. Stem Cells Dev. 2012, 21, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Campeau, P.M.; Rafei, M.; Boivin, M.N.; Sun, Y.; Grabowski, G.A.; Galipeau, J. Characterization of Gaucher disease bone marrow mesenchymal stromal cells reveals an altered inflammatory secretome. Blood 2009, 114, 3181–3190. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Lecourt, S.; Vanneaux, V.; Rapatel, C.; Boisgard, S.; Caillaud, C.; Boiret-Dupre, N.; Chomienne, C.; Marolleau, J.P.; Larghero, J.; et al. Glucocerebrosidase deficiency dramatically impairs human bone marrow haematopoiesis in an in vitro model of Gaucher disease. Br. J. Haematol. 2010, 150, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Taddei, T.H.; Dziura, J.; Chen, S.; Yang, R.; Hyogo, H.; Sullards, C.; Cohen, D.E.; Pastores, G.; Mistry, P.K. High incidence of cholesterol gallstone disease in type 1 Gaucher disease: Characterizing the biliary phenotype of type 1 Gaucher disease. J. Inherit. Metab. Dis. 2010, 33, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Ron, I.; Horowitz, M. ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum. Mol. Genet. 2005, 14, 2387–2398. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wang, H.; Zhu, D.; Hong, C.S.; Dmitriev, P.; Zhang, C.; Li, Y.; Ikejiri, B.; Brady, R.O.; Zhuang, Z. Mutant glucocerebrosidase in Gaucher disease recruits Hsp27 to the Hsp90 chaperone complex for proteasomal degradation. Proc. Natl. Acad. Sci. USA 2015, 112, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Reczek, D.; Schwake, M.; Schroder, J.; Hughes, H.; Blanz, J.; Jin, X.; Brondyk, W.; Van Patten, S.; Edmunds, T.; Saftig, P. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of β-glucocerebrosidase. Cell 2007, 131, 770–783. [Google Scholar]
- Berkovic, S.F.; Dibbens, L.M.; Oshlack, A.; Silver, J.D.; Katerelos, M.; Vears, D.F.; Lullmann-Rauch, R.; Blanz, J.; Zhang, K.W.; Stankovich, J.; et al. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am. J. Hum. Genet. 2008, 82, 673–684. [Google Scholar] [PubMed]
- Schroen, B.; Leenders, J.J.; van Erk, A.; Bertrand, A.T.; van Loon, M.; van Leeuwen, R.E.; Kubben, N.; Duisters, R.F.; Schellings, M.W.; Janssen, B.J.; et al. Lysosomal integral membrane protein 2 is a novel component of the cardiac intercalated disc and vital for load-induced cardiac myocyte hypertrophy. J. Exp. Med. 2007, 204, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Velayati, A.; DePaolo, J.; Gupta, N.; Choi, J.H.; Moaven, N.; Westbroek, W.; Goker-Alpan, O.; Goldin, E.; Stubblefield, B.K.; Kolodny, E.; et al. A mutation in SCARB2 is a modifier in Gaucher disease. Hum. Mutat. 2011, 32, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Jian, J.; Zhao, S.; Tian, Q.Y.; Liu, H.; Zhao, Y.; Chen, W.C.; Grunig, G.; Torres, P.A.; Wang, B.C.; Zeng, B.; et al. Association between progranulin and Gaucher disease. EBioMedicine 2016, 11, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Jian, J.; Tian, Q.Y.; Hettinghouse, A.; Zhao, S.; Liu, H.; Wei, J.; Grunig, G.; Zhang, W.; Setchell, K.D.; Sun, Y.; et al. Progranulin recruits HSP70 to β-Glucocerebrosidase and is therapeutic against Gaucher disease. EBioMedicine 2016, 13, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Petkau, T.L.; Leavitt, B.R. Progranulin in neurodegenerative disease. Trends Neurosci. 2014, 37, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Biegstraaten, M.; van Schaik, I.N.; Aerts, J.M.; Langeveld, M.; Mannens, M.M.; Bour, L.J.; Sidransky, E.; Tayebi, N.; Fitzgibbon, E.; Hollak, C.E. A monozygotic twin pair with highly discordant Gaucher phenotypes. Blood Cells Mol. Dis. 2011, 46, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Elstein, D.; Gellman, A.; Altarescu, G.; Abrahamov, A.; Hadas-Halpern, I.; Phillips, M.; Margalit, M.; Lebel, E.; Itzchaki, M.; Zimran, A. Disease severity in sibling pairs with type 1 Gaucher disease. J. Inherit. Metab. Dis. 2010, 33, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Valeiras, M.; Sidransky, E.; Tayebi, N. Lysosomal integral membrane protein-2: A new player in lysosome-related pathology. Mol. Genet. Metab. 2014, 111, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Aharon-Peretz, J.; Badarny, S.; Rosenbaum, H.; Gershoni-Baruch, R. Mutations in the glucocerebrosidase gene and Parkinson disease: Phenotype-genotype correlation. Neurology 2005, 65, 1460–1461. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.N.; Ross, B.M.; Wang, Y.; Mejia-Santana, H.; Harris, J.; Louis, E.D.; Cote, L.J.; Andrews, H.; Fahn, S.; Waters, C.; et al. Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology 2007, 69, 1270–1277. [Google Scholar] [CrossRef] [PubMed]
- Sato, C.; Morgan, A.; Lang, A.E.; Salehi-Rad, S.; Kawarai, T.; Meng, Y.; Ray, P.N.; Farrer, L.A.; St George-Hyslop, P.; Rogaeva, E. Analysis of the glucocerebrosidase gene in Parkinson’s disease. Mov. Disord. 2005, 20, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Patin, E.; Condroyer, C.; Leutenegger, A.L.; Lohmann, E.; Giladi, N.; Bar-Shira, A.; Belarbi, S.; Hecham, N.; Pollak, P.; et al. Parkinson’s disease-related LRRK2 G2019S mutation results from independent mutational events in humans. Hum. Mol. Genet. 2010, 19, 1998–2004. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Boot, B.; Locascio, J.J.; Jansen, I.E.; Winder-Rhodes, S.; Eberly, S.; Elbaz, A.; Brice, A.; Ravina, B.; van Hilten, J.J.; et al. Neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann. Neurol. 2016, 80, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Cilia, R.; Tunesi, S.; Marotta, G.; Cereda, E.; Siri, C.; Tesei, S.; Zecchinelli, A.L.; Canesi, M.; Mariani, C.B.; Meucci, N.; et al. Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann. Neurol. 2016, 80, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.L.; Gruschus, J.M.; Velayati, A.; Westbroek, W.; Goldin, E.; Moaven, N.; Sidransky, E.; Lee, J.C. α-synuclein interacts with Glucocerebrosidase providing a molecular link between Parkinson and Gaucher diseases. J. Biol. Chem. 2011, 286, 28080–28088. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain J. Neurol. 2014, 137, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.L.; Velayati, A.; Sidransky, E.; Lee, J.C. Membrane-bound α-synuclein interacts with glucocerebrosidase and inhibits enzyme activity. Mol. Genet. Metab. 2013, 108, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Gruschus, J.M.; Jiang, Z.; Yap, T.L.; Hill, S.A.; Grishaev, A.; Piszczek, G.; Sidransky, E.; Lee, J.C. Dissociation of glucocerebrosidase dimer in solution by its co-factor, saposin C. Biochem. Biophys. Res. Commun. 2015, 457, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Ron, I.; Rapaport, D.; Horowitz, M. Interaction between parkin and mutant glucocerebrosidase variants: A possible link between Parkinson disease and Gaucher disease. Hum. Mol. Genet. 2010, 19, 3771–3781. [Google Scholar] [CrossRef] [PubMed]
- Barkhuizen, M.; Anderson, D.G.; Grobler, A.F. Advances in GBA-associated Parkinson’s disease—Pathology, presentation and therapies. Neurochem. Int. 2016, 93, 6–25. [Google Scholar] [CrossRef] [PubMed]
- Cullen, V.; Sardi, S.P.; Ng, J.; Xu, Y.H.; Sun, Y.; Tomlinson, J.J.; Kolodziej, P.; Kahn, I.; Saftig, P.; Woulfe, J.; et al. Acid β-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alterα-synuclein processing. Ann. Neurol. 2011, 69, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.L.; Jiang, Z.; Heinrich, F.; Gruschus, J.M.; Pfefferkorn, C.M.; Barros, M.; Curtis, J.E.; Sidransky, E.; Lee, J.C. Structural features of membrane-bound glucocerebrosidase and α-synuclein probed by neutron reflectometry and fluorescence spectroscopy. J. Biol. Chem. 2015, 290, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Siebert, M.; Sidransky, E.; Westbroek, W. Glucocerebrosidase is shaking up the synucleinopathies. Brain J. Neurol. 2014, 137, 1304–1322. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.; Elstein, D.; Zimran, A.; Goker-Alpan, O. New directions in Gaucher disease. Hum. Mutat. 2016, 37, 1121–1136. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.L.; Gruschus, J.M.; Velayati, A.; Sidransky, E.; Lee, J.C. Saposin C protects glucocerebrosidase against α-synuclein inhibition. Biochemistry 2013, 52, 7161–7163. [Google Scholar] [CrossRef] [PubMed]
- Arends, M.; van Dussen, L.; Biegstraaten, M.; Hollak, C.E. Malignancies and monoclonal gammopathy in Gaucher disease; a systematic review of the literature. Br. J. Haematol. 2013, 161, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, B.E.; Weinreb, N.J.; Zimran, A.; Kacena, K.A.; Charrow, J.; Ward, E. Gaucher disease and cancer incidence: A study from the Gaucher Registry. Blood 2005, 105, 4569–4572. [Google Scholar] [CrossRef] [PubMed]
- Taddei, T.H.; Kacena, K.A.; Yang, M.; Yang, R.; Malhotra, A.; Boxer, M.; Aleck, K.A.; Rennert, G.; Pastores, G.M.; Mistry, P.K. The underrecognized progressive nature of N370S Gaucher disease and assessment of cancer risk in 403 patients. Am. J. Hematol. 2009, 84, 208–214. [Google Scholar] [CrossRef] [PubMed]
- De Fost, M.; Vom Dahl, S.; Weverling, G.J.; Brill, N.; Brett, S.; Haussinger, D.; Hollak, C.E. Increased incidence of cancer in adult Gaucher disease in Western Europe. Blood Cells Mol. Dis. 2006, 36, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Landgren, O.; Turesson, I.; Gridley, G.; Caporaso, N.E. Risk of malignant disease among 1525 adult male US Veterans with Gaucher disease. Arch. Intern. Med. 2007, 167, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Astudillo, L.; Therville, N.; Colacios, C.; Segui, B.; Andrieu-Abadie, N.; Levade, T. Glucosylceramidases and malignancies in mammals. Biochimie 2015, 125, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Braudeau, C.; Graveleau, J.; Rimbert, M.; Neel, A.; Hamidou, M.; Grosbois, B.; Besancon, A.; Giraudet, S.; Terrien, C.; Josien, R.; et al. Altered innate function of plasmacytoid dendritic cells restored by enzyme replacement therapy in Gaucher disease. Blood Cells Mol. Dis. 2013, 50, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Burstein, Y.; Zakuth, V.; Rechavi, G.; Spirer, Z. Abnormalities of cellular immunity and natural killer cells in Gaucher’s disease. J. Clin. Lab. Immunol. 1987, 23, 149–151. [Google Scholar] [PubMed]
- Pavlova, E.V.; Wang, S.Z.; Archer, J.; Dekker, N.; Aerts, J.M.; Karlsson, S.; Cox, T.M. B cell lymphoma and myeloma in murine Gaucher’s disease. J. Pathol. 2013, 231, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, E.V.; Archer, J.; Wang, S.; Dekker, N.; Aerts, J.M.; Karlsson, S.; Cox, T.M. Inhibition of UDP-glucosylceramide synthase in mice prevents Gaucher disease-associated B-cell malignancy. J. Pathol. 2015, 235, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Branagan, A.R.; Liu, J.; Boddupalli, C.S.; Mistry, P.K.; Dhodapkar, M.V. Clonal immunoglobulin against lysolipids in the origin of myeloma. N. Engl. J. Med. 2016, 374, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Medrano-Engay, B.; Irun, P.; Gervas-Arruga, J.; Andrade-Campos, M.; Andreu, V.; Alfonso, P.; Pocovi, M.; Giraldo, P. Iron homeostasis and infIammatory biomarker analysis in patients with type 1 Gaucher disease. Blood Cells Mol. Dis. 2014, 53, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Stirnemann, J.; Boutten, A.; Vincent, C.; Mekinian, A.; Heraoui, D.; Fantin, B.; Fain, O.; Mentre, F.; Belmatoug, N. Impact of imiglucerase on the serum glycosylated-ferritin level in Gaucher disease. Blood Cells Mol. Dis. 2011, 46, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Stirnemann, J.; Belmatoug, N.; Vincent, C.; Fain, O.; Fantin, B.; Mentre, F. Bone events and evolution of biologic markers in Gaucher disease before and during treatment. Arthritis Res. Ther. 2010, 12, R156. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E. Gaucher disease: Complexity in a “simple” disorder. Mol. Genet. Metab. 2004, 83, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Charrow, J.; Andersson, H.C.; Kaplan, P.; Kolodny, E.H.; Mistry, P.; Pastores, G.; Rosenbloom, B.E.; Scott, C.R.; Wappner, R.S.; Weinreb, N.J.; et al. The Gaucher registry: Demographics and disease characteristics of 1698 patients with Gaucher disease. Arch. Intern. Med. 2000, 160, 2835–2843. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, P.; Andersson, H.C.; Kacena, K.A.; Yee, J.D. The clinical and demographic characteristics of nonneuronopathic Gaucher disease in 887 children at diagnosis. Arch. Pediatrics Adolesc. Med. 2006, 160, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A.; Zimran, A.; Ida, H. Gaucher disease types 1 and 3: Phenotypic characterization of large populations from the ICGG Gaucher Registry. Am. J. Hematol. 2015, 90 (Suppl. S1), S12–S18. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.C.; Reinig, J.W.; Barranger, J.A.; Fink, J.; Shawker, T.H. Gaucher disease: Sonographic appearance of the spleen. Radiology 1986, 160, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Neudorfer, O.; Hadas-Halpern, I.; Elstein, D.; Abrahamov, A.; Zimran, A. Abdominal ultrasound findings mimicking hematological malignancies in a study of 218 Gaucher patients. Am. J. Hematol. 1997, 55, 28–34. [Google Scholar] [CrossRef]
- Regenboog, M.; Bohte, A.E.; Somers, I.; van Delden, O.M.; Maas, M.; Hollak, C.E. Imaging characteristics of focal splenic and hepatic lesions in type 1 Gaucher disease. Blood Cells Mol. Dis. 2016, 60, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, H. Hemorrhagic aspects of Gaucher disease. Rambam Maimonides Med. J. 2014, 5, e0039. [Google Scholar] [CrossRef] [PubMed]
- Gillis, S.; Hyam, E.; Abrahamov, A.; Elstein, D.; Zimran, A. Platelet function abnormalities in Gaucher disease patients. Am. J. Hematol. 1999, 61, 103–106. [Google Scholar] [CrossRef]
- Wenstrup, R.J.; Roca-Espiau, M.; Weinreb, N.J.; Bembi, B. Skeletal aspects of Gaucher disease: A review. Br. J. Radiol. 2002, 75 (Suppl. S1), A2–A12. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.A.; Hollak, C.E. The clinical spectrum and pathophysiology of skeletal complications in lysosomal storage disorders. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Zimran, A.; Bembi, B.; Kanis, J.; Reginster, J.Y.; Rizzoli, R.; Cooper, C.; Brandi, M.L. Gaucher disease and bone manifestations. Calcif. Tissue Int. 2014, 95, 477–494. [Google Scholar] [CrossRef] [PubMed]
- Yossipovitch, Z.H.; Herman, G.; Makin, M. Aseptic osteomyelitis in Gaucher’s disease. Isr. J. Med. Sci. 1965, 1, 531–536. [Google Scholar] [PubMed]
- Pastores, G.M.; Wallenstein, S.; Desnick, R.J.; Luckey, M.M. Bone density in Type 1 Gaucher disease. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 1996, 11, 1801–1807. [Google Scholar] [CrossRef] [PubMed]
- Deegan, P.B.; Pavlova, E.; Tindall, J.; Stein, P.E.; Bearcroft, P.; Mehta, A.; Hughes, D.; Wraith, J.E.; Cox, T.M. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine 2011, 90, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Poll, L.W.; Koch, J.A.; vom Dahl, S.; Loxtermann, E.; Sarbia, M.; Niederau, C.; Haussinger, D.; Modder, U. Extraosseous manifestation of Gaucher’s disease type I: MR and histological appearance. Eur. Radiol. 2000, 10, 1660–1663. [Google Scholar] [CrossRef] [PubMed]
- Zver, S.; Bracko, M.; Andoljsek, D. Primary bone angiosarcoma in a patient with Gaucher disease. Int. J. Hematol. 2010, 92, 374–377. [Google Scholar] [CrossRef]
- Kenan, S.; Abdelwahab, I.F.; Hermann, G.; Klein, M.; Pastores, G. Osteoblastoma of the humerus associated with type-I Gaucher’s disease. A case report. J. Bone Joint Surg. Br. Vol. 1996, 78, 702–705. [Google Scholar]
- Bembi, B.; Ciana, G.; Mengel, E.; Terk, M.R.; Martini, C.; Wenstrup, R.J. Bone complications in children with Gaucher disease. Br. J. Radiol. 2002, 75 (Suppl. S1), A37–A44. [Google Scholar] [CrossRef] [PubMed]
- Faden, M.A.; Krakow, D.; Ezgu, F.; Rimoin, D.L.; Lachman, R.S. The Erlenmeyer flask bone deformity in the skeletal dysplasias. Am. J. Med. Genet. Part A 2009, 149A, 1334–1345. [Google Scholar] [CrossRef] [PubMed]
- Amir, G.; Ron, N. Pulmonary pathology in Gaucher’s disease. Hum. Pathol. 1999, 30, 666–670. [Google Scholar] [CrossRef]
- Mistry, P.K.; Sirrs, S.; Chan, A.; Pritzker, M.R.; Duffy, T.P.; Grace, M.E.; Meeker, D.P.; Goldman, M.E. Pulmonary hypertension in type 1 Gaucher’s disease: Genetic and epigenetic determinants of phenotype and response to therapy. Mol. Genet. Metab. 2002, 77, 91–98. [Google Scholar] [CrossRef]
- Santamaria, F.; Parenti, G.; Guidi, G.; Filocamo, M.; Strisciuglio, P.; Grillo, G.; Farina, V.; Sarnelli, P.; Rizzolo, M.G.; Rotondo, A.; et al. Pulmonary manifestations of Gaucher disease: An increased risk for L444P homozygotes? Am. J. Respir. Critic. Care Med. 1998, 157, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Santoro, D.; Rosenbloom, B.E.; Cohen, A.H. Gaucher disease with nephrotic syndrome: Response to enzyme replacement therapy. Am. J. Kidney Dis. 2002, 40, E4. [Google Scholar] [CrossRef] [PubMed]
- Goldblatt, J.; Beighton, P. Cutaneous manifestations of Gaucher disease. Br. J. Dermatol. 1984, 111, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Raz, J.; Anteby, I.; Livni, N.; Benezra, D. Chronic uveitis in Gaucher’s disease. Ocular Immunol. Inflamm. 1993, 1, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Bruscolini, A.; Pirraglia, M.P.; Restivo, L.; Spinucci, G.; Abbouda, A. A branch retinal artery occlusion in a patient with Gaucher disease. Graefes Arch. Clin. Exp. Ophthalmol. 2012, 250, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Petrohelos, M.; Tricoulis, D.; Kotsiras, I.; Vouzoukos, A. Ocular manifestations of Gaucher’s disease. Am. J. Ophthalmol. 1975, 80, 1006–1010. [Google Scholar] [CrossRef]
- Roghi, A.; Poggiali, E.; Cassinerio, E.; Pedrotti, P.; Giuditta, M.; Milazzo, A.; Quattrocchi, G.; Cappellini, M.D. The role of cardiac magnetic resonance to assess the cardiac involvement in Gaucher type 1 patients: Morphological and functional evaluations. J. Cardiovasc. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Langeveld, M.; de Fost, M.; Aerts, J.M.; Sauerwein, H.P.; Hollak, C.E. Overweight, insulin resistance and type II diabetes in type I Gaucher disease patients in relation to enzyme replacement therapy. Blood Cells Mol. Dis. 2008, 40, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Elstein, D.; Rosenmann, E.; Reinus, C.; Paz, J.; Altarescu, G.; Zimran, A. Amyloidosis and gastric bleeding in a patient with Gaucher disease. J. Clin. Gastroenterol. 2003, 37, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Hrebicek, M.; Zeman, J.; Musilova, J.; Hodanova, K.; Renkema, G.H.; Veprekova, L.; Ledvinova, J.; Hrebicek, D.; Sokolova, J.; Aerts, J.M.; et al. A case of type I Gaucher disease with cardiopulmonary amyloidosis and chitotriosidase deficiency. Virchows Arch. Int. J. Pathol. 1996, 429, 305–309. [Google Scholar] [CrossRef]
- Hanash, S.M.; Rucknagel, D.L.; Heidelberger, K.P.; Radin, N.S. Primary amyloidosis associated with Gaucher’s disease. Ann. Intern. Med. 1978, 89, 639–641. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Dinur, T.; Quinn, T.; Sakanaka, K.; Levy, O.; Waters, C.; Fahn, S.; Dorovski, T.; Chung, W.K.; Pauciulo, M.; et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol. 2014, 71, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Bultron, G.; Kacena, K.; Pearson, D.; Boxer, M.; Yang, R.; Sathe, S.; Pastores, G.; Mistry, P.K. The risk of Parkinson’s disease in type 1 Gaucher disease. J. Inherit. Metab. Dis. 2010, 33, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Biegstraaten, M.; Mengel, E.; Marodi, L.; Petakov, M.; Niederau, C.; Giraldo, P.; Hughes, D.; Mrsic, M.; Mehta, A.; Hollak, C.E.; et al. Peripheral neuropathy in adult type 1 Gaucher disease: A 2-year prospective observational study. Brain J. Neurol. 2010, 133, 2909–2919. [Google Scholar] [CrossRef] [PubMed]
- Tajima, A.; Yokoi, T.; Ariga, M.; Ito, T.; Kaneshiro, E.; Eto, Y.; Ida, H. Clinical and genetic study of Japanese patients with type 3 Gaucher disease. Mol. Genet. Metab. 2009, 97, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Tylki-Szymanska, A.; Vellodi, A.; El-Beshlawy, A.; Cole, J.A.; Kolodny, E. Neuronopathic Gaucher disease: Demographic and clinical features of 131 patients enrolled in the International Collaborative Gaucher Group Neurological Outcomes Subregistry. J. Inherit. Metab. Dis. 2010, 33, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Kraoua, I.; Sedel, F.; Caillaud, C.; Froissart, R.; Stirnemann, J.; Chaurand, G.; Flodrops, H.; Tari, S.; Gourfinkel-An, I.; Mathieu, S.; et al. A French experience of type 3 Gaucher disease: Phenotypic diversity and neurological outcome of 10 patients. Brain Dev. 2011, 33, 131–139. [Google Scholar] [CrossRef]
- Abdelwahab, M.; Blankenship, D.; Schiffmann, R. Long-term follow-up and sudden unexpected death in Gaucher disease type 3 in Egypt. Neurol. Genet. 2016, 2, e55. [Google Scholar] [CrossRef] [PubMed]
- George, R.; McMahon, J.; Lytle, B.; Clark, B.; Lichtin, A. Severe valvular and aortic arch calcification in a patient with Gaucher’s disease homozygous for the D409H mutation. Clin. Genet. 2001, 59, 360–363. [Google Scholar] [CrossRef]
- Cindik, N.; Ozcay, F.; Suren, D.; Akkoyun, I.; Gokdemir, M.; Varan, B.; Alehan, F.; Ozbek, N.; Tokel, K. Gaucher disease with communicating hydrocephalus and cardiac involvement. Clin. Cardiol. 2010, 33, E26–E30. [Google Scholar] [CrossRef]
- Tamargo, R.J.; Velayati, A.; Goldin, E.; Sidransky, E. The role of saposin C in Gaucher disease. Mol. Genet. Metab. 2012, 106, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Mignot, C.; Doummar, D.; Maire, I.; De Villemeur, T.B.; French Type 2 Gaucher Disease Study Group. Type 2 Gaucher disease: 15 new cases and review of the literature. Brain Dev. 2006, 28, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Mignot, C.; Gelot, A.; De Villemeur, T.B. Gaucher disease. Handb. Clin. Neurol. 2013, 113, 1709–1715. [Google Scholar] [PubMed]
- Mignot, C.; Gelot, A.; Bessieres, B.; Daffos, F.; Voyer, M.; Menez, F.; Fallet Bianco, C.; Odent, S.; Le Duff, D.; Loget, P.; et al. Perinatal-lethal Gaucher disease. Am. J. Med. Genet. Part A 2003, 120A, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.F. Lysosomal storage diseases. Annu. Rev. Biochem. 1991, 60, 257–280. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Stirnemann, J.; Bourgne, C.; Pereira, B.; Pigeon, P.; Heraoui, D.; Froissart, R.; Rapatel, C.; Rose, C.; Belmatoug, N.; et al. The uptake of recombinant glucocerebrosidases by blood monocytes from type 1 Gaucher disease patients is variable. Br. J. Haematol. 2012, 157, 274–277. [Google Scholar] [CrossRef]
- Costello, R.; O’Callaghan, T.; Sebahoun, G. Gaucher disease and multiple myeloma. Leukemia Lymphoma 2006, 47, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Robak, T.; Urbanska-Rys, H.; Jerzmanowski, P.; Bartkowiak, J.; Liberski, P.; Kordek, R. Lymphoplasmacytic lymphoma with monoclonal gammopathy-related pseudo-Gaucher cell infiltration in bone marrow and spleen—Diagnostic and therapeutic dilemmas. Leukemia Lymphoma 2002, 43, 2343–2350. [Google Scholar]
- Yang, H.S.; Cho, K.S.; Park, T.S. Chronic myeloid leukemia with marked splenomegaly and pseudo-Gaucher cells. Blood Res. 2013, 48, 241. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.J.; Jones, R.D. Pseudo-Gaucher cells in myelodysplasia. J. Clin. Pathol. 1999, 52, 917–918. [Google Scholar] [CrossRef] [PubMed]
- Busarla, S.V.; Sadruddin, F.A.; Sohani, A.R. Pseudo-Gaucher cells in disseminated mycobacterial infection. Am. J. Hematol. 2013, 88, 155. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.; Wilder, S.; Horowitz, Z.; Reiner, O.; Gelbart, T.; Beutler, E. The human glucocerebrosidase gene and pseudogene: Structure and evolution. Genomics 1989, 4, 87–96. [Google Scholar] [CrossRef]
- Koprivica, V.; Stone, D.L.; Park, J.K.; Callahan, M.; Frisch, A.; Cohen, I.J.; Tayebi, N.; Sidransky, E. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am. J. Hum. Genet. 2000, 66, 1777–1786. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Kido, J.; Matsumoto, S.; Momosaki, K.; Mitsubuchi, H.; Shimazu, T.; Sugawara, K.; Endo, F.; Nakamura, K. Prenatal diagnosis of Gaucher disease using next-generation sequencing. Pediatr. Int. 2016, 58, 946–949. [Google Scholar] [CrossRef] [PubMed]
- Hollak, C.E.; Belmatoug, N.; Cole, J.A.; Vom Dahl, S.; Deegan, P.B.; Goldblatt, J.; Rosenbloom, B.; van Dussen, L.; Tylki-Szymanska, A.; Weinreb, N.J.; et al. Characteristics of type I Gaucher disease associated with persistent thrombocytopenia after treatment with imiglucerase for 4–5 years. Br. J. Haematol. 2012, 158, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Mitrovic, M.; Elezovic, I.; Miljic, P.; Suvajdzic, N. Acquired von Willebrand syndrome in patients with Gaucher disease. Blood Cells Mol. Dis. 2014, 52, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Spectre, G.; Roth, B.; Ronen, G.; Rosengarten, D.; Elstein, D.; Zimran, A.; Varon, D.; Revel-Vilk, S. Platelet adhesion defect in type I Gaucher Disease is associated with a risk of mucosal bleeding. Br. J. Haematol. 2011, 153, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Grosbois, B.; Rose, C.; Noel, E.; Serratrice Cde, R.; Dobbelaere, D.; Gressin, V.; Cherin, P.; Hartmann, A.; Javier, R.M.; Clerson, P.; et al. Gaucher disease and monoclonal gammopathy: A report of 17 cases and impact of therapy. Blood Cells Mol. Dis. 2009, 43, 138–139. [Google Scholar] [CrossRef] [PubMed]
- De Fost, M.; Out, T.A.; de Wilde, F.A.; Tjin, E.P.; Pals, S.T.; van Oers, M.H.; Boot, R.G.; Aerts, J.F.; Maas, M.; Vom Dahl, S.; et al. Immunoglobulin and free light chain abnormalities in Gaucher disease type I: Data from an adult cohort of 63 patients and review of the literature. Ann. Hematol. 2008, 87, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Brautbar, A.; Elstein, D.; Pines, G.; Abrahamov, A.; Zimran, A. Effect of enzyme replacement therapy on gammopathies in Gaucher disease. Blood Cells Mol. Dis. 2004, 32, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Hollak, C.E.; van Weely, S.; van Oers, M.H.; Aerts, J.M. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J. Clin. Investig. 1994, 93, 1288–1292. [Google Scholar] [CrossRef] [PubMed]
- Van Dussen, L.; Hendriks, E.J.; Groener, J.E.; Boot, R.G.; Hollak, C.E.; Aerts, J.M. Value of plasma chitotriosidase to assess non-neuronopathic Gaucher disease severity and progression in the era of enzyme replacement therapy. J. Inher. Metab. Dis. 2014, 37, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Bussink, A.P.; Verhoek, M.; Vreede, J.; Ghauharali-van der Vlugt, K.; Donker-Koopman, W.E.; Sprenger, R.R.; Hollak, C.E.; Aerts, J.M.; Boot, R.G. Common G102S polymorphism in chitotriosidase differentially affects activity towards 4-methylumbelliferyl substrates. FEBS J. 2009, 276, 5678–5688. [Google Scholar] [CrossRef] [PubMed]
- Bargagli, E.; Bennett, D.; Maggiorelli, C.; Di Sipio, P.; Margollicci, M.; Bianchi, N.; Rottoli, P. Human chitotriosidase: A sensitive biomarker of sarcoidosis. J. Clin. Immunol. 2013, 33, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, B.; Ghauharali-van der Vlugt, K.; Helmond, M.T.; Out, J.M.; Donker-Koopman, W.E.; Groener, J.E.; Boot, R.G.; Renkema, G.H.; van der Marel, G.A.; van Boom, J.H.; et al. Transglycosidase activity of chitotriosidase: Improved enzymatic assay for the human macrophage chitinase. J. Biol. Chem. 2003, 278, 40911–40916. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.A.; Ling, M.F.; Leung, J.; Shreffler, W.G.; Luster, A.D. Identification of human CCR8 as a CCL18 receptor. J. Exp. Med. 2013, 210, 1889–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonella, F.; Costabel, U. Biomarkers in connective tissue disease-associated interstitial lung disease. Semin. Respir. Crit. Care Med. 2014, 35, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Tsicopoulos, A.; Chang, Y.; Ait Yahia, S.; de Nadai, P.; Chenivesse, C. Role of CCL18 in asthma and lung immunity. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2013, 43, 716–722. [Google Scholar] [CrossRef]
- Boot, R.G.; Verhoek, M.; de Fost, M.; Hollak, C.E.; Maas, M.; Bleijlevens, B.; van Breemen, M.J.; van Meurs, M.; Boven, L.A.; Laman, J.D.; et al. Marked elevation of the chemokine CCL18/PARC in Gaucher disease: A novel surrogate marker for assessing therapeutic intervention. Blood 2004, 103, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Deegan, P.B.; Moran, M.T.; McFarlane, I.; Schofield, J.P.; Boot, R.G.; Aerts, J.M.; Cox, T.M. Clinical evaluation of chemokine and enzymatic biomarkers of Gaucher disease. Blood Cells Mol. Dis. 2005, 35, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.; Szer, J.; Stark, S.; Fletcher, J.M. Rapid, single-phase extraction of glucosylsphingosine from plasma: A universal screening and monitoring tool. Clin. Chim. Acta Int. J. Clin. Chem. 2015, 450, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Mirzaian, M.; Wisse, P.; Ferraz, M.J.; Gold, H.; Donker-Koopman, W.E.; Verhoek, M.; Overkleeft, H.S.; Boot, R.G.; Kramer, G.; Dekker, N.; et al. Mass spectrometric quantification of glucosylsphingosine in plasma and urine of type 1 Gaucher patients using an isotope standard. Blood Cells Mol. Dis. 2015, 54, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, V.; Chuang, W.L.; Liu, J.; Lischuk, A.; Kacena, K.; Lin, H.; Pastores, G.M.; Yang, R.; Keutzer, J.; Zhang, K.; et al. Glucosylsphingosine is a key biomarker of Gaucher disease. Am. J. Hematol. 2016, 91, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Mekinian, A.; Stirnemann, J.; Belmatoug, N.; Heraoui, D.; Fantin, B.; Fain, O.; Charpentier, A.; Rose, C. Ferritinemia during type 1 Gaucher disease: Mechanisms and progression under treatment. Blood Cells Mol. Dis. 2012, 49, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.; Yu, H.; Jain, D.; Mistry, P.K. Hyperferritinemia and iron overload in type 1 Gaucher disease. Am. J. Hematol. 2010, 85, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.; Kallemeijn, W.W.; Wegdam, W.; Joao Ferraz, M.; van Breemen, M.J.; Dekker, N.; Kramer, G.; Poorthuis, B.J.; Groener, J.E.; Cox-Brinkman, J.; et al. Biomarkers in the diagnosis of lysosomal storage disorders: Proteins, lipids, and inhibodies. J. Inher. Metab. Dis. 2011, 34, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.; Cappellini, M.D.; Berger, M.; Van Droogenbroeck, J.; de Fost, M.; Janic, D.; Marinakis, T.; Rosenbaum, H.; Villarubia, J.; Zhukovskaya, E.; et al. Recommendations for the management of the haematological and onco-haematological aspects of Gaucher disease. Br. J. Haematol. 2007, 138, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Sellos-Moura, M.; Barzegar, S.; Pan, L.; Shi, P.; Oommen, S.; Durant, J.; Ruiz, J.A. Development of a panel of highly sensitive, equivalent assays for detection of antibody responses to velaglucerase alfa or imiglucerase enzyme replacement therapy in patients with Gaucher disease. J. Immunol. Methods 2011, 373, 45–53. [Google Scholar] [CrossRef] [PubMed]
- De Fost, M.; Langeveld, M.; Franssen, R.; Hutten, B.A.; Groener, J.E.; de Groot, E.; Mannens, M.M.; Bikker, H.; Aerts, J.M.; Kastelein, J.J.; et al. Low HDL cholesterol levels in type I Gaucher disease do not lead to an increased risk of cardiovascular disease. Atherosclerosis 2009, 204, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Elstein, D.; Tiomkin, M.; Hadas-Halpern, I.; Zimran, A. Organ volume by computed tomography correlates with longitudinal axis on ultrasound in patients with Gaucher disease. Ultrasound Q. 2011, 27, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Vom Dahl, S.; Poll, L.; Di Rocco, M.; Ciana, G.; Denes, C.; Mariani, G.; Maas, M. Evidence-based recommendations for monitoring bone disease and the response to enzyme replacement therapy in Gaucher patients. Curr. Med. Res. Opin. 2006, 22, 1045–1064. [Google Scholar] [CrossRef] [PubMed]
- Maas, M.; van Kuijk, C.; Stoker, J.; Hollak, C.E.; Akkerman, E.M.; Aerts, J.F.; den Heeten, G.J. Quantification of bone involvement in Gaucher disease: MR imaging bone marrow burden score as an alternative to Dixon quantitative chemical shift MR imaging—Initial experience. Radiology 2003, 229, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Fedida, B.; Touraine, S.; Stirnemann, J.; Belmatoug, N.; Laredo, J.D.; Petrover, D. Bone marrow involvement in Gaucher disease at MRI: What long-term evolution can we expect under enzyme replacement therapy? Eur. Radiol. 2015, 25, 2969–2975. [Google Scholar] [CrossRef] [PubMed]
- Hollak, C.; Maas, M.; Akkerman, E.; den Heeten, A.; Aerts, H. Dixon quantitative chemical shift imaging is a sensitive tool for the evaluation of bone marrow responses to individualized doses of enzyme supplementation therapy in type 1 Gaucher disease. Blood Cells Mol. Dis. 2001, 27, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Maas, M.; Poll, L.W.; Terk, M.R. Imaging and quantifying skeletal involvement in Gaucher disease. Br. J. Radiol. 2002, 75 (Suppl. S1), A13–A24. [Google Scholar] [CrossRef]
- Mikosch, P.; Zitter, F.; Gallowitsch, H.J.; Wurtz, F.; Lind, P.; Mehta, A.B.; Hughes, D.A. Bone- and bone marrow scintigraphy in Gaucher disease type 1. Nukl. Nucl. Med. 2008, 47, N39–N43. [Google Scholar]
- Goker-Alpan, O. Therapeutic approaches to bone pathology in Gaucher disease: Past, present and future. Mol. Genet. Metab. 2011, 104, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Andersson, H.C.; Charrow, J.; Kaplan, P.; Mistry, P.; Pastores, G.M.; Prakash-Cheng, A.; Rosenbloom, B.E.; Scott, C.R.; Wappner, R.S.; Weinreb, N.J.; et al. Individualization of long-term enzyme replacement therapy for Gaucher disease. Genet. Med. 2005, 7, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Pastores, G.M.; Weinreb, N.J.; Aerts, H.; Andria, G.; Cox, T.M.; Giralt, M.; Grabowski, G.A.; Mistry, P.K.; Tylki-Szymanska, A. Therapeutic goals in the treatment of Gaucher disease. Sem. Hematol. 2004, 41, 4–14. [Google Scholar] [CrossRef]
- Beutler, E.; Demina, A.; Laubscher, K.; Garver, P.; Gelbart, T.; Balicki, D.; Vaughan, L. The clinical course of treated and untreated Gaucher disease. A study of 45 patients. Blood Cells Mol. Dis. 1995, 21, 86–108. [Google Scholar] [CrossRef] [PubMed]
- Zimran, A.; Hadas-Halpern, I.; Zevin, S.; Levy-Lahad, E.; Abrahamov, A. Low-dose high-frequency enzyme replacement therapy for very young children with severe Gaucher disease. Br. J. Haematol. 1993, 85, 783–786. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.L.; Rosenbloom, B.E.; Kay, A.C.; Garver, P.; Thurston, D.W.; Koziol, J.A.; Gelbart, T.; Beutler, E. A less costly regimen of alglucerase to treat Gaucher’s disease. N. Engl. J. Med. 1992, 327, 1632–1636. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Spearing, R.; Teague, L.; Robertson, P.; Blacklock, H. The outcome of clinical parameters in adults with severe Type I Gaucher disease using very low dose enzyme replacement therapy. Mol. Genet. Metab. 2007, 92, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.J.; Katz, K.; Kornreich, L.; Horev, G.; Frish, A.; Zaizov, R. Low-dose high-frequency enzyme replacement therapy prevents fractures without complete suppression of painful bone crises in patients with severe juvenile onset type I Gaucher disease. Blood Cells Mol. Dis. 1998, 24, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A.; Kacena, K.; Cole, J.A.; Hollak, C.E.; Zhang, L.; Yee, J.; Mistry, P.K.; Zimran, A.; Charrow, J.; vom Dahl, S. Dose-response relationships for enzyme replacement therapy with imiglucerase/alglucerase in patients with Gaucher disease type 1. Genet. Med. 2009, 11, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Charrow, J.; Scott, C.R. Long-term treatment outcomes in Gaucher disease. Am. J. Hematol. 2015, 90 (Suppl. S1), S19–S24. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.; Malhotra, A.; Haims, A.; Pastores, G.M.; Mistry, P.K. Focal splenic lesions in type I Gaucher disease are associated with poor platelet and splenic response to macrophage-targeted enzyme replacement therapy. J. Inherit. Metab. Dis. 2010, 33, 769–774. [Google Scholar] [CrossRef]
- Damiano, A.M.; Pastores, G.M.; Ware, J.E., Jr. The health-related quality of life of adults with Gaucher’s disease receiving enzyme replacement therapy: Results from a retrospective study. Qual. Life Res. Int. J. Qual. Life Asp. Treat. Care Rehabil. 1998, 7, 373–386. [Google Scholar] [CrossRef]
- Masek, B.J.; Sims, K.B.; Bove, C.M.; Korson, M.S.; Short, P.; Norman, D.K. Quality of life assessment in adults with type 1 Gaucher disease. Qual. Life Res. Int. J. Qual. Life Asp. Treatm. Care Rehabil. 1999, 8, 263–268. [Google Scholar] [CrossRef]
- Weinreb, N.; Barranger, J.; Packman, S.; Prakash-Cheng, A.; Rosenbloom, B.; Sims, K.; Angell, J.; Skrinar, A.; Pastores, G.M. Imiglucerase (Cerezyme) improves quality of life in patients with skeletal manifestations of Gaucher disease. Clin. Genet. 2007, 71, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Wenstrup, R.J.; Kacena, K.A.; Kaplan, P.; Pastores, G.M.; Prakash-Cheng, A.; Zimran, A.; Hangartner, T.N. Effect of enzyme replacement therapy with imiglucerase on BMD in type 1 Gaucher disease. J. Bone Miner. Res. 2007, 22, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Charrow, J.; Dulisse, B.; Grabowski, G.A.; Weinreb, N.J. The effect of enzyme replacement therapy on bone crisis and bone pain in patients with type 1 Gaucher disease. Clin. Genet. 2007, 71, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Weinreb, N.J.; Kaplan, P.; Cole, J.A.; Gwosdow, A.R.; Hangartner, T. Osteopenia in Gaucher disease develops early in life: Response to imiglucerase enzyme therapy in children, adolescents and adults. Blood Cells Mol. Dis. 2011, 46, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Deegan, P.; Vellodi, A.; Cole, J.A.; Yeh, M.; Weinreb, N.J. Timing of initiation of enzyme replacement therapy after diagnosis of type 1 Gaucher disease: Effect on incidence of avascular necrosis. Br. J. Haematol. 2009, 147, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.; Gonzalez, A.N.; Lopez, G.; Pedoeim, L.; Groden, C.; Sidransky, E. The clinical management of Type 2 Gaucher disease. Mol. Genet. Metab. 2015, 114, 110–122. [Google Scholar] [CrossRef]
- Elstein, D.; Hughes, D.; Goker-Alpan, O.; Stivel, M.; Baris, H.N.; Cohen, I.J.; Granovsky-Grisaru, S.; Samueloff, A.; Mehta, A.; Zimran, A. Outcome of pregnancies in women receiving velaglucerase alfa for Gaucher disease. J. Obstet. Gynaecol. Res. 2014, 40, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.; Hollak, C.E.; Boot, R.G.; Groener, J.E.; Maas, M. Substrate reduction therapy of glycosphingolipid storage disorders. J. Inher. Metab. Dis. 2006, 29, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Belmatoug, N.; Burlina, A.; Giraldo, P.; Hendriksz, C.J.; Kuter, D.J.; Mengel, E.; Pastores, G.M. Gastrointestinal disturbances and their management in miglustat-treated patients. J. Inherit. Metab. Dis. 2011, 34, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Lukina, E.; Watman, N.; Arreguin, E.A.; Banikazemi, M.; Dragosky, M.; Iastrebner, M.; Rosenbaum, H.; Phillips, M.; Pastores, G.M.; Rosenthal, D.I.; et al. A phase 2 study of eliglustat tartrate (Genz-112638), an oral substrate reduction therapy for Gaucher disease type 1. Blood 2010, 116, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Lukina, E.; Watman, N.; Dragosky, M.; Pastores, G.M.; Arreguin, E.A.; Rosenbaum, H.; Zimran, A.; Angell, J.; Ross, L.; Puga, A.C.; et al. Eliglustat, an investigational oral therapy for Gaucher disease type 1: Phase 2 trial results after 4 years of treatment. Blood Cells Mol. Dis. 2014, 53, 274–276. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.M.; Drelichman, G.; Cravo, R.; Balwani, M.; Burrow, T.A.; Martins, A.M.; Lukina, E.; Rosenbloom, B.; Ross, L.; Angell, J.; et al. Eliglustat compared with imiglucerase in patients with Gaucher’s disease type 1 stabilised on enzyme replacement therapy: A phase 3, randomised, open-label, non-inferiority trial. Lancet 2015, 385, 2355–2362. [Google Scholar] [CrossRef]
- Mistry, P.K.; Lukina, E.; Ben Turkia, H.; Amato, D.; Baris, H.; Dasouki, M.; Ghosn, M.; Mehta, A.; Packman, S.; Pastores, G.; et al. Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: The ENGAGE randomized clinical trial. JAMA 2015, 313, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Kamath, R.S.; Lukina, E.; Watman, N.; Dragosky, M.; Pastores, G.M.; Arreguin, E.A.; Rosenbaum, H.; Zimran, A.; Aguzzi, R.; Puga, A.C.; et al. Skeletal improvement in patients with Gaucher disease type 1: A phase 2 trial of oral eliglustat. Skelet. Radiol. 2014, 43, 1353–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belmatoug, N.; Di Rocco, M.; Fraga, C.; Giraldo, P.; Hughes, D.; Lukina, E.; Maison-Blanche, P.; Merkel, M.; Niederau, C.; Plckinger, U.; et al. Management and monitoring recommendations for the use of eliglustat in adults with type 1 Gaucher disease in Europe. Eur. J. Intern. Med. 2017, 37, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Ringden, O.; Groth, C.G.; Erikson, A.; Granqvist, S.; Mansson, J.E.; Sparrelid, E. Ten years’ experience of bone marrow transplantation for Gaucher disease. Transplantation 1995, 59, 864–870. [Google Scholar] [PubMed]
- Dunbar, C.E.; Kohn, D.B.; Schiffmann, R.; Barton, N.W.; Nolta, J.A.; Esplin, J.A.; Pensiero, M.; Long, Z.; Lockey, C.; Emmons, R.V.; et al. Retroviral transfer of the glucocerebrosidase gene into CD34+ cells from patients with Gaucher disease: In vivo detection of transduced cells without myeloablation. Hum. Gene Ther. 1998, 9, 2629–2640. [Google Scholar] [CrossRef] [PubMed]
- Dahl, M.; Doyle, A.; Olsson, K.; Mansson, J.E.; Marques, A.R.; Mirzaian, M.; Aerts, J.M.; Ehinger, M.; Rothe, M.; Modlich, U.; et al. Lentiviral gene therapy using cellular promoters cures type 1 Gaucher disease in mice. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martinez, A.; Beavan, M.; Gegg, M.E.; Chau, K.Y.; Whitworth, A.J.; Schapira, A.H. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci. Rep. 2016, 6, 31380. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G. Treating lysosomal storage diseases with pharmacological chaperones: From concept to clinics. EMBO Mol. Med. 2009, 1, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Maegawa, G.H.; Tropak, M.B.; Buttner, J.D.; Rigat, B.A.; Fuller, M.; Pandit, D.; Tang, L.; Kornhaber, G.J.; Hamuro, Y.; Clarke, J.T.; et al. Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J. Biol. Chem. 2009, 284, 23502–23516. [Google Scholar] [CrossRef] [PubMed]
- McNeill, A.; Magalhaes, J.; Shen, C.; Chau, K.Y.; Hughes, D.; Mehta, A.; Foltynie, T.; Cooper, J.M.; Abramov, A.Y.; Gegg, M.; et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain J. Neurol. 2014, 137, 1481–1495. [Google Scholar] [CrossRef] [PubMed]
- Kristinsson, S.Y.; Gridley, G.; Hoover, R.N.; Check, D.; Landgren, O. Long-term risks after splenectomy among 8,149 cancer-free American veterans: A cohort study with up to 27 years follow-up. Haematologica 2014, 99, 392–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, T.M.; Aerts, J.M.; Belmatoug, N.; Cappellini, M.D.; vom Dahl, S.; Goldblatt, J.; Grabowski, G.A.; Hollak, C.E.; Hwu, P.; Maas, M.; et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J. Inherit. Metab. Dis. 2008, 31, 319–336. [Google Scholar] [CrossRef] [PubMed]
- Baris, H.N.; Weisz Hubshman, M.; Bar-Sever, Z.; Kornreich, L.; Shkalim Zemer, V.; Cohen, I.J. Re-evaluation of bone pain in patients with type 1 Gaucher disease suggests that bone crises occur in small bones as well as long bones. Blood Cells Mol. Dis. 2015, 60, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Barton, N.W.; Brady, R.O.; Dambrosia, J.M.; Di Bisceglie, A.M.; Doppelt, S.H.; Hill, S.C.; Mankin, H.J.; Murray, G.J.; Parker, R.I.; Argoff, C.E.; et al. Replacement therapy for inherited enzyme deficiency—Macrophage-targeted glucocerebrosidase for Gaucher’s disease. N. Engl. J. Med. 1991, 324, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Vigan, M.; Stirnemann, J.; Caillaud, C.; Froissart, R.; Boutten, A.; Fantin, B.; Belmatoug, N.; Mentre, F. Modeling changes in biomarkers in Gaucher disease patients receiving enzyme replacement therapy using a pathophysiological model. Orphanet J. Rare Dis. 2014, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, N.J.; Goldblatt, J.; Villalobos, J.; Charrow, J.; Cole, J.A.; Kerstenetzky, M.; vom Dahl, S.; Hollak, C. Long-term clinical outcomes in type 1 Gaucher disease following 10 years of imiglucerase treatment. J. Inherit. Metab. Dis. 2013, 36, 543–553. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. https://doi.org/10.3390/ijms18020441
Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, Levade T, Astudillo L, Serratrice J, Brassier A, et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. International Journal of Molecular Sciences. 2017; 18(2):441. https://doi.org/10.3390/ijms18020441
Chicago/Turabian StyleStirnemann, Jérôme, Nadia Belmatoug, Fabrice Camou, Christine Serratrice, Roseline Froissart, Catherine Caillaud, Thierry Levade, Leonardo Astudillo, Jacques Serratrice, Anaïs Brassier, and et al. 2017. "A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments" International Journal of Molecular Sciences 18, no. 2: 441. https://doi.org/10.3390/ijms18020441
APA StyleStirnemann, J., Belmatoug, N., Camou, F., Serratrice, C., Froissart, R., Caillaud, C., Levade, T., Astudillo, L., Serratrice, J., Brassier, A., Rose, C., Billette de Villemeur, T., & Berger, M. G. (2017). A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. International Journal of Molecular Sciences, 18(2), 441. https://doi.org/10.3390/ijms18020441