Understanding the Molecular Genetics of Basal Cell Carcinoma

 , ,
, ,
Abstract
:1. Introduction
2. Established BCC-Associated Genes
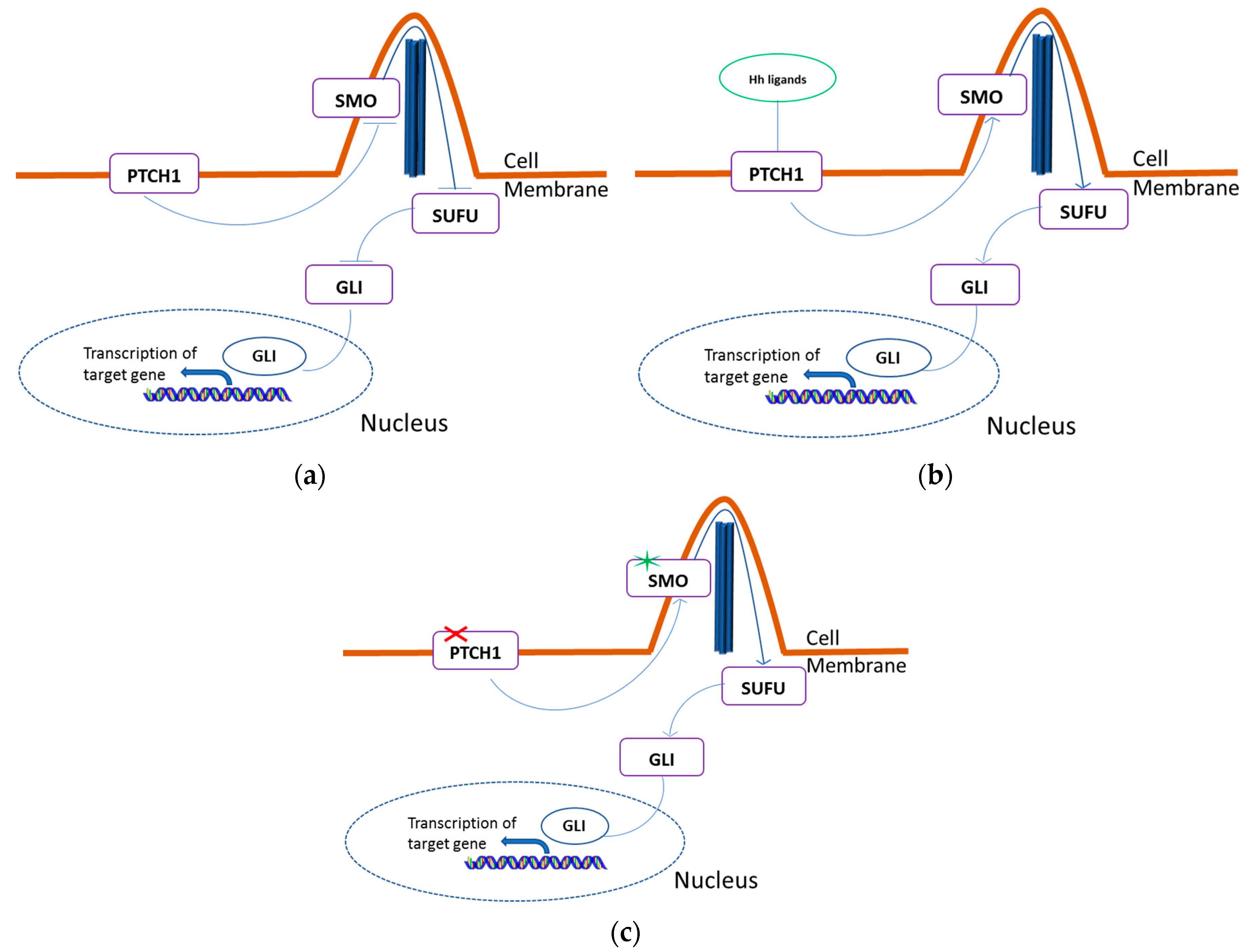
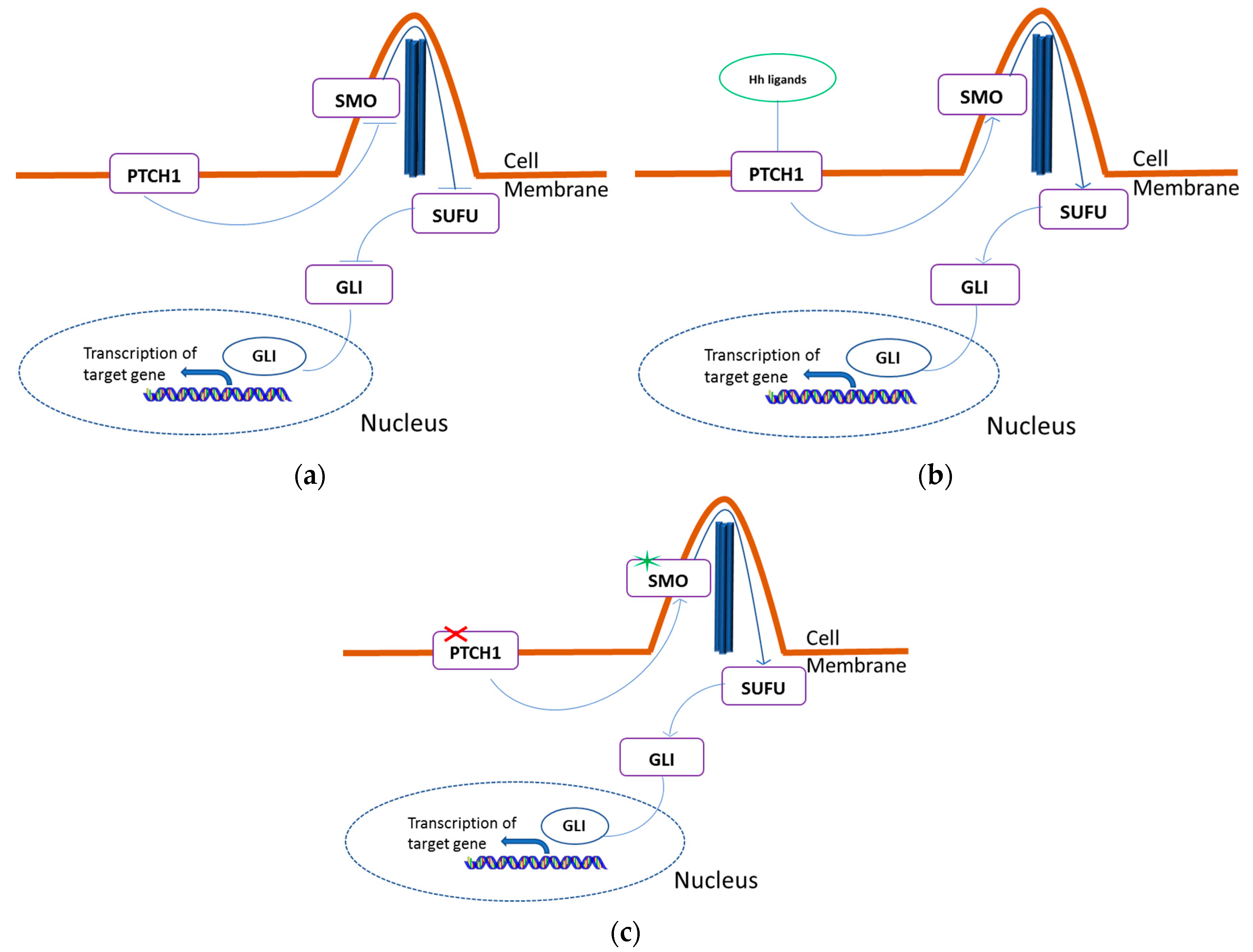
2.1. HH Pathway Genes
2.2. TP53 Gene
3. Novel BCC-Associated Genes
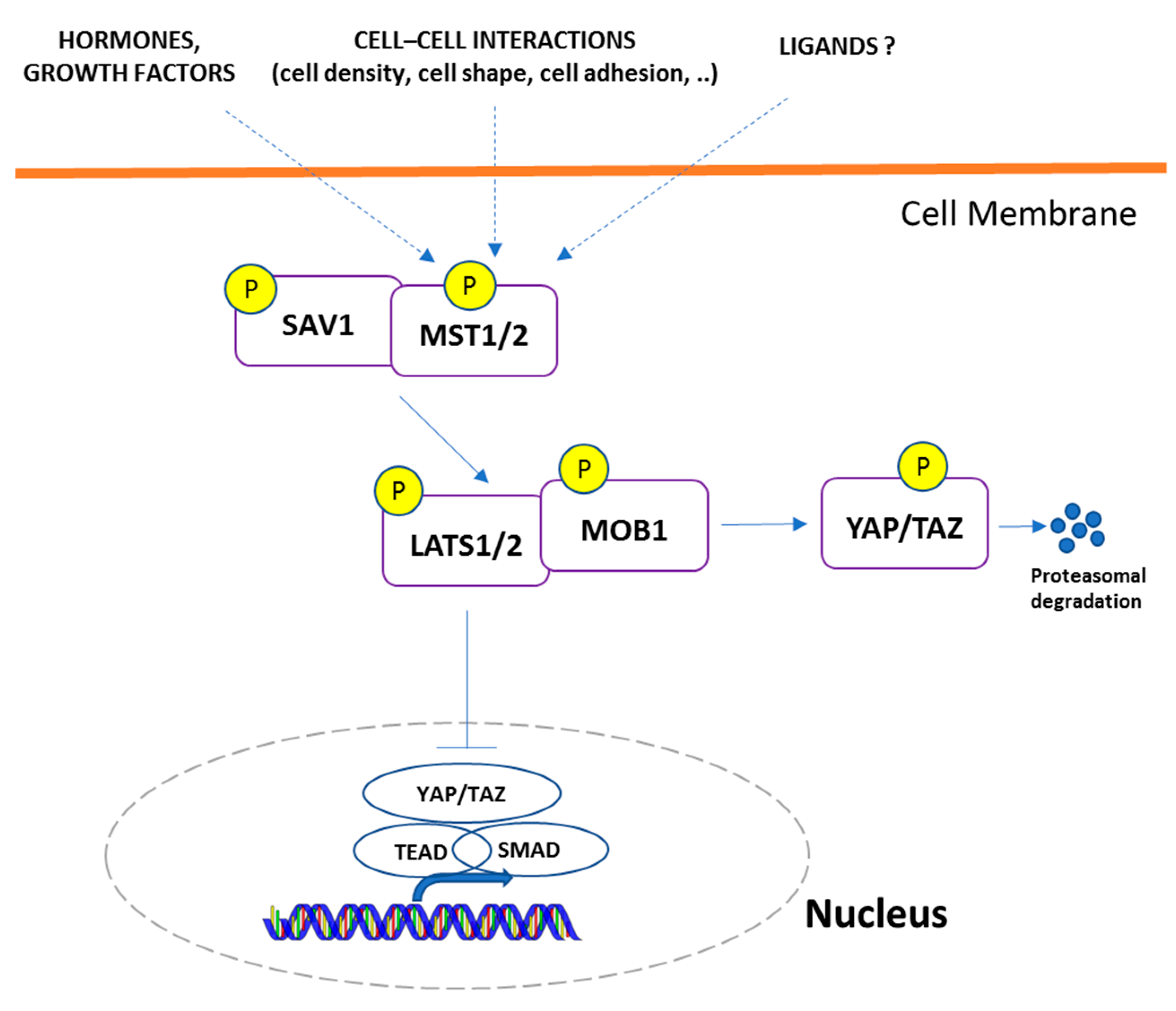
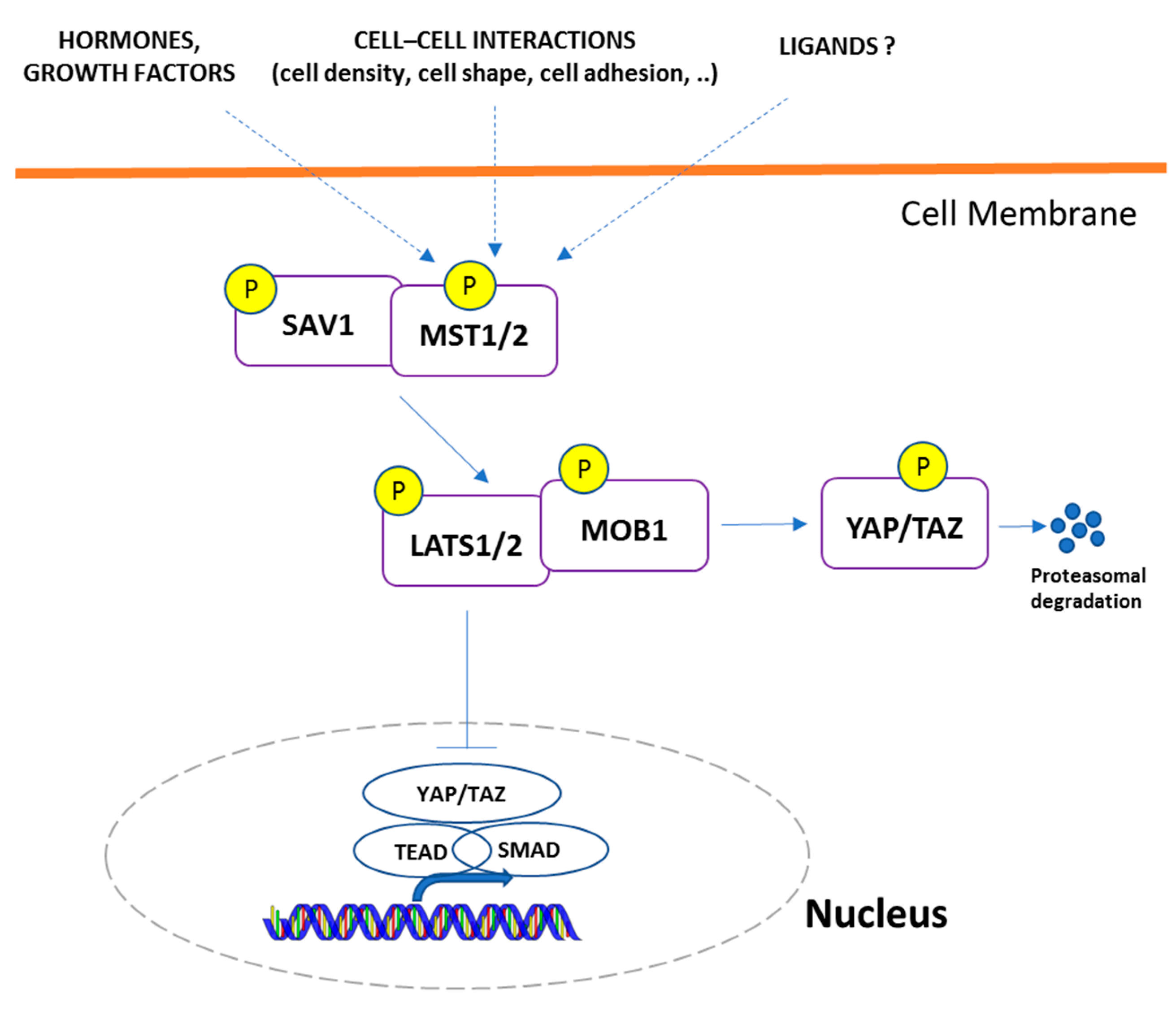
3.1. Hippo-YAP Signaling Genes
3.2. MYCN/FBXW7 Signaling
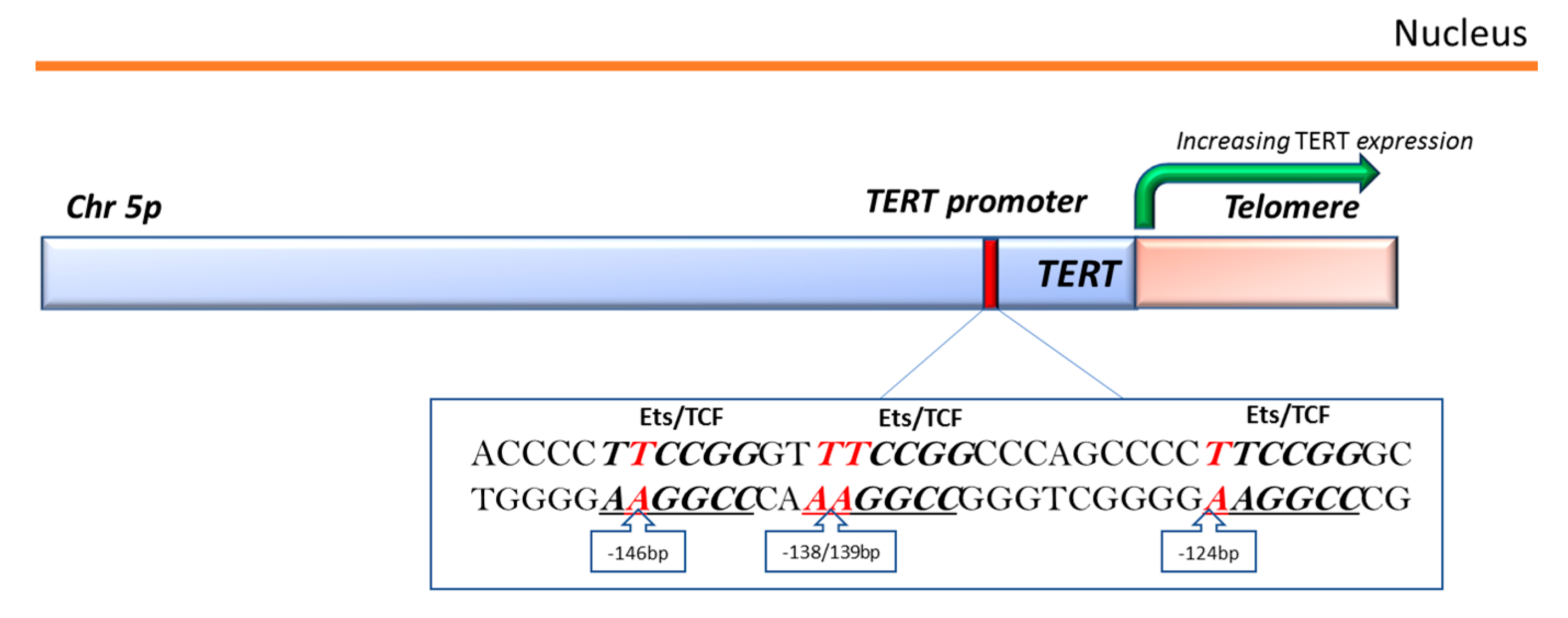
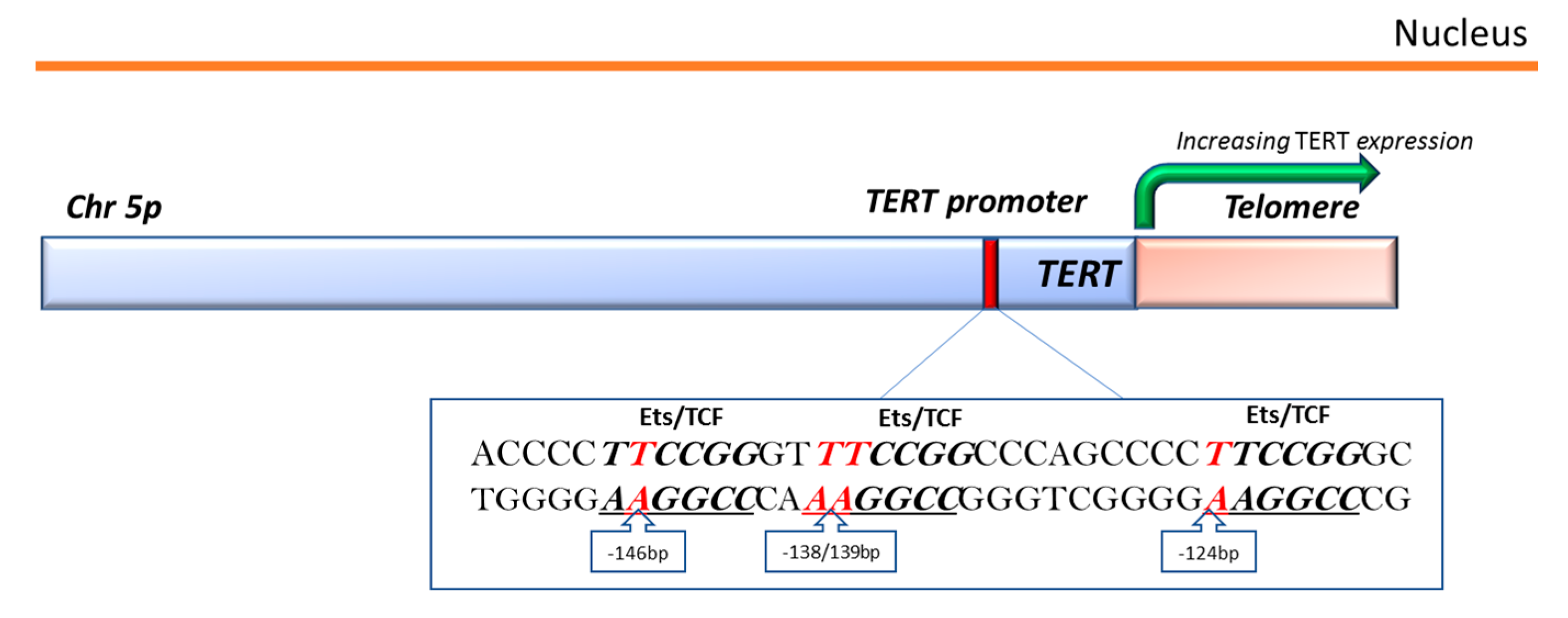
3.3. TERT-Promoter
3.4. DPH3-OXNAD1 Bidirectional Promoter
3.5. Other Potential BCC-Associated Genes
4. Molecular Therapy
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Puig, S.; Berrocal, A. Management of high-risk and advanced basal cell carcinoma. Clin. Transl. Oncol. 2015, 17, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Verkouteren, J.A.C.; Ramdas, K.H.R.; Wakkee, M.; Nijsten, T. Epidemiology of basal cell carcinoma: Scholarly review. Br. J. Dermatol. 2017, 177, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Flohil, S.C.; Seubring, I.; van Rossum, M.M.; Coebergh, J.W.; de Vries, E.; Nijsten, T. Trends in basal cell carcinoma incidence rates: A 37-year Dutch observational study. J. Investig. Dermatol. 2013, 133, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Deady, S.; Sharp, L.; Comber, H. Increasing skin cancer incidence in young; affluent; urban populations: A challenge for prevention. Br. J. Dermatol. 2014, 171, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Rubin, A.I.; Chen, E.H.; Ratner, D. Basal-cell carcinoma. N. Engl. J. Med. 2005, 353, 2262–2269. [Google Scholar] [CrossRef] [PubMed]
- White, J.C. Multiple benign cystic epitheliomas. J. Cutan. Genitourin. Dis. 1894, 12, 477–484. [Google Scholar]
- Jarisch, W. On the doctrine of skin tumors. Arch. Dermatol. Syphilol. 1894, 18, 162–222. [Google Scholar]
- Gorlin, R.J.; Goltz, R.W. Multiple nevoid basal cell epithelioma; jaw cysts and bifid rib: A syndrome. N. Engl. J. Med. 1960, 262, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Gailani, M.R.; Bale, S.J.; Leffell, D.J.; Di Giovanna, J.J.; Peck, G.L.; Poliak, S.; Drum, M.A.; Pastakia, B.; McBride, O.W.; Kase, R.; et al. Developmental defects in Gorlin syndrome related to a putative tumor suppressor gene on chromosome 9. Cell 1992, 69, 111–117. [Google Scholar] [CrossRef]
- Hahn, H.; Wicking, C.; Zaphiropoulous, P.G. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H., Jr.; et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef] [PubMed]
- Gailani, M.R.; Ståhle-Bäckdahl, M.; Leffell, D.J.; Glynn, M.; Zaphiropoulos, P.G.; Pressman, C.; Undén, A.B.; Dean, M.; Brash, D.E.; Bale, A.E.; et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat. Genet. 1996, 14, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Dahmane, N.; Lee, J.; Robins, P.; Heller, P.; Ruiz i Altaba, A. Activation of the transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature 1997, 389, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Murone, M.; Luoh, S.M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Aszterbaum, M.; Rothman, A.; Johnson, R.L.; Fisher, M.; Xie, J.; Bonifas, J.M.; Zhang, X.; Scott, M.P.; Epstein, E.H., Jr. Identification of mutations in the human PATCHED gene in sporadic basal cell carcinomas and in patients with the basal cell nevus syndrome. J. Investig. Dermatol. 1998, 11, 885–888. [Google Scholar] [CrossRef] [PubMed]
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Rev. Cancer 2008, 8, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ping, X.L.; Lee, P.K.; Wu, X.L.; Yao, Y.J.; Zhang, M.J.; Silvers, D.N.; Ratner, D.; Malhotra, R.; Peacocke, M.; et al. Role of PTCH and p53 genes in early-onset basal cell carcinoma. Am. J. Pathol. 2001, 158, 381–385. [Google Scholar] [CrossRef]
- Kim, M.Y.; Park, H.J.; Baek, S.C.; Byun, D.G.; Houh, D. Mutations of the p53 and PTCH gene in basal cell carcinomas: UV mutation signature and strand bias. J. Dermatol. Sci. 2002, 29, 1–9. [Google Scholar] [CrossRef]
- Jayaraman, S.S.; Rayhan, D.J.; Hazany, S.; Kolodney, M.S. Mutational landscape of basal cell carcinomas by whole-exome sequencing. J. Investig. Dermatol. 2014, 134, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Van der Schroeff, J.G.; Evers, L.M.; Boot, A.J.; Bos, J.L. Ras oncogene mutations in basal cell carcinomas and squamous cell carcinomas of human skin. J. Investig. Dermatol. 1990, 94, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Pierceall, W.E.; Goldberg, L.H.; Tainsky, M.A.; Mukhopadhyay, T.; Ananthaswamy, H.N. Ras gene mutation and amplification in human nonmelanoma skin cancers. Mol. Carcinog. 1991, 4, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 2016, 48, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Griewank, K.G.; Murali, R.; Schilling, B.; Schimming, T.; Möller, I.; Moll, I.; Schwamborn, M.; Sucker, A.; Zimmer, L.; Schadendorf, D.; et al. TERT promoter mutations are frequent in cutaneous basal cell carcinoma and squamous cell carcinoma. PLoS ONE 2013, 8, e80354. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.A.; Laughlin, T.S.; Rothberg, P.G. Mutations of the TERT promoter are common in basal cell carcinoma and squamous cell carcinoma. Mod. Pathol. 2014, 27, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Pópulo, H.; Boaventura, P.; Vinagre, J.; Batista, R.; Mendes, A.; Caldas, R.; Pardal, J.; Azevedo, F.; Honavar, M.; Guimarães, I.; et al. TERT promoter mutations in skin cancer: The effects of sun exposure and X-irradiation. J. Investig. Dermatol. 2014, 134, 2251–2257. [Google Scholar] [CrossRef] [PubMed]
- Denisova, E.; Heidenreich, B.; Nagore, E.; Rachakonda, P.S.; Hosen, I.; Akrap, I.; Traves, V.; García-Casado, Z.; López-Guerrero, J.A.; Requena, C.; et al. Frequent DPH3 promoter mutations in skin cancers. Oncotarget 2015, 6, 35922–35930. [Google Scholar] [PubMed]
- Ikehata, H.; Ono, T. The mechanisms of UV mutagenesis. J. Radiat. Res. 2011, 52, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, J.; Plaza, P.; Brettel, K. Repair of (6–4) Lesions in DNA by (6–4) Photolyase: 20 Years of Quest for the Photoreaction Mechanism. Photochem. Photobiol. 2017, 93, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.R. Ultraviolet radiation and skin cancer: Molecular mechanisms. J. Cutan. Pathol. 2005, 32, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Daya-Grosjean, L.; Couvé-Privat, S. Sonic hedgehog signaling in basal cell carcinomas. Cancer Lett. 2005, 225, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Justilien, V.; Fields, A.P. Molecular pathways: Novel approaches for improved therapeutic targeting of Hedgehog signaling in cancer stem cells. Clin. Cancer Res. 2015, 21, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Therond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Bangs, F.; Anderson, K.V. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028175. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, A.; Levesque, M.P.; Dummer, R.; Kabashima, K. Hedgehog signaling in basal cell carcinoma. J. Dermatol. Sci. 2015, 78, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. Hedgehog target genes: Mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Seto, M.; Ohta, M.; Asaoka, Y.; Ikenoue, T.; Tada, M.; Miyabayashi, K.; Mohri, D.; Tanaka, Y.; Ijichi, H.; Tateishi, K.; et al. Regulation of the hedgehog signaling by the mitogen-activated protein kinase cascade in gastric cancer. Mol. Carcinog. 2009, 48, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Rajurkar, M.; De Jesus-Monge, W.E.; Driscoll, D.R.; Appleman, V.A.; Huang, H.; Cotton, J.L.; Klimstra, D.S.; Zhu, L.J.; Simin, K.; Xu, L.; et al. The activity of GLI transcription factors is essential for Kras-induced pancreatic tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E1038–E1047. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, B.; Lu, Y.; Teng, K.Y.; Nuovo, G.; Li, X.; Shapiro, C.L.; Majumder, S. Hedgehog signaling is a novel therapeutic target in tamoxifen-resistant breast cancer aberrantly activated by PI3K/AKT pathway. Cancer Res. 2012, 72, 5048–5059. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.T.; Vanderbilt, D.B.; Lin, C.C.; Martin, K.H.; Brundage, K.M.; Ruppert, J.M. SOX9 inhibits beta-TrCP-mediated protein degradation to promote nuclear GLI1 expression and cancer stem cell properties. J. Cell Sci. 2015, 128, 1123–1138. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.; Caiping, S.; Qing, Z.; Xiaojing, W. Sonic hedgehog-GLI1 signals promote epithelial-mesenchymal transition in ovarian cancer by mediating PI3K/AKT pathway. Med. Oncol. 2015, 32, 368. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhu, G.; Huang, J.; Li, L.; Du, Y.; Gao, Y.; Wu, D.; Wang, X.; Hsieh, J.T.; He, D.; et al. Non-canonical GLI1/2 activation by PI3K/AKT signaling in renal cell carcinoma: A novel potential therapeutic target. Cancer Lett. 2016, 370, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.W. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers 2016, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Makinodan, E.; Marneros, A.G. Protein kinase a activation inhibits oncogenic sonic hedgehog signalling and suppresses basal cell carcinoma of the skin. Exp. Dermatol. 2012, 21, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Sheng, T.; Chi, S.; Zhang, X.; Xie, J. Regulation of GLI1 localization by the camp/protein kinase a signaling axis through a site near the nuclear localization signal. J. Biol. Chem. 2006, 281, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.; Ruiz i Altaba, A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 2009, 28, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Athar, M.; Li, C.; Kim, A.L.; Spiegelman, V.S.; Bickers, D.R. Sonic hedgehog signaling in Basal cell nevus syndrome. Cancer Res. 2014, 74, 4967–4975. [Google Scholar] [CrossRef] [PubMed]
- Reifenberger, J.; Wolter, M.; Knobbe, C.B.; Köhler, B.; Schönicke, A.; Scharwächter, C.; Kumar, K.; Blaschke, B.; Ruzicka, T.; Reifenberger, G. Somatic mutations in the PTCH; SMOH; SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol. 2005, 152, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Teh, M.T.; Blaydon, D.; Chaplin, T.; Foot, N.J.; Skoulakis, S.; Raghavan, M.; Harwood, C.A.; Proby, C.M.; Philpott, M.P.; Young, B.D.; et al. Genomewide single nucleotide polymorphism microarray mapping in basal cell carcinomas unveils uniparental disomy as a key somatic event. Cancer Res. 2005, 65, 8597–8603. [Google Scholar] [CrossRef] [PubMed]
- Santos, D.C.; Zaphiropoulos, P.G.; Neto, C.F.; Pimentel, E.R.; Sanches, J.A., Jr.; Ruiz, I.R. PTCH1 gene mutations in exon 17 and loss of heterozygosity on D9S180 microsatellite in sporadic and inherited human basal cell carcinomas. Int. J. Dermatol. 2011, 50, 838–843. [Google Scholar] [CrossRef] [PubMed]
- Gailani, M.R.; Leffell, D.J.; Ziegler, A.; Gross, E.G.; Brash, D.E.; Bale, A.E. Relationship between sunlight exposure and a key genetic alteration in basal cell carcinoma. J. Natl. Cancer Inst. 1996, 88, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.S.; Bu, D.F.; Li, X.Y.; Ma, Z.H.; Yang, Y.; Lin, Z.M.; Lu, F.M.; Tu, P.; Li, H. Unique features of PTCH1mutation spectrum in Chinese sporadic basal cellcarcinoma. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Lacour, J.P. Carcinogenesis of basal cell carcinomas: Genetics and molecular mechanisms. Br. J. Dermatol. 2002, 146 (Suppl. S61), 17–19. [Google Scholar] [CrossRef] [PubMed]
- Daya-Grosjean, L.; Sarasin, A. UV-specific mutations of the human patched gene in basal cell carcinomas from normal individuals and xeroderma pigmentosum patients. Mutat. Res. 2000, 450, 193–199. [Google Scholar] [CrossRef]
- Danaee, H.; Karagas, M.R.; Kelsey, K.T.; Perry, A.E.; Nelson, H.H. Allelic loss at Drosophila patched gene is highly prevalent in Basal and squamous cell carcinomas of the skin. J. Investig. Dermatol. 2006, 126, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Lassacher, A.; Quehenberger, F.; Kerl, H.; Wolf, P. UV fingerprints predominate in the PTCH mutation spectra of basal cell carcinomas independent of clinical phenotype. J. Investig. Dermatol. 2007, 127, 2872–2881. [Google Scholar] [CrossRef] [PubMed]
- Rady, P.; Scinicariello, F.; Wagner, R.F., Jr.; Tyring, S.K. p53 mutations in basal cell carcinomas. Cancer Res. 1992, 52, 3804–3806. [Google Scholar] [PubMed]
- Ziegler, A.; Leffell, D.J.; Kunala, S.; Sharma, H.W.; Gailani, M.; Simon, J.A.; Halperin, A.J.; Baden, H.P.; Shapiro, P.E.; Bale, A.E. Mutation hotspots due to sunlight in the p53 gene of nonmelanoma skin cancers. Proc. Natl. Acad. Sci. USA 1993, 90, 4216–4220. [Google Scholar] [CrossRef] [PubMed]
- Rosenstein, B.S.; Phelps, R.G.; Weinstock, M.A.; Bernstein, J.L.; Gordon, M.L.; Rudikoff, D.; Kantor, I.; Shelton, R.; Lebwohl, M.G. p53 mutations in basal cell carcinomas arising in routine users of sunscreens. Photochem. Photobiol. 1999, 70, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Bolshakov, S.; Walker, C.M.; Strom, S.S.; Selvan, M.S.; Clayman, G.L.; El-Naggar, A.; Lippman, S.M.; Kripke, M.L.; Ananthaswamy, H.N. p53 mutations in human aggressive and nonaggressive basal and squamous cell carcinomas. Clin. Cancer Res. 2003, 9, 228–324. [Google Scholar] [PubMed]
- Wang, Y.M.; Huang, Y.S.; Ma, Z.H.; Bu, D.F.; Wang, Y.; Tu, P.; Li, H. Frequency and features of TP53 mutation in 30 Chinese patients with sporadic basal cell carcinoma. Clin. Exp. Dermatol. 2014, 39, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Sarin, K.Y.; Li, J.R.; Yao, C.Y.; Urman, N.M.; Chang, A.L.S.; Tang, J.Y.; Oro, A.E. Rolling the Genetic Dice: Neutral and Deleterious Smoothened Mutations in Drug-Resistant Basal Cell Carcinoma. J. Investig. Dermatol. 2015, 135, 2138–2141. [Google Scholar] [CrossRef] [PubMed]
- Urman, N.M.; Mirza, A.; Atwood, S.X.; Whitson, R.J.; Sarin, K.Y.; Tang, J.Y.; Oro, A.E. Tumor-Derived Suppressor of Fused Mutations Reveal Hedgehog Pathway Interactions. PLoS ONE 2016, 11, e0168031. [Google Scholar] [CrossRef] [PubMed]
- Smyth, I.; Narang, M.A.; Evans, T.; Heimann, C.; Nakamura, Y.; Chenevix-Trench, G.; Pietsch, T.; Wicking, C.; Wainwright, B.J. Isolation and characterization of human patched 2 (PTCH2), a putative tumour suppressor gene inbasal cell carcinoma and medulloblastoma on chromosome 1p32. Hum. Mol. Genet. 1999, 8, 291–297. [Google Scholar] [CrossRef] [PubMed]
- De Zwaan, S.E.; Haass, N.K. Genetics of basal cell carcinoma. Australas. J. Dermatol. 2010, 51, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P. Cancer. p53; guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Strasser, A.; Kelly, G.L. Tumor-Suppressor Functions of the TP53 Pathway. Cold Spring Harb. Perspect. Med. 2016, 6, a026062. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, P. Non-melanoma skin cancer: What drives tumor development and progression? Carcinogenesis 2005, 26, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.Y.; Wang, J.; Mancianti, M.L.; Epstein, E.H., Jr. Basal cell carcinomas arise from hair follicle stem cells in Ptch1(+/−) mice. Cancer Cell 2011, 19, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, C.L.; Ananthaswamy, H.N. p53 and the pathogenesis of skin cancer. Toxicol. Appl. Pharmacol. 2007, 224, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Giglia-Mari, G.; Sarasin, A. TP53 mutations in human skin cancers. Hum. Mutat. 2003, 21, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Tornaletti, S.; Pfeifer, G.P. Slow repair of pyrimidine dimers at p53 mutation hotspots in skin cancer. Science 1994, 263, 1436–1438. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.L. The Hippo-YAP pathway in organ size control and tumorigenesis: An updated version. Genes Dev. 2010, 24, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Michaloglou, C.; Lehmann, W.; Martin, T.; Delaunay, C.; Hueber, A.; Barys, L.; Niu, H.; Billy, E.; Wartmann, M.; Ito, M.; et al. The tyrosine phosphatase PTPN14 is a negative regulator of YAP activity. PLoS ONE 2013, 8, e61916. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Chun, A.; Cheung, K.; Rashidi, B.; Yang, X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J. Biol. Chem. 2008, 283, 5496–5509. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Huang, J.; Wang, X.; Yuan, J.; Li, X.; Feng, L.; Park, J.I.; Chen, J. PTPN14 is required for the density-dependent control of YAP1. Genes Dev. 2012, 26, 1959–1971. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.E.; Li, Y.W.; Yang, N.; Shen, H.; Orillion, A.R.; Zhang, J. PTPN14 forms a complex with Kibra and LATS1 proteins and negatively regulates the YAP oncogenic function. J. Biol. Chem. 2014, 289, 23693–23700. [Google Scholar] [CrossRef] [PubMed]
- Overholtzer, M.; Zhang, J.; Smolen, G.A.; Muir, B.; Li, W.; Sgroi, D.C.; Deng, C.X.; Brugge, J.S.; Haber, D.A. Transforming properties of YAP; a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. USA 2006, 103, 12405–12410. [Google Scholar] [CrossRef] [PubMed]
- Schlegelmilch, K.; Mohseni, M.; Kirak, O.; Pruszak, J.; Rodriguez, J.R.; Zhou, D.; Kreger, B.T.; Vasioukhin, V.; Avruch, J.; Brummelkamp, T.J.; et al. Yap1 acts downstream of alpha-catenin to control epidermal proliferation. Cell 2011, 144, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pasolli, H.A.; Fuchs, E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc. Natl. Acad. Sci. USA 2011, 108, 2270–2275. [Google Scholar] [CrossRef] [PubMed]
- Tate, G.; Kishimoto, K.; Mitsuya, T. Biallelic alterations of the large tumor suppressor 1 (LATS1) gene in infiltrative; but not superficial; basal cell carcinomas in a Japanese patient with nevoid basal cell carcinoma syndrome. Med. Mol. Morphol. 2015, 48, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Hatton, B.A.; Knoepfler, P.S.; Kenney, A.M.; Rowitch, D.H.; de Alborán, I.M.; Olson, J.M.; Eisenman, R.N. N-myc is an essential downstream effector of Shh signaling during both normal and neoplastic cerebellar growth. Cancer Res. 2006, 66, 8655–8661. [Google Scholar] [CrossRef] [PubMed]
- Welcker, M.; Orian, A.; Jin, J.; Grim, J.E.; Harper, J.W.; Eisenman, R.N.; Clurman, B.E. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. USA 2004, 101, 9085–9090. [Google Scholar] [CrossRef] [PubMed]
- Freier, K.; Flechtenmacher, C.; Devens, F.; Hartschuh, W.; Hofele, C.; Lichter, P.; Joos, S. Recurrent NMYC copy number gain and high protein expression in basal cell carcinoma. Oncol. Rep. 2006, 15, 1141–1145. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, P.S.; Hosen, I.; de Verdier, P.J.; Fallah, M.; Heidenreich, B.; Ryk, C.; Wiklund, N.P.; Steineck, G.; Schadendorf, D.; Heimminki, K.; et al. TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc. Natl. Acad. Sci. USA 2013, 110, 17426–17431. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, B.; Kumar, R. TERT promoter mutations in telomere biology. Mutat. Res. 2017, 771, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, B.; Rachakonda, P.S.; Hosen, I.; Volz, F.; Hemminki, K.; Weyerbrock, A.; Kumar, R. TERT promoter mutations and telomere length in adult malignant gliomas and recurrences. Oncotarget 2015, 6, 10617–10633. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, B.; Rachakonda, P.S.; Hemminki, K.; Kumar, R. TERT promoter mutations in cancer development. Curr. Opin. Genet. Dev. 2014, 24, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shi, Y.; Ju, P.; Liu, R.; Yeo, S.P.; Xia, Y.; Owlanj, H.; Feng, Z. Silencing of diphthamide synthesis 3 (Dph3) reduces metastasis of murine melanoma. PLoS ONE 2012, 7, e49988. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, N.; Jacobsen, A.; Schultz, N.; Sander, C.; Lee, W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet. 2014, 46, 1160–1165. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, N.J.; Ny, L.; Nilsson, J.A.; Larsson, E. Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nat. Genet. 2014, 46, 1258–1263. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, B.; Brautigan, D.L. Protein phosphatase PP6 N terminal domain restricts G1 to S phase progression in human cancer cells. Cell Cycle 2007, 6, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Couzens, A.L.; Knight, J.D.; Kean, M.J.; Teo, G.; Weiss, A.; Dunham, W.H.; Lin, Z.Y.; Bagshaw, R.D.; Sicheri, F.; Pawson, T.; et al. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci. Signal. 2013, 6, rs15. [Google Scholar] [CrossRef] [PubMed]
- Hammond, D.; Zeng, K.; Espert, A.; Bastos, R.N.; Baron, R.D.; Gruneberg, U.; Barr, F.A. Melanoma-associated mutations in protein phosphatase 6 cause chromosome instability and DNA damage owing to dysregulated Aurora-A. J. Cell Sci. 2013, 126, 3429–3440. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Weiss, G.J.; Miller, W.H., Jr.; Gettinger, S.; Eigl, B.J.; Chang, A.L.; Dunbar, J.; Devens, S.; Faia, K.; Skliris, G.; et al. Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin. Cancer Res. 2013, 19, 2766–2774. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.J.; Messersmith, W.A.; Shaik, M.N.; Zheng, X.; McLachlan, K.R.; Cesari, R.; Courtney, R.; Levin, W.J.; El-Khoueiry, A.B. A phase I study of PF-04449913, an oral Hedgehog inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Amaral, T.; Garbe, C. Non-melanoma skin cancer: New and future synthetic drug treatments. Expert Opin. Pharmacother. 2017, 18, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Cucchi, D.; Occhione, M.A.; Gulino, A.; De Smaele, E. Hedgehog signaling pathway and its targets for treatment in basal cell carcinoma. J. Exp. Pharmacol. 2012, 4, 173–185. [Google Scholar] [PubMed]
- Basset-Seguin, N.; Hauschild, A.; Grob, J.J.; Kunstfeld, R.; Dréno, B.; Mortier, L.; Ascierto, P.A.; Licitra, L.; Dutriaux, C.; Thomas, L.; et al. Vismodegib in patients with advanced basal cell carcinoma (STEVIE): A pre-planned interim analysis of an international; open-label trial. Lancet Oncol. 2015, 16, 729–736. [Google Scholar] [CrossRef]
- Kish, T.; Corry, L. Sonidegib (Odomzo) for the Systemic Treatment of Adults with Recurrent, Locally Advanced Basal Cell Skin Cancer. Pharm. Ther. 2016, 41, 322–325. [Google Scholar]
- Brinkhuizen, T.; Reinders, M.G.; Van Geel, M.; Hendriksen, A.J.; Paulussen, A.D.; Winnepenninckx, V.J.; Keymeulen, K.B.; Soetekouw, P.M.; van Steensel, M.A. Acquired resistance to the Hedgehog pathway inhibitor vismodegib due to smoothened mutations in treatment of locally advanced basal cell carcinoma. J. Am. Acad. Dermatol. 2014, 71, 1005–1008. [Google Scholar] [CrossRef] [PubMed]
- Pricl, S.; Cortelazzi, B.; Dal Col, V.; Marson, D.; Laurini, E.; Fermeglia, M.; Licitra, L.; Pilotti, S.; Bossi, P.; Perrone, F. Smoothened (SMO) receptor mutations dictate resistance to vismodegib in basal cell carcinoma. Mol. Oncol. 2015, 9, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Gonnissen, A.; Isebaert, S.; Haustermans, K. Targeting the Hedgehog signaling pathway in cancer: Beyond Smoothened. Oncotarget 2015, 6, 13899–13913. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic analysis of Smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2010, 2, 51ra70. [Google Scholar] [CrossRef] [PubMed]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmaco-logical disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Brodowska, K.; Al-Moujahed, A.; Marmalidou, A.; Meyer Zu Horste, M.; Cichy, J.; Miller, J.W.; Gragoudas, E.; Vavvas, D.G. The clinically used photosensitizer Verteporfin (VP) inhibits YAP-TEAD andhuman retinoblastoma cell growth in vitro without light activation. Exp. Eye Res. 2014, 124, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, A.; Chaudhary, S.C.; Rana, M.; Elmets, C.A.; Athar, M. Basal cell carcinoma pathogenesis and therapy involvinghedgehog signaling and beyond. Mol. Carcinog. 2017, 56, 2543–2557. [Google Scholar] [CrossRef] [PubMed]
- Glaser, R.; Andridge, R.; Yang, E.V.; Shana’ah, A.Y.; Di Gregorio, M.; Chen, M.; Johnson, S.L.; De Renne, L.A.; Lambert, D.R.; Jewell, S.D.; et al. Tumor site immune markers associated with risk for subsequent basal cell carcinomas. PLoS ONE 2011, 6, e25160. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.A.; Bishop, G.A.; Lowes, M.A.; Cooke, B.; Barnetson, R.S.; Halliday, G.M. Cytokine profiles in spontaneously regressing basal cell carcinomas. Br. J. Dermatol. 2000, 143, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Kaporis, H.G.; Guttman-Yassky, E.; Lowes, M.A.; Haider, A.S.; Fuentes-Duculan, J.; Darabi, K.; Whynot-Ertelt, J.; Khatcherian, A.; Cardinale, I.; Novitskaya, I.; et al. Human basal cell carcinoma is associated with Foxp3+ T cells in a Th2 dominant microenvironment. J. Investig. Dermatol. 2007, 127, 2391–2398. [Google Scholar] [CrossRef] [PubMed]
- Elamin, I.; Zecević, R.D.; Vojvodić, D.; Medenica, L.; Pavlović, M.D. Cytokine concentrations in basal cell carcinomas of different histological types and localization. Acta Dermatovenerol. Alp. Pannonica Adriat. 2008, 17, 55–59. [Google Scholar] [PubMed]
- Pellegrini, C.; Orlandi, A.; Costanza, G.; Di Stefani, A.; Piccioni, A.; Di Cesare, A.; Chiricozzi, A.; Ferlosio, A.; Peris, K.; Fargnoli, M.C. Expression of IL-23/Th17-related cytokines in basal cell carcinoma and in the response to medical treatments. PLoS ONE 2017, 12, e0183415. [Google Scholar] [CrossRef] [PubMed]
- Nardinocchi, L.; Sonego, G.; Passarelli, F.; Avitabile, S.; Scarponi, C.; Failla, C.M.; Simoni, S.; Albanesi, C.; Cavani, A. Interleukin-17 and interleukin-2 promote tumor progression in human nonmelanoma skin cancer. Eur. J. Immunol. 2015, 45, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Leidner, R.; Stankevich, E.; Piening, B.; Bifulco, C.; Lowy, I.; Fury, M.G. Responses of metastatic basal cell and cutaneous squamous cell carcinomas to anti-PD1 monoclonal antibody REGN2810. J. Immunother. Cancer 2016, 4, 70. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Eichstaedt, J.; Möbus, L.; Kähler, K.; Weichenthal, M.; Schwarz, T.; Weidinger, S. Regression of melanoma metastases and multiple non-melanoma skin cancers in xeroderma pigmentosum by the PD1-antibody pembrolizumab. Eur. J. Cancer 2017, 77, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Lilo, M.T.; Ogurtsova, A.; Esandrio, J.; Xu, H.; Brothers, P.; Schollenberger, M.; Sharfman, W.H.; Taube, J.M. Basal cell carcinoma: PD-L1/PD-1 checkpoint expression and tumor regression after PD-1 blockade. J. Immunother. Cancer 2017, 5, 23. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Gene | Nr. of Samples Analyzed | Mutations (%) | LOH (%) | References |
|---|---|---|---|---|
| PTCH1 | 37 | 32.4 | 24.3 | [12] |
| 55 | - | 66.7 | [51] | |
| 26 a | 11.8 | 38.2 | [15] | |
| 24 | 54.2 | - | [11] | |
| 15 | 40.0 | 53.3 | [18] | |
| 31 | 54.8 | 43.5 | [52] | |
| 42 | 66.7 | 52.6 | [19] * | |
| 14 b | 64.3 | 92.8 | [49] | |
| 12 b | 8.3 | 40.0 | [50] | |
| 12 | 75.0 | 41.7 | [48] | |
| 293 c | 73.0 | 55.0 | [22] * | |
| TP53 | 14 | 50.0 | na | [57] |
| 18 d | 61.1 | 5.5 | [51] | |
| 27 | 56.0 | na | [58] | |
| 20 | 35.0 | na | [59] | |
| 24 | 45.8 | na | [11] | |
| 15 | 33.0 | - | [18] | |
| 50 e | 66.0 | na | [60] | |
| 98 f | 37.7 | na | [60] | |
| 42 | 40.5 | 7.9 | [48] | |
| 30 | 20.0 | na | [61] | |
| 12 | 66.7 | na | [19] | |
| 293 | 61.0 | 17.0 | [22] * | |
| SMO | 47 | 6.38 | na | [14] |
| 42 | 9.5 | na | [48] | |
| 293 | 20.0 | na | [22] * | |
| SUFU | 42 | 2.4 | na | [48] |
| 293 | 8.0 | 5.0 | [22] * | |
| NOTCH1 | 12 | 50.0 | na | [19] * |
| 293 | 29.0 | 2.7 | [22] * | |
| NOTCH2 | 12 | 66.7 | na | [19] * |
| 293 | 26.0 | na | [22] * | |
| LATS1 | 293 | 16.0 | 4.0 | [22] * |
| LATS1 | 293 | 12.0 | 5.0 | [22] * |
| PPP6C | 293 | 15.0 | 46.0 | [22] * |
| STK19 | 293 | 10.0 | na | [22] * |
| MYCN | 293 | 30.0 | na | [22] * |
| ARID1A | 293 | 26.0 | 3.0 | [22] * |
| PTPN14 | 293 | 22.0 | 5.0 | [22] * |
| CASP8 | 293 | 11.0 | 3.0 | [22] * |
| CSMD1 | 12 | 91.7 | na | [19] * |
| DPP10 | 12 | 75.0 | na | [19] * |
| CSMD2 | 12 | 66.7 | na | [19] * |
| CSMD3 | 12 | 58.3 | na | [19] * |
| PREX2 | 12 | 58.3 | na | [19] * |
| DCC | 12 | 50.0 | na | [19] * |
| GRIN2A | 12 | 50.0 | na | [19] * |
| TERT-promoter | 32 | 56.2 | na | [23] |
| 42 g | 73.8 | na | [24] | |
| 196 h | 38.8 | na | [25] | |
| 137 | 65.0 | na | [26] | |
| DPH3/OXNAD1 promoter | 137 | 41.6 | na | [26] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellegrini, C.; Maturo, M.G.; Di Nardo, L.; Ciciarelli, V.; Gutiérrez García-Rodrigo, C.; Fargnoli, M.C. Understanding the Molecular Genetics of Basal Cell Carcinoma. Int. J. Mol. Sci. 2017, 18, 2485. https://doi.org/10.3390/ijms18112485
Pellegrini C, Maturo MG, Di Nardo L, Ciciarelli V, Gutiérrez García-Rodrigo C, Fargnoli MC. Understanding the Molecular Genetics of Basal Cell Carcinoma. International Journal of Molecular Sciences. 2017; 18(11):2485. https://doi.org/10.3390/ijms18112485
Chicago/Turabian StylePellegrini, Cristina, Maria Giovanna Maturo, Lucia Di Nardo, Valeria Ciciarelli, Carlota Gutiérrez García-Rodrigo, and Maria Concetta Fargnoli. 2017. "Understanding the Molecular Genetics of Basal Cell Carcinoma" International Journal of Molecular Sciences 18, no. 11: 2485. https://doi.org/10.3390/ijms18112485