Sphingosine-1-Phosphate Receptor 1, Expressed in Myeloid Cells, Slows Diet-Induced Atherosclerosis and Protects against Macrophage Apoptosis in Ldlr KO Mice


Abstract
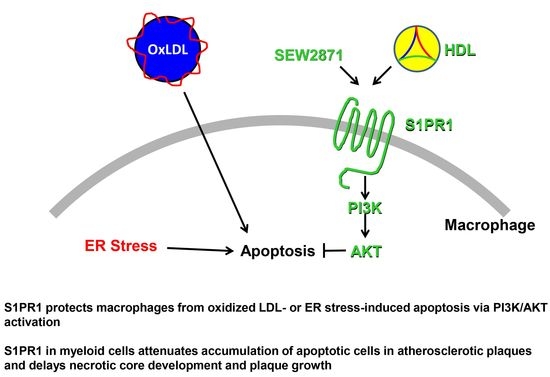
:
1. Introduction
2. Results
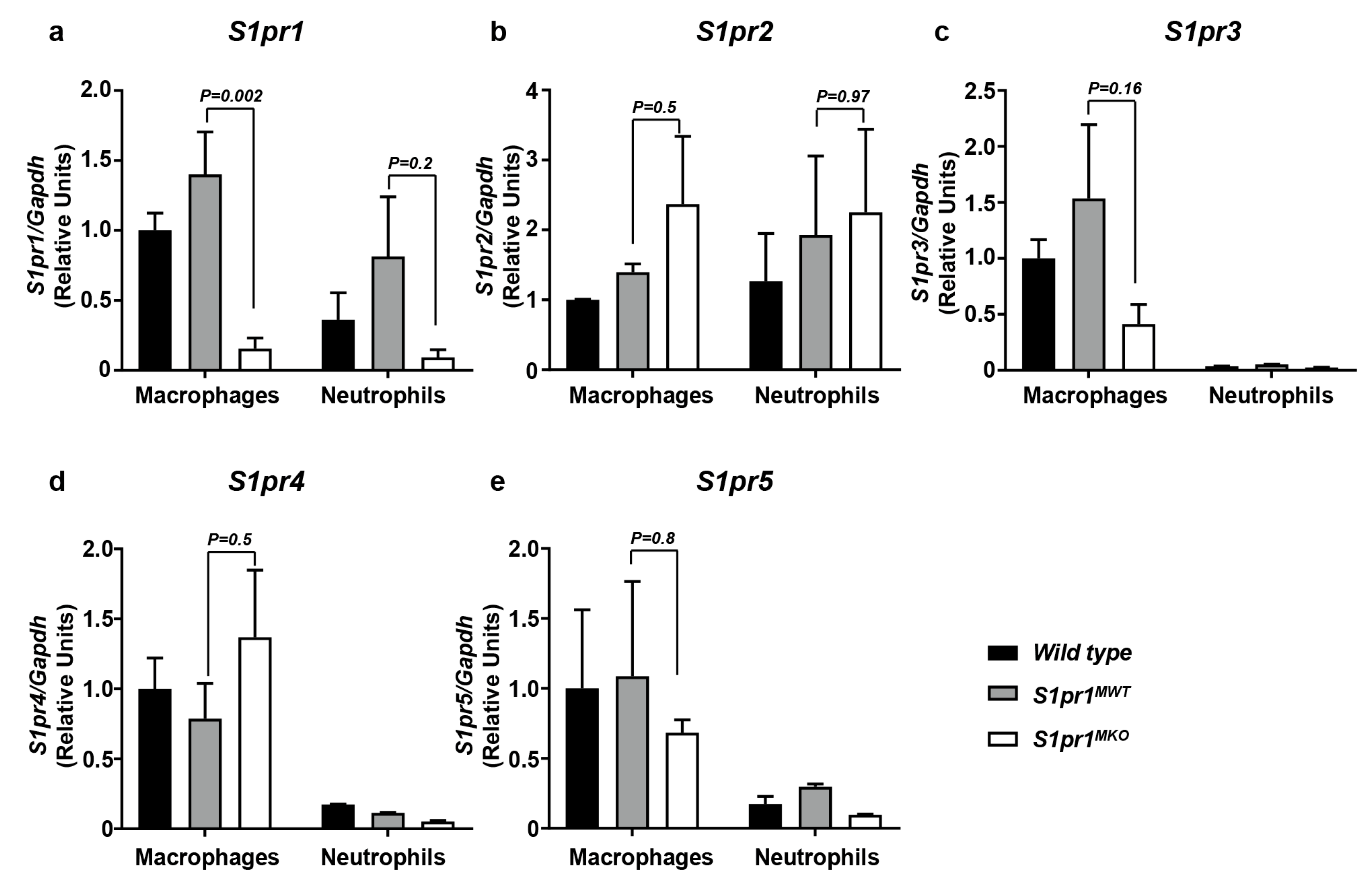
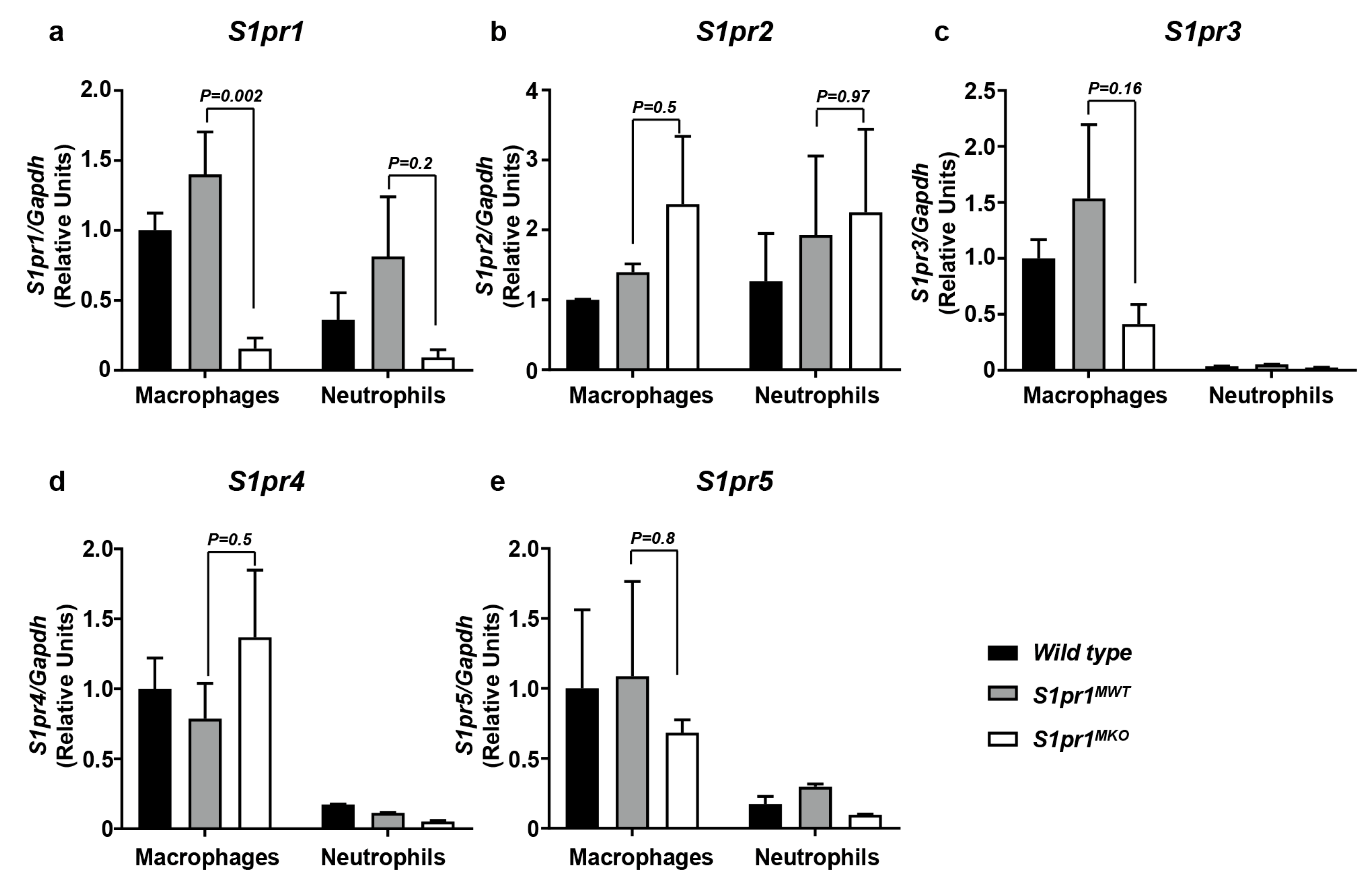
2.1. Selective Inactivation of S1PR1 in Myeloid Cells
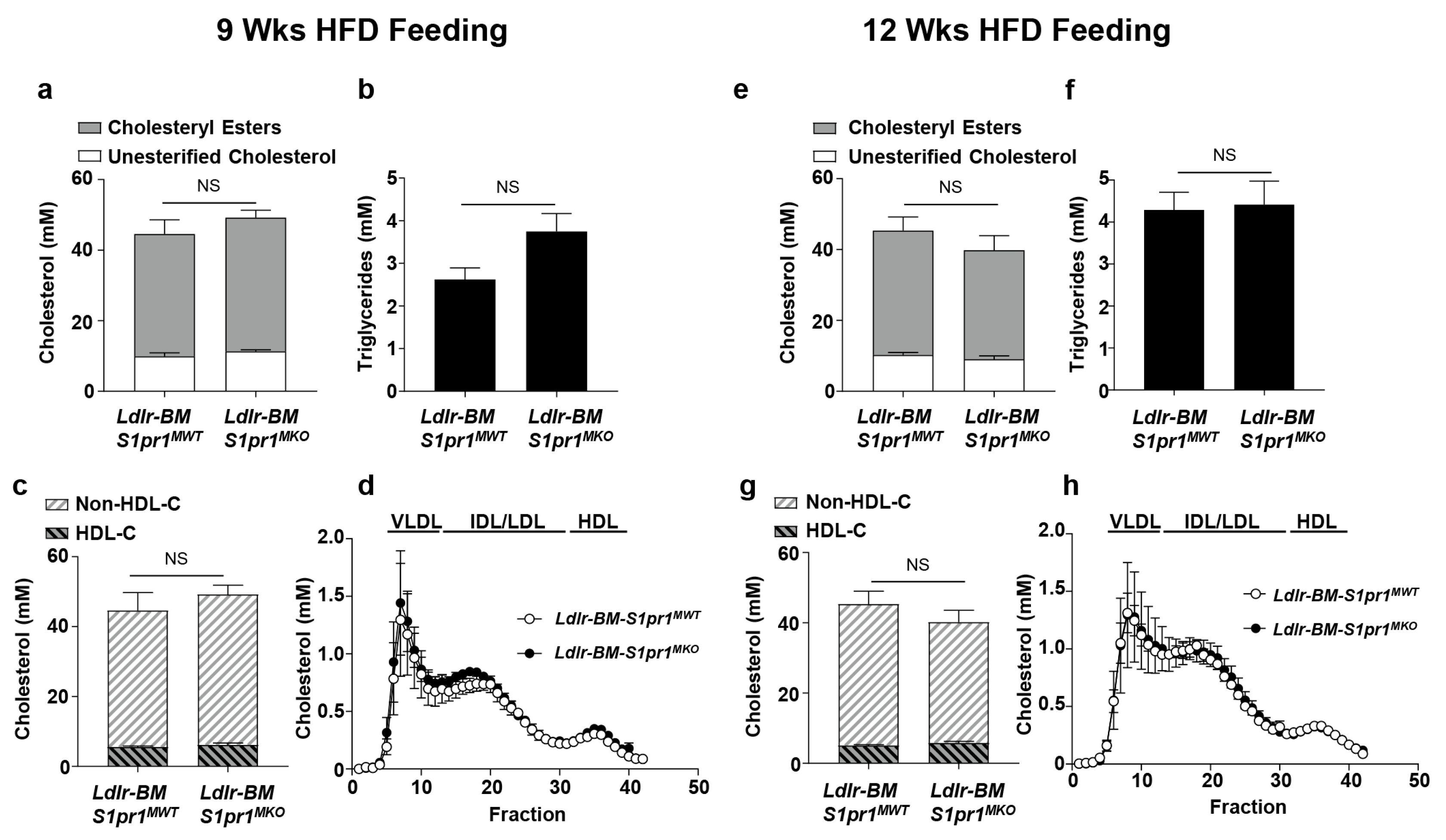
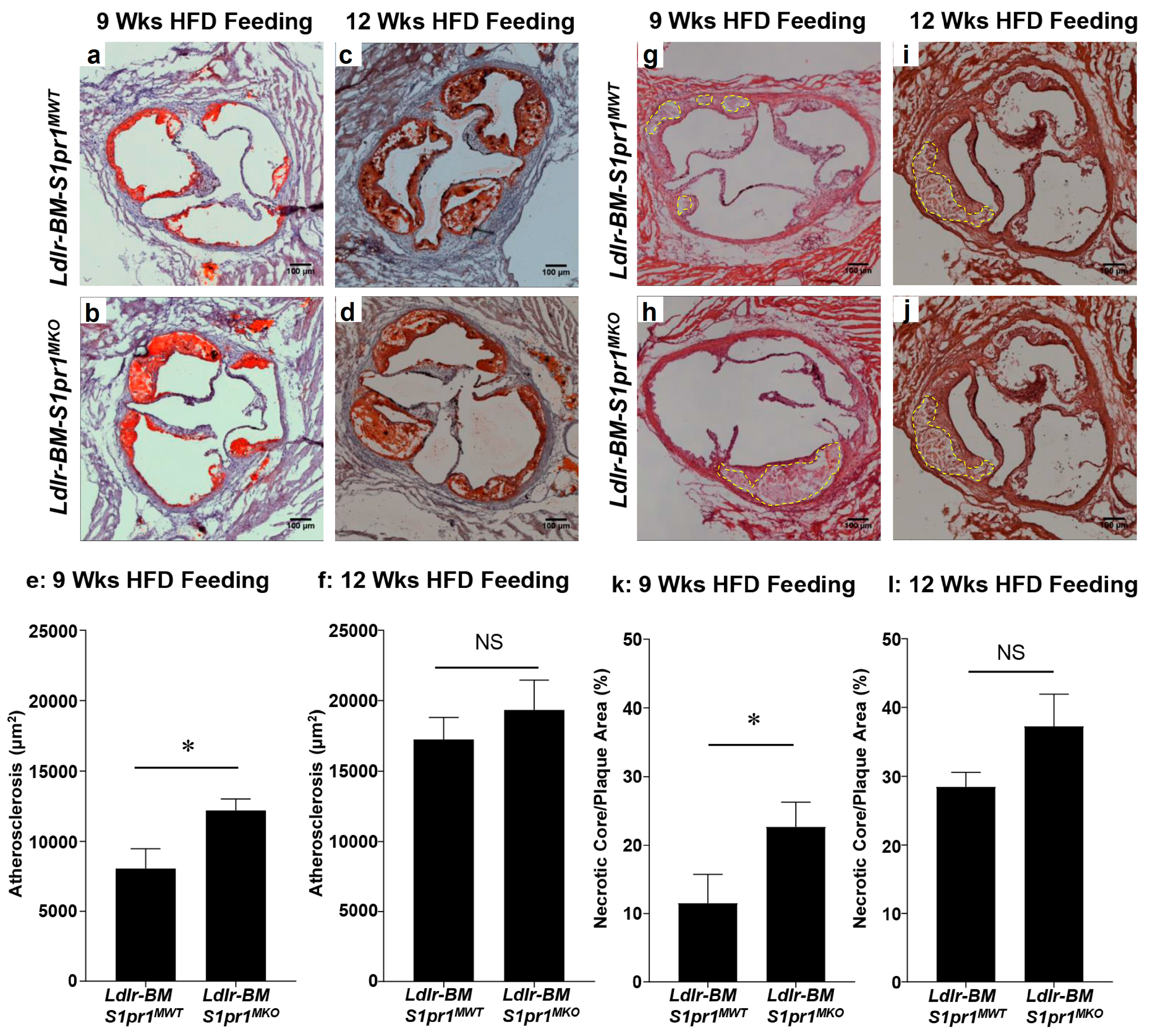
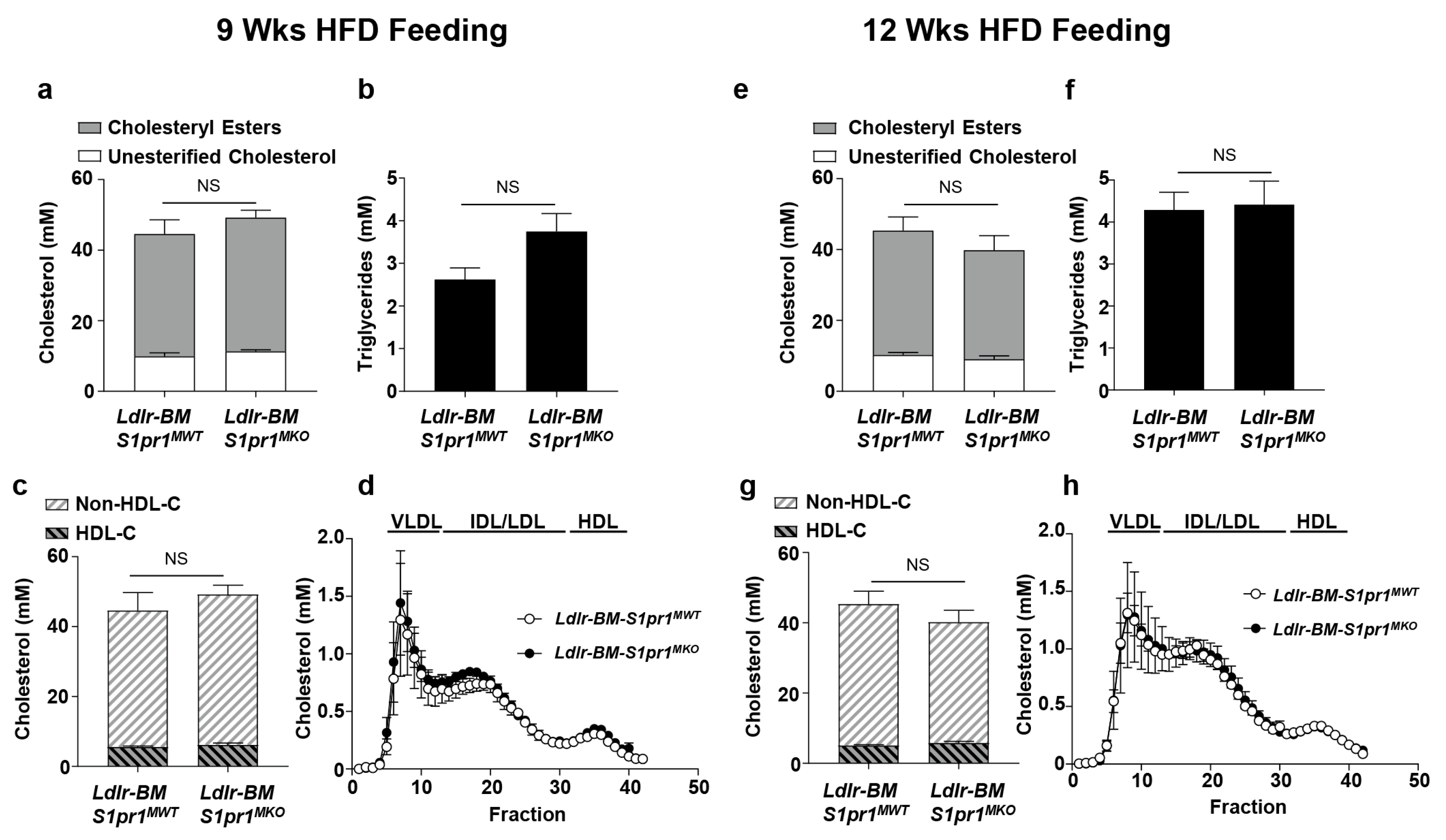
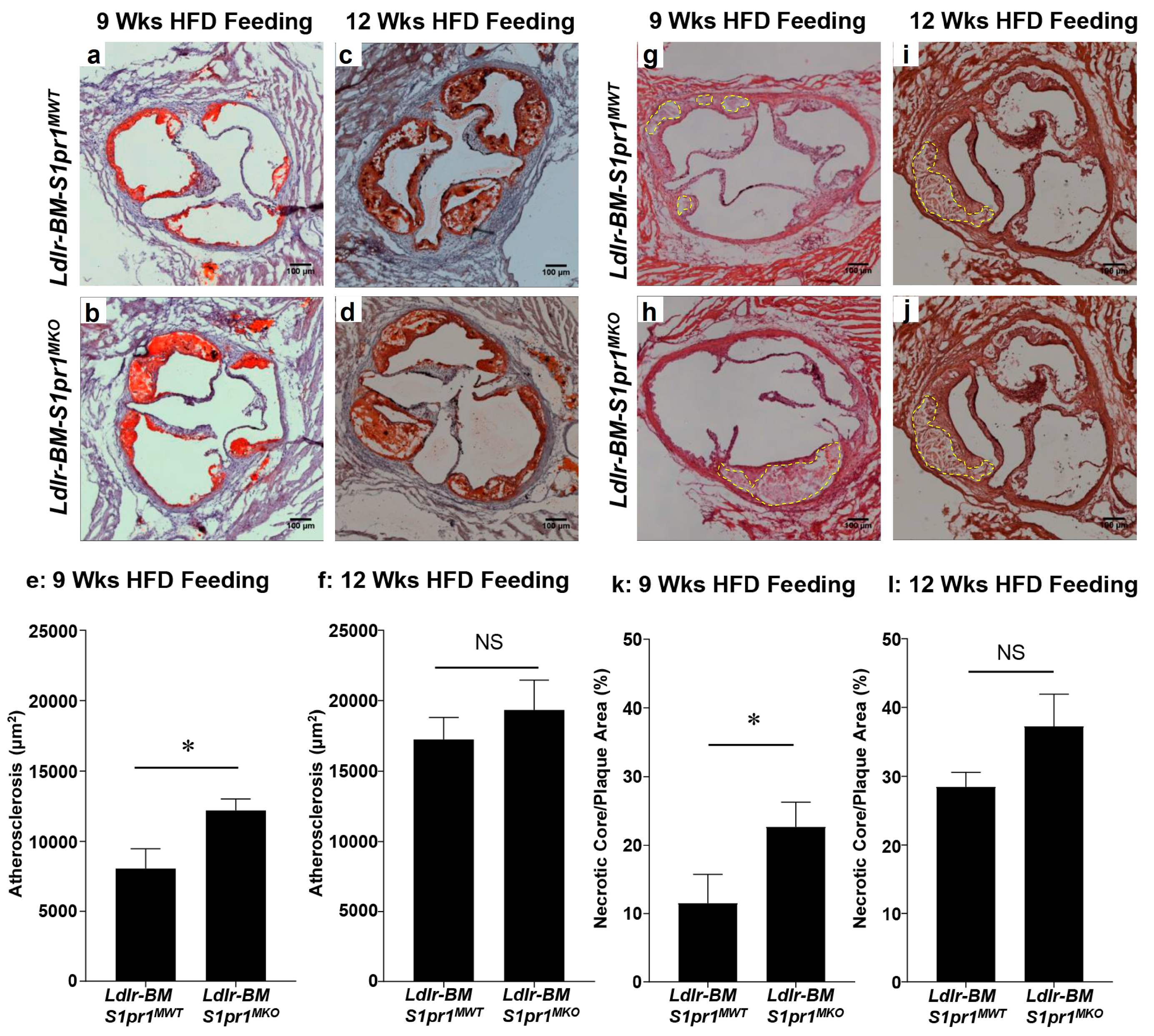
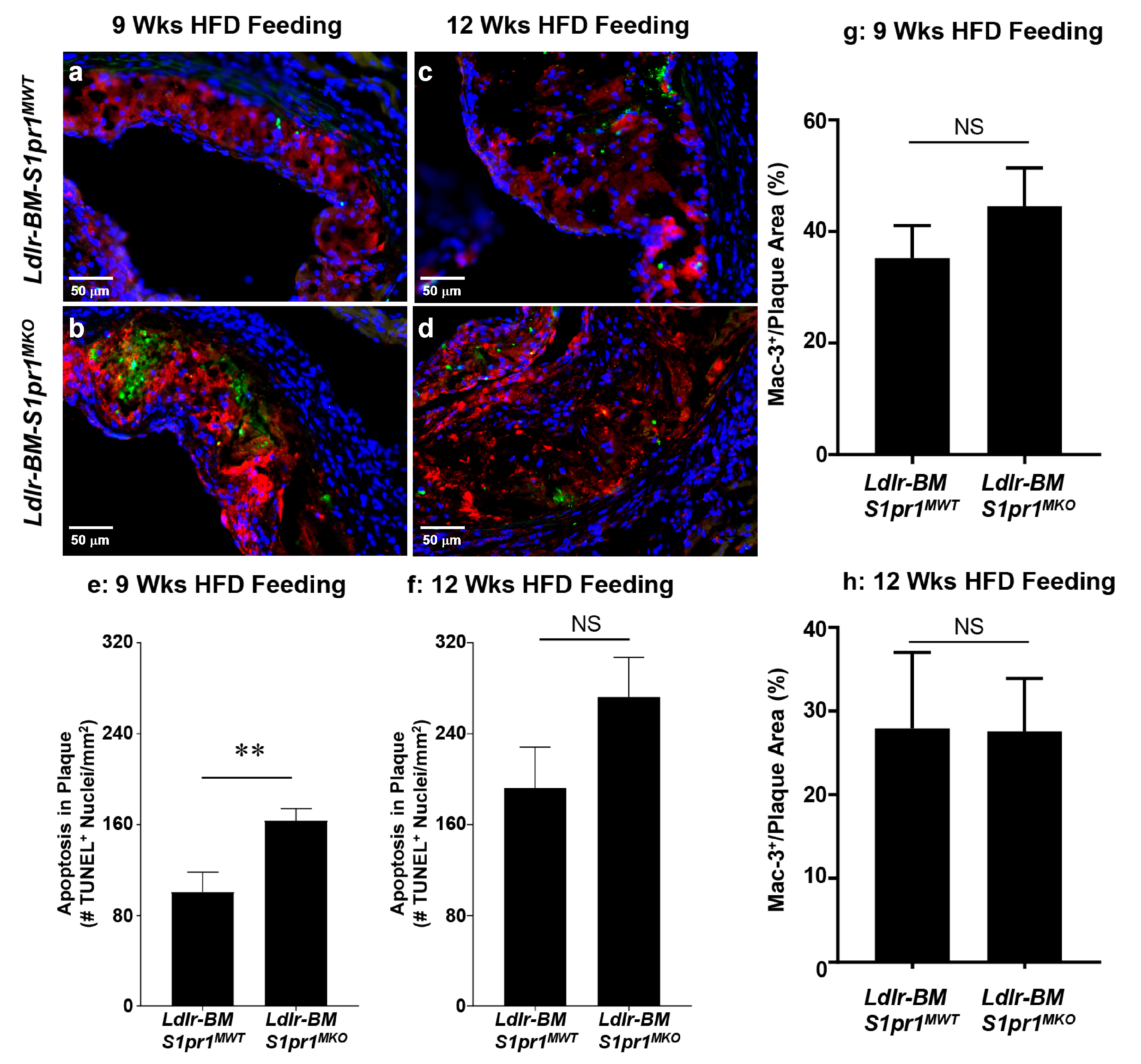
2.2. Effect of Myeloid Selective S1PR1 Deficiency on HF Diet-Induced Atherosclerosis in BM Transplanted Ldlr KO Mice
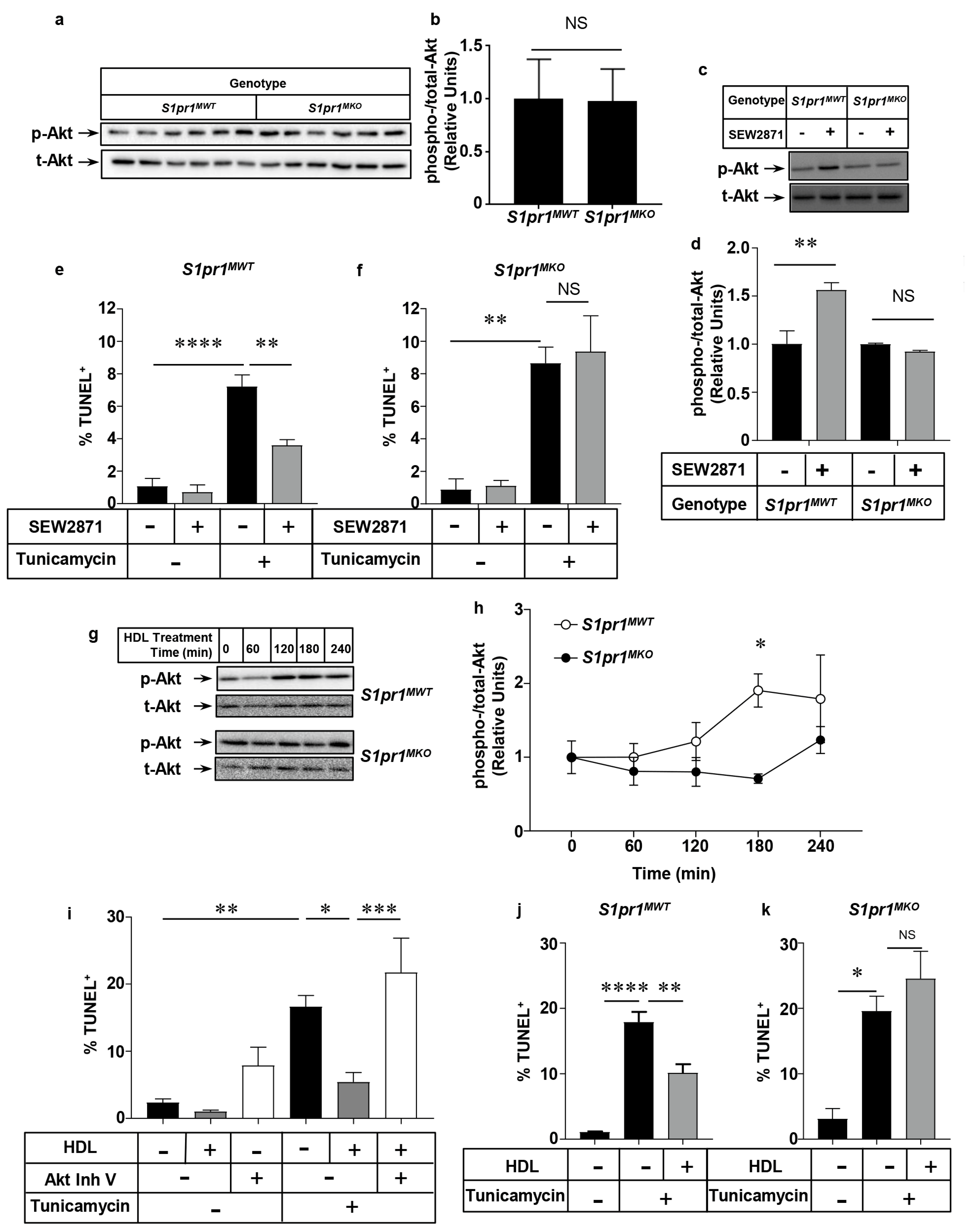
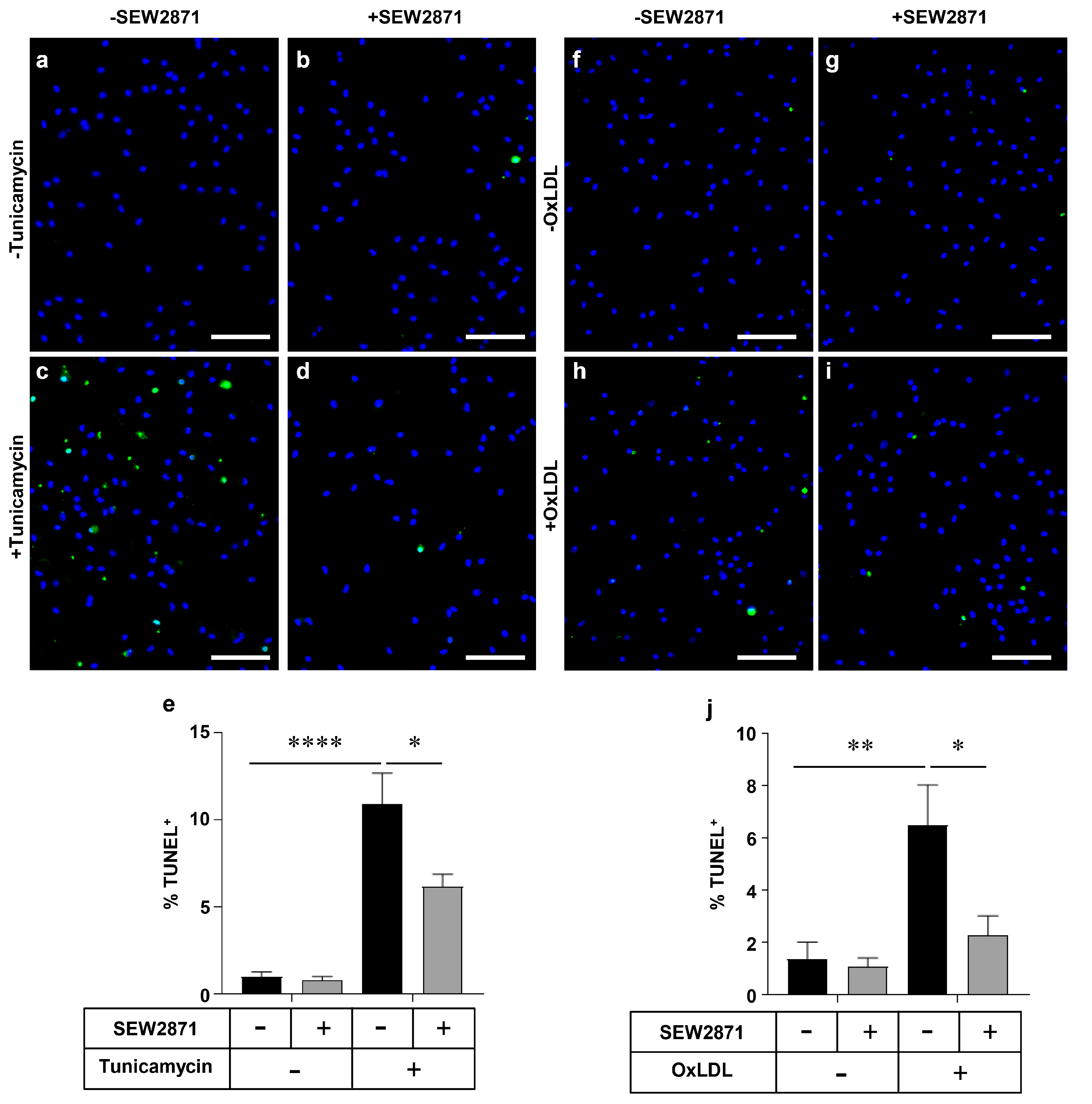
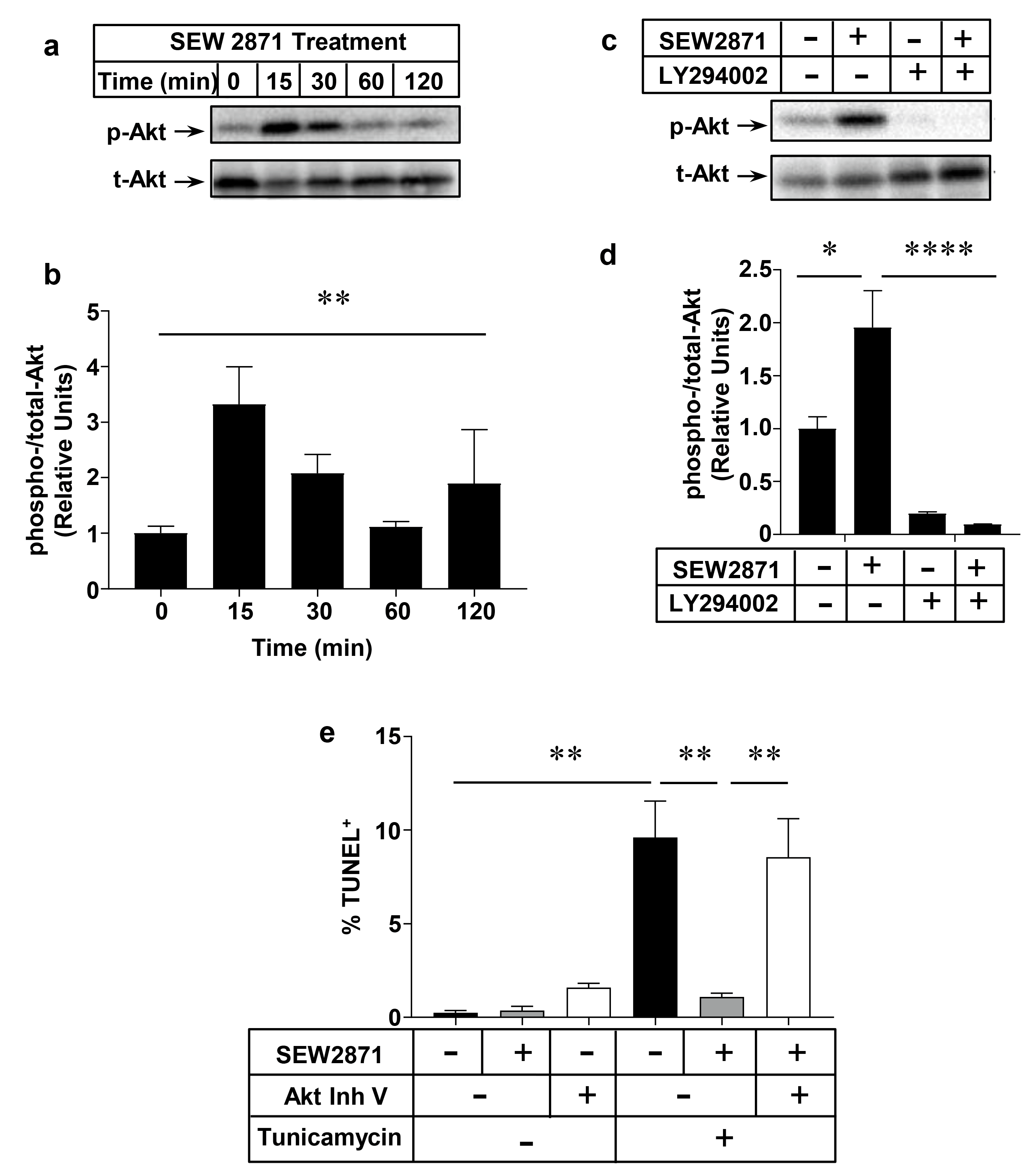
2.3. SEW2871, a Selective S1PR1 Agonist, Protects Macrophages against Apoptosis
2.4. Role of the PI3K/Akt Pathway in S1PR1 Agonist-Mediated Protection against Macrophage Apoptosis
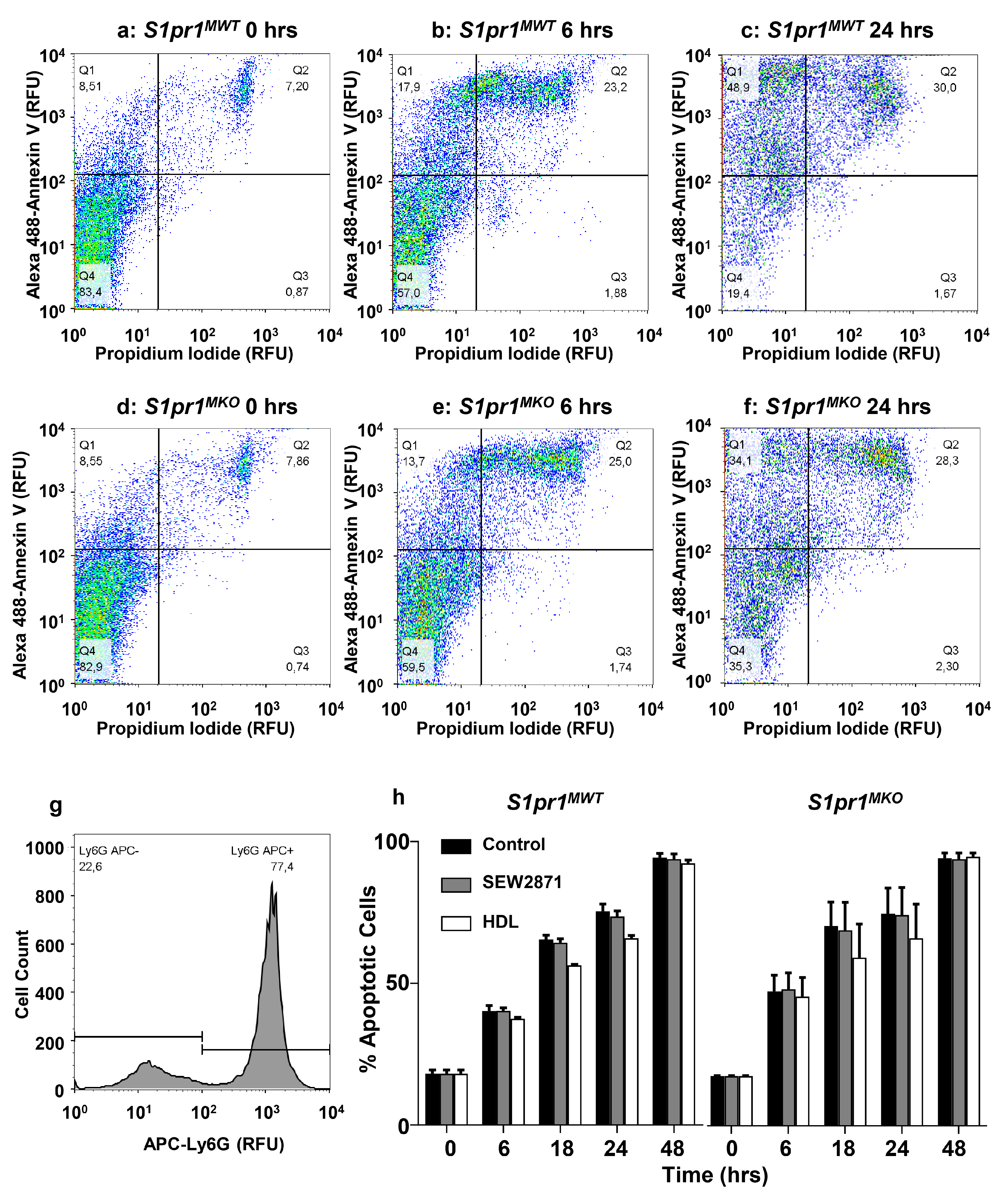
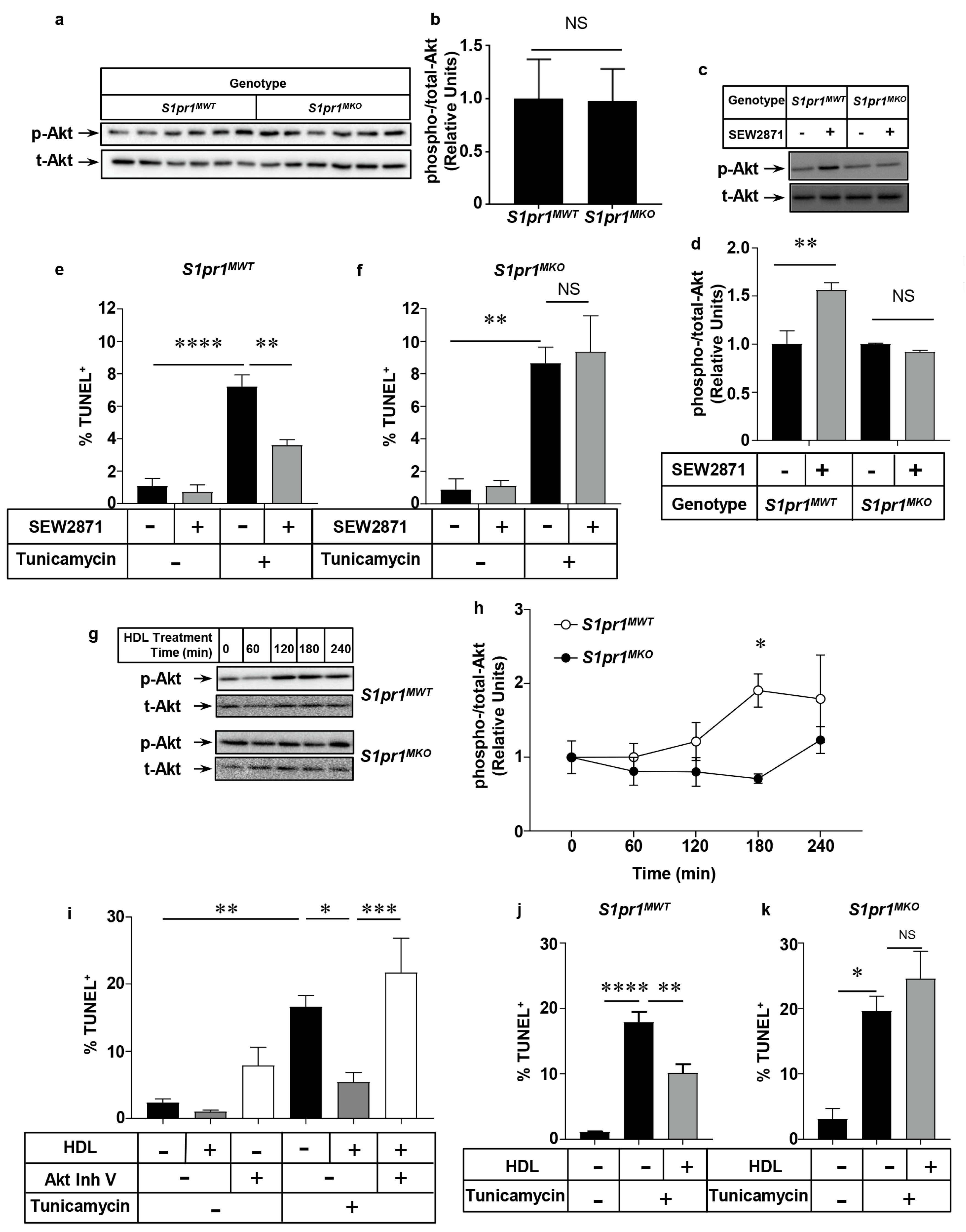
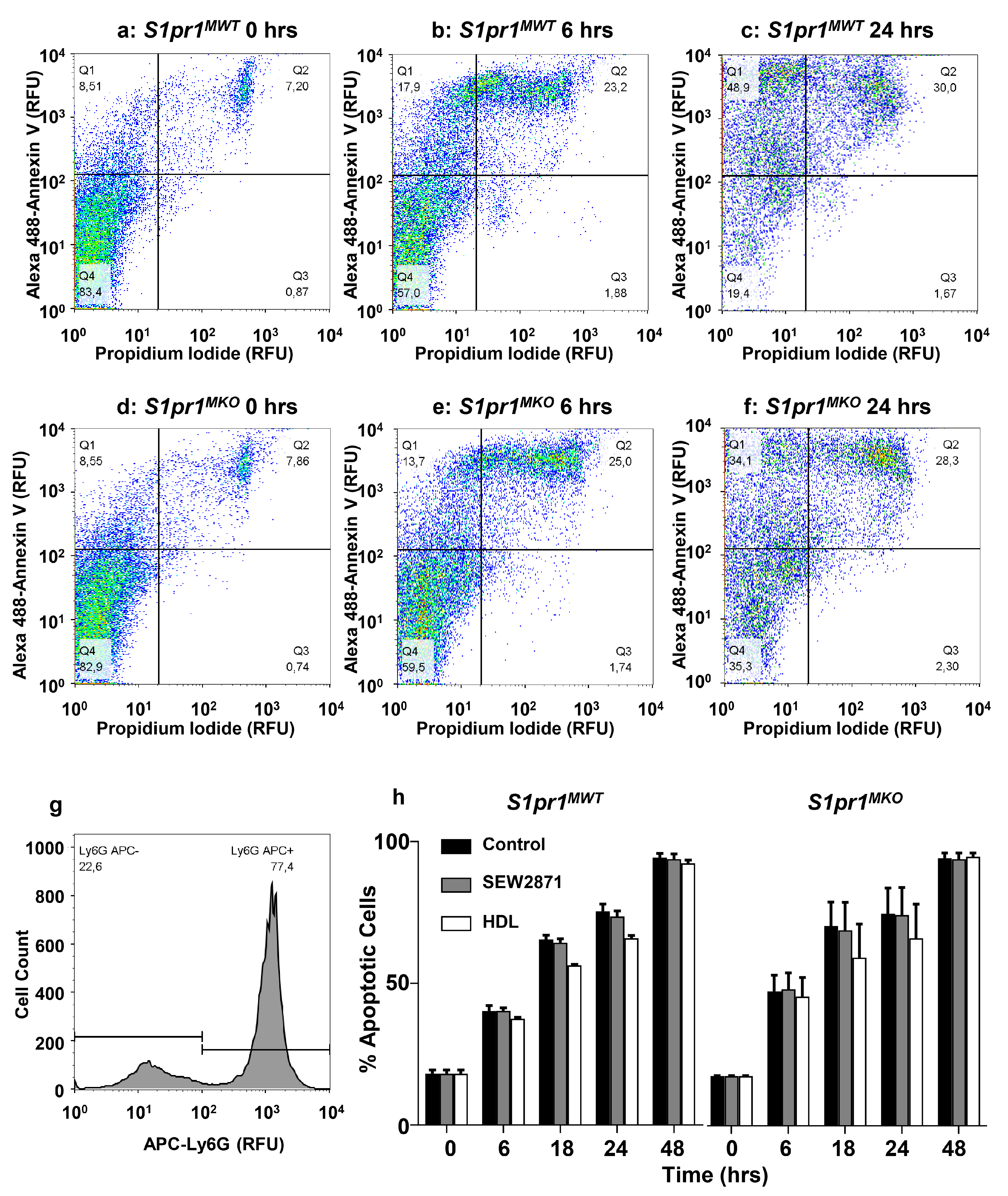
2.5. Effect of Macrophage S1PR1 Deficiency on Protection against Apoptosis
2.6. HDL Protects Macrophages from Apoptosis in an Akt and S1PR1 Dependent Manner
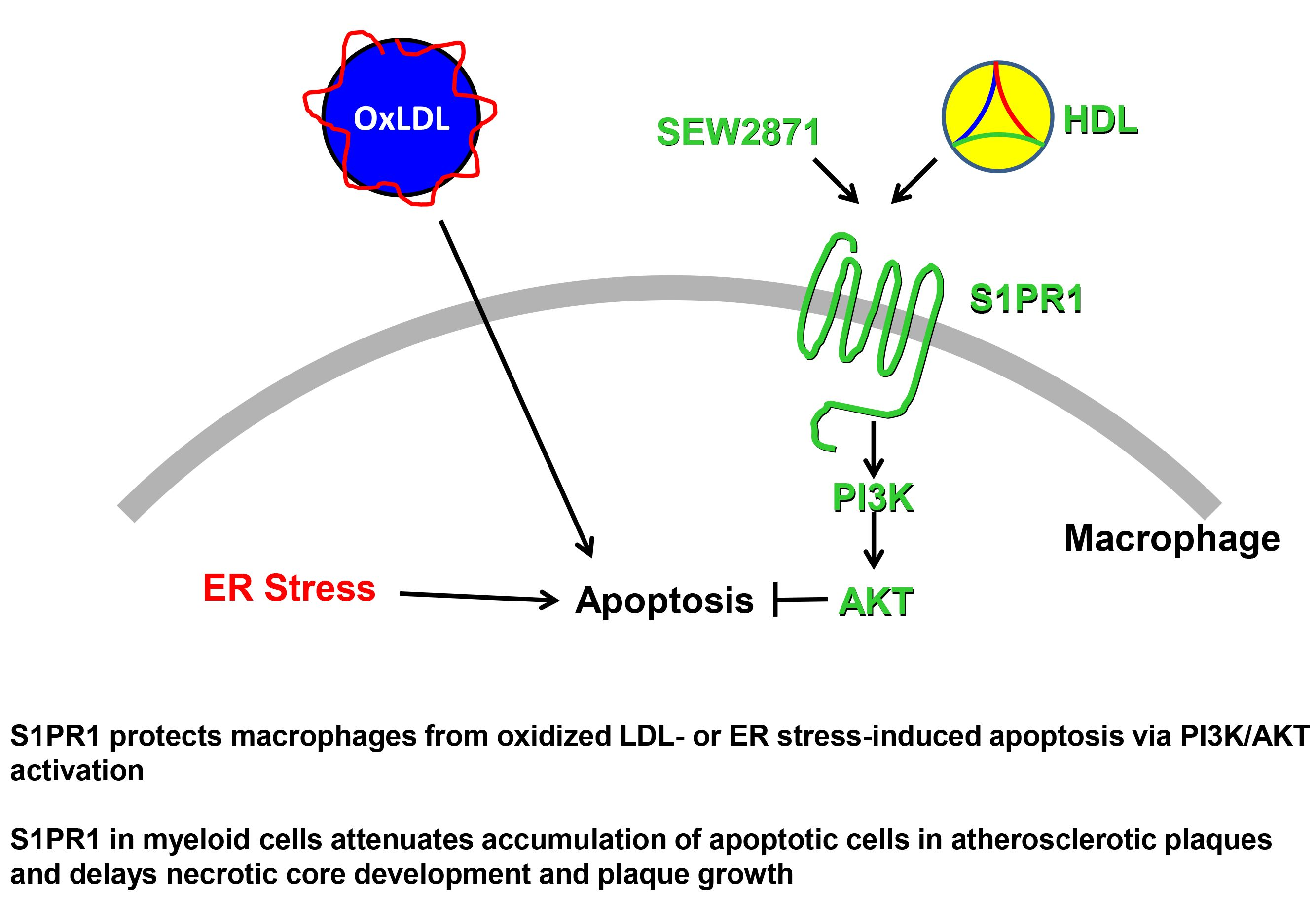
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Mice
4.3. BM Transplantation
4.4. Plasma Lipids
4.5. Histology
4.6. Cell Preparation, Culture and Treatment
4.7. RT-PCR
4.8. Apoptosis
4.9. Electrophoresis and Immunoblotting
4.10. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Apo | apolipoprotein |
| BM | bone marrow |
| DAPI | 4′,6′-diamidino-2-phenylindole |
| ER | endoplasmic reticulum |
| HDL | high density lipoprotein |
| HF | high fat |
| JAK2 | Janus kinase 2 |
| KO | knockout |
| LDL | low density lipoprotein |
| OxLDL | oxidized LDL |
| PI3K | phosphatidylinositol-3-kinase |
| S1P | sphingosine-1-phosphate |
| S1PR | sphingosine-1-phosphate receptor |
| SR-B1 | scavenger receptor class B type 1 |
| STAT3 | signal transducer and activator of transcription 3 |
| TUNEL | terminal deoxynucleotidyl transferase dUTP nick end labeling |
References
- Gimbrone, M.A., Jr.; Topper, J.N.; Nagel, T.; Anderson, K.R.; Garcia-Cardena, G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann. N. Y. Acad. Sci. 2000, 902, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Geng, Y.J.; Aikawa, M.; Schoenbeck, U.; Mach, F.; Clinton, S.K.; Sukhova, G.K.; Lee, R.T. Macrophages and atherosclerotic plaque stability. Curr. Opin. Lipidol. 1996, 7, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Macrophage apoptosis in atherosclerosis: Consequences on plaque progression and the role of endoplasmic reticulum stress. Antioxid. Redox Signal. 2009, 11, 2333–2339. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.; Trigatti, B.L. Macrophage Apoptosis and Necrotic Core Development in Atherosclerosis: A Rapidly Advancing Field with Clinical Relevance to Imaging and Therapy. Can. J. Cardiol. 2017, 33, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Kavurma, M.M.; Rayner, K.J.; Karunakaran, D. The walking dead: Macrophage inflammation and death in atherosclerosis. Curr. Opin. Lipidol. 2017, 28, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2010, 10, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, D.; Geoffrion, M.; Wei, L.; Gan, W.; Richards, L.; Shangari, P.; DeKemp, E.M.; Beanlands, R.A.; Perisic, L.; Maegdefessel, L.; et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci. Adv. 2016, 2, e1600224. [Google Scholar] [CrossRef] [PubMed]
- Maiolino, G.; Rossitto, G.; Caielli, P.; Bisogni, V.; Rossi, G.P.; Calo, L.A. The role of oxidized low-density lipoproteins in atherosclerosis: The myths and the facts. Med. Inflamm. 2013, 2013, 714653. [Google Scholar] [CrossRef] [PubMed]
- Martinet, W.; Kockx, M.M. Apoptosis in atherosclerosis: Focus on oxidized lipids and inflammation. Curr. Opin. Lipidol. 2001, 12, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.T.; Parthasarathy, S.; Fong, L.G.; Steinberg, D. Oxidatively modified low density lipoproteins: A potential role in recruitment and retention of monocyte/macrophages during atherogenesis. Proc. Natl. Acad. Sci. USA 1987, 84, 2995–2998. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Werstuck, G.H.; Lhotak, S.; de Koning, A.B.; Sood, S.K.; Hossain, G.S.; Moller, J.; Ritskes-Hoitinga, M.; Falk, E.; Dayal, S.; et al. Association of multiple cellular stress pathways with accelerated atherosclerosis in hyperhomocysteinemic apolipoprotein E-deficient mice. Circulation 2004, 110, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lhotak, S.; Hilditch, B.A.; Austin, R.C. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation 2005, 111, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Myoishi, M.; Hao, H.; Minamino, T.; Watanabe, K.; Nishihira, K.; Hatakeyama, K.; Asada, Y.; Okada, K.; Ishibashi-Ueda, H.; Gabbiani, G.; et al. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation 2007, 116, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Lhotak, S.; Zhou, J.; Austin, R.C. Immunohistochemical detection of the unfolded protein response in atherosclerotic plaques. Methods Enzymol. 2011, 489, 23–46. [Google Scholar] [PubMed]
- Khan, M.I.; Pichna, B.A.; Shi, Y.; Bowes, A.J.; Werstuck, G.H. Evidence supporting a role for endoplasmic reticulum stress in the development of atherosclerosis in a hyperglycaemic mouse model. Antioxid. Redox Signal. 2009, 11, 2289–2298. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.X.; Tabas, I. The UPR in atherosclerosis. Semin. Immunopathol. 2013, 35, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, T.; Hla, T. Structural and functional characteristics of S1P receptors. J. Cell. Biochem. 2004, 92, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Weigert, A.; Johann, A.M.; von Knethen, A.; Schmidt, H.; Geisslinger, G.; Brune, B. Apoptotic cells promote macrophage survival by releasing the antiapoptotic mediator sphingosine-1-phosphate. Blood 2006, 108, 1635–1642. [Google Scholar] [CrossRef] [PubMed]
- Weigert, A.; Weis, N.; Brune, B. Regulation of macrophage function by sphingosine-1-phosphate. Immunobiology 2009, 214, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Bot, M.; Van Veldhoven, P.P.; de Jager, S.C.; Johnson, J.; Nijstad, N.; Van Santbrink, P.J.; Westra, M.M.; Van Der Hoeven, G.; Gijbels, M.J.; Muller-Tidow, C.; et al. Hematopoietic sphingosine 1-phosphate lyase deficiency decreases atherosclerotic lesion development in LDL-receptor deficient mice. PLoS ONE 2013, 8, e63360. [Google Scholar] [CrossRef] [PubMed]
- Poti, F.; Ceglarek, U.; Burkhardt, R.; Simoni, M.; Nofer, J.R. SKI-II—A sphingosine kinase 1 inhibitor—Exacerbates atherosclerosis in low-density lipoprotein receptor-deficient (LDL-R-/-) mice on high cholesterol diet. Atherosclerosis 2015, 240, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Nofer, J.R.; Bot, M.; Brodde, M.; Taylor, P.J.; Salm, P.; Brinkmann, V.; van Berkel, T.; Assmann, G.; Biessen, E.A. FTY720, a synthetic sphingosine 1 phosphate analogue, inhibits development of atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 2007, 115, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Poti, F.; Costa, S.; Bergonzini, V.; Galletti, M.; Pignatti, E.; Weber, C.; Simoni, M.; Nofer, J.R. Effect of sphingosine 1-phosphate (S1P) receptor agonists FTY720 and CYM5442 on atherosclerosis development in LDL receptor deficient (LDL-R(-)/(-)) mice. Vasc. Pharmacol. 2012, 57, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Poti, F.; Gualtieri, F.; Sacchi, S.; Weissen-Plenz, G.; Varga, G.; Brodde, M.; Weber, C.; Simoni, M.; Nofer, J.R. KRP-203, sphingosine 1-phosphate receptor type 1 agonist, ameliorates atherosclerosis in LDL-R-/- mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Galvani, S.; Sanson, M.; Blaho, V.A.; Swendeman, S.L.; Obinata, H.; Conger, H.; Dahlback, B.; Kono, M.; Proia, R.L.; Smith, J.D.; et al. HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci. Signal. 2015, 8, ra79. [Google Scholar] [CrossRef] [PubMed]
- Skoura, A.; Michaud, J.; Im, D.S.; Thangada, S.; Xiong, Y.; Smith, J.D.; Hla, T. Sphingosine-1-phosphate receptor-2 function in myeloid cells regulates vascular inflammation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Allende, M.L.; Yamashita, T.; Proia, R.L. G-protein-coupled receptor S1P1 acts within endothelial cells to regulate vascular maturation. Blood 2003, 102, 3665–3667. [Google Scholar] [CrossRef] [PubMed]
- Clausen, B.E.; Burkhardt, C.; Reith, W.; Renkawitz, R.; Forster, I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999, 8, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Sanchez, L.; Espinosa-Luna, J.E.; Chavez-Rueda, K.; Legorreta-Haquet, M.V.; Montoya-Diaz, E.; Blanco-Favela, F. Innate immune system cells in atherosclerosis. Arch. Med. Res. 2014, 45, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rotzius, P.; Thams, S.; Soehnlein, O.; Kenne, E.; Tseng, C.N.; Bjorkstrom, N.K.; Malmberg, K.J.; Lindbom, L.; Eriksson, E.E. Distinct infiltration of neutrophils in lesion shoulders in ApoE-/- mice. Am. J. Pathol. 2010, 177, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Im, D.S. Pharmacological tools for lysophospholipid GPCRs: Development of agonists and antagonists for LPA and S1P receptors. Acta Pharmacol. Sin. 2010, 31, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Al-Jarallah, A.; Chen, X.; Gonzalez, L.; Trigatti, B.L. High density lipoprotein stimulated migration of macrophages depends on the scavenger receptor class B, type I, PDZK1 and Akt1 and is blocked by sphingosine 1 phosphate receptor antagonists. PLoS ONE 2014, 9, e106487. [Google Scholar] [CrossRef] [PubMed]
- Christoffersen, C.; Obinata, H.; Kumaraswamy, S.B.; Galvani, S.; Ahnstrom, J.; Sevvana, M.; Egerer-Sieber, C.; Muller, Y.A.; Hla, T.; Nielsen, L.B.; et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc. Natl. Acad. Sci. USA 2011, 108, 9613–9618. [Google Scholar] [CrossRef] [PubMed]
- Poti, F.; Simoni, M.; Nofer, J.R. Atheroprotective role of high-density lipoprotein (HDL)-associated sphingosine-1-phosphate (S1P). Cardiovasc. Res. 2014, 103, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Christoffersen, C.; Jauhiainen, M.; Moser, M.; Porse, B.; Ehnholm, C.; Boesl, M.; Dahlback, B.; Nielsen, L.B. Effect of apolipoprotein M on high density lipoprotein metabolism and atherosclerosis in low density lipoprotein receptor knock-out mice. J. Biol. Chem. 2008, 283, 1839–1847. [Google Scholar] [CrossRef] [PubMed]
- Graler, M.H. Targeting sphingosine 1-phosphate (S1P) levels and S1P receptor functions for therapeutic immune interventions. Cell. Physiol. Biochem. 2010, 26, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Sato, K.; Kuwabara, A.; Tomura, H.; Ishiwara, M.; Kobayashi, I.; Ui, M.; Okajima, F. Sphingosine 1-phosphate may be a major component of plasma lipoproteins responsible for the cytoprotective actions in human umbilical vein endothelial cells. J. Biol. Chem. 2001, 276, 31780–31785. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Sato, K.; Malchinkhuu, E.; Tomura, H.; Tamama, K.; Kuwabara, A.; Murakami, M.; Okajima, F. High-density lipoprotein stimulates endothelial cell migration and survival through sphingosine 1-phosphate and its receptors. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1283–1288. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Tomura, H.; Mogi, C.; Kuwabara, A.; Damirin, A.; Ishizuka, T.; Sekiguchi, A.; Ishiwara, M.; Im, D.S.; Sato, K.; et al. Role of scavenger receptor class B type I and sphingosine 1-phosphate receptors in high density lipoprotein-induced inhibition of adhesion molecule expression in endothelial cells. J. Biol. Chem. 2006, 281, 37457–37467. [Google Scholar] [CrossRef] [PubMed]
- Rodrigueza, W.V.; Thuahnai, S.T.; Temel, R.E.; Lund-Katz, S.; Phillips, M.C.; Williams, D.L. Mechanism of scavenger receptor class B type I-mediated selective uptake of cholesteryl esters from high density lipoprotein to adrenal cells. J. Biol. Chem. 1999, 274, 20344–20350. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.M.; Murao, K.; Imachi, H.; Yu, X.; Abe, H.; Yamauchi, A.; Niimi, M.; Miyauchi, A.; Wong, N.C.; Ishida, T. A mutant high-density lipoprotein receptor inhibits proliferation of human breast cancer cells. Cancer Res. 2004, 64, 1515–1521. [Google Scholar] [CrossRef] [PubMed]
- Danilo, C.; Gutierrez-Pajares, J.L.; Mainieri, M.A.; Mercier, I.; Lisanti, M.P.; Frank, P.G. Scavenger receptor class B type I regulates cellular cholesterol metabolism and cell signaling associated with breast cancer development. Breast Cancer Res. BCR 2013, 15, R87. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Zhao, D.; Schouteden, S.; Sorci-Thomas, M.G.; Van Veldhoven, P.P.; Eggermont, K.; Liu, G.; Verfaillie, C.M.; Feng, Y. Regulation of high-density lipoprotein on hematopoietic stem/progenitor cells in atherosclerosis requires scavenger receptor type BI expression. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1900–1909. [Google Scholar] [CrossRef] [PubMed]
- Mineo, C.; Shaul, P.W. HDL stimulation of endothelial nitric oxide synthase: A novel mechanism of HDL action. Trends Cardiovasc. Med. 2003, 13, 226–231. [Google Scholar] [CrossRef]
- Tan, J.T.; Prosser, H.C.; Dunn, L.L.; Vanags, L.Z.; Ridiandries, A.; Tsatralis, T.; Leece, L.; Clayton, Z.E.; Yuen, S.C.; Robertson, S.; et al. High-Density Lipoproteins Rescue Diabetes-Impaired Angiogenesis via Scavenger Receptor Class B Type I. Diabetes 2016, 65, 3091–3103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ahmed, A.M.; McFarlane, N.; Capone, C.; Boreham, D.R.; Truant, R.; Igdoura, S.A.; Trigatti, B.L. Regulation of SR-BI-mediated selective lipid uptake in Chinese hamster ovary-derived cells by protein kinase signaling pathways. J. Lipid Res. 2007, 48, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Feuerborn, R.; Becker, S.; Poti, F.; Nagel, P.; Brodde, M.; Schmidt, H.; Christoffersen, C.; Ceglarek, U.; Burkhardt, R.; Nofer, J.R. High density lipoprotein (HDL)-associated sphingosine 1-phosphate (S1P) inhibits macrophage apoptosis by stimulating STAT3 activity and survivin expression. Atherosclerosis 2017, 257, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Gan, W.; Liu, Z.; Shen, Z.; Wang, J.; Shi, R.; Liu, Y.; Liu, Y.; Jiang, M.; Zhang, Z.; Wu, Y. Erythropoeitin Signaling in Macrophages Promotes Dying Cell Clearance and Immune Tolerance. Immunity 2016, 44, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Sukocheva, O.; Wadham, C.; Gamble, J.; Xia, P. Sphingosine-1-phosphate receptor 1 transmits estrogens’ effects in endothelial cells. Steroids 2015, 104, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Qian, J.; Xie, X.; Lin, L.; Ma, J.; Huang, Z.; Fu, M.; Zou, Y.; Ge, J. High density lipoprotein cholesterol promotes the proliferation of bone-derived mesenchymal stem cells via binding scavenger receptor-B type I and activation of PI3K/Akt, MAPK/ERK1/2 pathways. Mol. Cell. Biochem. 2012, 371, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Osaki, M.; Oshimura, M.; Ito, H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Bruno, B.; Giaccone, L.; Rotta, M.; Anderson, K.; Boccadoro, M. Novel targeted drugs for the treatment of multiple myeloma: From bench to bedside. Leukemia 2005, 19, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Park, J.; Han, S.W.; Im, S.A.; Kim, T.Y.; Oh, D.Y.; Bang, Y.J. NVP-BKM120, a novel PI3K inhibitor, shows synergism with a STAT3 inhibitor in human gastric cancer cells harboring KRAS mutations. Int. J. Oncol. 2012, 40, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Park, K.W.; Nozell, S.E.; Benveniste, E.N. Protective role of STAT3 in NMDA and glutamate-induced neuronal death: Negative regulatory effect of SOCS3. PLoS ONE 2012, 7, e50874. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wang, H.; Guan, H.; Shi, S.; Li, Y.; Wu, X.; Li, N.; Yang, C.; Bai, X.; Cai, W.; et al. IL10 inhibits starvation-induced autophagy in hypertrophic scar fibroblasts via cross talk between the IL10-IL10R-STAT3 and IL10-AKT-mTOR pathways. Cell Death Dis. 2016, 7, e2133. [Google Scholar] [CrossRef] [PubMed]
- Covey, S.D.; Krieger, M.; Wang, W.; Penman, M.; Trigatti, B.L. Scavenger receptor class B type I-mediated protection against atherosclerosis in LDL receptor-negative mice involves its expression in bone marrow-derived cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1589–1594. [Google Scholar] [CrossRef] [PubMed]
- Rigotti, A.; Trigatti, B.L.; Penman, M.; Rayburn, H.; Herz, J.; Krieger, M. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc. Natl. Acad. Sci. USA 1997, 94, 12610–12615. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.; Dadoo, O.; Serkis, V.; Abutouk, D.; MacDonald, M.; Dhingani, N.; Macri, J.; Igdoura, S.A.; Trigatti, B.L. The effects of diet on occlusive coronary artery atherosclerosis and myocardial infarction in scavenger receptor class B, type 1/low-density lipoprotein receptor double knockout mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2394–2403. [Google Scholar] [CrossRef] [PubMed]
- Swamydas, M.; Luo, Y.; Dorf, M.E.; Lionakis, M.S. Isolation of Mouse Neutrophils. Curr. Protoc. Immunol. 2015, 110, 3–20. [Google Scholar] [PubMed]
- Trigatti, B.L.; Mangroo, D.; Gerber, G.E. Photoaffinity labeling and fatty acid permeation in 3T3-L1 adipocytes. J. Biol. Chem. 1991, 266, 22621–22625. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Forward Primer Sequence | Reverse Primer Sequence |
|---|---|---|
| S1pr1 | 5′-ACT TTG CGA GTG AGC TG-3′ | 5′-AGT GAG CCT TCA GTT ACA GC-3′ |
| S1pr2 | 5′-TTC TGG AGG GTA ACA CAG TGG T-3′ | 5′-ACA CCC TTT GTA TCA AGT GGC A-3′ |
| S1pr3 | 5′-TGG TGT GCG GCT GTC TAG TCA A-3′ | 5′-CAC AGC AAG CAG ACC TCC AGA-3′ |
| S1pr4 | 5′-AAC CAA AGA TGT CAG CCA GG-3′ | 5′-GCA GAA GTCT CCA CGT CCT C-3′ |
| S1pr5 | 5′-GTC GTC CAC TGG AGC ACT G-3′ | 5′-GTA CAC CAA ATG CCC AGC TT-3′ |
| Gapdh | 5′-ACC ACA GTC CAT GCC ATC AC-3′ | 5′-TCC ACC ACC CTG TTG CTG TA-3′ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez, L.; Qian, A.S.; Tahir, U.; Yu, P.; Trigatti, B.L. Sphingosine-1-Phosphate Receptor 1, Expressed in Myeloid Cells, Slows Diet-Induced Atherosclerosis and Protects against Macrophage Apoptosis in Ldlr KO Mice. Int. J. Mol. Sci. 2017, 18, 2721. https://doi.org/10.3390/ijms18122721
Gonzalez L, Qian AS, Tahir U, Yu P, Trigatti BL. Sphingosine-1-Phosphate Receptor 1, Expressed in Myeloid Cells, Slows Diet-Induced Atherosclerosis and Protects against Macrophage Apoptosis in Ldlr KO Mice. International Journal of Molecular Sciences. 2017; 18(12):2721. https://doi.org/10.3390/ijms18122721
Chicago/Turabian StyleGonzalez, Leticia, Alexander S. Qian, Usama Tahir, Pei Yu, and Bernardo L. Trigatti. 2017. "Sphingosine-1-Phosphate Receptor 1, Expressed in Myeloid Cells, Slows Diet-Induced Atherosclerosis and Protects against Macrophage Apoptosis in Ldlr KO Mice" International Journal of Molecular Sciences 18, no. 12: 2721. https://doi.org/10.3390/ijms18122721