Translational Aspects of Sphingolipid Metabolism in Renal Disorders

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Podocytes
1.1. Renal Glomerular Podocyte Role and Function
1.2. Podocyte Injury
2. Sphingolipids
2.1. Overview of Sphingolipid Metabolism
2.2. Sphingolipids in Health and Disease
2.2.1. Sphingolipids and Cancer Treatment
2.2.2. Sphingolipids and Inflammatory Responses
2.2.3. Sphingolipids and Insulin Resistance
3. Sphingolipids in Podocyte Injury
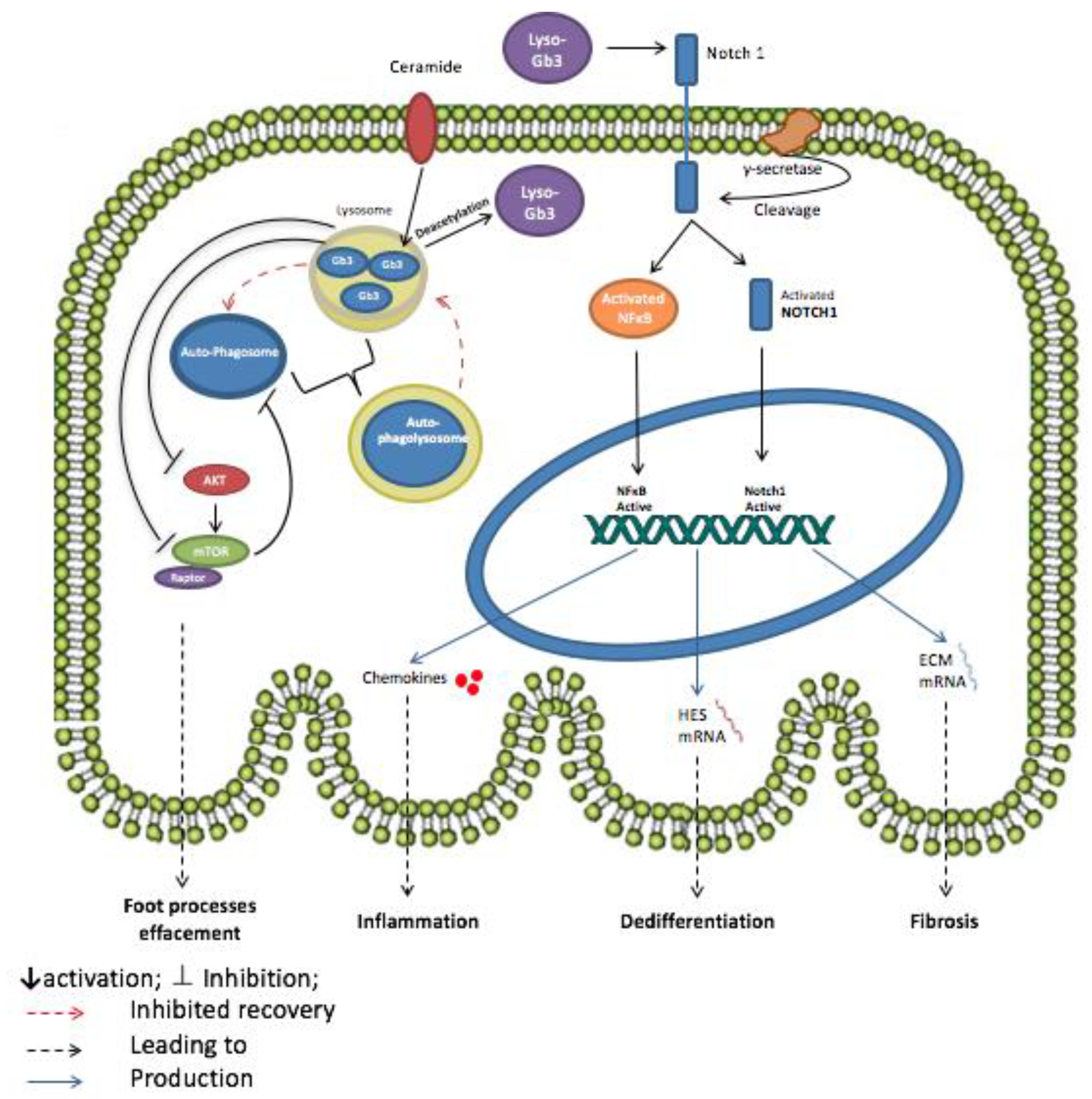
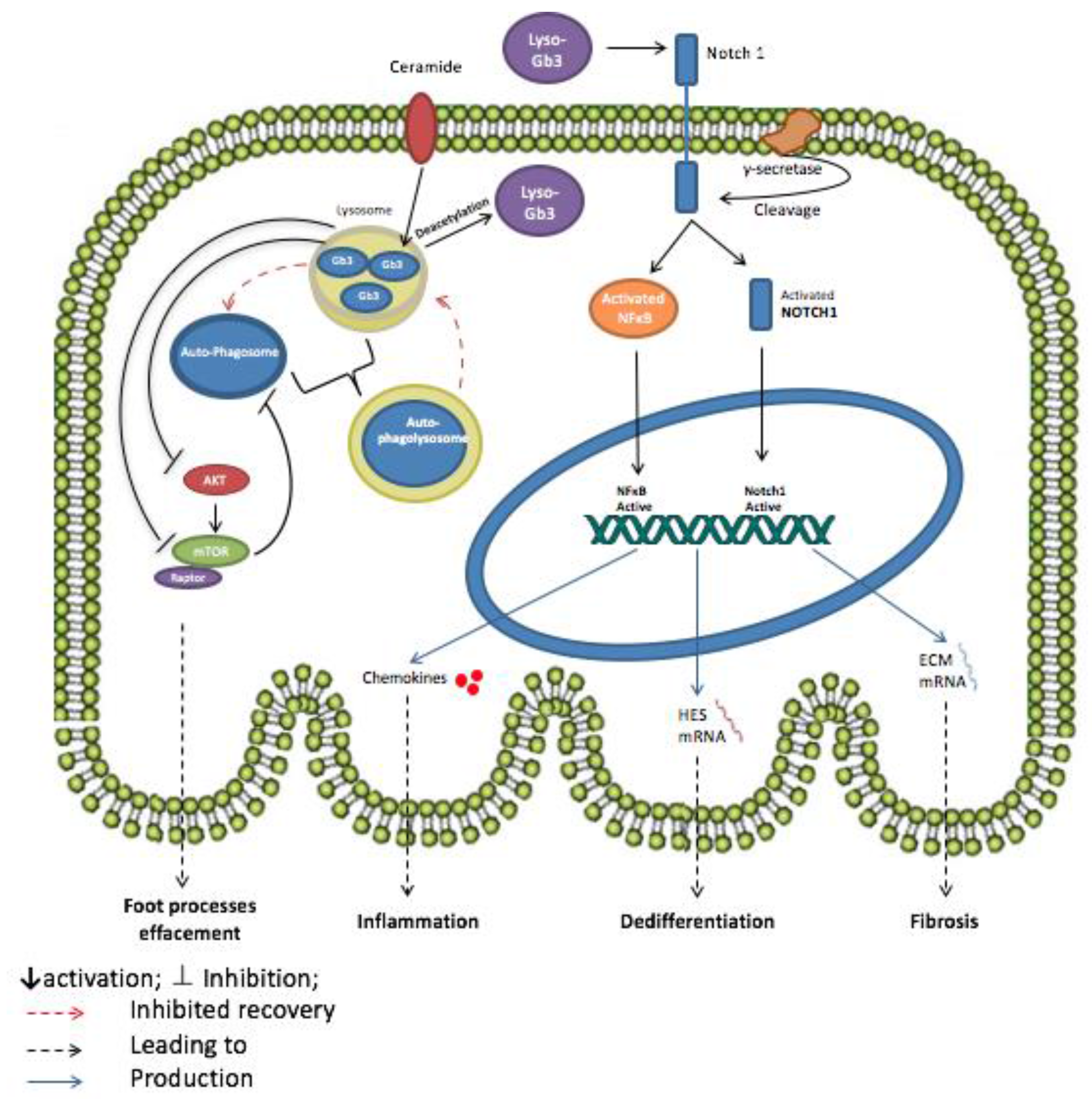
3.1. Globotriaosylceramide (Gb3)
3.2. The Sphingomyelinase-Pathway
3.3. Sphingosine-1-Phosphate
4. Sphingolipids as Potential Therapeutic Targets
5. Conslusions
Acknowledgments
Conflicts of Interest
References
- Pavenstadt, H.; Kriz, W.; Kretzler, M. Cell biology of the glomerular podocyte. Physiol. Rev. 2003, 83, 253–307. [Google Scholar] [CrossRef] [PubMed]
- Merscher, S.; Fornoni, A. Podocyte pathology and nephropathy—Sphingolipids in glomerular diseases. Front. Endocrinol. 2014, 5, 127. [Google Scholar] [CrossRef] [PubMed]
- Cellesi, F.; Li, M.; Rastaldi, M.P. Podocyte injury and repair mechanisms. Curr. Opin. Nephrol. Hypertens. 2015, 24, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Nagata, M. Podocyte injury and its consequences. Kidney Int. 2016, 89, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Futerman, A.H.; Hannun, Y.A. The complex life of simple sphingolipids. EMBO Rep. 2004, 5, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Kollmeyer, J.; Symolon, H.; Momin, A.; Munter, E.; Wang, E.; Kelly, S.; Allegood, J.C.; Liu, Y.; Peng, Q.; et al. Ceramides and other bioactive sphingolipid backbones in health and disease: Lipidomic analysis, metabolism and roles in membrane structure, dynamics, signaling and autophagy. Biochim. Biophys. Acta 2006, 1758, 1864–1884. [Google Scholar] [CrossRef] [PubMed]
- Kolesnick, R.N.; Kronke, M. Regulation of ceramide production and apoptosis. Annu. Rev. Physiol. 1998, 60, 643–665. [Google Scholar] [CrossRef] [PubMed]
- Luberto, C.; Hannun, Y.A. Sphingolipid metabolism in the regulation of bioactive molecules. Lipids 1999, 34, S5–S11. [Google Scholar] [CrossRef] [PubMed]
- Pettus, B.J.; Bielawska, A.; Spiegel, S.; Roddy, P.; Hannun, Y.A.; Chalfant, C.E. Ceramide kinase mediates cytokine- and calcium ionophore-induced arachidonic acid release. J. Biol. Chem. 2003, 278, 38206–38213. [Google Scholar] [CrossRef] [PubMed]
- Pettus, B.J.; Kroesen, B.J.; Szulc, Z.M.; Bielawska, A.; Bielawski, J.; Hannun, Y.A.; Busman, M. Quantitative measurement of different ceramide species from crude cellular extracts by normal-phase high-performance liquid chromatography coupled to atmospheric pressure ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2004, 18, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, Y.H.; Hannun, Y.A. Translational aspects of sphingolipid metabolism. Trends Mol. Med. 2007, 13, 327–336. [Google Scholar] [CrossRef] [PubMed]
- El Alwani, M.; Wu, B.X.; Obeid, L.M.; Hannun, Y.A. Bioactive sphingolipids in the modulation of the inflammatory response. Pharmacol. Ther. 2006, 112, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Ogretmen, B. Sphingolipids in cancer: Regulation of pathogenesis and therapy. FEBS Lett. 2006, 580, 5467–5476. [Google Scholar] [CrossRef] [PubMed]
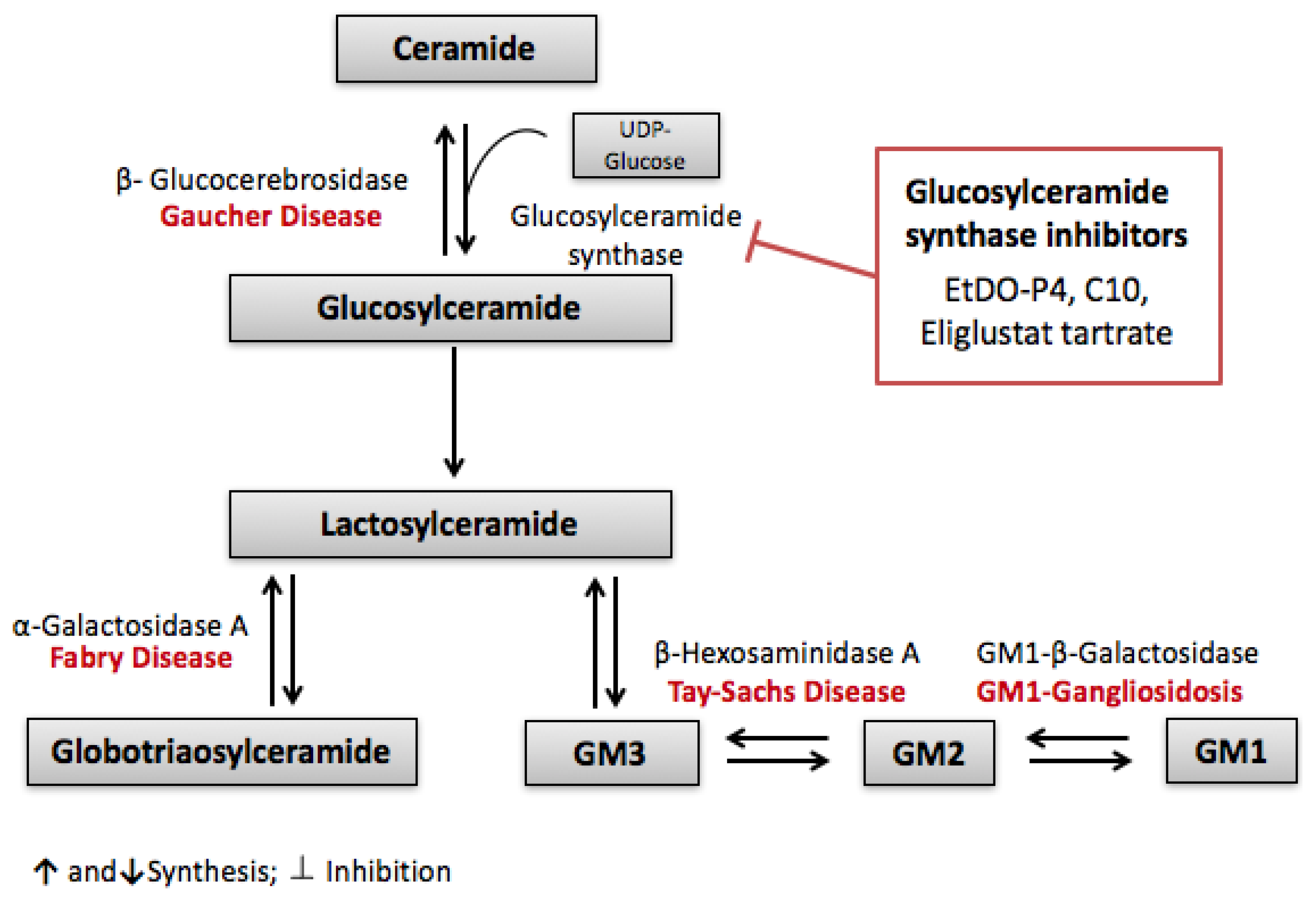
- Kolter, T.; Sandhoff, K. Sphingolipid metabolism diseases. Biochim. Biophys. Acta 2006, 1758, 2057–2079. [Google Scholar] [CrossRef] [PubMed]
- Riboni, L.; Viani, P.; Bassi, R.; Prinetti, A.; Tettamanti, G. The role of sphingolipids in the process of signal transduction. Prog. Lipid Res. 1997, 36, 153–195. [Google Scholar] [CrossRef]
- Kolesnick, R.N.; Goni, F.M.; Alonso, A. Compartmentalization of ceramide signaling: Physical foundations and biological effects. J. Cell. Physiol. 2000, 184, 285–300. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. The Ceramide-centric universe of lipid-mediated cell regulation: Stress encounters of the lipid kind. J. Biol. Chem. 2002, 277, 25847–25850. [Google Scholar] [CrossRef] [PubMed]
- Kroesen, B.J.; Jacobs, S.; Pettus, B.J.; Sietsma, H.; Kok, J.W.; Hannun, Y.A.; de Leij, L.F. BcR-induced apoptosis involves differential regulation of C16 and C24-ceramide formation and sphingolipid-dependent activation of the proteasome. J. Biol. Chem. 2003, 278, 14723–14731. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Luberto, C. Lipid metabolism: Ceramide transfer protein adds a new dimension. Curr. Biol. 2004, 14, R163–R165. [Google Scholar] [CrossRef] [PubMed]
- Sundararaj, K.P.; Wood, R.E.; Ponnusamy, S.; Salas, A.M.; Szulc, Z.; Bielawska, A.; Obeid, L.M.; Hannun, Y.A.; Ogretmen, B. Rapid shortening of telomere length in response to ceramide involves the inhibition of telomere binding activity of nuclear glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 2004, 279, 6152–6162. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Kumagai, K.; Yasuda, S.; Miura, Y.; Kawano, M.; Fukasawa, M.; Nishijima, M. Molecular machinery for non-vesicular trafficking of ceramide. Nature 2003, 426, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Obeid, L.M.; Linardic, C.M.; Karolak, L.A.; Hannun, Y.A. Programmed cell death induced by ceramide. Science 1993, 259, 1769–1771. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Abe, A.; Shayman, J.A. Ceramide formation during heat shock: A potential mediator of α B-crystallin transcription. Proc. Natl. Acad. Sci. USA 1995, 92, 12275–12279. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, O.M.; Discher, D.J.; Bishopric, N.H.; Webster, K.A. Rapid activation of neutral sphingomyelinase by hypoxia-reoxygenation of cardiac myocytes. Circ. Res. 2000, 86, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Merrill, A.H., Jr.; Wang, E. Biosynthesis of long-chain (sphingoid) bases from serine by LM cells. Evidence for introduction of the 4-trans-double bond after de novo biosynthesis of N-acylsphinganine(s). J. Biol. Chem. 1986, 261, 3764–3769. [Google Scholar] [PubMed]
- Merrill, A.H., Jr.; Nixon, D.W.; Williams, R.D. Activities of serine palmitoyltransferase (3-ketosphinganine synthase) in microsomes from different rat tissues. J. Lipid Res. 1985, 26, 617–622. [Google Scholar] [PubMed]
- Perry, D.K.; Carton, J.; Shah, A.K.; Meredith, F.; Uhlinger, D.J.; Hannun, Y.A. Serine palmitoyltransferase regulates de novo ceramide generation during etoposide-induced apoptosis. J. Biol. Chem. 2000, 275, 9078–9084. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, K.; Yasuda, S.; Okemoto, K.; Nishijima, M.; Kobayashi, S.; Hanada, K. CERT mediates intermembrane transfer of various molecular species of ceramides. J. Biol. Chem. 2005, 280, 6488–6495. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Ridgway, N.D. Molecular mechanisms and regulation of ceramide transport. Biochim. Biophys. Acta 2005, 1734, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [PubMed]
- Tettamanti, G.; Bassi, R.; Viani, P.; Riboni, L. Salvage pathways in glycosphingolipid metabolism. Biochimie 2003, 85, 423–437. [Google Scholar] [CrossRef]
- Levy, M.; Futerman, A.H. Mammalian ceramide synthases. IUBMB Life 2010, 62, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Pewzner-Jung, Y.; Ben-Dor, S.; Futerman, A.H. When do Lasses (longevity assurance genes) become CerS (ceramide synthases)?: Insights into the regulation of ceramide synthesis. J. Biol. Chem. 2006, 281, 25001–25005. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.; van Echten-Deckert, G. Conversion of dihydroceramide to ceramide occurs at the cytosolic face of the endoplasmic reticulum. FEBS Lett. 1997, 416, 153–155. [Google Scholar] [CrossRef]
- Marchesini, N.; Hannun, Y.A. Acid and neutral sphingomyelinases: Roles and mechanisms of regulation. Biochem. Cell Biol. 2004, 82, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Abenoza, P.; Sibley, R.K. Farber’s disease: A fine structural study. Ultrastruct. Pathol. 1987, 11, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Bar, J.; Linke, T.; Ferlinz, K.; Neumann, U.; Schuchman, E.H.; Sandhoff, K. Molecular analysis of acid ceramidase deficiency in patients with Farber disease. Hum. Mutat. 2001, 17, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Ferlinz, K.; Kopal, G.; Bernardo, K.; Linke, T.; Bar, J.; Breiden, B.; Neumann, U.; Lang, F.; Schuchman, E.H.; Sandhoff, K. Human acid ceramidase: Processing, glycosylation, and lysosomal targeting. J. Biol. Chem. 2001, 276, 35352–35360. [Google Scholar] [CrossRef] [PubMed]
- El Bawab, S.; Bielawska, A.; Hannun, Y.A. Purification and characterization of a membrane-bound nonlysosomal ceramidase from rat brain. J. Biol. Chem. 1999, 274, 27948–27955. [Google Scholar] [CrossRef] [PubMed]
- Romiti, E.; Meacci, E.; Tani, M.; Nuti, F.; Farnararo, M.; Ito, M.; Bruni, P. Neutral/alkaline and acid ceramidase activities are actively released by murine endothelial cells. Biochem. Biophys. Res. Commun. 2000, 275, 746–751. [Google Scholar] [CrossRef] [PubMed]
- Tani, M.; Okino, N.; Mori, K.; Tanigawa, T.; Izu, H.; Ito, M. Molecular cloning of the full-length cDNA encoding mouse neutral ceramidase. A novel but highly conserved gene family of neutral/alkaline ceramidases. J. Biol. Chem. 2000, 275, 11229–11234. [Google Scholar] [CrossRef] [PubMed]
- El Bawab, S.; Roddy, P.; Qian, T.; Bielawska, A.; Lemasters, J.J.; Hannun, Y.A. Molecular cloning and characterization of a human mitochondrial ceramidase. J. Biol. Chem. 2000, 275, 21508–21513. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, Y.; Okino, N.; Tani, M.; Ito, M. Molecular cloning and characterization of a secretory neutral ceramidase of Drosophila melanogaster. J. Biochem. 2002, 132, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, Y.; Tani, M.; Okino, N.; Iida, H.; Ito, M. Molecular cloning and functional analysis of zebrafish neutral ceramidase. J. Biol. Chem. 2004, 279, 44012–44022. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Xu, R.; Bielawska, A.; Szulc, Z.M.; Obeid, L.M. Cloning and characterization of a Saccharomyces cerevisiae alkaline ceramidase with specificity for dihydroceramide. J. Biol. Chem. 2000, 275, 31369–31378. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Xu, R.; Bielawska, A.; Obeid, L.M. Cloning of an alkaline ceramidase from Saccharomyces cerevisiae. An enzyme with reverse (CoA-independent) ceramide synthase activity. J. Biol. Chem. 2000, 275, 6876–6884. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.; van Echten-Deckert, G.; Rother, J.; Sandhoff, K.; Wang, E.; Merrill, A.H., Jr. Characterization of ceramide synthesis. A dihydroceramide desaturase introduces the 4,5-trans-double bond of sphingosine at the level of dihydroceramide. J. Biol. Chem. 1997, 272, 22432–22437. [Google Scholar] [CrossRef] [PubMed]
- Mitsutake, S.; Tani, M.; Okino, N.; Mori, K.; Ichinose, S.; Omori, A.; Iida, H.; Nakamura, T.; Ito, M. Purification, characterization, molecular cloning, and subcellular distribution of neutral ceramidase of rat kidney. J. Biol. Chem. 2001, 276, 26249–26259. [Google Scholar] [CrossRef] [PubMed]
- Tani, M.; Okino, N.; Mitsutake, S.; Tanigawa, T.; Izu, H.; Ito, M. Purification and characterization of a neutral ceramidase from mouse liver. A single protein catalyzes the reversible reaction in which ceramide is both hydrolyzed and synthesized. J. Biol. Chem. 2000, 275, 3462–3468. [Google Scholar] [CrossRef] [PubMed]
- Merrill, A.H., Jr.; Stevens, V.L. Modulation of protein kinase C and diverse cell functions by sphingosine—A pharmacologically interesting compound linking sphingolipids and signal transduction. Biochim. Biophys. Acta 1989, 1010, 131–139. [Google Scholar] [CrossRef]
- Ma, Y.; Pitson, S.; Hercus, T.; Murphy, J.; Lopez, A.; Woodcock, J. Sphingosine activates protein kinase A type II by a novel cAMP-independent mechanism. J. Biol. Chem. 2005, 280, 26011–26017. [Google Scholar] [CrossRef] [PubMed]
- Pettus, B.J.; Chalfant, C.E.; Hannun, Y.A. Ceramide in apoptosis: An overview and current perspectives. Biochim. Biophys. Acta 2002, 1585, 114–125. [Google Scholar] [CrossRef]
- Rosen, H.; Goetzl, E.J. Sphingosine 1-phosphate and its receptors: An autocrine and paracrine network. Nat. Rev. Immunol. 2005, 5, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Olivera, A.; Rosenfeldt, H.M.; Bektas, M.; Wang, F.; Ishii, I.; Chun, J.; Milstien, S.; Spiegel, S. Sphingosine kinase type 1 induces G12/13-mediated stress fiber formation, yet promotes growth and survival independent of G protein-coupled receptors. J. Biol. Chem. 2003, 278, 46452–46460. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell. Biol. 2003, 4, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Darroch, P.; Wan, K.F.; Kong, K.C.; Ktistakis, N.; Pyne, N.J.; Pyne, S. Regulation of cell survival by lipid phosphate phosphatases involves the modulation of intracellular phosphatidic acid and sphingosine 1-phosphate pools. Biochem. J. 2005, 391 Pt 1, 25–32. [Google Scholar] [CrossRef]
- Schumann, J.; Grevot, A.; Ledieu, D.; Wolf, A.; Schubart, A.; Piaia, A.; Sutter, E.; Cote, S.; Beerli, C.; Pognan, F.; et al. Reduced Activity of Sphingosine-1-Phosphate Lyase Induces Podocyte-related Glomerular Proteinuria, Skin Irritation, and Platelet Activation. Toxicol. Pathol. 2015, 43, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Lovric, S.; Goncalves, S.; Gee, H.Y.; Oskouian, B.; Srinivas, H.; Choi, W.I.; Shril, S.; Ashraf, S.; Tan, W.; Rao, J.; et al. Mutations in sphingosine-1-phosphate lyase cause nephrosis with ichthyosis and adrenal insufficiency. J. Clin. Investig. 2017, 127, 912–928. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M.; Kono, K.; Liu, H.; Shimizugawa, T.; Minekura, H.; Spiegel, S.; Kohama, T. Ceramide kinase, a novel lipid kinase. Molecular cloning and functional characterization. J. Biol. Chem. 2002, 277, 23294–23300. [Google Scholar] [CrossRef] [PubMed]
- Pettus, B.J.; Bielawska, A.; Subramanian, P.; Wijesinghe, D.S.; Maceyka, M.; Leslie, C.C.; Evans, J.H.; Freiberg, J.; Roddy, P.; Hannun, Y.A.; et al. Ceramide 1-phosphate is a direct activator of cytosolic phospholipase A2. J. Biol. Chem. 2004, 279, 11320–11326. [Google Scholar] [CrossRef] [PubMed]
- Simanshu, D.K.; Kamlekar, R.K.; Wijesinghe, D.S.; Zou, X.; Zhai, X.; Mishra, S.K.; Molotkovsky, J.G.; Malinina, L.; Hinchcliffe, E.H.; Chalfant, C.E.; et al. Non-vesicular trafficking by a ceramide-1-phosphate transfer protein regulates eicosanoids. Nature 2013, 500, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Kitano, M.; Hla, T.; Sekiguchi, M.; Kawahito, Y.; Yoshimura, R.; Miyazawa, K.; Iwasaki, T.; Sano, H.; Saba, J.D.; Tam, Y.Y. Sphingosine 1-phosphate/sphingosine 1-phosphate receptor 1 signaling in rheumatoid synovium: Regulation of synovial proliferation and inflammatory gene expression. Arthritis Rheumatol. 2006, 54, 7427–7453. [Google Scholar] [CrossRef] [PubMed]
- Mietla, J.A.; Wijesinghe, D.S.; Hoeferlin, L.A.; Shultz, M.D.; Natarajan, R.; Fowler, A.A., 3rd; Chalfant, C.E. Characterization of eicosanoid synthesis in a genetic ablation model of ceramide kinase. J. Lipid Res. 2013, 54, 1834–1847. [Google Scholar] [CrossRef] [PubMed]
- Hankins, J.L.; Fox, T.E.; Barth, B.M.; Unrath, K.A.; Kester, M. Exogenous ceramide-1-phosphate reduces lipopolysaccharide (LPS)-mediated cytokine expression. J. Biol. Chem. 2011, 286, 44357–44366. [Google Scholar] [CrossRef] [PubMed]
- Ogretmen, B.; Hannun, Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004, 4, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Radin, N.S. Killing tumours by ceramide-induced apoptosis: A critique of available drugs. Biochem. J. 2003, 371, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Jatoi, A.; Suman, V.J.; Schaefer, P.; Block, M.; Loprinzi, C.; Roche, P.; Garneau, S.; Morton, R.; Stella, P.J.; Alberts, S.R.; et al. A phase II study of topical ceramides for cutaneous breast cancer. Breast Cancer Res. Treat. 2003, 80, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Seelan, R.S.; Qian, C.; Yokomizo, A.; Bostwick, D.G.; Smith, D.I.; Liu, W. Human acid ceramidase is overexpressed but not mutated in prostate cancer. Genes Chromosom. Cancer 2000, 29, 137–146. [Google Scholar] [CrossRef]
- Raisova, M.; Goltz, G.; Bektas, M.; Bielawska, A.; Riebeling, C.; Hossini, A.M.; Eberle, J.; Hannun, Y.A.; Orfanos, C.E.; Geilen, C.C. Bcl-2 overexpression prevents apoptosis induced by ceramidase inhibitors in malignant melanoma and HaCaT keratinocytes. FEBS Lett. 2002, 516, 47–52. [Google Scholar] [CrossRef]
- Selzner, M.; Bielawska, A.; Morse, M.A.; Rudiger, H.A.; Sindram, D.; Hannun, Y.A.; Clavien, P.A. Induction of apoptotic cell death and prevention of tumor growth by ceramide analogues in metastatic human colon cancer. Cancer Res. 2001, 61, 1233–1240. [Google Scholar] [PubMed]
- Song, M.; Zang, W.; Zhang, B.; Cao, J.; Yang, G. GCS overexpression is associated with multidrug resistance of human HCT-8 colon cancer cells. J. Exp. Clin. Cancer Res. 2012, 31, 23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, J.; Qiu, Z.; Gao, P.; Wu, X.; Zhou, G. Co-suppression of MDR1 (multidrug resistance 1) and GCS (glucosylceramide synthase) restores sensitivity to multidrug resistance breast cancer cells by RNA interference (RNAi). Cancer Biol. Ther. 2009, 8, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Chai, L.; McLaren, R.P.; Byrne, A.; Chuang, W.L.; Huang, Y.; Dufault, M.R.; Pacheco, J.; Madhiwalla, S.; Zhang, X.; Zhang, M.; et al. The chemosensitizing activity of inhibitors of glucosylceramide synthase is mediated primarily through modulation of P-gp function. Int. J. Oncol. 2011, 38, 701–711. [Google Scholar] [PubMed]
- Baran, Y.; Bielawski, J.; Gunduz, U.; Ogretmen, B. Targeting glucosylceramide synthase sensitizes imatinib-resistant chronic myeloid leukemia cells via endogenous ceramide accumulation. J. Cancer Res. Clin. Oncol. 2011, 137, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Xie, K.M.; Yang, G.Q.; Mu, H.J.; Yin, Y.; Zhang, B.; Xie, P. The effect of glucosylceramide synthase on P-glycoprotein function in K562/AO2 leukemia drug-resistance cell line. Int. J. Hematol. 2011, 93, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Futerman, A.H.; Pagano, R.E. Determination of the intracellular sites and topology of glucosylceramide synthesis in rat liver. Biochem. J. 1991, 280 Pt 2, 295–302. [Google Scholar] [CrossRef]
- Jeckel, D.; Karrenbauer, A.; Burger, K.N.; van Meer, G.; Wieland, F. Glucosylceramide is synthesized at the cytosolic surface of various Golgi subfractions. J. Cell Biol. 1992, 117, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.P.; Buronfosse, A.; Combaret, V.; Pedron, S.; Fertil, B.; Portoukalian, J. Gangliosides protect human melanoma cells from ionizing radiation-induced clonogenic cell death. Glycoconj. J. 1996, 13, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Gouaze, V.; Liu, Y.Y.; Prickett, C.S.; Yu, J.Y.; Giuliano, A.E.; Cabot, M.C. Glucosylceramide synthase blockade down-regulates P-glycoprotein and resensitizes multidrug-resistant breast cancer cells to anticancer drugs. Cancer Res. 2005, 65, 3861–3867. [Google Scholar] [CrossRef] [PubMed]
- Visentin, B.; Vekich, J.A.; Sibbald, B.J.; Cavalli, A.L.; Moreno, K.M.; Matteo, R.G.; Garland, W.A.; Lu, Y.; Yu, S.; Hall, H.S.; et al. Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell 2006, 9, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, J.; Erwin, P.A.; Dantas, A.P.; Chen, H.; Michel, T. VEGF induces S1P1 receptors in endothelial cells: Implications for cross-talk between sphingolipid and growth factor receptors. Proc. Natl. Acad. Sci. USA 2003, 100, 10664–10669. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Hirabayashi, T.; Shimizu, M.; Murayama, T. Ceramide-1-phosphate activates cytosolic phospholipase A2α directly and by PKC pathway. Biochem. Pharmacol. 2006, 71, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Chalfant, C.E.; Spiegel, S. Sphingosine 1-phosphate and ceramide 1-phosphate: Expanding roles in cell signaling. J. Cell Sci. 2005, 118, 4605–4612. [Google Scholar] [CrossRef] [PubMed]
- Pettus, B.J.; Kitatani, K.; Chalfant, C.E.; Taha, T.A.; Kawamori, T.; Bielawski, J.; Obeid, L.M.; Hannun, Y.A. The coordination of prostaglandin E2 production by sphingosine-1-phosphate and ceramide-1-phosphate. Mol. Pharmacol. 2005, 68, 330–335. [Google Scholar] [CrossRef] [PubMed]
- MacKinnon, A.C.; Buckley, A.; Chilvers, E.R.; Rossi, A.G.; Haslett, C.; Sethi, T. Sphingosine kinase: A point of convergence in the action of diverse neutrophil priming agents. J. Immunol. 2002, 169, 6394–6400. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, F.B.; Pang, S.J.; Melendez, A.J. Anaphylatoxin signaling in human neutrophils. A key role for sphingosine kinase. J. Biol. Chem. 2004, 279, 44802–44811. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Munoz, A.; Kong, J.Y.; Salh, B.; Steinbrecher, U.P. Ceramide-1-phosphate blocks apoptosis through inhibition of acid sphingomyelinase in macrophages. J. Lipid Res. 2004, 45, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Munoz, A.; Kong, J.; Salh, B.; Steinbrecher, U.P. Sphingosine-1-phosphate inhibits acid sphingomyelinase and blocks apoptosis in macrophages. FEBS Lett. 2003, 539, 56–60. [Google Scholar] [CrossRef]
- Krumholz, H.M.; Ross, J.S.; Presler, A.H.; Egilman, D.S. What have we learnt from Vioxx? BMJ 2007, 334, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro, M.; Zhou, Y.T.; Levi, M.; Unger, R.H. Fatty acid-induced beta cell apoptosis: A link between obesity and diabetes. Proc. Natl. Acad. Sci. USA 1998, 95, 2498–2502. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H.; Orci, L. Lipoapoptosis: Its mechanism and its diseases. Biochim. Biophys. Acta 2002, 1585, 202–212. [Google Scholar] [CrossRef]
- Turinsky, J.; O’Sullivan, D.M.; Bayly, B.P. 1,2-Diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. J. Biol. Chem. 1990, 265, 16880–16885. [Google Scholar] [PubMed]
- Adams, J.M., 2nd; Pratipanawatr, T.; Berria, R.; Wang, E.; DeFronzo, R.A.; Sullards, M.C.; Mandarino, L.J. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 2004, 53, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Eid, S.; Boutary, S.; Braych, K.; Sabra, R.; Massaad, C.; Hamdy, A.; Rashid, A.; Moodad, S.; Block, K.; Gorin, Y. mTORC2 Signaling Regulates Nox4-Induced Podocyte Depletion in Diabetes. Antioxid. Redox Signal. 2016, 25, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Summers, S.A. Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Arch. Biochem. Biophys. 2003, 419, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Knotts, T.A.; Wang, L.P.; Li, G.; Dobrowsky, R.T.; Florant, G.L.; Summers, S.A. A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J. Biol. Chem. 2003, 278, 10297–10303. [Google Scholar] [CrossRef] [PubMed]
- Summers, S.A.; Garza, L.A.; Zhou, H.; Birnbaum, M.J. Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol. Cell. Biol. 1998, 18, 5457–5464. [Google Scholar] [CrossRef] [PubMed]
- Teruel, T.; Hernandez, R.; Lorenzo, M. Ceramide mediates insulin resistance by tumor necrosis factor-α in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes 2001, 50, 2563–2571. [Google Scholar] [CrossRef] [PubMed]
- Summers, S.A. Ceramides in insulin resistance and lipotoxicity. Prog. Lipid Res. 2006, 45, 42–72. [Google Scholar] [CrossRef] [PubMed]
- Stratford, S.; Hoehn, K.L.; Liu, F.; Summers, S.A. Regulation of insulin action by ceramide: Dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J. Biol. Chem. 2004, 279, 36608–36615. [Google Scholar] [CrossRef] [PubMed]
- Stratford, S.; DeWald, D.B.; Summers, S.A. Ceramide dissociates 3′-phosphoinositide production from pleckstrin homology domain translocation. Biochem. J. 2001, 354 Pt 2, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Tejada, T.; Catanuto, P.; Ijaz, A.; Santos, J.V.; Xia, X.; Sanchez, P.; Sanabria, N.; Lenz, O.; Elliot, S.J.; Fornoni, A. Failure to phosphorylate AKT in podocytes from mice with early diabetic nephropathy promotes cell death. Kidney Int. 2008, 73, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Welsh, G.I.; Hale, L.J.; Eremina, V.; Jeansson, M.; Maezawa, Y.; Lennon, R.; Pons, D.A.; Owen, R.J.; Satchell, S.C.; Miles, M.J.; et al. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 2010, 12, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Fornoni, A. Proteinuria, the podocyte, and insulin resistance. N. Engl. J. Med. 2010, 363, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Cowart, L.A. Sphingolipids: Players in the pathology of metabolic disease. Trends Endocrinol. Metab. 2009, 20, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Fall, B.; Scott, C.R.; Mauer, M.; Shankland, S.; Pippin, J.; Jefferson, J.A.; Wallace, E.; Warnock, D.; Najafian, B. Urinary Podocyte Loss Is Increased in Patients with Fabry Disease and Correlates with Clinical Severity of Fabry Nephropathy. PLoS ONE 2016, 11, e0168346. [Google Scholar] [CrossRef] [PubMed]
- Liebau, M.C.; Braun, F.; Hopker, K.; Weitbrecht, C.; Bartels, V.; Muller, R.U.; Brodesser, S.; Saleem, M.A.; Benzing, T.; Schermer, B.; et al. Dysregulated autophagy contributes to podocyte damage in Fabry’s disease. PLoS ONE 2013, 8, e63506. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Nino, M.D.; Carpio, D.; Sanz, A.B.; Ruiz-Ortega, M.; Mezzano, S.; Ortiz, A. Lyso-Gb3 activates Notch1 in human podocytes. Hum. Mol. Genet. 2015, 24, 5720–5732. [Google Scholar] [CrossRef] [PubMed]
- Askari, H.; Kaneski, C.R.; Semino-Mora, C.; Desai, P.; Ang, A.; Kleiner, D.E.; Perlee, L.T.; Quezado, M.; Spollen, L.E.; Wustman, B.A.; et al. Cellular and tissue localization of globotriaosylceramide in Fabry disease. Virchows Arch. 2007, 451, 823–834. [Google Scholar] [CrossRef] [PubMed]
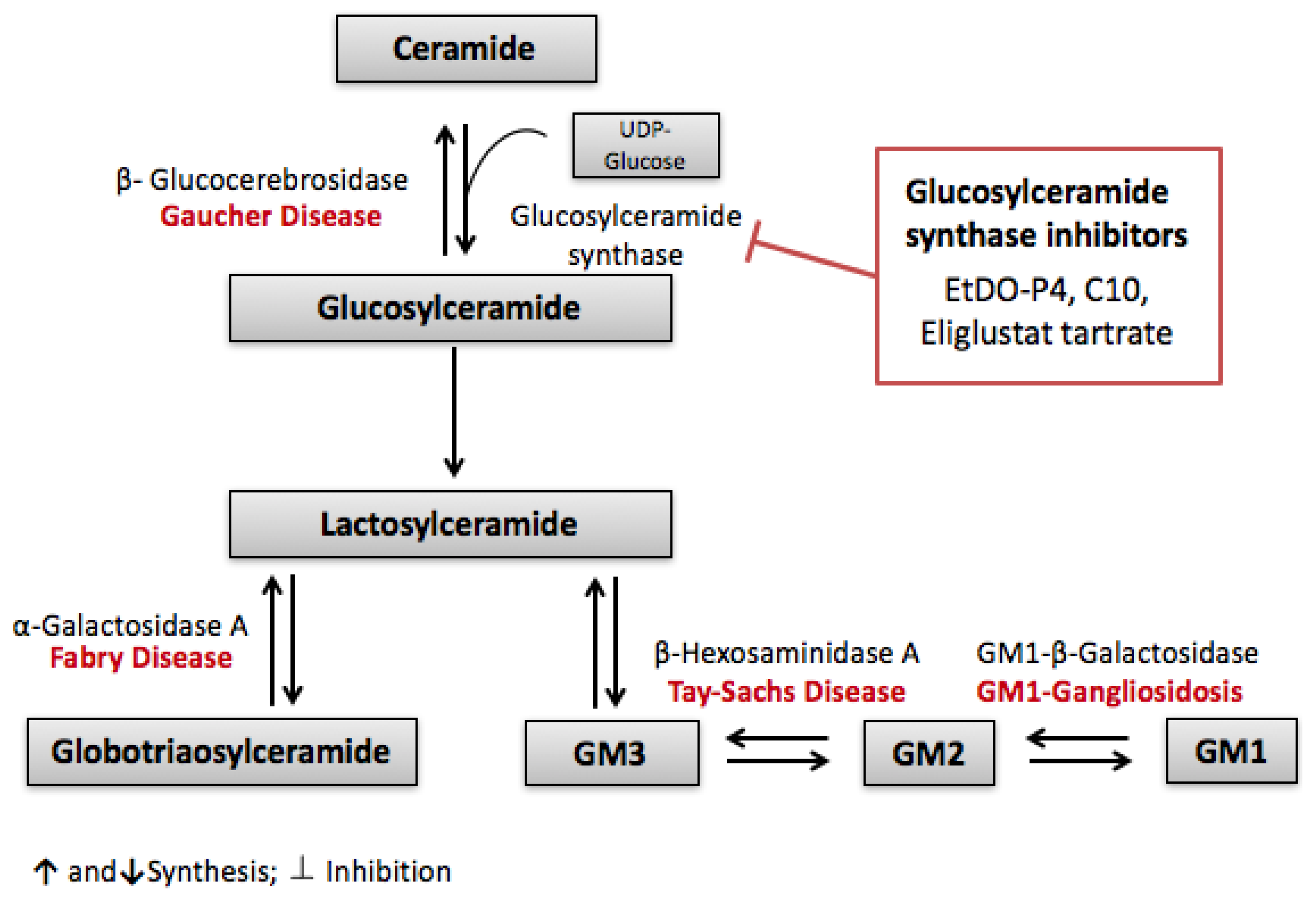
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Oliveira, J.P.; Waldek, S.; Warnock, D.G.; Cianciaruso, B.; Wanner, C.; Fabry, R. Nephropathy in males and females with Fabry disease: Cross-sectional description of patients before treatment with enzyme replacement therapy. Nephrol. Dial. Transplant. 2008, 23, 1600–1607. [Google Scholar] [CrossRef] [PubMed]
- Terryn, W.; Cochat, P.; Froissart, R.; Ortiz, A.; Pirson, Y.; Poppe, B.; Serra, A.; Van Biesen, W.; Vanholder, R.; Wanner, C. Fabry nephropathy: Indications for screening and guidance for diagnosis and treatment by the European Renal Best Practice. Nephrol. Dial. Transplant. 2013, 28, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Waldek, S.; Banikazemi, M.; Bushinsky, D.A.; Charrow, J.; Desnick, R.J.; Lee, P.; Loew, T.; Vedder, A.C.; Abichandani, R.; et al. Sustained, long-term renal stabilization after 54 months of agalsidase beta therapy in patients with Fabry disease. J. Am. Soc. Nephrol. 2007, 18, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Fernandez, B.; Ortiz, A.; Gomez-Guerrero, C.; Egido, J. Therapeutic approaches to diabetic nephropathy—Beyond the RAS. Nat. Rev. Nephrol. 2014, 10, 325–346. [Google Scholar] [CrossRef] [PubMed]
- Goni, F.M.; Alonso, A. Effects of ceramide and other simple sphingolipids on membrane lateral structure. Biochim. Biophys. Acta 2009, 1788, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Sanz, A.B.; Sanchez-Nino, M.D.; Ramos, A.M.; Moreno, J.A.; Santamaria, B.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. NF-κB in renal inflammation. J. Am. Soc. Nephrol. 2010, 21, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Sirin, Y.; Susztak, K. Notch in the kidney: Development and disease. J. Pathol. 2012, 226, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Niranjan, T.; Bielesz, B.; Gruenwald, A.; Ponda, M.P.; Kopp, J.B.; Thomas, D.B.; Susztak, K. The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat. Med. 2008, 14, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Bielesz, B.; Sirin, Y.; Si, H.; Niranjan, T.; Gruenwald, A.; Ahn, S.; Kato, H.; Pullman, J.; Gessler, M.; Haase, V.H.; et al. Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J. Clin. Investig. 2010, 120, 4040–4054. [Google Scholar] [CrossRef] [PubMed]
- Van Blitterswijk, W.J.; van der Luit, A.H.; Veldman, R.J.; Verheij, M.; Borst, J. Ceramide: Second messenger or modulator of membrane structure and dynamics? Biochem. J. 2003, 369 Pt 2, 199–211. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Becker, K.A.; Gulbins, E. Ceramide-enriched membrane domains—Structure and function. Biochim. Biophys. Acta 2009, 1788, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Fornoni, A.; Sageshima, J.; Wei, C.; Merscher-Gomez, S.; Aguillon-Prada, R.; Jauregui, A.N.; Li, J.; Mattiazzi, A.; Ciancio, G.; Chen, L.; et al. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci. Transl. Med. 2011, 3, 85ra46. [Google Scholar] [CrossRef] [PubMed]
- Boini, K.M.; Xia, M.; Abais, J.M.; Xu, M.; Li, C.X.; Li, P.L. Acid sphingomyelinase gene knockout ameliorates hyperhomocysteinemic glomerular injury in mice lacking cystathionine-beta-synthase. PLoS ONE 2012, 7, e45020. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.W.; Bennett, P.H.; Nelson, R.G. Podocyte number predicts long-term urinary albumin excretion in Pima Indians with Type II diabetes and microalbuminuria. Diabetologia 1999, 42, 1341–1344. [Google Scholar] [CrossRef] [PubMed]
- Steffes, M.W.; Schmidt, D.; McCrery, R.; Basgen, J.M. Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int. 2001, 59, 2104–2113. [Google Scholar] [CrossRef] [PubMed]
- Verzola, D.; Gandolfo, M.T.; Ferrario, F.; Rastaldi, M.P.; Villaggio, B.; Gianiorio, F.; Giannoni, M.; Rimoldi, L.; Lauria, F.; Miji, M.; et al. Apoptosis in the kidneys of patients with type II diabetic nephropathy. Kidney Int. 2007, 72, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- White, K.E.; Bilous, R.W.; Marshall, S.M.; El Nahas, M.; Remuzzi, G.; Piras, G.; De Cosmo, S.; Viberti, G. Podocyte number in normotensive type 1 diabetic patients with albuminuria. Diabetes 2002, 51, 3083–3089. [Google Scholar] [CrossRef] [PubMed]
- Pagtalunan, M.E.; Miller, P.L.; Jumping-Eagle, S.; Nelson, R.G.; Myers, B.D.; Rennke, H.G.; Coplon, N.S.; Sun, L.; Meyer, T.W. Podocyte loss and progressive glomerular injury in type II diabetes. J. Clin. Investig. 1997, 99, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Kremer, G.J.; Atzpodien, W.; Schnellbacher, E. Plasma glycosphingolipids in diabetics and normals. Klin. Wochenschr. 1975, 53, 637–638. [Google Scholar] [CrossRef] [PubMed]
- Haus, J.M.; Kashyap, S.R.; Kasumov, T.; Zhang, R.; Kelly, K.R.; Defronzo, R.A.; Kirwan, J.P. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 2009, 58, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Gorska, M.; Dobrzyn, A.; Baranowski, M. Concentrations of sphingosine and sphinganine in plasma of patients with type 2 diabetes. Med. Sci. Monit. 2005, 11, CR35–CR38. [Google Scholar] [PubMed]
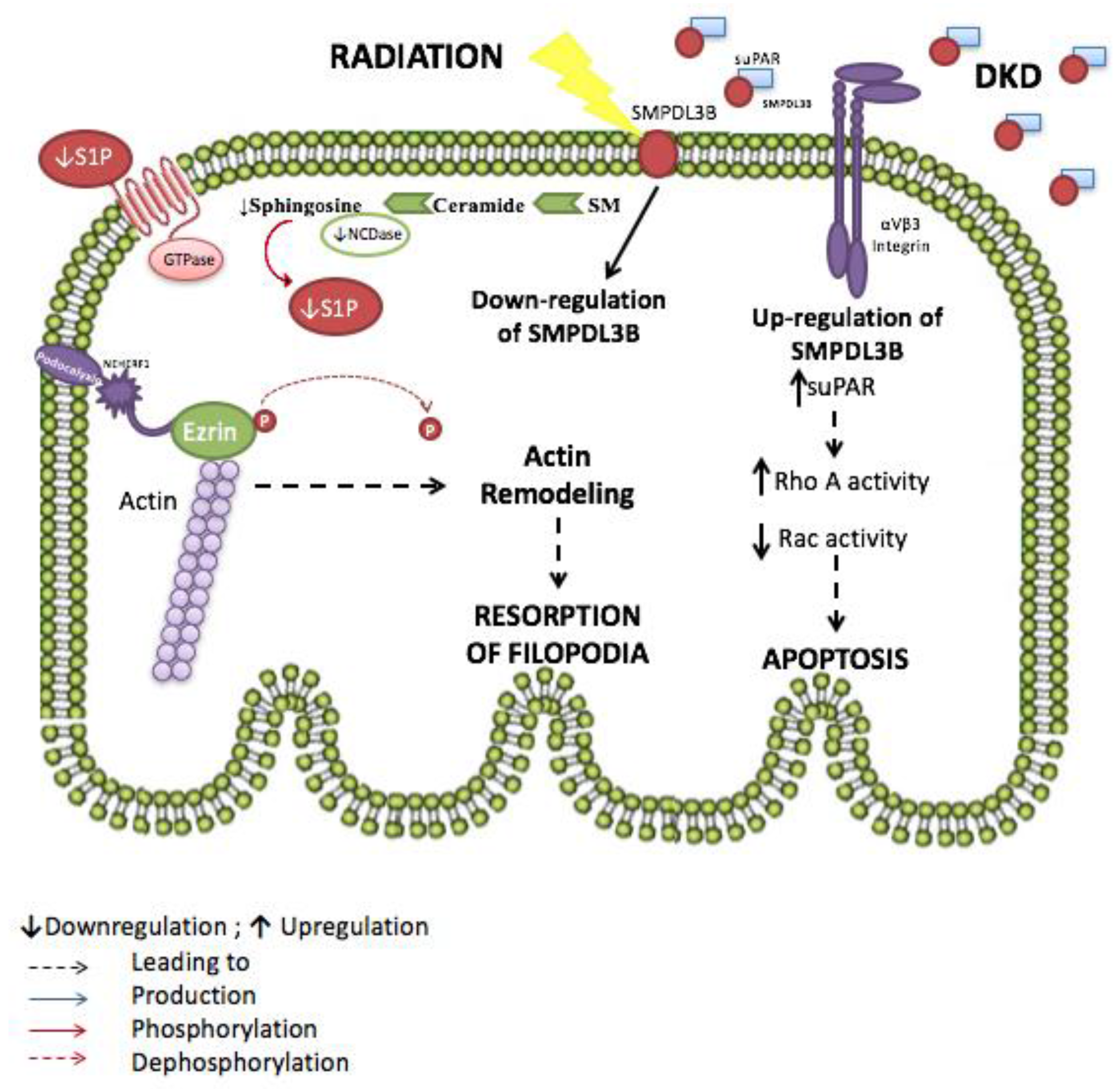
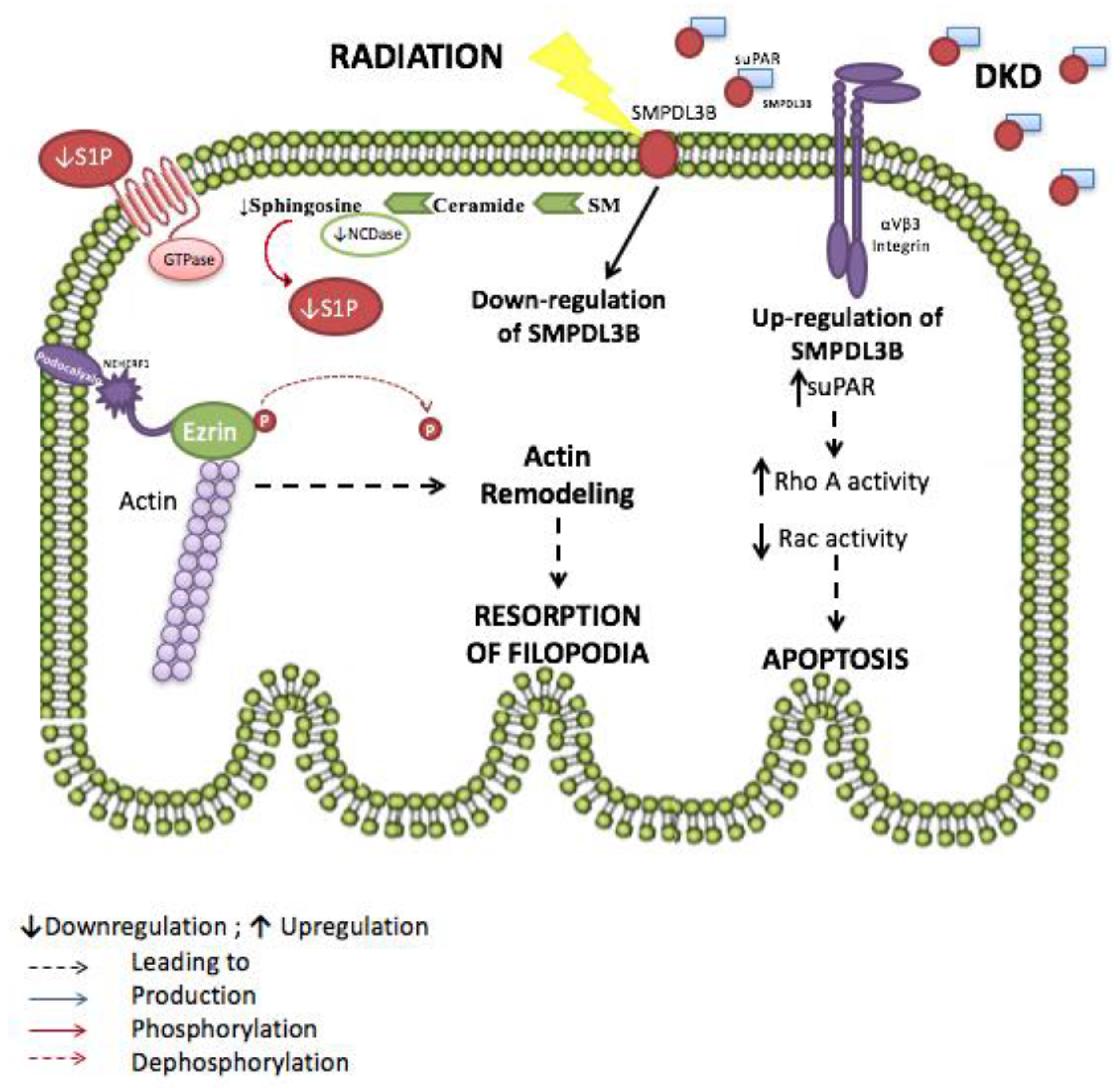
- Yoo, T.H.; Pedigo, C.E.; Guzman, J.; Correa-Medina, M.; Wei, C.; Villarreal, R.; Mitrofanova, A.; Leclercq, F.; Faul, C.; Li, J.; et al. Sphingomyelinase-like phosphodiesterase 3b expression levels determine podocyte injury phenotypes in glomerular disease. J. Am. Soc. Nephrol. 2015, 26, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Brunskill, E.W.; Potter, S.S. Changes in the gene expression programs of renal mesangial cells during diabetic nephropathy. BMC Nephrol. 2012, 13, 70. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, S.; Takahashi-Fujigasaki, J.; Kanazawa, Y.; Matoba, K.; Kawanami, D.; Yokota, T.; Iwamoto, T.; Tajima, N.; Manome, Y.; Utsunomiya, K. Sphingosine-1-phosphate induces differentiation of cultured renal tubular epithelial cells under Rho kinase activation via the S1P2 receptor. Clin. Exp. Nephrol. 2014, 18, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Samad, F.; Hester, K.D.; Yang, G.; Hannun, Y.A.; Bielawski, J. Altered adipose and plasma sphingolipid metabolism in obesity: A potential mechanism for cardiovascular and metabolic risk. Diabetes 2006, 55, 2579–2587. [Google Scholar] [CrossRef] [PubMed]
- Kitiyakara, C.; Eggers, P.; Kopp, J.B. Twenty-one-year trend in ESRD due to focal segmental glomerulosclerosis in the United States. Am. J. Kidney Dis. 2004, 44, 815–825. [Google Scholar] [CrossRef]
- Baum, M.A. Outcomes after renal transplantation for FSGS in children. Pediatr Transplant 2004, 8, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Hubsch, H.; Montane, B.; Abitbol, C.; Chandar, J.; Shariatmadar, S.; Ciancio, G.; Burke, G.; Miller, J.; Strauss, J.; Zilleruelo, G. Recurrent focal glomerulosclerosis in pediatric renal allografts: The Miami experience. Pediatr. Nephrol. 2005, 20, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Senggutuvan, P.; Cameron, J.S.; Hartley, R.B.; Rigden, S.; Chantler, C.; Haycock, G.; Williams, D.G.; Ogg, C.; Koffman, G. Recurrence of focal segmental glomerulosclerosis in transplanted kidneys: Analysis of incidence and risk factors in 59 allografts. Pediatr. Nephrol. 1990, 4, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Moller, C.C.; Altintas, M.M.; Li, J.; Schwarz, K.; Zacchigna, S.; Xie, L.; Henger, A.; Schmid, H.; Rastaldi, M.P.; et al. Modification of kidney barrier function by the urokinase receptor. Nat. Med. 2008, 14, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Lukashev, M.; Simon, D.I.; Bodary, S.C.; Rosenberg, S.; Doyle, M.V.; Chapman, H.A. Regulation of integrin function by the urokinase receptor. Science 1996, 273, 1551–1555. [Google Scholar] [CrossRef] [PubMed]
- Blasi, F.; Carmeliet, P. uPAR: A versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol. 2002, 3, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Trachtman, H.; Li, J.; Dong, C.; Friedman, A.L.; Gassman, J.J.; McMahan, J.L.; Radeva, M.; Heil, K.M.; Trautmann, A.; et al. Circulating suPAR in two cohorts of primary FSGS. J. Am. Soc. Nephrol. 2012, 23, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Merscher-Gomez, S.; Guzman, J.; Pedigo, C.E.; Lehto, M.; Aguillon-Prada, R.; Mendez, A.; Lassenius, M.I.; Forsblom, C.; Yoo, T.; Villarreal, R.; et al. Cyclodextrin protects podocytes in diabetic kidney disease. Diabetes 2013, 62, 3817–3827. [Google Scholar] [CrossRef] [PubMed]
- Ruotsalainen, V.; Ljungberg, P.; Wartiovaara, J.; Lenkkeri, U.; Kestila, M.; Jalanko, H.; Holmberg, C.; Tryggvason, K. Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc. Natl. Acad. Sci. USA 1999, 96, 7962–7967. [Google Scholar] [CrossRef] [PubMed]
- Cassady, J.R. Clinical radiation nephropathy. Int J. Radiat. Oncol. Biol. Phys. 1995, 31, 1249–1256. [Google Scholar] [CrossRef]
- Dawson, L.A.; Kavanagh, B.D.; Paulino, A.C.; Das, S.K.; Miften, M.; Li, X.A.; Pan, C.; Ten Haken, R.K.; Schultheiss, T.E. Radiation-associated kidney injury. Int J. Radiat. Oncol. Biol. Phys. 2010, 76 (Suppl. 3), S108–S115. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.Y.; May, K.S.; Iyer, R.V.; Chandrasekhar, R.; Wilding, G.E.; McCloskey, S.A.; Khushalani, N.I.; Yendamuri, S.S.; Gibbs, J.F.; Fakih, M.; et al. Renal atrophy secondary to chemoradiotherapy of abdominal malignancies. Int J. Radiat. Oncol. Biol. Phys. 2010, 78, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.P.; Robbins, M.E. Radiation nephropathy. Semin. Nephrol. 2003, 23, 486–499. [Google Scholar] [CrossRef]
- Sera, N.; Hida, A.; Imaizumi, M.; Nakashima, E.; Akahoshi, M. The association between chronic kidney disease and cardiovascular disease risk factors in atomic bomb survivors. Radiat. Res. 2013, 179, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Mitrofanova, A.; Bielawski, J.; Yang, Y.; Marples, B.; Fornoni, A.; Zeidan, Y.H. Sphingomyelinase-like phosphodiesterase 3b mediates radiation-induced damage of renal podocytes. FASEB J. 2017, 31, 771–780. [Google Scholar] [CrossRef] [PubMed]
- United States Renal Data System. The USRDS and its products. Am. J. Kidney Dis. 1998, 32 (Suppl. 1), S20–S37. [Google Scholar]
- Raine, A.E.; Margreiter, R.; Brunner, F.P.; Ehrich, J.H.; Geerlings, W.; Landais, P.; Loirat, C.; Mallick, N.P.; Selwood, N.H.; Tufveson, G.; et al. Report on management of renal failure in Europe, XXII 1991. Nephrol. Dial. Transplant. 1992, 7 (Suppl. 2), 7–35. [Google Scholar] [PubMed]
- Herzog, C.A.; Ma, J.Z.; Collins, A.J. Poor long-term survival after acute myocardial infarction among patients on long-term dialysis. N. Engl. J. Med. 1998, 339, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Kasiske, B.L.; Guijarro, C.; Massy, Z.A.; Wiederkehr, M.R.; Ma, J.Z. Cardiovascular disease after renal transplantation. J. Am. Soc. Nephrol. 1996, 7, 158–165. [Google Scholar] [PubMed]
- Splaver, A.; Lamas, G.A.; Hennekens, C.H. Homocysteine and cardiovascular disease: Biological mechanisms, observational epidemiology, and the need for randomized trials. Am. Heart J. 2004, 148, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Homocysteine Studies, C. Homocysteine and risk of ischemic heart disease and stroke: A meta-analysis. JAMA 2002, 288, 2015–2022. [Google Scholar] [CrossRef]
- Hultberg, B.; Andersson, A.; Arnadottir, M. Reduced, free and total fractions of homocysteine and other thiol compounds in plasma from patients with renal failure. Nephron 1995, 70, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, D.E.; Gupta, V.J. Sulphr containing amino acids in chronic renal failure with particular reference to homocystine and cysteine-homocysteine mixed disulphide. Eur. J. Clin. Investig. 1979, 9, 301–307. [Google Scholar] [CrossRef]
- Van Guldener, C.; Robinson, K. Homocysteine and renal disease. Semin. Thromb. Hemost. 2000, 26, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Werstuck, G.H.; Lentz, S.R.; Dayal, S.; Hossain, G.S.; Sood, S.K.; Shi, Y.Y.; Zhou, J.; Maeda, N.; Krisans, S.K.; Malinow, M.R.; et al. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J. Clin. Investig. 2001, 107, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Castro, R.; Rivera, I.; Blom, H.J.; Jakobs, C.; Tavares de Almeida, I. Homocysteine metabolism, hyperhomocysteinaemia and vascular disease: An overview. J. Inherit. Metab. Dis. 2006, 29, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Xia, M.; Li, N.; Zhang, C.; Tang, L.; Li, P.L. Contribution of guanine nucleotide exchange factor Vav2 to hyperhomocysteinemic glomerulosclerosis in rats. Hypertension 2009, 53, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Ingram, A.J.; Krepinsky, J.C.; James, L.; Austin, R.C.; Tang, D.; Salapatek, A.M.; Thai, K.; Scholey, J.W. Activation of mesangial cell MAPK in response to homocysteine. Kidney Int. 2004, 66, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Zhang, A.Y.; Li, N.; Muh, R.W.; Fillet, M.; Renert, A.F.; Li, P.L. Inhibition of ceramide-redox signaling pathway blocks glomerular injury in hyperhomocysteinemic rats. Kidney Int. 2006, 70, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Sen, U.; Basu, P.; Abe, O.A.; Givvimani, S.; Tyagi, N.; Metreveli, N.; Shah, K.S.; Passmore, J.C.; Tyagi, S.C. Hydrogen sulfide ameliorates hyperhomocysteinemia-associated chronic renal failure. Am. J. Physiol. Ren. Physiol. 2009, 297, F410–F419. [Google Scholar] [CrossRef] [PubMed]
- Boini, K.M.; Xia, M.; Li, C.; Zhang, C.; Payne, L.P.; Abais, J.M.; Poklis, J.L.; Hylemon, P.B.; Li, P.L. Acid Sphingomyelinase Gene Deficiency Ameliorates the Hyperhomocysteinemia-Induced Glomerular Injury in Mice. Am. J. Pathol. 2011, 179, 2210–2219. [Google Scholar] [CrossRef] [PubMed]
- Eichholtz, T.; Jalink, K.; Fahrenfort, I.; Moolenaar, W.H. The bioactive phospholipid lysophosphatidic acid is released from activated platelets. Biochem. J. 1993, 291 Pt 3, 677–680. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. Sphingolipid metabolites: Members of a new class of lipid second messengers. J. Membr. Biol. 1995, 146, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, O.; Simon, M.F.; Viode, C.; Rugani, N.; Leballe, F.; Ragab, A.; Fournie, B.; Sarda, L.; Chap, H. Secretory phospholipase A2 generates the novel lipid mediator lysophosphatidic acid in membrane microvesicles shed from activated cells. Cell 1995, 80, 919–927. [Google Scholar] [CrossRef]
- Le Stunff, H.; Milstien, S.; Spiegel, S. Generation and metabolism of bioactive sphingosine-1-phosphate. J. Cell. Biochem. 2004, 92, 882–899. [Google Scholar] [CrossRef] [PubMed]
- Hla, T. Signaling and biological actions of sphingosine 1-phosphate. Pharmacol. Res. 2003, 47, 401–407. [Google Scholar] [CrossRef]
- Mills, G.B.; Moolenaar, W.H. The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer 2003, 3, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Ruban, E.L.; Ferro, R.; Arifin, S.A.; Falasca, M. Lysophosphatidylinositol: A novel link between ABC transporters and G-protein-coupled receptors. Biochem. Soc. Trans. 2014, 42, 1372–1377. [Google Scholar] [CrossRef] [PubMed]
- Anliker, B.; Chun, J. Lysophospholipid G protein-coupled receptors. J. Biol. Chem. 2004, 279, 20555–20558. [Google Scholar] [CrossRef] [PubMed]
- Siess, W.; Tigyi, G. Thrombogenic and atherogenic activities of lysophosphatidic acid. J. Cell Biochem. 2004, 92, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Karliner, J.S. Mechanisms of cardioprotection by lysophospholipids. J. Cell Biochem. 2004, 92, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Rosen, H. Regulation of immunity by lysosphingolipids and their G protein-coupled receptors. J. Clin. Investig. 2004, 114, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Nava, V.E.; Lacana, E.; Poulton, S.; Liu, H.; Sugiura, M.; Kono, K.; Milstien, S.; Kohama, T.; Spiegel, S. Functional characterization of human sphingosine kinase-1. FEBS Lett. 2000, 473, 81–84. [Google Scholar] [CrossRef]
- Brindley, D.N.; English, D.; Pilquil, C.; Buri, K.; Ling, Z.C. Lipid phosphate phosphatases regulate signal transduction through glycerolipids and sphingolipids. Biochim. Biophys. Acta 2002, 1582, 33–44. [Google Scholar] [CrossRef]
- Awad, A.S.; Rouse, M.D.; Khutsishvili, K.; Huang, L.; Bolton, W.K.; Lynch, K.R.; Okusa, M.D. Chronic sphingosine 1-phosphate 1 receptor activation attenuates early-stage diabetic nephropathy independent of lymphocytes. Kidney Int. 2011, 79, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.S.; Ye, H.; Huang, L.; Li, L.; Foss, F.W.; Macdonald, T.L.; Lynch, K.R.; Okusa, M.D. Selective sphingosine 1-phosphate 1 receptor activation reduces ischemia-reperfusion injury in mouse kidney. Am. J. Physiol. Ren. Physiol. 2006, 290, F1516–F1524. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Hadjidemetriou, I.; Maharaj, A.; Meimaridou, E.; Buonocore, F.; Saleem, M.; Hurcombe, J.; Bierzynska, A.; Barbagelata, E.; Bergada, I. Sphingosine-1-phosphate lyase mutations cause primary adrenal insufficiency and steroid-resistant nephrotic syndrome. J. Clin. Investig. 2017, 127, 942–953. [Google Scholar] [CrossRef] [PubMed]
- Genter, M.B.; Van Veldhoven, P.P.; Jegga, A.G.; Sakthivel, B.; Kong, S.; Stanley, K.; Witte, D.P.; Ebert, C.L.; Aronow, B.J. Microarray-based discovery of highly expressed olfactory mucosal genes: Potential roles in the various functions of the olfactory system. Physiol. Genom. 2003, 16, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Serra, M.; Saba, J.D. Sphingosine 1-phosphate lyase, a key regulator of sphingosine 1-phosphate signaling and function. Adv. Enzym. Regul. 2010, 50, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Schwab, S.R.; Pereira, J.P.; Matloubian, M.; Xu, Y.; Huang, Y.; Cyster, J.G. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science 2005, 309, 1735–1739. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Anada, Y.; Tani, M.; Ikeda, M.; Sano, T.; Kihara, A.; Igarashi, Y. Lack of sphingosine 1-phosphate-degrading enzymes in erythrocytes. Biochem. Biophys. Res. Commun. 2007, 357, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Yatomi, Y.; Yamamura, S.; Ruan, F.; Igarashi, Y. Sphingosine 1-phosphate induces platelet activation through an extracellular action and shares a platelet surface receptor with lysophosphatidic acid. J. Biol. Chem. 1997, 272, 5291–5297. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Q.; Kramer, J.; Moran, L.; O‘Neill, E.; Nouraldeen, A.; Oravecz, T.; Wilson, A.G. Pharmacokinetic/pharmacodynamic modelling of 2-acetyl-4(5)-tetrahydroxybutyl imidazole-induced peripheral lymphocyte sequestration through increasing lymphoid sphingosine 1-phosphate. Xenobiotica 2010, 40, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Billich, A.; Baumruker, T.; Beerli, C.; Bigaud, M.; Bruns, C.; Calzascia, T.; Isken, A.; Kinzel, B.; Loetscher, E.; Metzler, B.; et al. Partial deficiency of sphingosine-1-phosphate lyase confers protection in experimental autoimmune encephalomyelitis. PLoS ONE 2013, 8, e59630. [Google Scholar] [CrossRef] [PubMed]
- Bagdanoff, J.T.; Donoviel, M.S.; Nouraldeen, A.; Carlsen, M.; Jessop, T.C.; Tarver, J.; Aleem, S.; Dong, L.; Zhang, H.; Boteju, L.; et al. Inhibition of sphingosine 1-phosphate lyase for the treatment of rheumatoid arthritis: discovery of (E)-1-(4-((1R,2S,3R)-1,2,3,4-tetrahydroxybutyl)-1H-imidazol-2-yl)ethanone oxime (LX2931) and (1R,2S,3R)-1-(2-(isoxazol-3-yl)-1H-imidazol-4-yl)butane-1,2,3,4-tetraol (LX2932). J. Med. Chem. 2010, 53, 8650–8662. [Google Scholar] [PubMed]
- Vogel, P.; Donoviel, M.S.; Read, R.; Hansen, G.M.; Hazlewood, J.; Anderson, S.J.; Sun, W.; Swaffield, J.; Oravecz, T. Incomplete inhibition of sphingosine 1-phosphate lyase modulates immune system function yet prevents early lethality and non-lymphoid lesions. PLoS ONE 2009, 4, e4112. [Google Scholar] [CrossRef] [PubMed]
- Weiler, S.; Braendlin, N.; Beerli, C.; Bergsdorf, C.; Schubart, A.; Srinivas, H.; Oberhauser, B.; Billich, A. Orally active 7-substituted (4-benzylphthalazin-1-yl)-2-methylpiperazin-1-yl]nicotinonitriles as active-site inhibitors of sphingosine 1-phosphate lyase for the treatment of multiple sclerosis. J. Med. Chem. 2014, 57, 5074–5084. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.M.; Stablein, D.M.; Munoz, R.; Hebert, D.; McDonald, R.A. Contributions of the Transplant Registry: The 2006 Annual Report of the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS). Pediatr. Transplant. 2007, 11, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, F.; Heeringa, S.F. Specific podocin mutations determine age of onset of nephrotic syndrome all the way into adult life. Kidney Int. 2009, 75, 669–671. [Google Scholar] [CrossRef] [PubMed]
- Weiner, G.J. Rituximab: Mechanism of action. Semin. Hematol. 2010, 47, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Perosa, F.; Favoino, E.; Caragnano, M.A.; Dammacco, F. Generation of biologically active linear and cyclic peptides has revealed a unique fine specificity of rituximab and its possible cross-reactivity with acid sphingomyelinase-like phosphodiesterase 3b precursor. Blood 2006, 107, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Emer, J.J.; Claire, W. Rituximab: A review of dermatological applications. J. Clin. Aesthet. Dermatol. 2009, 2, 29–37. [Google Scholar] [PubMed]
- Neufeld, E.F. Enzyme replacement therapy—A brief history. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Thurberg, B.L.; Rennke, H.; Colvin, R.B.; Dikman, S.; Gordon, R.E.; Collins, A.B.; Desnick, R.J.; O‘Callaghan, M. Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int. 2002, 62, 1933–1946. [Google Scholar] [CrossRef] [PubMed]
- Najafian, B.; Tondel, C.; Svarstad, E.; Sokolovkiy, A.; Smith, K.; Mauer, M. One Year of Enzyme Replacement Therapy Reduces Globotriaosylceramide Inclusions in Podocytes in Male Adult Patients with Fabry Disease. PLoS ONE 2016, 11, e0152812. [Google Scholar] [CrossRef] [PubMed]
- Tøndel, C.; Bostad, L.; Larsen, K.K.; Hirth, A.; Vikse, B.E.; Houge, G.; Svarstad, E. Agalsidase Benefits Renal Histology in Young Patients with Fabry Disease. J. Am. Soc. Nephrol. 2012, 24, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Radin, N.S. Treatment of Gaucher disease with an enzyme inhibitor. Glycoconj. J. 1996, 13, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, R.H.; Platt, F.M. Substrate reduction therapy for glycosphingolipid storage disorders. Expert Opin. Investig. Drugs 2001, 10, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, B.E.; Weinreb, N.J. Gaucher disease: A comprehensive review. Crit Rev. Oncog. 2013, 18, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Shayman, J. ELIGLUSTAT TARTRATE: Glucosylceramide Synthase Inhibitor Treatment of Type 1 Gaucher Disease. Drugs Future 2010, 35, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Abe, A.; Arend, L.J.; Lee, L.; Lingwood, C.; Brady, R.O.; Shayman, J.A. Glycosphingolipid depletion in fabry disease lymphoblasts with potent inhibitors of glucosylceramide synthase. Kidney Int. 2000, 57, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Abe, A.; Gregory, S.; Lee, L.; Killen, P.D.; Brady, R.O.; Kulkarni, A.; Shayman, J.A. Reduction of globotriaosylceramide in Fabry disease mice by substrate deprivation. J. Clin. Investig. 2000, 105, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.; Sun, Y.; Bangari, D.S.; Budman, E.; Park, H.; Nietupski, J.B.; Allaire, A.; Cromwell, M.A.; Wang, B.; Grabowski, G.A.; et al. CNS-accessible Inhibitor of Glucosylceramide Synthase for Substrate Reduction Therapy of Neuronopathic Gaucher Disease. Mol. Ther. 2016, 24, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, M.F.; Santos, J.I.; Alves, S. Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders. Int. J. Mol. Sci. 2016, 17, 1065. [Google Scholar] [CrossRef] [PubMed]
- Dupre, T.V.; Doll, M.A.; Shah, P.P.; Sharp, C.N.; Siow, D.; Megyesi, J.; Shayman, J.; Bielawska, A.; Bielawski, J.; Beverly, L.J.; et al. Inhibiting glucosylceramide synthase exacerbates cisplatin-induced acute kidney injury. J. Lipid Res. 2017, 58, 1439–1452. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abou Daher, A.; El Jalkh, T.; Eid, A.A.; Fornoni, A.; Marples, B.; Zeidan, Y.H. Translational Aspects of Sphingolipid Metabolism in Renal Disorders. Int. J. Mol. Sci. 2017, 18, 2528. https://doi.org/10.3390/ijms18122528
Abou Daher A, El Jalkh T, Eid AA, Fornoni A, Marples B, Zeidan YH. Translational Aspects of Sphingolipid Metabolism in Renal Disorders. International Journal of Molecular Sciences. 2017; 18(12):2528. https://doi.org/10.3390/ijms18122528
Chicago/Turabian StyleAbou Daher, Alaa, Tatiana El Jalkh, Assaad A. Eid, Alessia Fornoni, Brian Marples, and Youssef H. Zeidan. 2017. "Translational Aspects of Sphingolipid Metabolism in Renal Disorders" International Journal of Molecular Sciences 18, no. 12: 2528. https://doi.org/10.3390/ijms18122528
APA StyleAbou Daher, A., El Jalkh, T., Eid, A. A., Fornoni, A., Marples, B., & Zeidan, Y. H. (2017). Translational Aspects of Sphingolipid Metabolism in Renal Disorders. International Journal of Molecular Sciences, 18(12), 2528. https://doi.org/10.3390/ijms18122528