RNA Sequencing and Coexpression Analysis Reveal Key Genes Involved in α-Linolenic Acid Biosynthesis in Perilla frutescens Seed

 ,
,
Abstract
:1. Introduction
2. Results
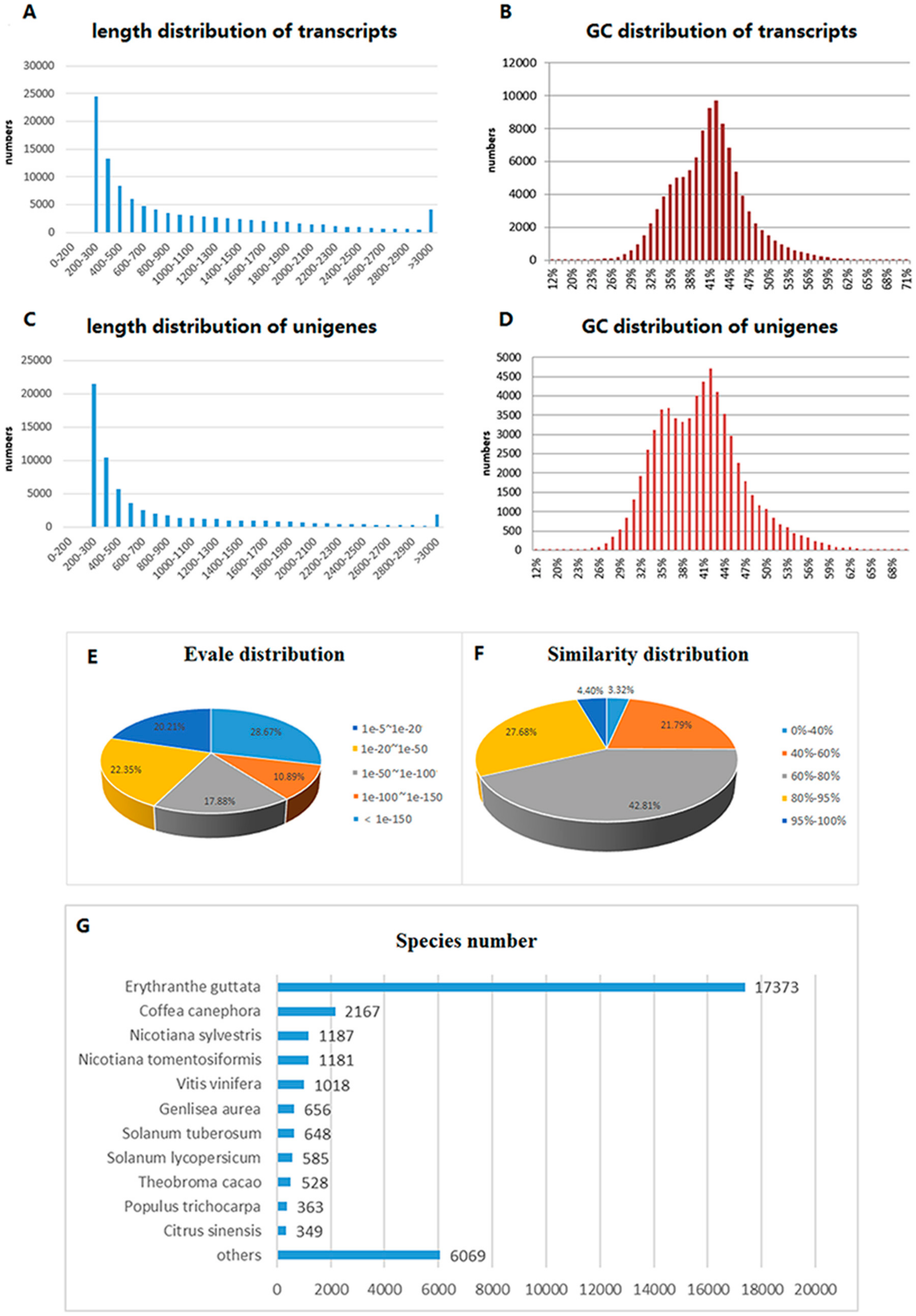
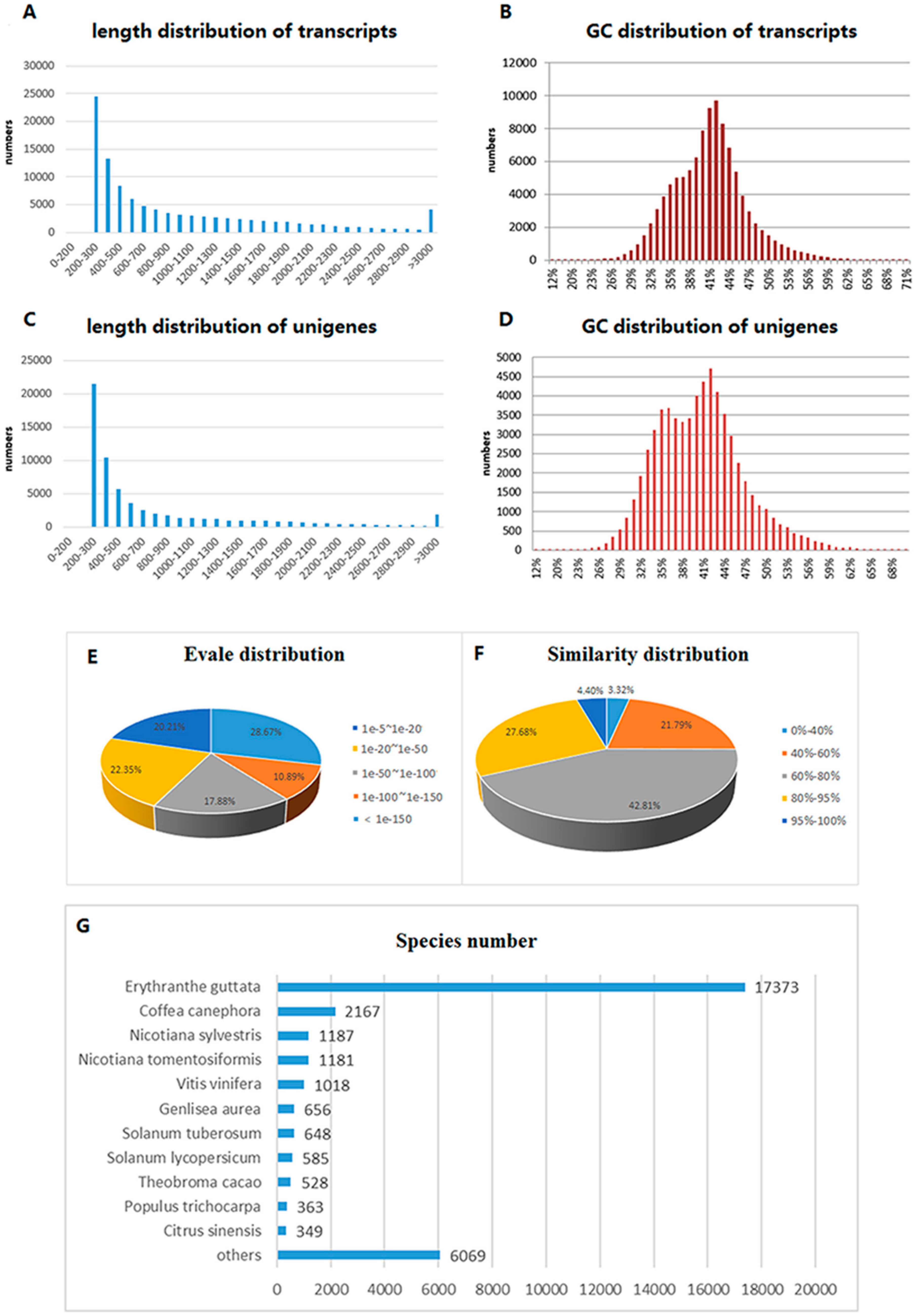
2.1. Transcriptome Sequencing and Assembly
2.2. Functional Annotation and Classification
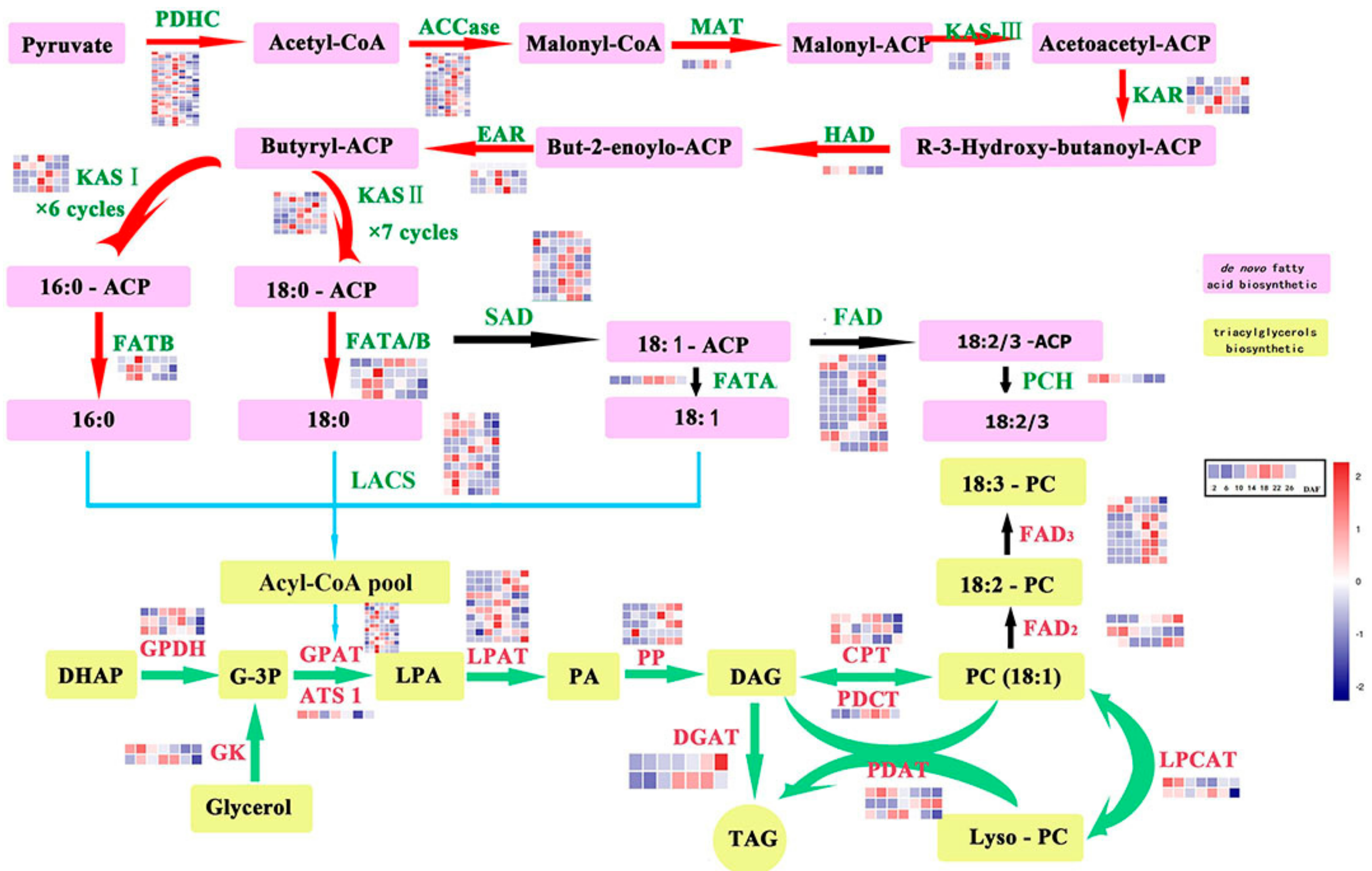
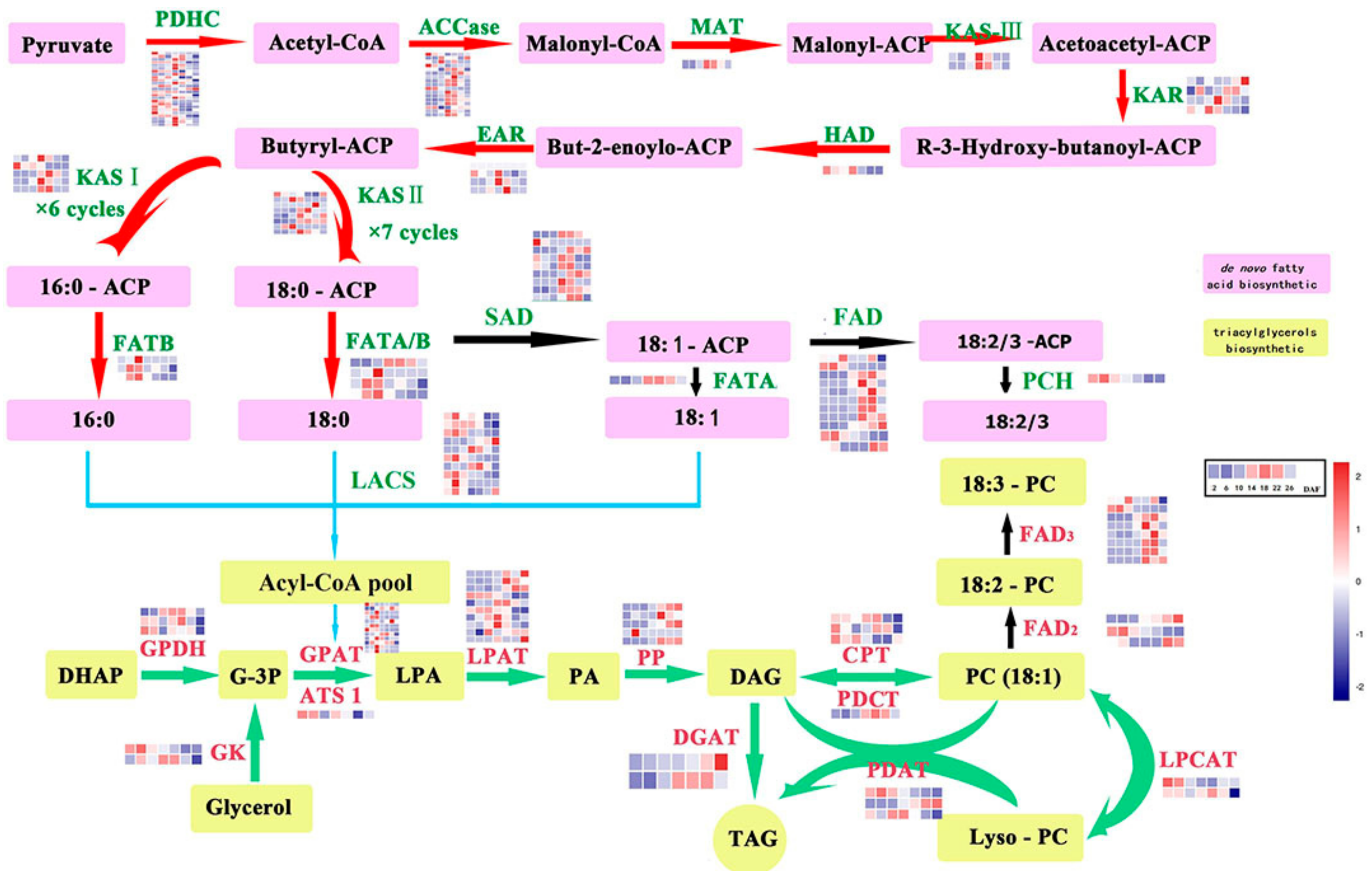
2.3. Genes Related to Lipid Biosynthesis in Perilla Seed
2.4. Differentially Expressed Genes (DEGs)
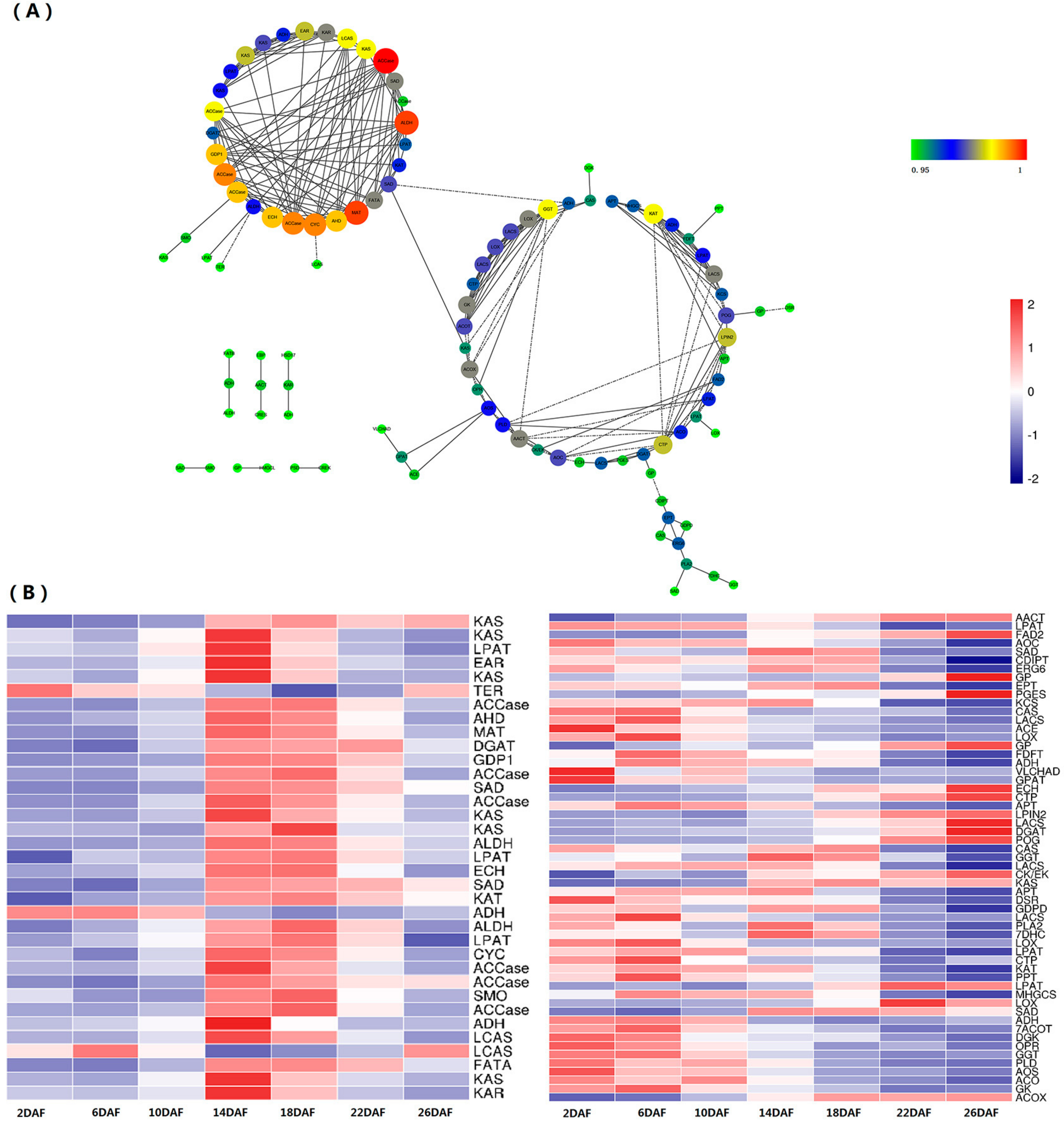
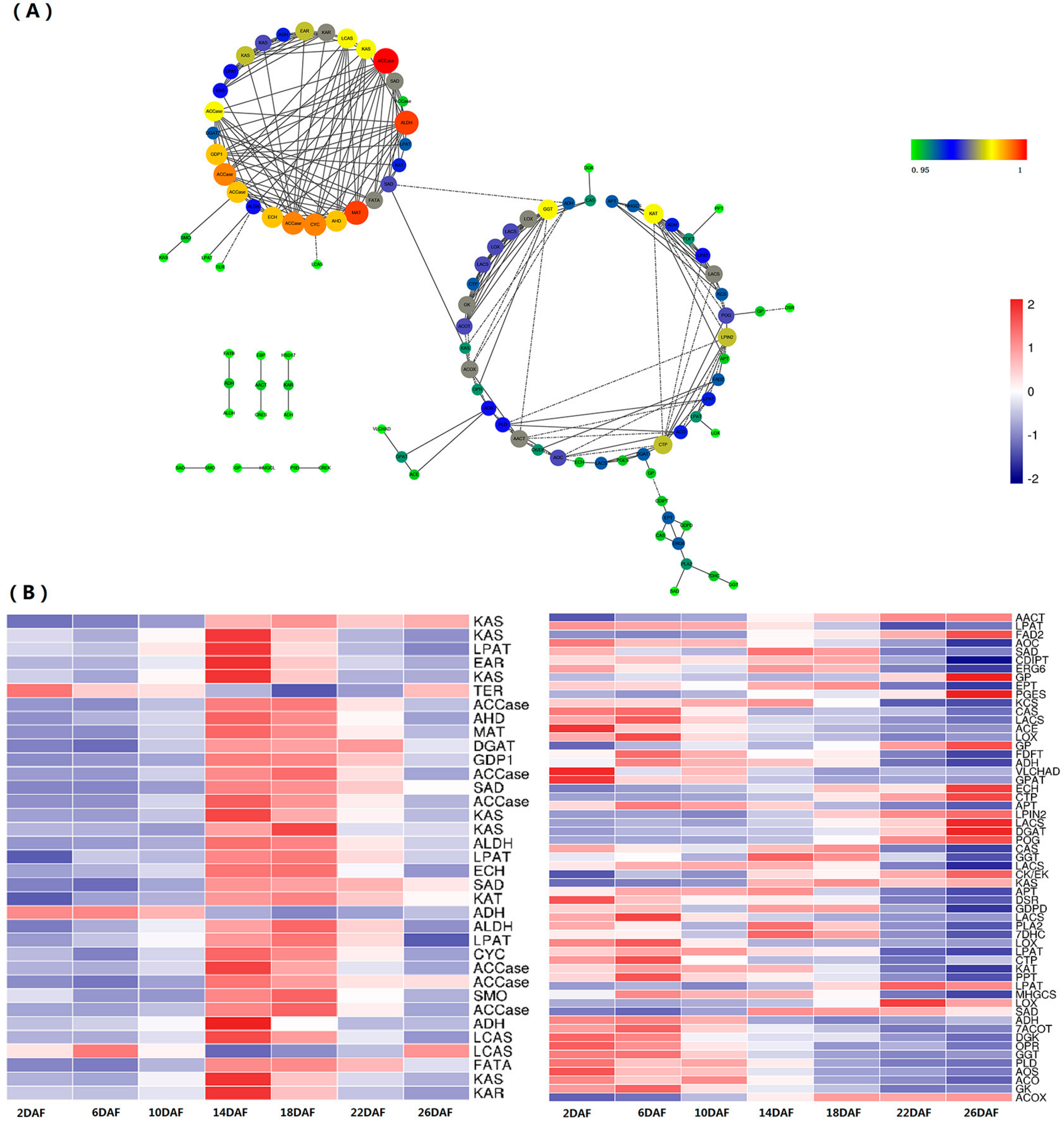
2.5. Coexpression of Lipid Metabolism Genes
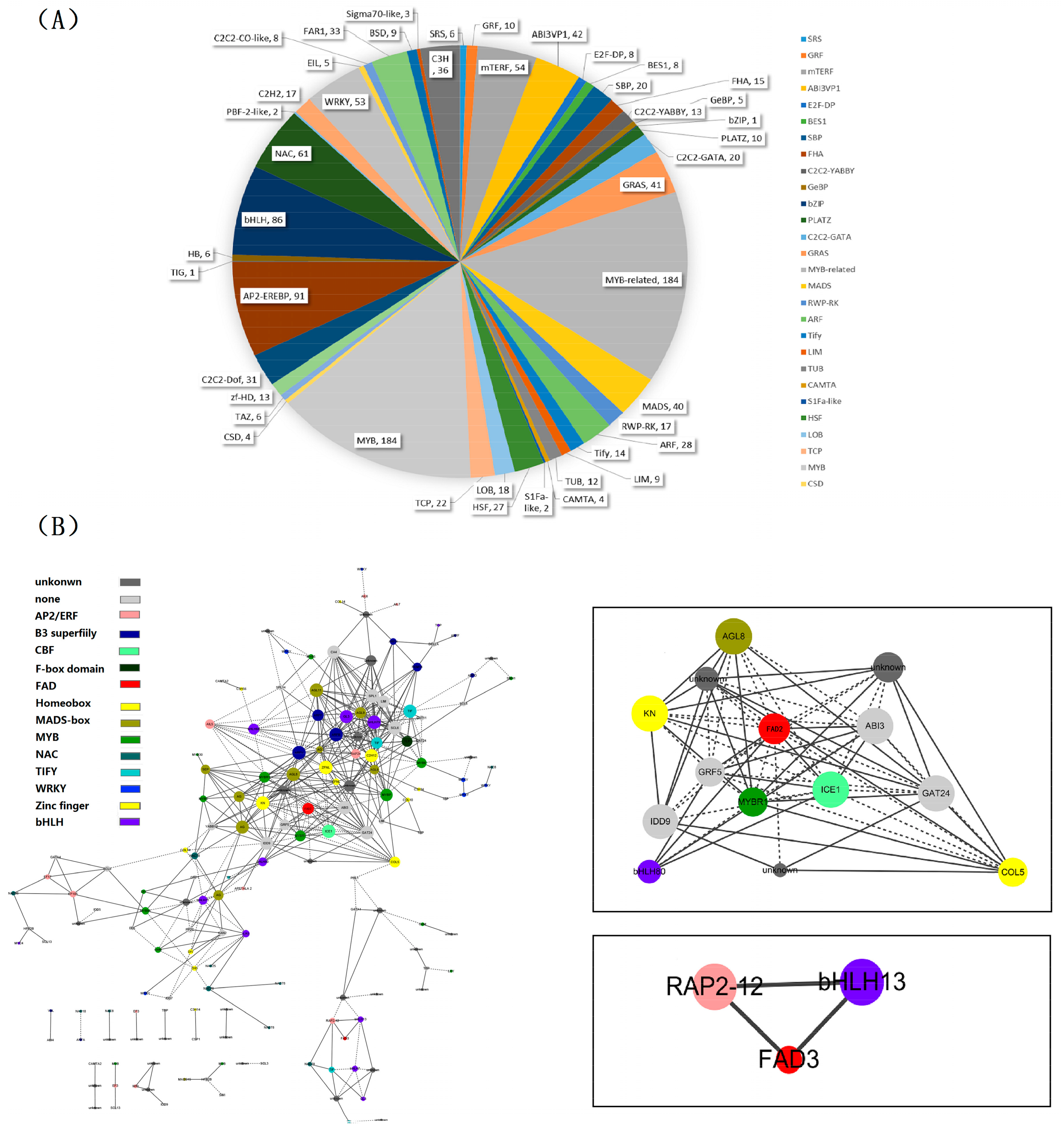
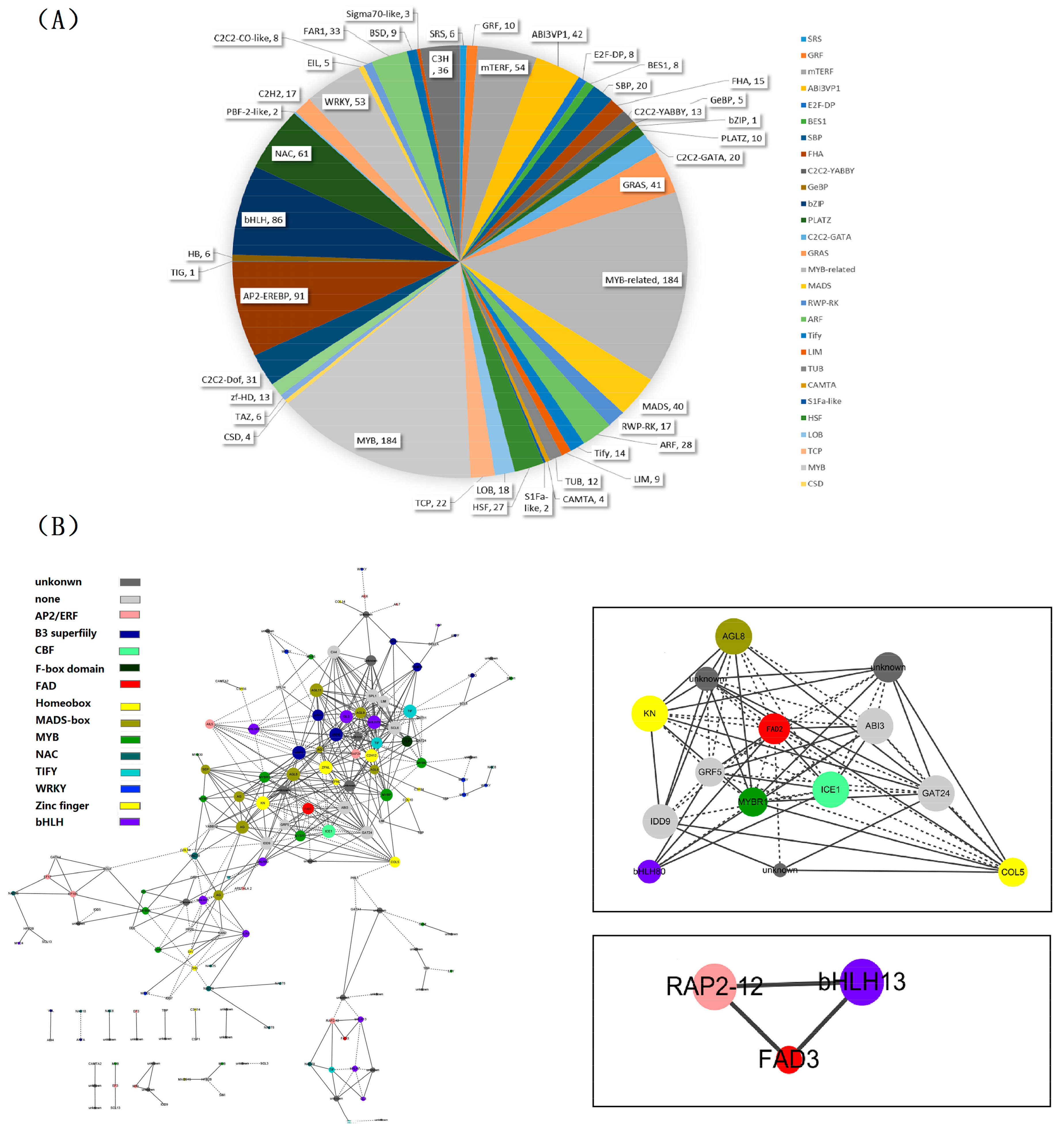
2.6. Identification of Transcription Factor Families
2.7. Identification and Functional Annotation of the Genes Coexpressing with Perilla Fatty Acid Desaturase Genes
2.8. The qRT-PCR Analysis of the Lipid Synthesis Genes in the Perilla
3. Discussion and Conclusions
4. Materials and Methods
4.1. Plant Materials, Library Construction, and Transcriptome Sequencing
4.2. Data Filtering and De Novo Assembly
4.3. Functional Annotation of Unigenes
4.4. Identification of Differentially Expressed Genes
4.5. Transcription Factor Identification and Co-Expression with Fatty Acid Desaturase
4.6. Quantitative Real-Time PCR Analysis
Supplementary Materials
Authors Contributors:
Acknowledgments
Conflicts of Interest
References
- Peiretti, P.G. Fatty Acid Content and Chemical Composition of Vegetative Parts of Perilla (Perilla frutescens L.) after Different Growth Lengths. Res. J. Med. Plants 2011, 5, 72–78. [Google Scholar] [CrossRef]
- Sa, K.J.; Choi, S.H.; Ueno, M.; Park, K.C.; Park, Y.J.; Ma, K.H.; Lee, J.K. Identification of genetic variations of cultivated and weedy types of perilla, species in korea and japan using morphological and ssr markers. Genes Genom. 2013, 35, 649–659. [Google Scholar] [CrossRef]
- Asif, M. Health effects of omega-3,6,9 fatty acids: Perilla frutescens, is a good example of plant oils. Orient. Pharm. Exp. Med. 2011, 11, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Akond, M.; Liu, S.; Boney, M.; Kantartzi, S.K.; Meksem, K.; Bellaloui, N.; Lightfoot, D.A.; Kassen, M.A. Identification of Quantitative Trait Loci (QTL) Underlying Protein, Oil, and Five Major Fatty Acids’ Contents in Soybean. Am. J. Plant Sci. 2014, 5, 158–167. [Google Scholar] [CrossRef]
- Ramos, M.J.; Fernández, C.M.; Casas, A.; Rodríguez, L.; Pérez, A. Influence of fatty acid composition of raw materials on biodiesel properties. Bioresour. Technol. 2009, 100, 261. [Google Scholar] [CrossRef] [PubMed]
- Connor, W.E. Importance of n-3 fatty acids in health and disease. Am. J. Clin. Nutr. 2000, 71, 171S. [Google Scholar] [PubMed]
- Schuchardt, J.P.; Huss, M.; Stauss-Grabo, M.; Hahn, A. Significance of long-chain polyunsaturated fatty acids (PUFAs) for the development and behaviour of children. Eur. J. Pediatr. 2010, 169, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Janssen, C.I.; Kiliaan, A.J. Long-chain polyunsaturated fatty acids (LCPUFA) from genesis to senescence: The influence of LCPUFA on neural development, aging, and neurodegeneration. Prog. Lipid Res. 2014, 53, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.; Bandhiwal, N.; Shah, N.; Kant, C.; Gaur, R.; Bhatia, S. Global Transcriptome analysis of developing chickpea (Cicer arietinum L.) seeds. Front. Plant Sci. 2014, 5, 698. [Google Scholar] [CrossRef] [PubMed]
- Severin, A.J.; Woody, J.L.; Bolon, Y.; Joseph, B.; Diers, B.W.; Farmer, A.D.; Muehlbauer, G.J.; Nelson, R.T.; Grant, D.; Specht, E.J.; et al. RNA-Seq Atlas of Glycine max: A guide to the soybean transcriptome. BMC Plant Biol. 2010, 10, 160. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.E.; Kim, M.Y.; Shim, S.; Li, J.; Lee, S. Gene expression profiling for seed protein and oil synthesis during early seed development in soybean. Genes Genom. 2015, 37, 409–418. [Google Scholar] [CrossRef]
- Yin, D.; Wang, Y.; Zhang, X.; Li, H.; Lu, X.; Zhang, J.; Zhang, W.; Chen, S. De novo assembly of the peanut (Arachis hypogaea L.) seed transcriptome revealed candidate unigenes for oil accumulation pathways. PLoS ONE 2013, 8, e73767. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Kayam, G.; Faigenboimdoron, A.; Clevenger, J.; Oziasakins, P.; Hovav, R. Gene expression profiling during seed-filling process in peanut with emphasis on oil biosynthesis networks. Plant Sci. 2016, 248, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Dussert, S.; Guerin, C.; Andersson, M.; Joët, T.; Tranbarger, T.J.; Pizot, M.; Sarah, G.; Omore, A.; Durand-Gasselin, T.; Morcillo, F. Comparative transcriptome analysis of three oil palm fruit and seed tissues that differ in oil content and fatty acid composition. Plant Physiol. 2013, 162, 1337–1358. [Google Scholar] [CrossRef] [PubMed]
- Venglat, P.; Xiang, D.; Qiu, S.; Stone, S.L.; Tibiche, C.; Cram, D.; Alting-mees, M.; Nowak, J.; Cloutier, S.; Deyholos, M.; et al. Gene expression analysis of flax seed development. BMC Plant Biol. 2011, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- Fatima, T.; Snyder, C.L.; Schroeder, W.; Cram, D.; Datla, R.; Wishart, D.S.; Weselake, R.; Krishna, P. Fatty acid composition of developing sea buckthorn (Hippophae rhamnoides L.) berry and the transcriptome of the mature seed. PLoS ONE 2012, 7, e34099. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Liu, X.; Yiu, S.; Lim, B.L. De novo assembly and characterization of Camelina sativa transcriptome by paired-end sequencing. BMC Genom. 2013, 14, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Xu, R.; Wang, R.; Liu, A. Transcriptome analysis of Sacha Inchi (Plukenetia volubilis L.) seeds at two developmental stages. BMC Genom. 2012, 13, 716. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, L.; Shu, Q.; Wu, J.; Chen, L.; Shao, S.; Yin, D. Fatty acid composition of developing tree peony (Paeonia section Moutan DC.) seeds and transcriptome analysis during seed development. BMC Genom. 2015, 16, 208. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.U.; Lee, K.; Shim, D.; Lee, J.H.; Chen, G.Q.; Hwang, S. Transcriptome analysis and identification of genes associated with ω-3 fatty acid biosynthesis in Perilla frutescens (L.) var. frutescens. BMC Genom. 2016, 17, 474. [Google Scholar] [CrossRef] [PubMed]
- Yonekura-Sakakibara, K.; Saito, K. Transcriptome Coexpression Analysis Using ATTED-II for Integrated Transcriptomic/Metabolomic Analysis. Methods Mol. Biol. 2013, 1011, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Willsey, A.J.; Sanders, S.J.; Li, M.; Dong, S.; Tebbenkamp, A.T.; Muhle, R.A.; Reilly, S.K.; Lin, L.; Fertuzinhos, S.; Miller, J.A.; et al. Coexpression Networks Implicate Human Midfetal Deep Cortical Projection Neurons in the Pathogenesis of Autism. Cell 2013, 155, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, N.; Chothani, S.P.; Harris, L.; Dimitrova, N. Identifying RNAseq-based coding-noncoding co-expression interactions in breast cancer. In Proceedings of the 2013 IEEE International Workshop on Genomic Signal Processing and Statistics, Houston, TX, USA, 17–19 November 2013; pp. 11–14. [Google Scholar] [CrossRef]
- Du, J.; Wang, S.; He, C.; Zhou, B.; Ruan, Y.L.; Shou, H. Identification of regulatory networks and hub genes controlling soybean seed set and size using RNA sequencing analysis. J. Exp. Bot. 2017, 68, 1955. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Liu, G.; Huang, Z.; Duan, W.; Tan, H.; Li, Y.; Hou, X. Temperature expression patterns of genes and their coexpression with LncRNAs revealed by RNA-Seq in non-heading Chinese cabbage. BMC Genom. 2016, 17, 297. [Google Scholar] [CrossRef] [PubMed]
- Iorizzo, M.; Ellison, S.; Senalik, D.; Zeng, P.; Satapoomin, P.; Huang, J.; Bowman, M.J.; Lovene, M.; Sanseverion, W.; Cavagnaro, P.F.; et al. A high-quality carrot genome assembly provides new insights into carotenoid accumulation and asterid genome evolution. Nat. Genet. 2016, 48, 657. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Jiang, Y.; Ma, C.; Silvestri, G.; Bosinger, S.E.; Li, B.; Jong, A.; Zhou, Y.; Huang, S. Coexpression Network Analysis of Benign and Malignant Phenotypes of SIV-Infected Sooty Mangabey and Rhesus Macaque. PLoS ONE 2016, 11, e0156170. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhu, H.; Zhou, L.; Li, J.; Zhao, L.; Wu, S.; Wang, J.; Liu, W.; Chen, Z. Genes related to the very early stage of ConA-induced fulminant hepatitis: A gene-chip-based study in a mouse model. BMC Genom. 2010, 11, 240. [Google Scholar] [CrossRef] [PubMed]
- Filteau, M.; Pavey, S.A.; Stcyr, J.; Bernatchez, L. Gene Coexpression Networks Reveal Key Drivers of Phenotypic Divergence in Lake Whitefish. Mol. Biol. Evol. 2013, 30, 1384–1396. [Google Scholar] [CrossRef] [PubMed]
- Hirai, M.Y.; Sugiyama, K.; Sawada, Y.; Tohge, T.; Obayashi, T.; Suzuki, A.; Araki, R.; Sakural, N.; Suzuki, H.; Aoki, K.; et al. Omics-based identification of Arabidopsis Myb transcription factors regulating aliphatic glucosinolate biosynthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 6478. [Google Scholar] [CrossRef] [PubMed]
- Yonekura-Sakakibara, K.; Tohge, T.; Matsuda, F.; Nakabayashi, R.; Takayama, H.; Niida, R.; Watanabe-Takahashi, A.; Inoue, E.; Saito, K. Comprehensive flavonol profiling and transcriptome coexpression analysis leading to decoding gene-metabolite correlations in Arabidopsis. Plant Cell 2008, 20, 2160–2176. [Google Scholar] [CrossRef] [PubMed]
- Yonekura-Sakakibara, K.; Tohge, T.; Niida, R.; Saito, K. Identification of a flavonol 7-Orhamnosyltransferase gene determining flavonoid pattern in Arabidopsis by transcriptome coexpression analysis and reverse genetics. J. Biol. Chem. 2007, 282, 14932–14941. [Google Scholar] [CrossRef] [PubMed]
- Albinsky, D.; Sawada, Y.; Kuwahara, A.; Nagano, M.; Hirai, A.; Saito, K.; Hirai, M.Y. Widely targeted metabolomics and coexpression analysis as tools to identify genes involved in the side-chain elongation steps of aliphatic glucosinolate biosynthesis. Amino Acids 2010, 39, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.C.; Tai, S.S.; Peng, C.C.; Tzen, J.T. Identification of Three Novel Unique Proteins in Seed Oil Bodies of Sesame. Plant Cell Physiol. 1998, 39, 935. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.U.; Hsieh, K.; Ratnayake, C.; Huang, A.H. A novel group of oleosins is present inside the pollen of Arabidopsis. J. Biol. Chem. 2002, 277, 22677. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.H. Oleosins and oil bodies in seeds and other organs. Plant Physiol. 1996, 110, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Vindigni, J.D.; Wien, F.; Giuliani, A.; Erpapazoqlou, Z.; Tache, R.; Jaqic, F.; Chardot, T.; Gohon, Y.; Forissard, M. Fold of an oleosin targeted to cellular oil bodies. Biochim. Biophys. Acta 2013, 1828, 1881–1888. [Google Scholar] [CrossRef] [PubMed]
- Le, B.H.; Cheng, C.; Bui, A.Q.; Wagmaister, J.A.; Henry, K.F.; Pelletier, J.; Kwong, L.; Belmonte, M.; Kirkbride, R.; Horvath, S.; et al. Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc. Natl. Acad. Sci. USA 2010, 107, 8063–8070. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.Y.; Weselake, R.J. Gene coexpression clusters and putative regulatory elements underlying seed storage reserve accumulation in arabidopsis. BMC Genom. 2011, 12, 286. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhong, X.J.; He, J.; Jiang, M.Y.; Yu, X.F.; Li, X. Identification and characterization of AP2/ERF transcription factors in moso bamboo (Phyllostachys edulis). Mol. Biol. 2016, 50, 785. [Google Scholar] [CrossRef]
- An, D.H.; Michung, S. Overexpression of Arabidopsis WRI1 enhanced seed mass and storage oil content in Camelina sativa. Plant Biotechnol. Rep. 2015, 9, 137–148. [Google Scholar] [CrossRef]
- Baud, S.; Wuillème, S.; To, A.; Rochat, C.; Lepiniec, L. Role of wrinkled1 in the transcriptional regulation of glycolytic and fatty acid biosynthetic genes in arabidopsis. Plant J. 2009, 60, 933. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Kim, J.K.; Shin, J.S.; Suh, M.C. The SebHLH transcription factor mediates trans-activation of the SeFAD2 gene promoter through binding to E-and G-box elements. Plant Mol. Biol. 2007, 64, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Fan, H.J.; Ling, H.Q. Genome-wide identification and characterization of the bHLH, gene family in tomato. Front. Plant Sci. 2015, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J. Analysis and expression of the cotton gene for the Δ-12 fatty acid desaturase 2-4 (FAD2-4). Ph.D. Thesis, University of North Texas, Denton, TX, USA, August 2003. [Google Scholar]
- Makkena, S.; Labm, R.S. The bhlh transcription factor spatula is a key regulator of organ size in arabidopsis thaliana. Plant Signal. Behav. 2013, 8, e24140. [Google Scholar] [CrossRef] [PubMed]
- Duan, P.; Ni, S.; Wang, J.; Zhang, B.; Xu, R.; Wang, Y.; Chen, H.; Zhu, X.; Li, Y. Regulation of osgrf4 by osmir396 controls grain size and yield in rice. Nat. Plants 2015, 2, 15203. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2013, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; Garcíagómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. Clusterprofiler: An r package for comparing biological themes among gene clusters. OMICS 2012, 16, 284. [Google Scholar] [CrossRef] [PubMed]
- Ariani, A.; Gepts, P. Genome-wide identification and characterization of aquaporin gene family in common bean (Phaseolus vulgaris L.). Mol. Genet. Genom. 2015, 290, 1771. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M. D; Mccarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L.P. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Smoot, M.; Ono, K.; Ideker, T.; Maere, S. PiNGO: A Cytoscape plugin to find candidate genes in biological networks. Bioinformatics 2011, 27, 1030–1031. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Total Reads | Total Bases | GC Content | Q20 | Q30 |
|---|---|---|---|---|---|
| 2DAF | 18,094,914 | 2,261,864,250 | 46.64% | 94.64% | 89.76% |
| 6DAF | 17,885,482 | 2,235,685,250 | 47.88% | 94.36% | 89.70% |
| 10DAF | 17,075,076 | 2,134,384,500 | 49.31% | 94.38% | 89.77% |
| 14DAF | 16,747,764 | 2,093,470,500 | 49.61% | 94.25% | 89.58% |
| 18DAF | 14,929,122 | 1,866,140,250 | 51.42% | 93.70% | 88.05% |
| 22DAF | 20,751,538 | 2,593,942,250 | 52.52% | 93.74% | 88.71% |
| 26DAF | 21,517,792 | 2,689,724,000 | 47.89% | 95.39% | 91.21% |
| Total | 127,001,688 | 15,875,211,000 |
| Item | Total Number (bp) | N50 (bp) | Median Length (bp) | Average Length (bp) | Total Length (bp) |
|---|---|---|---|---|---|
| Transcripts | 104,638 | 1608 | 600 | 968 | 101,378,085 |
| Unigenes | 64,156 | 1417 | 402 | 777 | 49,883,108 |
| Database | Account | Percentage c |
|---|---|---|
| NR a | 32,132 | 50.08% |
| KEGG classified unigenes | 10,904 | 17.00% |
| COG classified unigenes | 8654 | 14.47% |
| GO classified unigenes | 22,263 | 34.70% |
| Blast_hit b | 31,287 | 48.77% |
| Pfam classified unigenes | 19,340 | 30.15% |
| Eggnog classified unigenes | 9425 | 14.69% |
| TmHMM classified unigenes | 6719 | 10.47% |
| SignalP classified unigenes | 2354 | 3.67% |
| All annotated unigenes | 39,760 | 61.97% |
| All | 64,156 | 100.00% |
| 2DAF | 6DAF | 10DAF | 14DAF | 18DAF | 22DAF | 26DAF | |
|---|---|---|---|---|---|---|---|
| 2DAF | 0 | ||||||
| 6DAF | 137↑ 263↓ | 0 | |||||
| 10DAF | 876↑ 1316↓ | 294↑ 558↓ | 0 | ||||
| 14DAF | 1755↑ 1399↓ | 1352↑ 1227↓ | 353↑ 368↓ | 0 | |||
| 18DAF | 3022↑ 1813↓ | 2370↑ 1583↓ | 2001↑ 955↓ | 257↑ 85↓ | 0 | ||
| 22DAF | 3666↑ 2031↓ | 3188↑ 1816↓ | 2941↑ 1363↓ | 1350↑ 674↓ | 47↑ 83↓ | 0 | |
| 26DAF | 4751↑ 2221↓ | 4730↑ 2119↓ | 4445↑ 1692↓ | 2615↑ 1268↓ | 1253↑ 949↓ | 360↑ 122↓ | 0 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Song, C.; Song, L.; Shang, Z.; Yang, S.; Zhang, D.; Sun, W.; Shen, Q.; Zhao, D. RNA Sequencing and Coexpression Analysis Reveal Key Genes Involved in α-Linolenic Acid Biosynthesis in Perilla frutescens Seed. Int. J. Mol. Sci. 2017, 18, 2433. https://doi.org/10.3390/ijms18112433
Zhang T, Song C, Song L, Shang Z, Yang S, Zhang D, Sun W, Shen Q, Zhao D. RNA Sequencing and Coexpression Analysis Reveal Key Genes Involved in α-Linolenic Acid Biosynthesis in Perilla frutescens Seed. International Journal of Molecular Sciences. 2017; 18(11):2433. https://doi.org/10.3390/ijms18112433
Chicago/Turabian StyleZhang, Tianyuan, Chi Song, Li Song, Zhiwei Shang, Sen Yang, Dong Zhang, Wei Sun, Qi Shen, and Degang Zhao. 2017. "RNA Sequencing and Coexpression Analysis Reveal Key Genes Involved in α-Linolenic Acid Biosynthesis in Perilla frutescens Seed" International Journal of Molecular Sciences 18, no. 11: 2433. https://doi.org/10.3390/ijms18112433
APA StyleZhang, T., Song, C., Song, L., Shang, Z., Yang, S., Zhang, D., Sun, W., Shen, Q., & Zhao, D. (2017). RNA Sequencing and Coexpression Analysis Reveal Key Genes Involved in α-Linolenic Acid Biosynthesis in Perilla frutescens Seed. International Journal of Molecular Sciences, 18(11), 2433. https://doi.org/10.3390/ijms18112433