
Single-Nucleotide Polymorphism of PPARγ, a Protein at the Crossroads of Physiological and Pathological Processes

 , , and
, , and
Abstract
:1. Introduction
2. Results
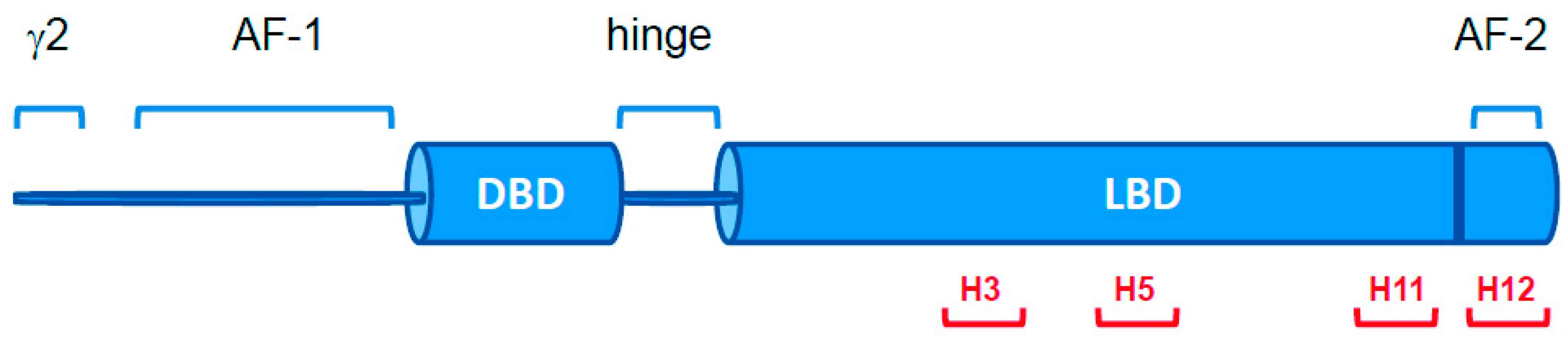
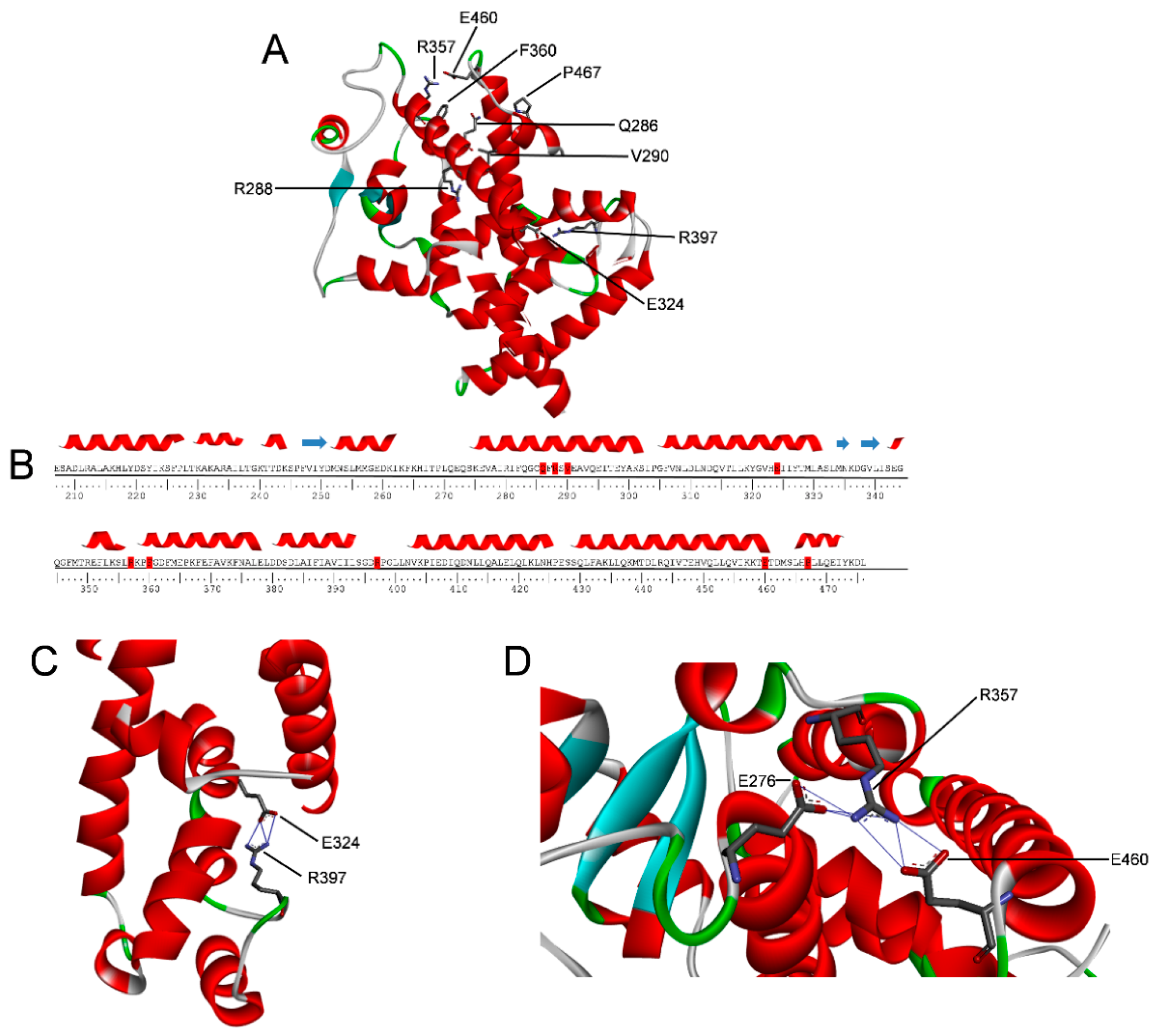
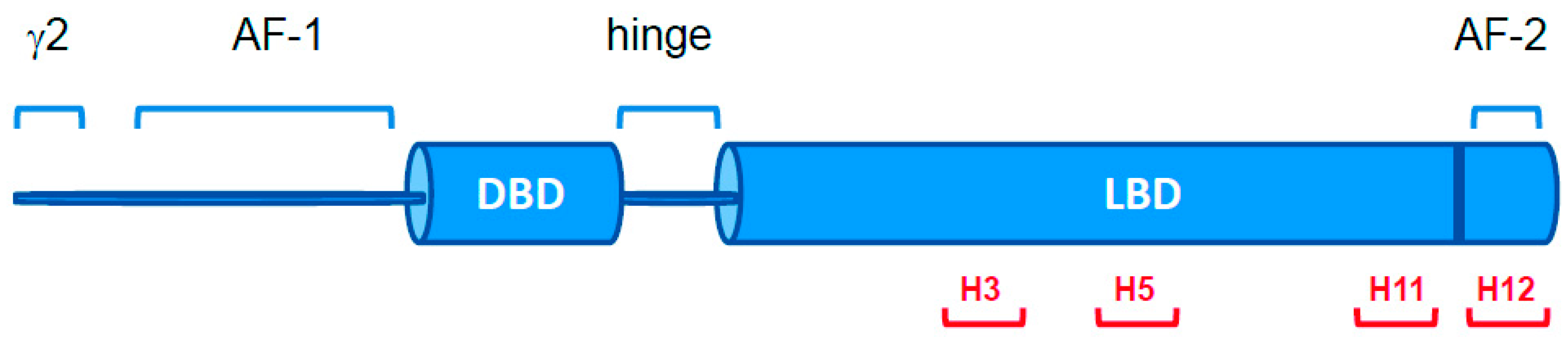
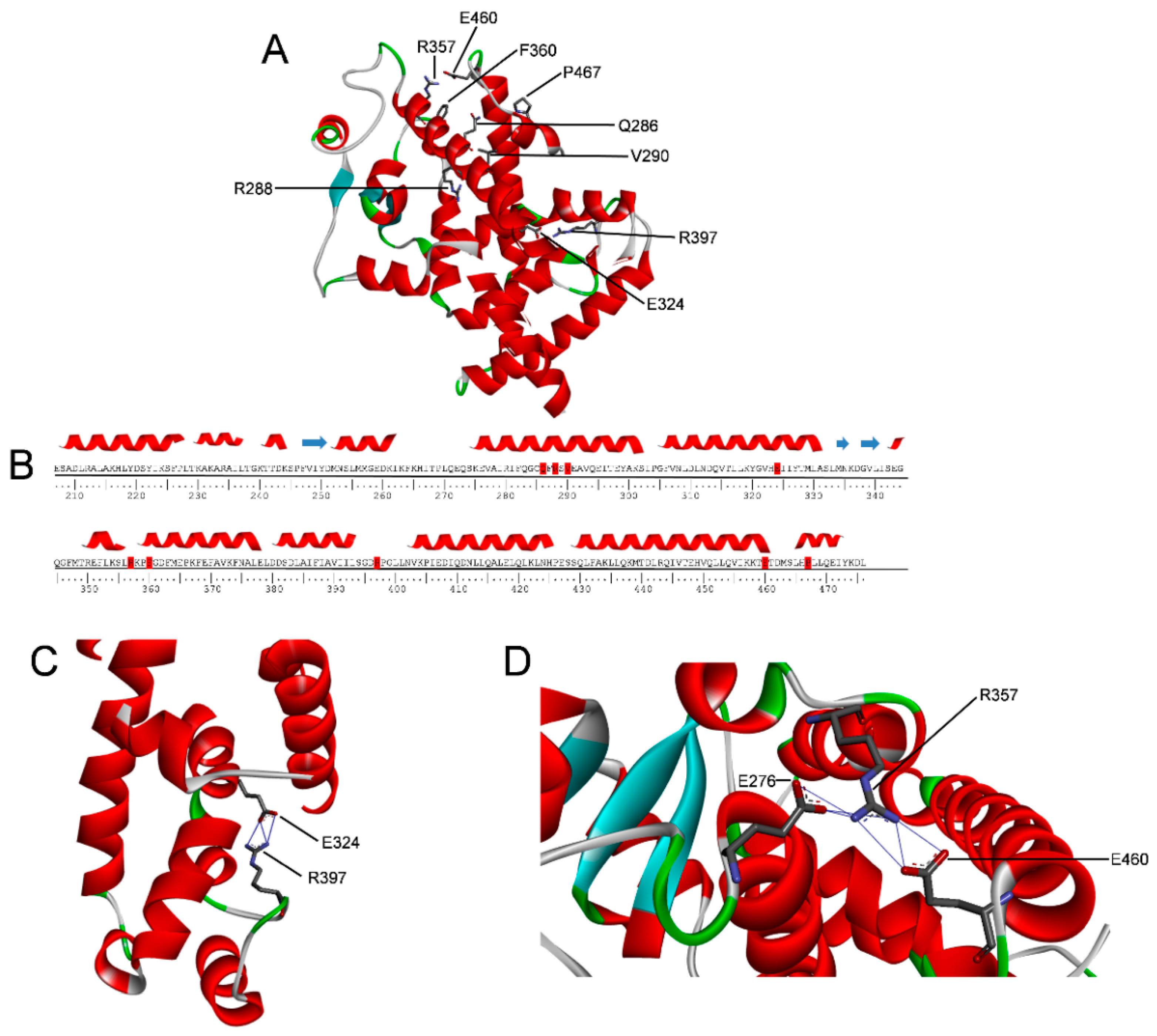
2.1. PPARγ Variants
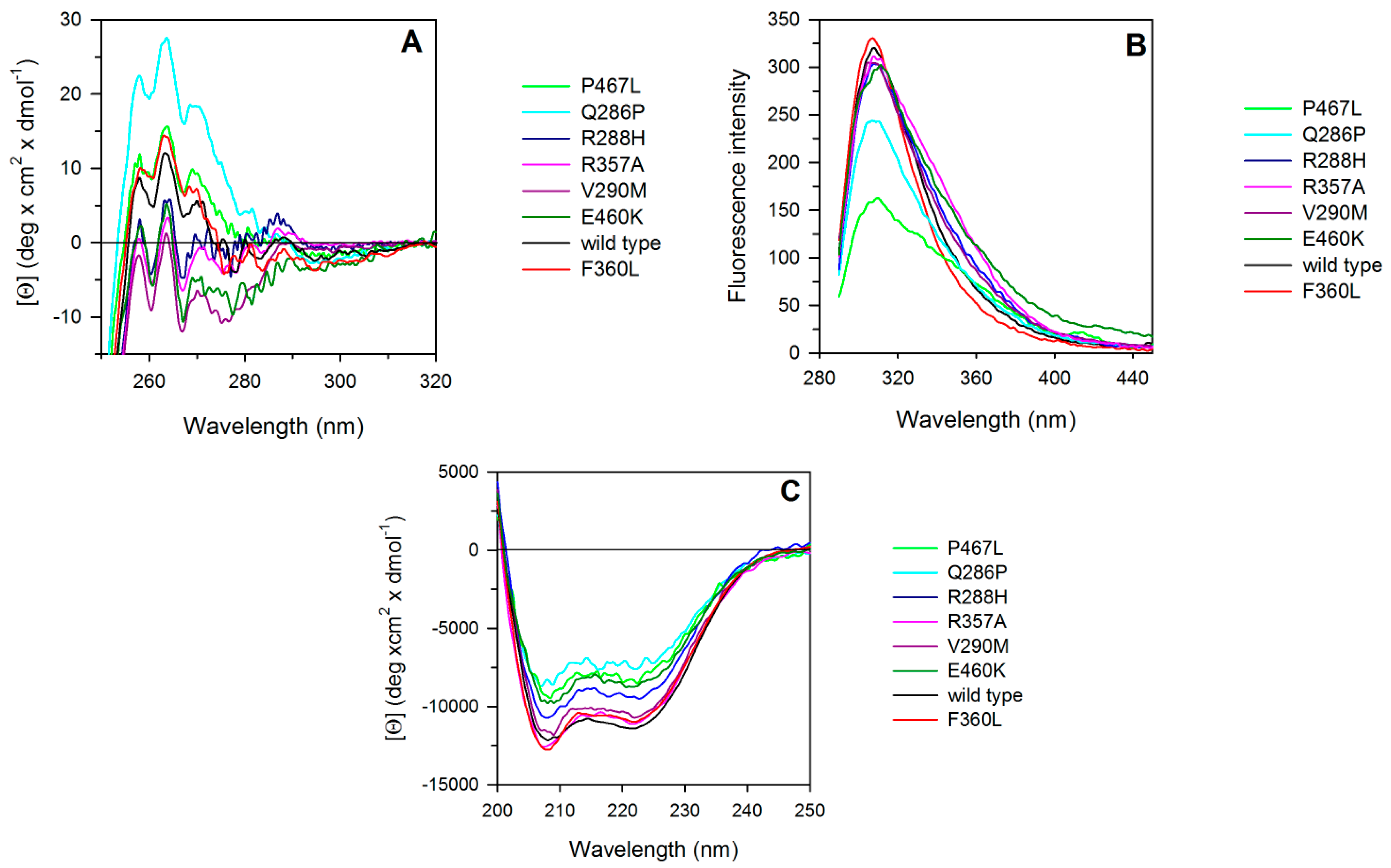
2.2. Spectroscopic Characterization of PPARγ Wild Type and Variants
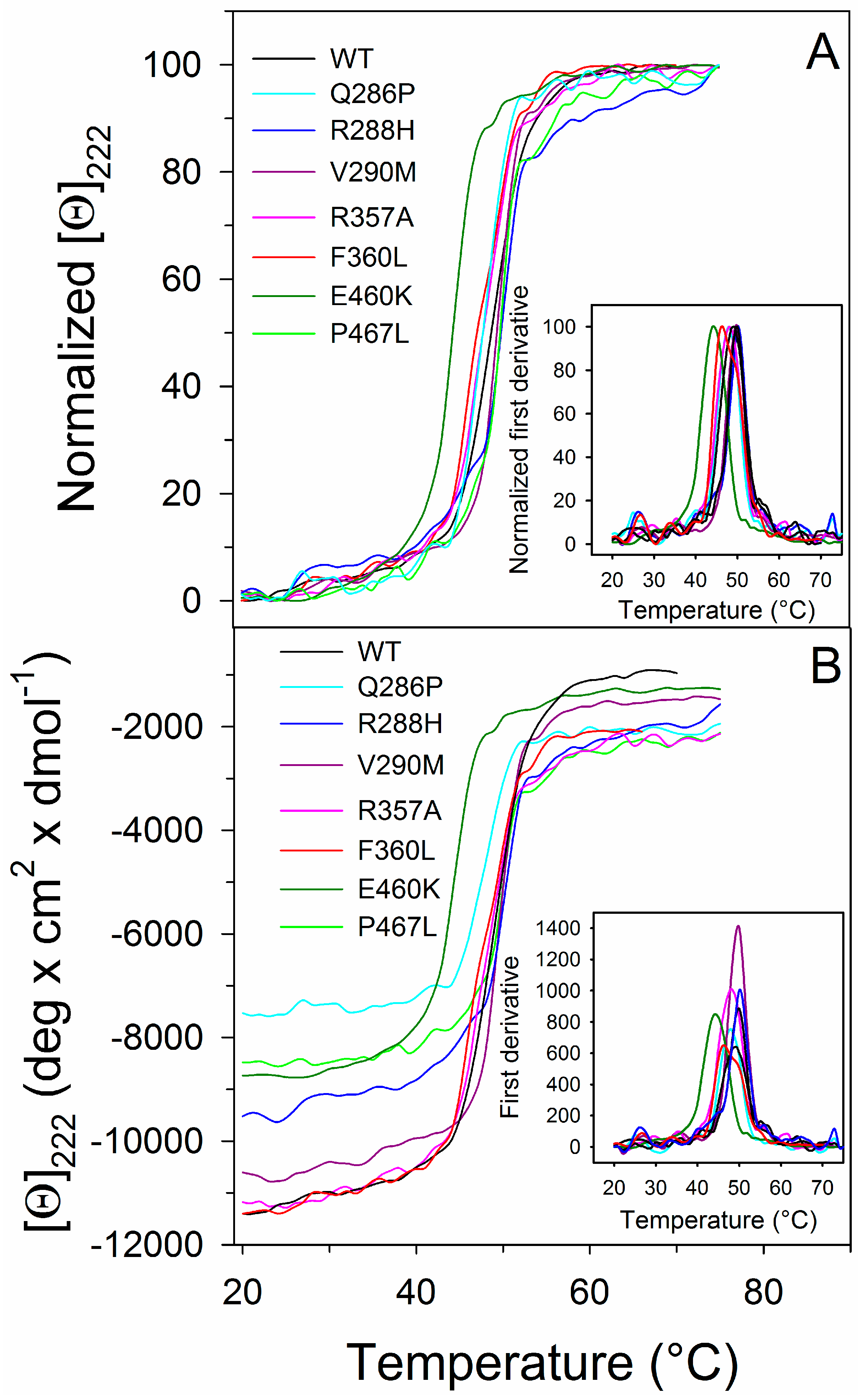
2.3. Thermal Unfolding
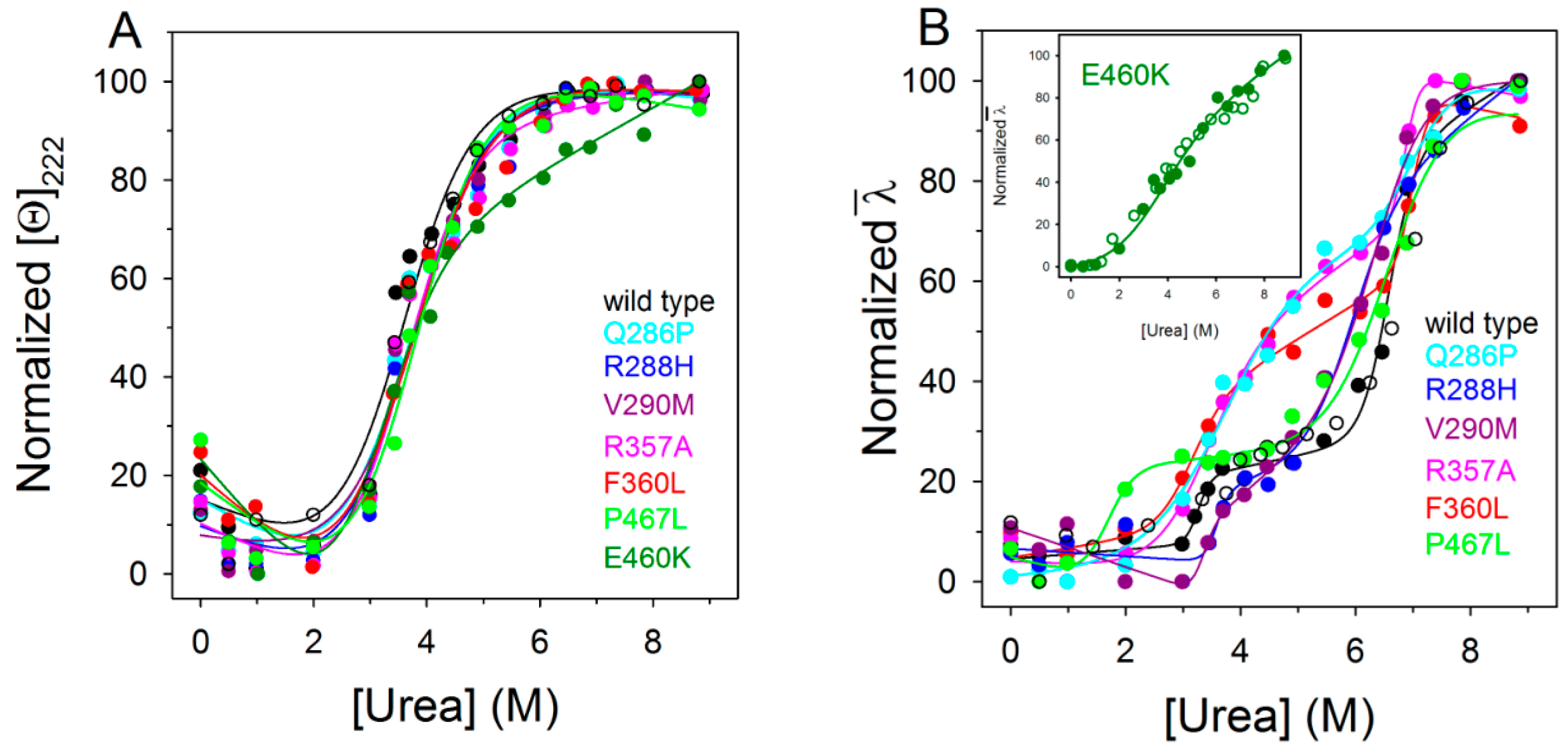
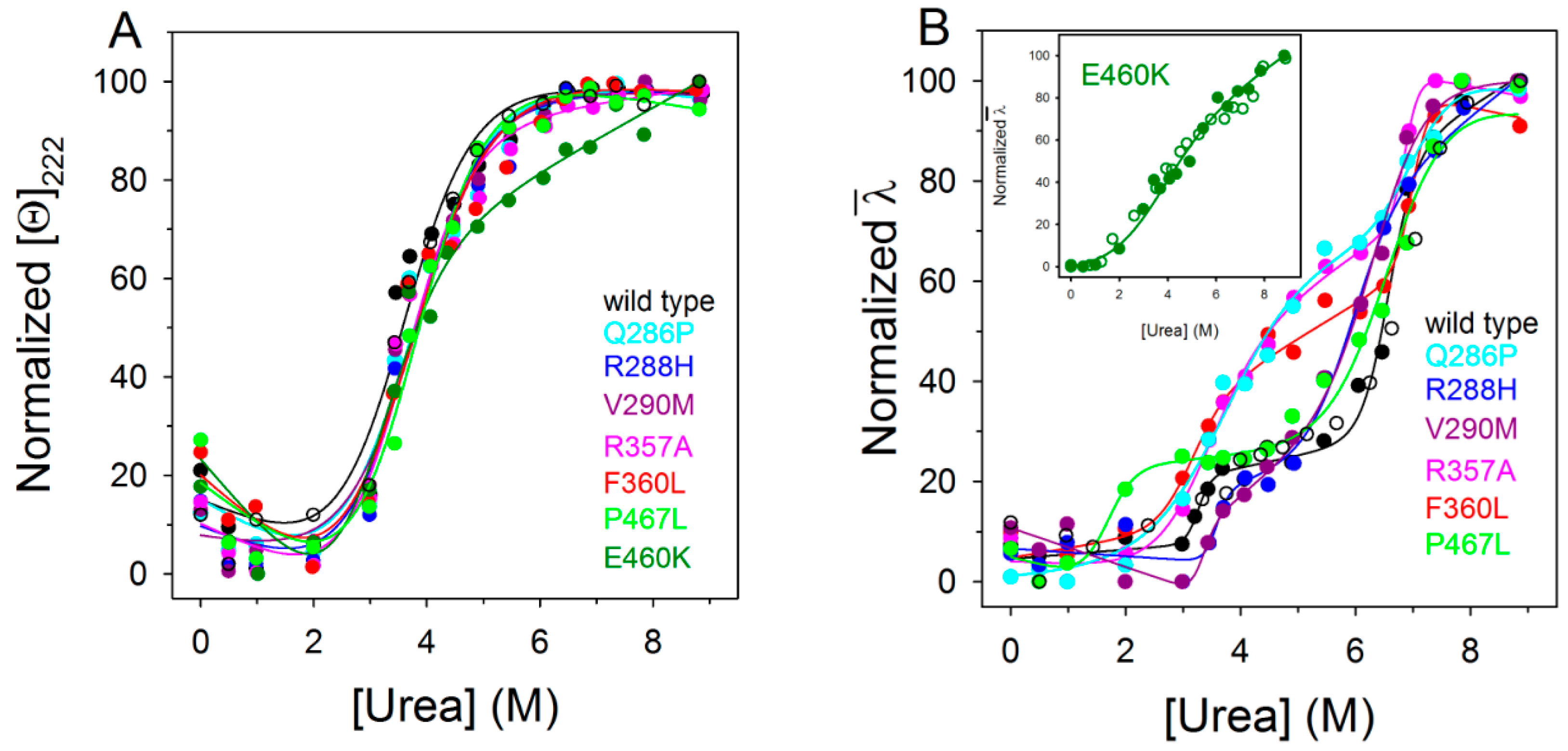
2.4. Urea-Induced Equilibrium Unfolding Transitions
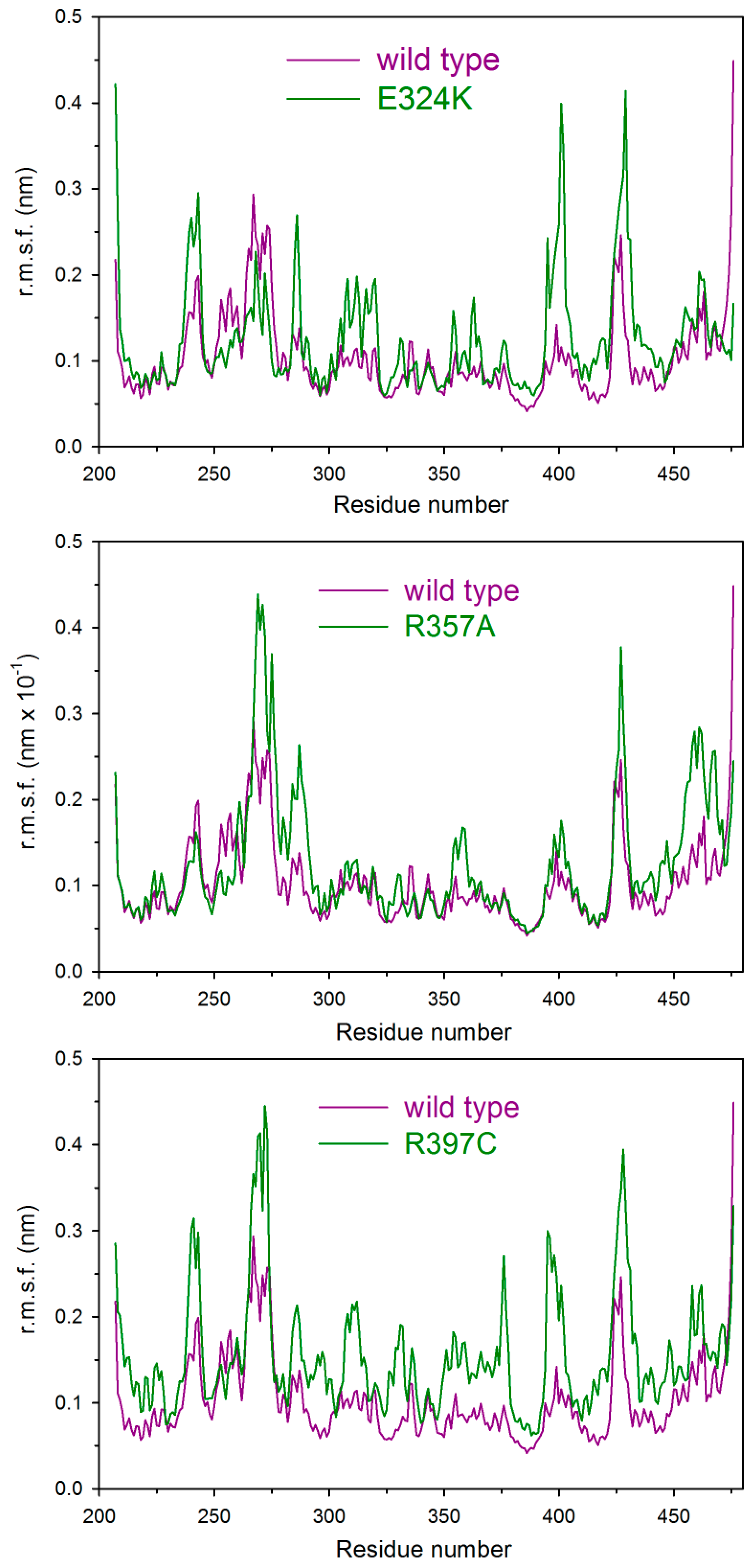
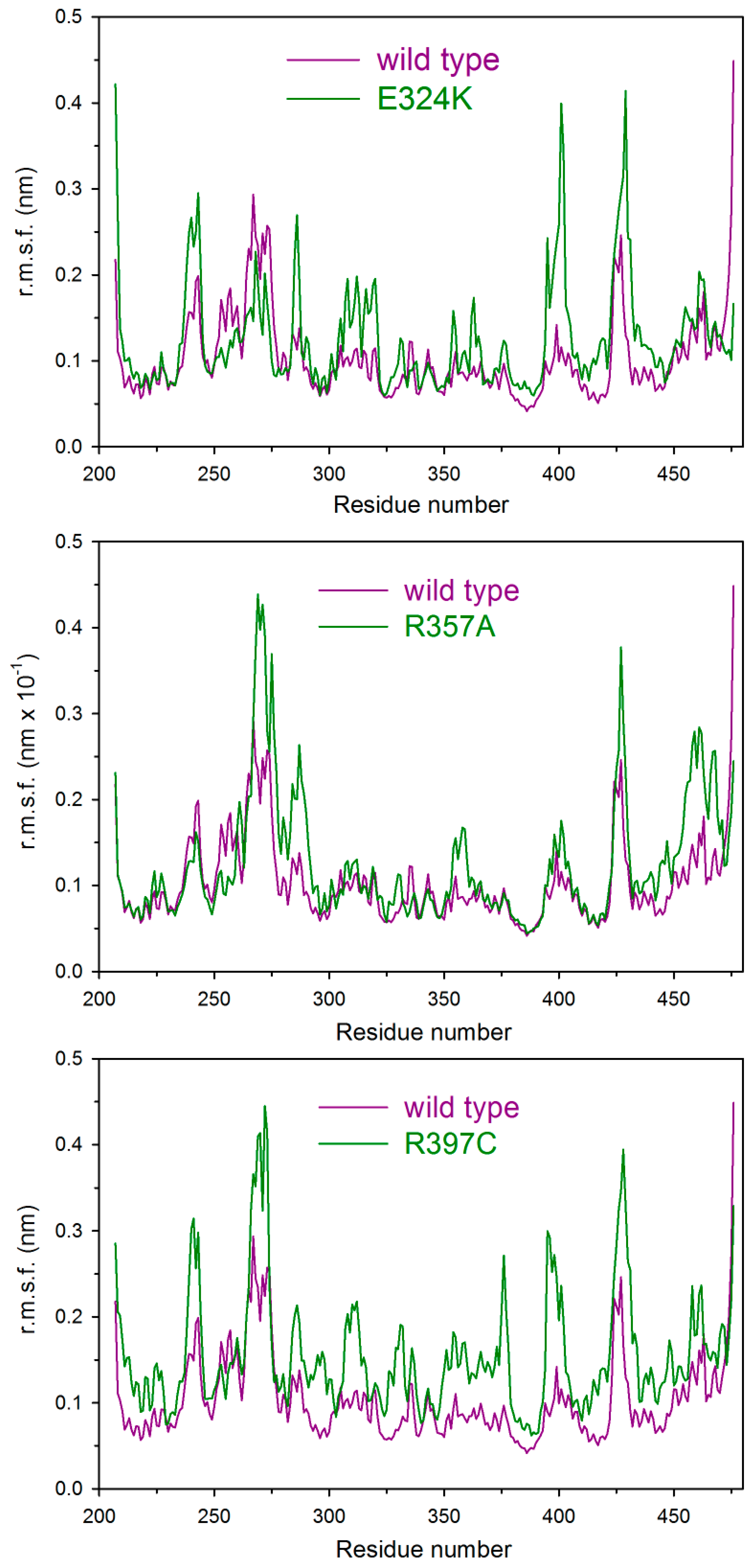
2.5. Molecular Dynamics
2.6. Transcription Activity
3. Discussion
4. Materials and Methods
4.1. Plasmids and Site-Directed Mutagenesis
4.2. Protein Preparation
4.3. Cell Culture and Transfections
4.4. Spectroscopic Measurements
4.5. Urea-Induced Equilibrium Unfolding
4.6. Thermal Denaturation Experiments
4.7. Data Analysis
4.8. Molecular Dynamics Simulations
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DTT | Dithiothreitol |
| LBD | Ligand-Binding Domain |
| MD | Molecular Dynamics |
| nsSNPs | Non-synonymous single-nucleotide polymorphisms |
| PPARγ | Peroxisome Proliferator-Activated Receptor γ |
References
- Sauer, S. Ligands for the nuclear peroxisome proliferator-activated receptor γ. Trends Pharmacol. Sci. 2015, 36, 688–704. [Google Scholar] [CrossRef] [PubMed]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-γ. Nature 1998, 395, 137–143. [Google Scholar] [PubMed]
- Chandra, V.; Huang, P.; Hamuro, Y.; Raghuram, S.; Wang, Y.; Burris, T.P.; Rastinejad, F. Structure of the intact PPAR-γ-RXR-nuclear receptor complex on DNA. Nature 2008, 456, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARs in health and disease. Nature 2000, 405, 421–424. [Google Scholar] [PubMed]
- Michalik, L.; Wahli, W. Involvement of PPAR nuclear receptors in tissue injury and wound repair. J. Clin. Investig. 2006, 116, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Anghel, S.I.; Wahli, W. Fat poetry: A kingdom for PPARγ. Cell. Res. 2007, 17, 486–511. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Barish, G.D.; Wang, Y.X. PPARs and the complex journey to obesity. Nat. Med. 2004, 10, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Semple, R.K.; Chatterjee, V.K.; O’Rahilly, S. PPARγ and human metabolic disease. J. Clin. Investig. 2006, 116, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xu, J.; Yu, X.; Yang, R.; Han, Z.C. Peroxisome proliferator-activated receptor γ in malignant diseases. Crit. Rev. Oncol. Hematol. 2006, 58, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mandard, S.; Patsouris, D. Nuclear control of the inflammatory response in mammals by peroxisome proliferator-activated receptors. PPAR Res. 2013, 2013, 613864. [Google Scholar] [CrossRef] [PubMed]
- Lehrke, M.; Lazar, M.A. The many faces of PPARγ. Cell. 2005, 123, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Spiegelman, B.M. Fat and beyond: The diverse biology of PPARγ. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Sarraf, P.; Mueller, E.; Smith, W.M.; Wright, H.M.; Kum, J.B.; Aaltonen, L.A.; de la Chapelle, A.; Spiegelman, B.M.; Eng, C. Loss-of-function mutations in PPARγ associated with human colon cancer. Mol. Cell 1999, 3, 799–804. [Google Scholar] [CrossRef]
- Garcia-Vallvé, S.; Guasch, L.; Tomas-Hernández, S.; del Bas, J.M.; Ollendorff, V.; Arola, L.; Pujadas, G.; Mulero, M. Peroxisome proliferator-activated receptor γ (PPARγ) and ligand choreography: Newcomers take the stage. J. Med. Chem. 2015, 58, 5381–5394. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Song, J.; Park, K.W. The multifaceted factor peroxisome proliferator-activated receptor γ (PPARγ) in metabolism, immunity, and cancer. Arch. Pharm. Res. 2015, 38, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.H.; Niu, T.; Ma, Y.; You, N.C.; Song, Y.; Sobel, E.M.; Hsu, Y.H.; Balasubramanian, R.; Qiao, Y.; Tinker, L.; et al. Common genetic variants in peroxisome proliferator-activated receptor-γ (PPARG) and type 2 diabetes risk among Women’s Health Initiative postmenopausal women. J. Clin. Endocrinol. Metab. 2013, 98, E600–E604. [Google Scholar] [CrossRef] [PubMed]
- Walkey, C.J.; Spiegelman, B.M. A functional peroxisome proliferator-activated receptor-γ ligand-binding domain is not required for adipogenesis. J. Biol. Chem. 2008, 283, 24290–24294. [Google Scholar] [CrossRef] [PubMed]
- Barroso, I.; Gurnell, M.; Crowley, V.E.; Agostini, M.; Schwabe, J.W.; Soos, M.A.; Maslen, G.L.; Williams, T.D.; Lewis, H.; Schafer, A.J.; et al. Dominant negative mutations in human PPARγ associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 1999, 402, 880–883. [Google Scholar] [PubMed]
- Savage, D.B.; Tan, G.D.; Acerini, C.L.; Jebb, S.A.; Agostini, M.; Gurnell, M.; Williams, R.L.; Umpleby, A.M.; Thomas, E.L.; Bell, J.D.; et al. Human metabolic syndrome resulting from dominant-negative mutations in the nuclear receptor peroxisome proliferator-activated receptor-γ. Diabetes 2003, 52, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Meirhaeghe, A.; Amouyel, P. Impact of genetic variation of PPARγ in humans. Mol. Genet. Metab. 2004, 83, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Schoenmakers, E.; Mitchell, C.; Szatmari, I.; Savage, D.; Smith, A.; Rajanayagam, O.; Semple, R.; Luan, J.; Bath, L.; et al. Non-DNA binding, dominant-negative, human PPARγ mutations cause lipodystrophic insulin resistance. Cell Metab. 2006, 4, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, G.D.; Savage, D.B.; Fielding, B.A.; Collins, J.; Hodson, L.; Humphreys, S.M.; O’Rahilly, S.; Chatterjee, K.; Frayn, K.N.; Karpe, F. Fatty acid metabolism in patients with PPARγ mutations. J. Clin. Endocrinol. Metab. 2008, 93, 4462–4470. [Google Scholar] [CrossRef] [PubMed]
- Jeninga, E.H.; Gurnell, M.; Kalkhoven, E. Functional implications of genetic variation in human PPARγ. Trends Endocrinol. Metab. 2009, 20, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Visser, M.E.; Kropman, E.; Kranendonk, M.E.; Koppen, A.; Hamers, N.; Stroes, E.S.; Kalkhoven, E.; Monajemi, H. Characterisation of non-obese diabetic patients with marked insulin resistance identifies a novel familial partial lipodystrophy-associated PPARγ mutation (Y151C). Diabetologia 2011, 54, 1639–1644. [Google Scholar] [CrossRef] [PubMed]
- Lori, C.; Pasquo, A.; Montanari, R.; Capelli, D.; Consalvi, V.; Chiaraluce, R.; Cervoni, L.; Loiodice, F.; Laghezza, A.; Aschi, M.; et al. Structural basis of the transactivation deficiency of the human PPARγ F360L mutant associated with familial partial lipodystrophy. Acta. Crystallogr. D Biol. Crystallogr. 2014, 70, 1965–1976. [Google Scholar] [CrossRef] [PubMed]
- Puhl, A.; Webb, P.; Polikarpov, I. Structural dataset for the PPARγ V290M mutant. Data Brief. 2016, 7, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Pasquo, A.; Consalvi, V.; Knapp, S.; Alfano, I.; Ardini, M.; Stefanini, S.; Chiaraluce, R. Structural stability of human protein tyrosine phosphatase ρ catalytic domain:effect of point mutations. PLoS ONE 2012, 7, e32555. [Google Scholar] [CrossRef] [PubMed]
- Lori, C.; Lantella, A.; Pasquo, A.; Alexander, L.T.; Knapp, S.; Chiaraluce, R.; Consalvi, V. Effect of single amino acid substitution observed in cancer on Pim-1 kinase thermodynamic stability and structure. PLoS ONE 2013, 8, e64824. [Google Scholar] [CrossRef] [PubMed]
- Casadio, R.; Vassura, M.; Tiwari, S.; Fariselli, P.; Martelli, P.L. Correlating disease-related mutations to their effect on protein stability: A large-scale analysis of the human proteome. Hum. Mutat. 2011, 32, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Thusberg, J.; Olatubosun, A.; Vihinen, M. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 2011, 32, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Vihinen, M. Performance of protein stability predictors. Hum. Mutat. 2010, 31, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Kucukkal, T.G.; Petukh, M.; Li, L.; Alexov, E. Structural and physico-chemical effects of disease and non-disease nsSNPs on proteins. Curr. Opin. Struct. Biol. 2015, 32, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Allali-Hassani, A.; Wasney, G.A.; Chau, I.; Hong, B.S.; Senisterra, G.; Loppnau, P.; Shi, Z.; Moult, J.; Edwards, A.M.; Arrowsmith, C.H.; et al. A survey of proteins encoded by non-synonymous single nucleotide polymorphisms reveals a significant fraction with altered stability and activity. Biochem. J. 2009, 424, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Miteva, M.A.; Wang, L.; Alexov, E. Analyzing effects of naturally occurring missense mutations. Comput. Math. Methods Med. 2012, 2012, 805827. [Google Scholar] [CrossRef] [PubMed]
- Yates, C.M.; Sternberg, M.J. Proteins and domains vary in their tolerance of non-synonymous single nucleotide polymorphisms (nsSNPs). J. Mol. Biol. 2013, 425, 1274–1286. [Google Scholar] [CrossRef] [PubMed]
- Ormond, K.E.; Wheeler, M.T.; Hudgins, L.; Klein, T.E.; Butte, A.J.; Altman, R.B.; Ashley, E.A.; Greely, H.T. Challenges in the clinical application of whole-genome sequencing. Lancet 2010, 375, 1749–1751. [Google Scholar] [CrossRef]
- Kucukkal, T.G.; Yang, Y.; Chapman, S.C.; Cao, W.; Alexov, E. Computational and experimental approaches to reveal the effects of single nucleotide polymorphisms with respect to disease diagnostics. Int. J. Mol. Sci. 2014, 15, 9670–9717. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2011, 39, D945–D950. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Ma, L.; Liu, Y.; Qu, H. Pedican: An online gene for pediatric cancers with literature evidence. Sci. Rep. 2015, 5, 11435. [Google Scholar] [CrossRef] [PubMed]
- Lori, L.; Pasquo, A.; Lori, C.; Petrosino, M.; Chiaraluce, R.; Tallant, C.; Knapp, S.; Consalvi, V. Effect of BET missense mutations on bromodomain function, inhibitor binding and stability. PLoS ONE. 2016, 11, e0159180. [Google Scholar] [CrossRef] [PubMed]
- Choy, N.; Raussens, V.; Narayanaswami, V. Inter-molecular coiled-coil formation in human apolipoprotein E C-terminal domain. J. Mol. Biol. 2003, 334, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Kiss, R.S.; Weers, P.M.; Narayanaswami, V.; Cohen, J.; Kay, C.M.; Ryan, R.O. Structure-guided protein engineering modulates helix bundle exchangeable apolipoprotein properties. J. Biol. Chem. 2003, 278, 21952–21959. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.K.; Pace, C.N.; Scholtz, J.M. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995, 4, 2138–2148. [Google Scholar] [CrossRef] [PubMed]
- Geierhaas, C.D.; Nickson, A.A.; Lindorff-Larsen, K.; Clarke, J.; Vendruscolo, M. BPPred: A computational tool to predict biophysical quantities of proteins. Protein Sci. 2007, 16, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Auton, M.; Holthauzen, L.M.; Bolen, D.W. Anatomy of energetic changes accompanying urea-induced protein denaturation. Proc. Natl. Acad. Sci. USA 2007, 104, 15317–15322. [Google Scholar] [CrossRef] [PubMed]
- Pinelli, A.; Godio, C.; Laghezza, A.; Mitro, N.; Fracchiolla, G.; Tortorella, V.; Lavecchia, A.; Novellino, E.; Fruchart, J.C.; Staels, B.; et al. Synthesis, biological evaluation, and molecular modeling investigation of new chiral fibrates with PPARα and PPARγ agonist activity. J. Med. Chem. 2005, 48, 5509–5519. [Google Scholar] [CrossRef] [PubMed]
- Fariselli, P.; Martelli, P.L.; Savojardo, C.; Casadio, R. INPS: Predicting the impact of non-synonymous variations on protein stability from sequence. Bioinformatics 2015, 31, 2816–2821. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, Y.; Rost, B. Correlating protein function and stability through the analysis of single amino acid substitutions. BMC Bioinformatics 2009, 10, S8. [Google Scholar] [CrossRef] [PubMed]
- Fernald, G.H.; Capriotti, E.; Daneshjou, R.; Karczewski, K.J.; Altman, R.B. Bioinformatics challenges for personalized medicine. Bioinformatics 2011, 27, 1741–1748. [Google Scholar] [CrossRef]
- Stefl, S.; Nishi, H.; Petukh, M.; Panchenko, A.R.; Alexov, E. molecular mechanisms of disease-causing missense mutations. J. Mol. Biol. 2013, 425, 3919–3936. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Gutierrez, M.P.; Roszer, T.; Ricote, M. Biology and therapeutic applications of peroxisome proliferator-activated receptors. Curr. Top. Med. Chem. 2012, 12, 548–584. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Shah, Y.M.; Gonzalez, F.J. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat. Rev. Cancer 2012, 12, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.P.; Moras, D. Structural studies on nuclear receptors. Cell. Mol. Life. Sci. 2000, 57, 1748–1769. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Wu, J.S.; Tsai, H.D.; Huang, C.Y.; Chen, J.J.; Sun, G.Y.; Lin, T.N. Peroxisome proliferator-activated receptor γ (PPAR-γ) and neurodegenerative disorders. Mol. Neurobiol. 2012, 46, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.P.; Akiyama, T.E.; Meinke, P.T. PPARs: Therapeutic targets for metabolic disease. Trends Pharmacol. Sci. 2005, 26, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Pochetti, G.; Mitro, N.; Lavecchia, A.; Gilardi, F.; Besker, N.; Scotti, E.; Aschi, M.; Re, N.; Fracchiolla, G.; Laghezza, A.; et al. Structural insight into peroxisome proliferator-activated receptor γ binding of two ureidofibrate-like enantiomers by molecular dynamics, cofactor interaction analysis, and site-directed mutagenesis. J. Med. Chem. 2010, 53, 4354–4366. [Google Scholar] [CrossRef] [PubMed]
- Kallenberger, B.C.; Love, J.D.; Chatterjee, V.K.; Schwabe, J.W. A dynamic mechanism of nuclear receptor activation and its perturbation in a human disease. Nat. Struct. Biol. 2003, 10, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Pochetti, G.; Godio, C.; Mitro, N.; Caruso, D.; Galmozzi, A.; Scurati, S.; Loiodice, F.; Fracchiolla, G.; Tortorella, P.; Laghezza, A.; et al. Insights into the mechanism of partial agonism: Crystal structures of the peroxisome proliferator-activated receptor γ ligand-binding domain in the complex with two enantiomeric ligands. J. Biol. Chem. 2007, 282, 17314–17324. [Google Scholar] [CrossRef] [PubMed]
- Raspe´, E.; Madsen, L.; Lefebvre, A.M.; Leitersdorf, I.; Gelman, L.; Peinado-Onsurbe, J.; Dallongeville, J.; Fruchart, J.C.; Berge, R.; Staels, B. Modulation of rat liver apolipoprotein gene expression and serum lipid levels by tetradecylthioacetic acid (TTA) via PPARalpha activation. J. Lipid Res. 1999, 40, 2099–2110. [Google Scholar] [PubMed]
- Gill, S.C.; von Hippel, P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989, 182, 319–326. [Google Scholar] [CrossRef]
- Benjwal, S.; Verma, S.; Rohm, K.H.; Gursky, O. Monitoring protein aggregation during thermal unfolding in circular dichroism experiments. Protein Sci. 2006, 15, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Royer, C.A.; Mann, C.J.; Matthews, C.R. Resolution of the fluorescence equilibrium unfolding profile of Trp aporepressor using single tryptophan mutants. Protein Sci. 1993, 2, 1844–1852. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.M.; Bolen, D.W. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry 1988, 27, 8063–8068. [Google Scholar] [CrossRef] [PubMed]
- Rowling, P.J.; Cook, R.; Itzhaki, L.S. Toward classification of BRCA1 missense variants using a biophysical approach. J. Biol. Chem. 2010, 285, 20080–20087. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; van der Spoel, D. Gromacs 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhor, G.; Mark, A.E.; Berendsen, H.J.C. Gromacs: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. Gromacs 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Minicozzi, V.; Chiaraluce, R.; Consalvi, V.; Giordano, C.; Narcisi, C.; Punzi, P.; Rossi, G.C.; Morante, S. Computational and experimental studies on β-sheet breakers targeting Amyloid-β1–40 fibrils. J. Biol. Chem. 2014, 289, 11242–11252. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, M.G.; Minicozzi, V.; Foderà, V.; Militello, V.; Vetri, V.; Morante, S.; Leone, M. Thioflavin-T templates amyloid-β(1–40) conformation and aggregation pathway. Biophys. Chem. 2015, 206, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD-Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PPARγ | Tm (°C) | ∆GH2O (kcal/mol) | m (kcal/mol/M) | [Urea]0.5 (M) |
|---|---|---|---|---|
| Wild type | 49.5 | 3.37 ± 0.15 | 0.95 ± 0.05 | 3.16 |
| Q286P | 48.0 | 3.07 ± 0.15 | 0.84 ± 0.05 | 3.65 |
| R288H | 50.0 | 3.43 ± 0.12 | 0.94 ± 0.04 | 3.65 |
| V290M | 49.5 | 3.40 ± 0.10 | 0.89 ± 0.03 | 3.82 |
| R357A | 48.0 | 3.56 ± 0.10 | 1.00 ± 0.03 | 3.56 |
| F360L | 46.5 | 2.97 ± 0.10 | 0.83 ± 0.03 | 3.58 |
| P467L | 50.0 | 3.48 ± 0.11 | 0.93 ± 0.04 | 3.74 |
| E460K | 44.0 | 3.20 ± 0.16 | 1.07 ± 0.06 | 3.00 |
| PPARγ | mI–N (kcal/mol/M) | D50I–N (M) | ΔGH2OI–N (kcal/mol) | mU–I (kcal/mol/M) | D50U–I (M) | ΔGH2OU–I (kcal/mol) |
|---|---|---|---|---|---|---|
| Wild type | 5.30 ± 0.64 | 3.26 ± 0.09 | 17.27 | 2.47 ± 0.32 | 6.56 ± 0.04 | 16.20 |
| Q286P | 0.85 ± 0.10 | 3.73 ± 0.07 | 3.17 | 1.53 ± 0.18 | 7.00 ± 0.11 | 10.76 |
| R288H | 5.24 ± 0.52 | 3.57 ± 0.09 | 18.71 | 1.42 ± 0.17 | 5.89 ± 0.07 | 8.36 |
| V290M | 4.96 ± 0.64 | 3.32 ± 0.18 | 16.47 | 1.29 ± 0.14 | 6.41 ± 0.05 | 8.27 |
| R357A | 1.21 ± 0.16 | 3.31 ± 0.06 | 4.01 | 4.71 ± 0.50 | 6.88 ± 0.04 | 32.40 |
| F360L | 1.87 ± 0.22 | 3.09 ± 0.14 | 5.79 | 3.71 ± 0.48 | 6.86 ± 0.06 | 25.45 |
| P467L | 2.48 ± 0.24 | 1.66 ± 0.24 | 4.12 | 1.13 ± 0.16 | 6.58 ± 0.08 | 7.43 |
| System | Backbone r.m.s.d. (nm) | Gyration Radius (nm) | H3–H12 Distance (nm) | H12 Subportion (280–287) Distance (nm) |
|---|---|---|---|---|
| wild type | 0.28 (0.02) | 1.96 (0.01) | 1.14 (0.05) | 1.45 (0.05) |
| Q286P | 0.26 (0.01) | 1.96 (0.01) | 1.00 (0.05) | 1.42 (0.06) |
| R288H | 0.26 (0.02) | 1.95 (0.01) | 1.2 (0.1) | 1.46 (0.06) |
| V290M | 0.28 (0.02) | 1.95 (0.01) | 1.15 (0.05) | 1.40 (0.03) |
| E324K | 0.28 (0.02) | 1.94 (0.01) | 1.05 (0.04) | 1.39 (0.04) |
| R357A | 0.39 (0.02) | 1.92 (0.01) | 1.3 (0.1) | 1.5 (0.1) |
| F360L | 0.28 (0.02) | 1.98 (0.01) | 1.44 (0.06) | 1.60 (0.06) |
| R397C | 0.38 (0.03) | 1.98 (0.01) | 1.26 (0.05) | 1.45 (0.08 |
| E460K | 0.29 (0.02) | 1.97 (0.01) | 1.10 (0.06) | 1.42 (0.05) |
| P467L | 0.28 (0.02) | 1.95 (0.02) | 1.13 (0.06) | 1.41 (0.05) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrosino, M.; Lori, L.; Pasquo, A.; Lori, C.; Consalvi, V.; Minicozzi, V.; Morante, S.; Laghezza, A.; Giorgi, A.; Capelli, D.; et al. Single-Nucleotide Polymorphism of PPARγ, a Protein at the Crossroads of Physiological and Pathological Processes. Int. J. Mol. Sci. 2017, 18, 361. https://doi.org/10.3390/ijms18020361
Petrosino M, Lori L, Pasquo A, Lori C, Consalvi V, Minicozzi V, Morante S, Laghezza A, Giorgi A, Capelli D, et al. Single-Nucleotide Polymorphism of PPARγ, a Protein at the Crossroads of Physiological and Pathological Processes. International Journal of Molecular Sciences. 2017; 18(2):361. https://doi.org/10.3390/ijms18020361
Chicago/Turabian StylePetrosino, Maria, Laura Lori, Alessandra Pasquo, Clorinda Lori, Valerio Consalvi, Velia Minicozzi, Silvia Morante, Antonio Laghezza, Alessandra Giorgi, Davide Capelli, and et al. 2017. "Single-Nucleotide Polymorphism of PPARγ, a Protein at the Crossroads of Physiological and Pathological Processes" International Journal of Molecular Sciences 18, no. 2: 361. https://doi.org/10.3390/ijms18020361