Reactive Oxygen Species and NOX Enzymes Are Emerging as Key Players in Cutaneous Wound Repair


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
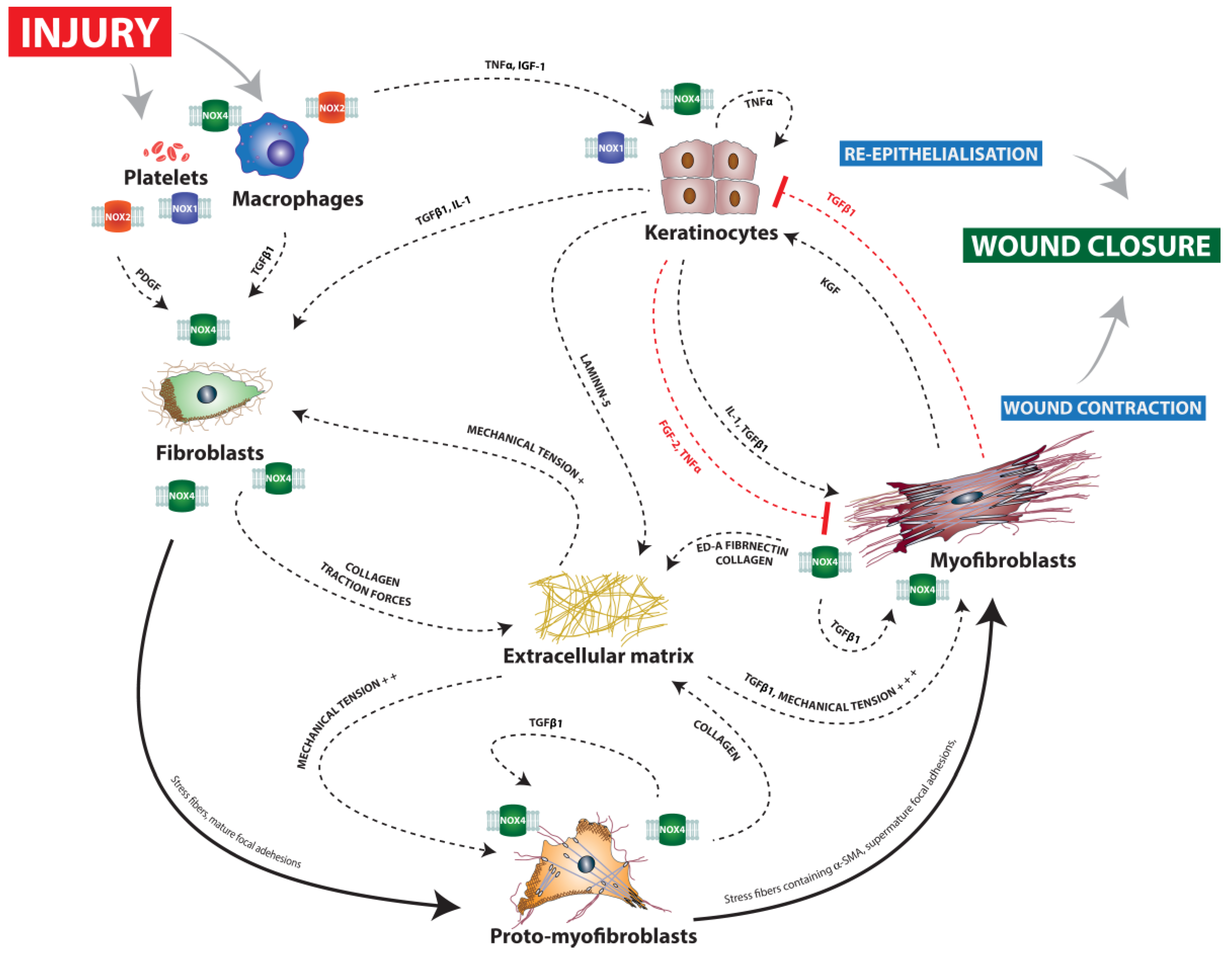
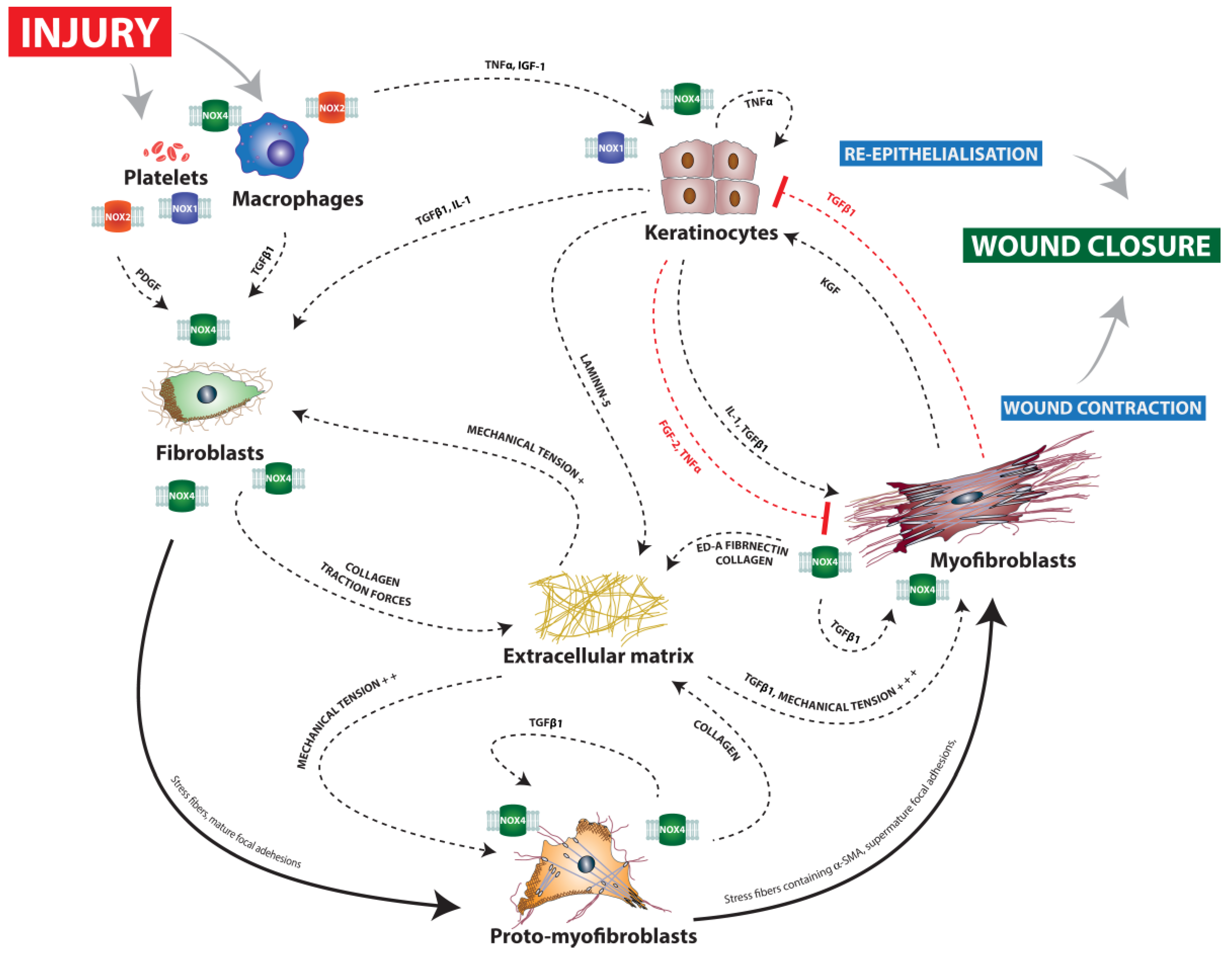
2. The Role of ROS and Redox Signaling during the Coagulation Phase
3. The Role of ROS and Redox Signaling during the Inflammation Phase
4. The Role of ROS and Redox Signaling during the Proliferation Phase
4.1. Angiogenesis
4.2. Re-Epithelialization
4.3. Wound Contraction
5. The Role of ROS and Redox Signaling during the Maturation Phase
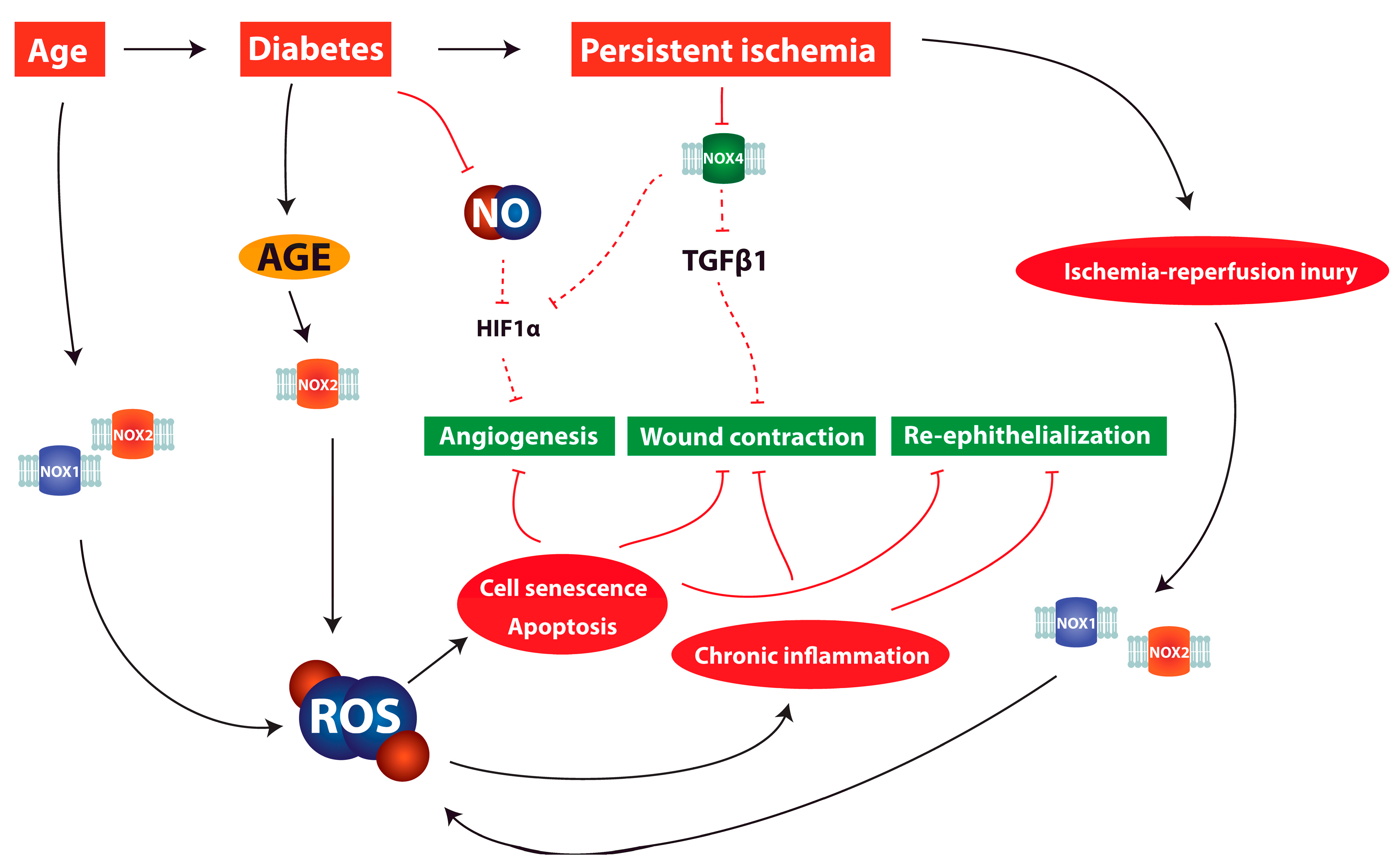
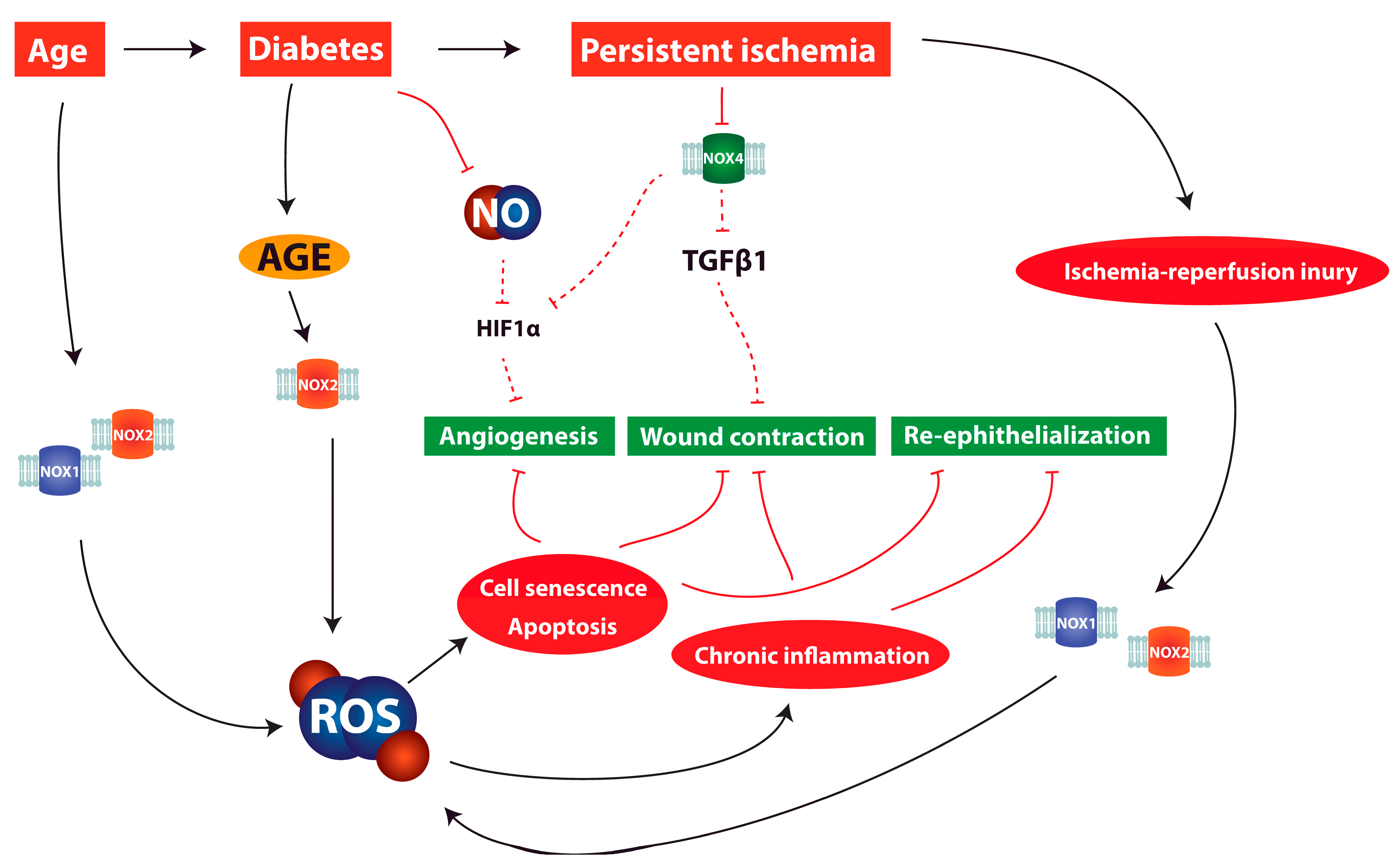
6. ROS and Redox Signaling in Chronic Wounds
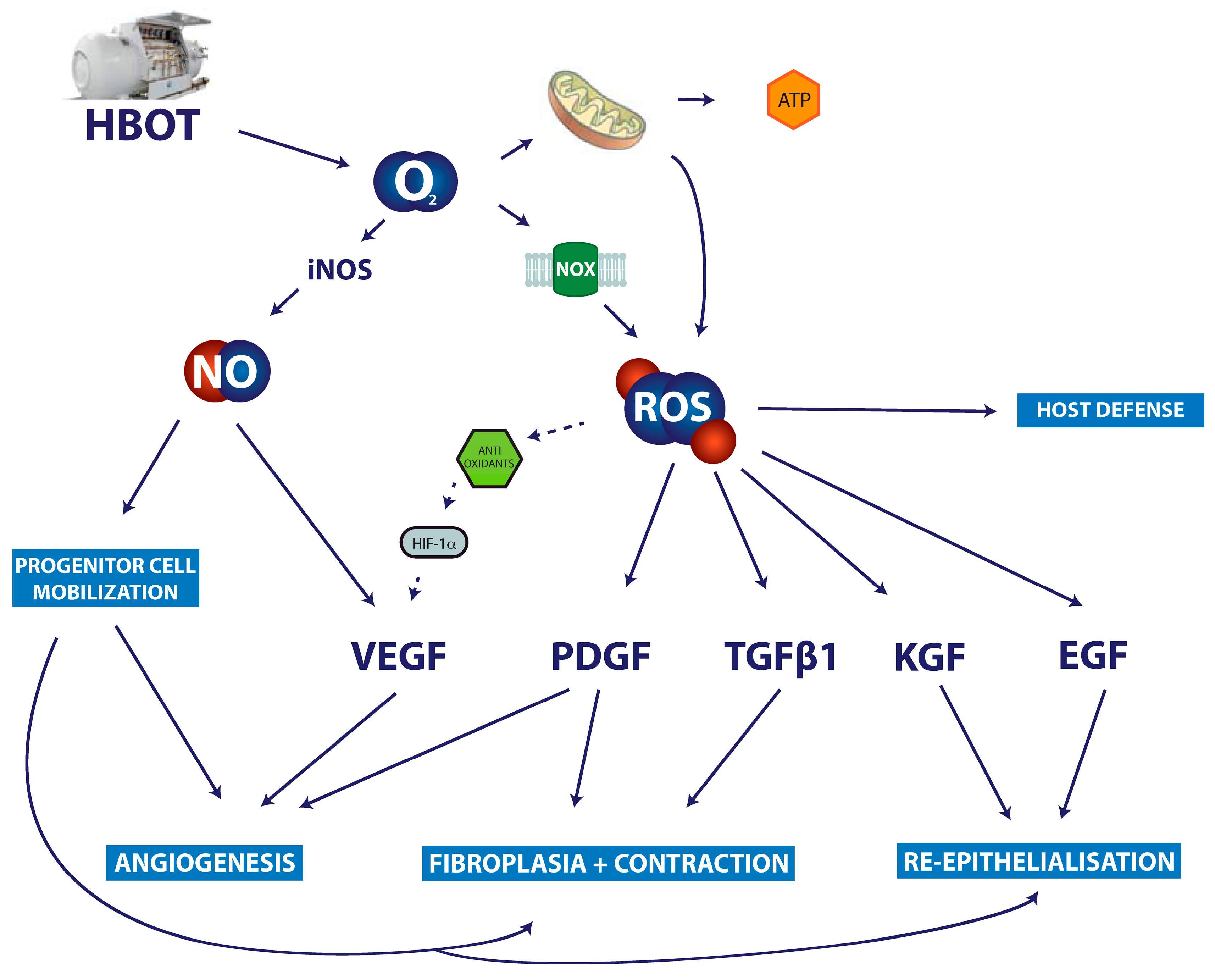
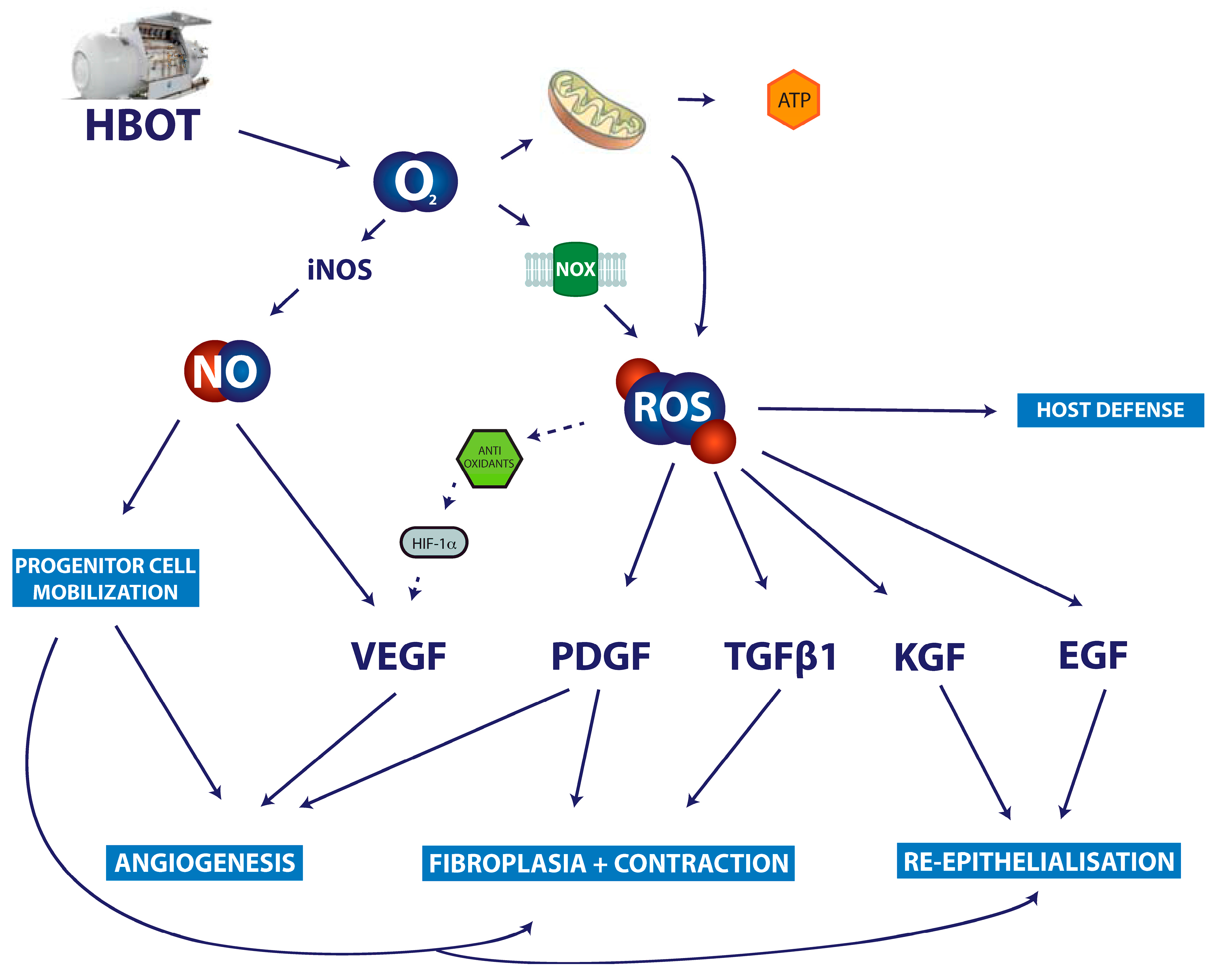
7. Discussion and Clinical Relevance
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hunt, T.K.; Zederfeldt, B.; Goldstick, T.K. Oxygen and healing. Am. J. Surg. 1969, 118, 521–525. [Google Scholar] [CrossRef]
- Schreml, S.; Szeimies, R.M.; Prantl, L.; Karrer, S.; Landthaler, M.; Babilas, P. Oxygen in acute and chronic wound healing. Br. J. Dermatol. 2010, 163, 257–268. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Dinger, B.; Sanders, K.; Hoidal, J.; Obeso, A.; Stensaas, L.; Fidone, S.; Gonzalez, C. Effect of p47phox gene deletion on ros production and oxygen sensing in mouse carotid body chemoreceptor cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L916–L924. [Google Scholar] [CrossRef] [PubMed]
- Schugart, R.C.; Friedman, A.; Zhao, R.; Sen, C.K. Wound angiogenesis as a function of tissue oxygen tension: A mathematical model. Proc. Natl. Acad. Sci. USA 2008, 105, 2628–2633. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Friedman, A.; Sen, C.K. A mathematical model of ischemic cutaneous wounds. Proc. Natl. Acad. Sci. USA 2009, 106, 16782–16787. [Google Scholar] [CrossRef] [PubMed]
- Tandara, A.A.; Mustoe, T.A. Oxygen in wound healing—More than a nutrient. World J. Surg. 2004, 28, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Roy, S. Redox signals in wound healing. Biochim. Biophys. Acta 2008, 1780, 1348–1361. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ros-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Beckman, K.B.; Ames, B.N. The free radical theory of aging matures. Physiol. Rev. 1998, 78, 547–581. [Google Scholar] [PubMed]
- Inoguchi, T.; Sonta, T.; Tsubouchi, H.; Etoh, T.; Kakimoto, M.; Sonoda, N.; Sato, N.; Sekiguchi, N.; Kobayashi, K.; Sumimoto, H.; et al. Protein kinase c-dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: Role of vascular NAD(P)H oxidase. J. Am. Soc. Nephrol. 2003, 14, S227–S232. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Khanna, S.; Nallu, K.; Hunt, T.K.; Sen, C.K. Dermal wound healing is subject to redox control. Mol. Ther. 2006, 13, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K. The general case for redox control of wound repair. Wound Repair Regen. 2003, 11, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R. Physiological roles of xanthine oxidoreductase. Drug Metab. Rev. 2004, 36, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Soneja, A.; Drews, M.; Malinski, T. Role of nitric oxide, nitroxidative and oxidative stress in wound healing. Pharmacol. Rep. 2005, 57, 108–119. [Google Scholar] [PubMed]
- Rathore, R.; Zheng, Y.M.; Niu, C.F.; Liu, Q.H.; Korde, A.; Ho, Y.S.; Wang, Y.X. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ros-pkcepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.B.; Maguire, J.J.; Mahdavian, M.; Wicke, C.; Marcocci, L.; Scheuenstuhl, H.; Chang, M.; Le, A.X.; Hopf, H.W.; Hunt, T.K. Wound hypoxia and acidosis limit neutrophil bacterial killing mechanisms. Arch. Surg. 1997, 132, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Stadelmann, W.K.; Digenis, A.G.; Tobin, G.R. Physiology and healing dynamics of chronic cutaneous wounds. Am. J. Surg. 1998, 176, 26S–38S. [Google Scholar] [CrossRef]
- Schafer, M.; Werner, S. Oxidative stress in normal and impaired wound repair. Pharmacol. Res. 2008, 58, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A.; Brandes, R.P.; Bassus, S.; Kronemann, N.; Kirchmaier, C.M.; Busse, R.; Schini-Kerth, V.B. Oxidative stress and expression of p22phox are involved in the up-regulation of tissue factor in vascular smooth muscle cells in response to activated platelets. FASEB J. 2000, 14, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Sulciner, D.J.; Gutkind, J.S.; Irani, K.; Goldschmidt-Clermont, P.J.; Finkel, T. Regulation of reactive-oxygen-species generation in fibroblasts by Rac1. Biochem. J. 1996, 318 Pt 2, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Gregg, D.; de Carvalho, D.D.; Kovacic, H. Integrins and coagulation: A role for ros/redox signaling? Antioxid. Redox Signal. 2004, 6, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A. Redox regulation of the coagulation cascade. Antioxid. Redox Signal. 2005, 7, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Herkert, O.; Djordjevic, T.; BelAiba, R.S.; Gorlach, A. Insights into the redox control of blood coagulation: Role of vascular NADPH oxidase-derived reactive oxygen species in the thrombogenic cycle. Antioxid. Redox Signal. 2004, 6, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Penn, M.S.; Patel, C.V.; Cui, M.Z.; DiCorleto, P.E.; Chisolm, G.M. Ldl increases inactive tissue factor on vascular smooth muscle cell surfaces: Hydrogen peroxide activates latent cell surface tissue factor. Circulation 1999, 99, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Salvemini, D.; Botting, R. Modulation of platelet function by free radicals and free-radical scavengers. Trends Pharmacol. Sci. 1993, 14, 36–42. [Google Scholar] [CrossRef]
- Herkert, O.; Diebold, I.; Brandes, R.P.; Hess, J.; Busse, R.; Gorlach, A. NADPH oxidase mediates tissue factor-dependent surface procoagulant activity by thrombin in human vascular smooth muscle cells. Circulation 2002, 105, 2030–2036. [Google Scholar] [CrossRef] [PubMed]
- Krotz, F.; Sohn, H.Y.; Gloe, T.; Zahler, S.; Riexinger, T.; Schiele, T.M.; Becker, B.F.; Theisen, K.; Klauss, V.; Pohl, U. NAD(P)H oxidase-dependent platelet superoxide anion release increases platelet recruitment. Blood 2002, 100, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.S.; Sung, J.Y.; Kim, O.S.; Kim, Y.J.; Hur, K.C.; Kazlauskas, A.; Rhee, S.G. Platelet-derived growth factor-induced H2O2 production requires the activation of phosphatidylinositol 3-kinase. J. Biol. Chem. 2000, 275, 10527–10531. [Google Scholar] [CrossRef] [PubMed]
- Greer, N.; Foman, N.; Dorrian, J.; Fitzgerald, P.; MacDonald, R.; Rutks, I.; Wilt, T. Advanced wound care therapies for non-healing diabetic, venous, and arterial ulcers: A systematic review. Ann. Int. Med. 2013, 159, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Brooker, R.J. Genetics: Analysis & Principles, 4th ed.; McGraw-Hill: New York, NY, USA, 2012; p. 85. [Google Scholar]
- Eckert, J.W.; Abramson, S.L.; Starke, J.; Brandt, M.L. The surgical implications of chronic granulomatous disease. Am. J. Surg. 1995, 169, 320–323. [Google Scholar] [CrossRef]
- Deffert, C.; Cachat, J.; Krause, K.H. Phagocyte NADPH oxidase, chronic granulomatous disease and mycobacterial infections. Cell. Microbiol. 2014, 16, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, V.B.; Eiserich, J.P.; Chumley, P.H.; Jablonsky, M.J.; Krishna, N.R.; Kirk, M.; Barnes, S.; Darley-Usmar, V.M.; Freeman, B.A. Nitration of unsaturated fatty acids by nitric oxide-derived reactive nitrogen species peroxynitrite, nitrous acid, nitrogen dioxide, and nitronium ion. Chem. Res. Toxicol. 1999, 12, 83–92. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira-Marques, V.; Cyrne, L.; Marinho, H.S.; Antunes, F. A quantitative study of NF-κ activation by H2O2: Relevance in inflammation and synergy with TNF-α. J. Immunol. 2007, 178, 3893–3902. [Google Scholar] [CrossRef] [PubMed]
- Klyubin, I.V.; Kirpichnikova, K.M.; Gamaley, I.A. Hydrogen peroxide-induced chemotaxis of mouse peritoneal neutrophils. Eur. J. Cell Biol. 1996, 70, 347–351. [Google Scholar] [PubMed]
- Nakamura, H.; Herzenberg, L.A.; Bai, J.; Araya, S.; Kondo, N.; Nishinaka, Y.; Herzenberg, L.A.; Yodoi, J. Circulating thioredoxin suppresses lipopolysaccharide-induced neutrophil chemotaxis. Proc. Natl. Acad. Sci. USA 2001, 98, 15143–15148. [Google Scholar] [CrossRef] [PubMed]
- Fraticelli, A.; Serrano, C.V., Jr.; Bochner, B.S.; Capogrossi, M.C.; Zweier, J.L. Hydrogen peroxide and superoxide modulate leukocyte adhesion molecule expression and leukocyte endothelial adhesion. Biochim. Biophys. Acta 1996, 1310, 251–259. [Google Scholar] [CrossRef]
- Ogura, M.; Kitamura, M. Oxidant stress incites spreading of macrophages via extracellular signal-regulated kinases and p38 mitogen-activated protein kinase. J. Immunol. 1998, 161, 3569–3574. [Google Scholar] [PubMed]
- Lu, H.; Youker, K.; Ballantyne, C.; Entman, M.; Smith, C.W. Hydrogen peroxide induces lfa-1-dependent neutrophil adherence to cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H835–H842. [Google Scholar] [PubMed]
- Haddad, J.J.; Saade, N.E.; Safieh-Garabedian, B. Redox regulation of TNF-α biosynthesis: Augmentation by irreversible inhibition of gamma-glutamylcysteine synthetase and the involvement of an IkappaB-alpha/NF-kappaB-independent pathway in alveolar epithelial cells. Cell. Signal. 2002, 14, 211–218. [Google Scholar] [CrossRef]
- Lin, Z.Q.; Kondo, T.; Ishida, Y.; Takayasu, T.; Mukaida, N. Essential involvement of IL-6 in the skin wound-healing process as evidenced by delayed wound healing in IL-6-deficient mice. J. Leukoc. Biol. 2003, 73, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Levigne, D.; Modarressi, A.; Krause, K.H.; Pittet-Cuenod, B. NADPH oxidase 4 deficiency leads to impaired wound repair and reduced dityrosine-crosslinking, but does not affect myofibroblast formation. Free Radic. Biol. Med. 2016, 96, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Bedard, K.; Sorce, S.; Hinz, B.; Dubois-Dauphin, M.; Krause, K.H. NOX4 expression in human microglia leads to constitutive generation of reactive oxygen species and to constitutive IL-6 expression. J. Innate Immun. 2009, 1, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lan, T.; Zhang, C.; Zeng, C.; Hou, J.; Yang, Z.; Zhang, M.; Liu, J.; Liu, B. Reciprocal activation between IL-6/stat3 and NOX4/Akt signalings promotes proliferation and survival of non-small cell lung cancer cells. Oncotarget 2015, 6, 1031–1048. [Google Scholar] [CrossRef] [PubMed]
- Ashino, H.; Shimamura, M.; Nakajima, H.; Dombou, M.; Kawanaka, S.; Oikawa, T.; Iwaguchi, T.; Kawashima, S. Novel function of ascorbic acid as an angiostatic factor. Angiogenesis 2003, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.J.; Venema, R.C. Quercetin inhibits enos, microtubule polymerization, and mitotic progression in bovine aortic endothelial cells. J. Nutr. 2006, 136, 1178–1184. [Google Scholar] [PubMed]
- Lin, M.T.; Yen, M.L.; Lin, C.Y.; Kuo, M.L. Inhibition of vascular endothelial growth factor-induced angiogenesis by resveratrol through interruption of src-dependent vascular endothelial cadherin tyrosine phosphorylation. Mol. Pharmacol. 2003, 64, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Polytarchou, C.; Papadimitriou, E. Antioxidants inhibit angiogenesis in vivo through down-regulation of nitric oxide synthase expression and activity. Free Radic. Res. 2004, 38, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Nehls, V.; Herrmann, R. The configuration of fibrin clots determines capillary morphogenesis and endothelial cell migration. Microvasc. Res. 1996, 51, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, L.; Shu, B.; Tang, J.; Zhang, L.; Xie, J.; Qi, S.; Xu, Y. Granulocyte/macrophage colony-stimulating factor influences angiogenesis by regulating the coordinated expression of VEGF and the ang/tie system. PLoS ONE 2014, 9, e92691. [Google Scholar] [CrossRef] [PubMed]
- Singer, A.J.; Clark, R.A. Cutaneous wound healing. N. Engl. J. Med. 1999, 341, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Battegay, E.J.; Rupp, J.; Iruela-Arispe, L.; Sage, E.H.; Pech, M. PDGF-BB modulates endothelial proliferation and angiogenesis in vitro via PDGF beta-receptors. J. Cell Biol. 1994, 125, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Suri, C.; Jones, P.F.; Patan, S.; Bartunkova, S.; Maisonpierre, P.C.; Davis, S.; Sato, T.N.; Yancopoulos, G.D. Requisite role of angiopoietin-1, a ligand for the tie2 receptor, during embryonic angiogenesis. Cell 1996, 87, 1171–1180. [Google Scholar] [CrossRef]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.F.; Yeo, K.T.; Berse, B.; Yeo, T.K.; Senger, D.R.; Dvorak, H.F.; van de Water, L. Expression of vascular permeability factor (vascular endothelial growth factor) by epidermal keratinocytes during wound healing. J. Exp. Med. 1992, 176, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Berse, B.; Brown, L.F.; Van de Water, L.; Dvorak, H.F.; Senger, D.R. Vascular permeability factor (vascular endothelial growth factor) gene is expressed differentially in normal tissues, macrophages, and tumors. Mol. Biol. Cell 1992, 3, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Khanna, S.; Babior, B.M.; Hunt, T.K.; Ellison, E.C.; Roy, S. Oxidant-induced vascular endothelial growth factor expression in human keratinocytes and cutaneous wound healing. J. Biol. Chem. 2002, 277, 33284–33290. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.; Hunt, T.K.; Hussain, M.Z. Hydrogen peroxide stimulates macrophage vascular endothelial growth factor release. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2357–H2363. [Google Scholar] [PubMed]
- Kuroki, M.; Voest, E.E.; Amano, S.; Beerepoot, L.V.; Takashima, S.; Tolentino, M.; Kim, R.Y.; Rohan, R.M.; Colby, K.A.; Yeo, K.T.; et al. Reactive oxygen intermediates increase vascular endothelial growth factor expression in vitro and in vivo. J. Clin. Investig. 1996, 98, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- Cecarini, V.; Gee, J.; Fioretti, E.; Amici, M.; Angeletti, M.; Eleuteri, A.M.; Keller, J.N. Protein oxidation and cellular homeostasis: Emphasis on metabolism. Biochim. Biophys. Acta 2007, 1773, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Gengrinovitch, S.; Berman, B.; David, G.; Witte, L.; Neufeld, G.; Ron, D. Glypican-1 is a VEGF165 binding proteoglycan that acts as an extracellular chaperone for VEGF165. J. Biol. Chem. 1999, 274, 10816–10822. [Google Scholar] [CrossRef] [PubMed]
- Maulik, N. Redox regulation of vascular angiogenesis. Antioxid. Redox Signal. 2002, 4, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Abid, M.R.; Tsai, J.C.; Spokes, K.C.; Deshpande, S.S.; Irani, K.; Aird, W.C. Vascular endothelial growth factor induces manganese-superoxide dismutase expression in endothelial cells by a rac1-regulated NADPH oxidase-dependent mechanism. FASEB J. 2001, 15, 2548–2550. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Castresana, M.R.; Newman, W.H. Reactive oxygen and NF-κ in VEGF-induced migration of human vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2001, 285, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Tang, Y.; Fukai, T.; Dikalov, S.I.; Ma, Y.; Fujimoto, M.; Quinn, M.T.; Pagano, P.J.; Johnson, C.; Alexander, R.W. Novel role of gp91(phox)-containing NAD(P)H oxidase in vascular endothelial growth factor-induced signaling and angiogenesis. Circ. Res. 2002, 91, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M. Redox signaling in angiogenesis: Role of NADPH oxidase. Cardiovasc. Res. 2006, 71, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Arbiser, J.L.; Petros, J.; Klafter, R.; Govindajaran, B.; McLaughlin, E.R.; Brown, L.F.; Cohen, C.; Moses, M.; Kilroy, S.; Arnold, R.S.; et al. Reactive oxygen generated by NOX1 triggers the angiogenic switch. Proc. Natl. Acad. Sci. USA 2002, 99, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E.; Gu, J.; Schau, M.; Bunn, H.F. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7987–7992. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.E.; Berra, E.; Gothie, E.; Roux, D.; Pouyssegur, J. P42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (hif-1alpha) and enhance the transcriptional activity of hif-1. J. Biol. Chem. 1999, 274, 32631–32637. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.F.; Detmar, M.; Claffey, K.; Nagy, J.A.; Feng, D.; Dvorak, A.M.; Dvorak, H.F. Vascular permeability factor/vascular endothelial growth factor: A multifunctional angiogenic cytokine. EXS 1997, 79, 233–269. [Google Scholar] [PubMed]
- Mittal, M.; Roth, M.; Konig, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Ismail, S.; Sturrock, A.; Wu, P.; Cahill, B.; Norman, K.; Huecksteadt, T.; Sanders, K.; Kennedy, T.; Hoidal, J. NOX4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: The role of autocrine production of transforming growth factor-{beta}1 and insulin-like growth factor binding protein-3. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L489–L499. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex iii stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed]
- Minet, E.; Michel, G.; Mottet, D.; Raes, M.; Michiels, C. Transduction pathways involved in hypoxia-inducible factor-1 phosphorylation and activation. Free Radic. Biol. Med. 2001, 31, 847–855. [Google Scholar] [CrossRef]
- Bonello, S.; Zahringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Gorlach, A. Reactive oxygen species activate the hif-1alpha promoter via a functional NFκB site. Arterioscler. Thrombosis Vasc. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Petry, A.; Hess, J.; Gorlach, A. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol. Biol. Cell 2010, 21, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Peshavariya, H.M.; Chan, E.C.; Liu, G.S.; Jiang, F.; Dusting, G.J. Transforming growth factor-beta1 requires NADPH oxidase 4 for angiogenesis in vitro and in vivo. J. Cell. Mol. Med. 2014, 18, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Lambeth, J.D. NOX4: A hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Madlener, M.; Pfeilschifter, J.; Werner, S. Induction of inducible nitric oxide synthase and its corresponding tetrahydrobiopterin-cofactor-synthesizing enzyme GTP-cyclohydrolase i during cutaneous wound repair. J. Investig. Dermatol. 1998, 111, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Kolb, N.; Werner, E.R.; Pfeilschifter, J. Coordinated induction of inducible nitric oxide synthase and GTP-cyclohydrolase i is dependent on inflammatory cytokines and interferon-gamma in hacat keratinocytes: Implications for the model of cutaneous wound repair. J. Investig. Dermatol. 1998, 111, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Brune, B.; Zhou, J. Nitric oxide and superoxide: Interference with hypoxic signaling. Cardiovasc. Res. 2007, 75, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Stallmeyer, B.; Kampfer, H.; Kolb, N.; Pfeilschifter, J. Nitric oxide triggers enhanced induction of vascular endothelial growth factor expression in cultured keratinocytes (hacat) and during cutaneous wound repair. FASEB J. 1999, 13, 2002–2014. [Google Scholar] [PubMed]
- Raja; Sivamani, K.; Garcia, M.S.; Isseroff, R.R. Wound re-epithelialization: Modulating keratinocyte migration in wound healing. Front. Biosci. 2007, 12, 2849–2868. [Google Scholar] [CrossRef] [PubMed]
- Stanley, A.; Hynes, A.; Brakebusch, C.; Quondamatteo, F. Rho GTPases and NOX dependent ros production in skin. Is there a connection? Histol. Histopathol. 2012, 27, 1395–1406. [Google Scholar] [PubMed]
- Nam, H.J.; Park, Y.Y.; Yoon, G.; Cho, H.; Lee, J.H. Co-treatment with hepatocyte growth factor and TGF-β1 enhances migration of hacat cells through NADPH oxidase-dependent ros generation. Exp. Mol. Med. 2010, 42, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Misner, B.J.; Chiu, R.J.; Meyskens, F.L., Jr. Redox effector factor-1, combined with reactive oxygen species, plays an important role in the transformation of jb6 cells. Carcinogenesis 2007, 28, 2382–2390. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.M.; Ziegler, T.R.; Johnson, J.M.; Gu, L.; Hansen, J.M.; Jones, D.P. Selective protection of nuclear thioredoxin-1 and glutathione redox systems against oxidation during glucose and glutamine deficiency in human colonic epithelial cells. Free Radic. Biol. Med. 2007, 42, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Leoni, G.; Alam, A.; Neumann, P.A.; Lambeth, J.D.; Cheng, G.; McCoy, J.; Hilgarth, R.S.; Kundu, K.; Murthy, N.; Kusters, D.; et al. Annexin a1, formyl peptide receptor, and NOX1 orchestrate epithelial repair. J. Clin. Investig. 2013, 123, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Haase, I.; Evans, R.; Pofahl, R.; Watt, F.M. Regulation of keratinocyte shape, migration and wound epithelialization by IGF-1- and EGF-dependent signalling pathways. J. Cell Sci. 2003, 116, 3227–3238. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Peng, T.; Du, J.; Sukhanov, S.; Li, Y.; Itabe, H.; Parthasarathy, S.; Delafontaine, P. A redox-sensitive pathway mediates oxidized ldl-induced downregulation of insulin-like growth factor-1 receptor. J. Lipid Res. 2005, 46, 1266–1277. [Google Scholar] [CrossRef] [PubMed]
- Hober, S.; Lundstrom Ljung, J.; Uhlen, M.; Nilsson, B. Insulin-like growth factors i and ii are unable to form and maintain their native disulfides under in vivo redox conditions. FEBS Lett. 1999, 443, 271–276. [Google Scholar] [CrossRef]
- Madigan, M.C.; McEnaney, R.M.; Shukla, A.J.; Hong, G.; Kelley, E.E.; Tarpey, M.M.; Gladwin, M.; Zuckerbraun, B.S.; Tzeng, E. Xanthine oxidoreductase function contributes to normal wound healing. Mol. Med. 2015, 21, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Bugge, T.H.; Kombrinck, K.W.; Flick, M.J.; Daugherty, C.C.; Danton, M.J.; Degen, J.L. Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell 1996, 87, 709–719. [Google Scholar] [CrossRef]
- Grange, L.; Nguyen, M.V.; Lardy, B.; Derouazi, M.; Campion, Y.; Trocme, C.; Paclet, M.H.; Gaudin, P.; Morel, F. NAD(P)H oxidase activity of NOX4 in chondrocytes is both inducible and involved in collagenase expression. Antioxid. Redox Signal. 2006, 8, 1485–1496. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, P.F.; Ziprin, P.; Peck, D.H.; Darzi, A.W. Hypoxia increases reepithelialization via an alphavbeta6-dependent pathway. Wound Repair Regen. 2005, 13, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Lund, L.R.; Green, K.A.; Stoop, A.A.; Ploug, M.; Almholt, K.; Lilla, J.; Nielsen, B.S.; Christensen, I.J.; Craik, C.S.; Werb, Z.; et al. Plasminogen activation independent of uPA and tPA maintains wound healing in gene-deficient mice. EMBO J. 2006, 25, 2686–2697. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.O.; Park, S.J.; Yoon, S.Y.; Yun, C.H.; Chung, A.S. Sustained production of H2O2 activates pro-matrix metalloproteinase-2 through receptor tyrosine kinases/phosphatidylinositol 3-kinase/NF-κB pathway. J. Biol. Chem. 2002, 277, 30271–30282. [Google Scholar] [CrossRef] [PubMed]
- Stief, T.W. Oxidized fibrin stimulates the activation of pro-urokinase and is the preferential substrate of human plasmin. Blood Coagul. Fibrinolysis 1993, 4, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Goldkorn, T.; Balaban, N.; Matsukuma, K.; Chea, V.; Gould, R.; Last, J.; Chan, C.; Chavez, C. EGF-receptor phosphorylation and signaling are targeted by H2O2 redox stress. Ame. J. Respir. Cell Mol. Biol. 1998, 19, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Peus, D.; Vasa, R.A.; Meves, A.; Pott, M.; Beyerle, A.; Squillace, K.; Pittelkow, M.R. H2O2 is an important mediator of uvb-induced EGF-receptor phosphorylation in cultured keratinocytes. J. Investig. Dermatol. 1998, 110, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Ono, I.; Gunji, H.; Zhang, J.Z.; Maruyama, K.; Kaneko, F. Studies on cytokines related to wound healing in donor site wound fluid. J. Dermatol. Sci. 1995, 10, 241–245. [Google Scholar] [CrossRef]
- Antoniades, H.N.; Galanopoulos, T.; Neville-Golden, J.; Kiritsy, C.P.; Lynch, S.E. Expression of growth factor and receptor mrnas in skin epithelial cells following acute cutaneous injury. Am. J. Pathol. 1993, 142, 1099–1110. [Google Scholar] [PubMed]
- Kairuz, E.; Upton, Z.; Dawson, R.A.; Malda, J. Hyperbaric oxygen stimulates epidermal reconstruction in human skin equivalents. Wound Repair Regen. 2007, 15, 266–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivekananda, J.; Lin, A.; Coalson, J.J.; King, R.J. Acute inflammatory injury in the lung precipitated by oxidant stress induces fibroblasts to synthesize and release transforming growth factor-alpha. J. Biol. Chem. 1994, 269, 25057–25061. [Google Scholar] [PubMed]
- Kopp, J.; Wang, G.Y.; Kulmburg, P.; Schultze-Mosgau, S.; Huan, J.N.; Ying, K.; Seyhan, H.; Jeschke, M.D.; Kneser, U.; Bach, A.D.; et al. Accelerated wound healing by in vivo application of keratinocytes overexpressing KGF. Mol. Ther. 2004, 10, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Marchese, C.; Maresca, V.; Cardinali, G.; Belleudi, F.; Ceccarelli, S.; Bellocci, M.; Frati, L.; Torrisi, M.R.; Picardo, M. Uvb-induced activation and internalization of keratinocyte growth factor receptor. Oncogene 2003, 22, 2422–2431. [Google Scholar] [CrossRef] [PubMed]
- Higton, D.I.; James, D.W. The force of contraction of full-thickness wounds of rabbit skin. Br. J. Surg. 1964, 51, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, H.P.; Hunt, T.K. Collagen organization critical role in wound contraction. Adv. Wound Care 2012, 1, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Gabbiani, G.; Ryan, G.B.; Majno, G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 1971, 27, 549–550. [Google Scholar] [CrossRef] [PubMed]
- Klingberg, F.; Hinz, B.; White, E.S. The myofibroblast matrix: Implications for tissue repair and fibrosis. J. Pathol. 2013, 229, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. Formation and function of the myofibroblast during tissue repair. J. Investig. Dermatol. 2007, 127, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Desmouliere, A.; Geinoz, A.; Gabbiani, F.; Gabbiani, G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Donnelly, S.C.; Peng, T.; Bucala, R.; Metz, C.N. Peripheral blood fibrocytes: Differentiation pathway and migration to wound sites. J. Immunol. 2001, 166, 7556–7562. [Google Scholar] [CrossRef] [PubMed]
- Direkze, N.C.; Forbes, S.J.; Brittan, M.; Hunt, T.; Jeffery, R.; Preston, S.L.; Poulsom, R.; Hodivala-Dilke, K.; Alison, M.R.; Wright, N.A. Multiple organ engraftment by bone-marrow-derived myofibroblasts and fibroblasts in bone-marrow-transplanted mice. Stem Cells 2003, 21, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Tokura, Y. Epithelial-mesenchymal transition in the skin. J. Dermatol. Sci. 2011, 61, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Dulauroy, S.; Di Carlo, S.E.; Langa, F.; Eberl, G.; Peduto, L. Lineage tracing and genetic ablation of adam12(+) perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat. Med. 2012, 18, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, P.; Betsholtz, C. Not all myofibroblasts are alike: Revisiting the role of PDGF-A and PDGF-B using PDGF-targeted mice. Curr. Opin. Nephrol. Hypertens. 1998, 7, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Desmouliere, A.; Rubbia-Brandt, L.; Grau, G.; Gabbiani, G. Heparin induces alpha-smooth muscle actin expression in cultured fibroblasts and in granulation tissue myofibroblasts. Lab. Investig. 1992, 67, 716–726. [Google Scholar] [PubMed]
- Rubbia-Brandt, L.; Sappino, A.P.; Gabbiani, G. Locally applied GM-CSF induces the accumulation of alpha-smooth muscle actin containing myofibroblasts. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1991, 60, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.; Grose, R. Regulation of wound healing by growth factors and cytokines. Physiol. Rev. 2003, 83, 835–870. [Google Scholar] [PubMed]
- Hinz, B.; Gabbiani, G. Mechanisms of force generation and transmission by myofibroblasts. Curr. Opin. Biotechnol. 2003, 14, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Mastrangelo, D.; Iselin, C.E.; Chaponnier, C.; Gabbiani, G. Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am. J. Pathol. 2001, 159, 1009–1020. [Google Scholar] [CrossRef]
- Wipff, P.J.; Hinz, B. Myofibroblasts work best under stress. J. Bodyw. Mov. Ther. 2009, 13, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Vedrenne, N.; Coulomb, B.; Danigo, A.; Bonte, F.; Desmouliere, A. The complex dialogue between (myo)fibroblasts and the extracellular matrix during skin repair processes and ageing. Pathol. Biol. 2012, 60, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.Y.; Wu, L.C.; Dai, T.; Chen, S.Y.; Wang, A.Y.; Lin, K.; Lin, D.M.; Yang, J.Q.; Cheng, B.; Zhang, L.; et al. NADPH oxidase-2 is a key regulator of human dermal fibroblasts: A potential therapeutic strategy for the treatment of skin fibrosis. Exp. Dermatol. 2014, 23, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Carnesecchi, S.; Deffert, C.; Donati, Y.; Basset, O.; Hinz, B.; Preynat-Seauve, O.; Guichard, C.; Arbiser, J.L.; Banfi, B.; Pache, J.C.; et al. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid. Redox Signal. 2011, 15, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Gauldie, J.; Bonniaud, P.; Sime, P.; Ask, K.; Kolb, M. TGF-beta, Smad3 and the process of progressive fibrosis. Biochem. Soc. Trans. 2007, 35, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, R.A.; Leof, E.B. Tgf-beta signaling: A tale of two responses. J. Cell. Biochem. 2007, 102, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. How cells read TGF-beta signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.; Hill, C.S. How the smads regulate transcription. Int. J. Biochem. Cell Biol. 2008, 40, 383–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Matsuura, I.; He, D.; Liu, F. Transforming growth factor-{beta}-inducible phosphorylation of Smad3. J. Biol. Chem. 2009, 284, 9663–9673. [Google Scholar] [CrossRef] [PubMed]
- Sturrock, A.; Cahill, B.; Norman, K.; Huecksteadt, T.P.; Hill, K.; Sanders, K.; Karwande, S.V.; Stringham, J.C.; Bull, D.A.; Gleich, M.; et al. Transforming growth factor-beta1 induces NOX4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L661–L673. [Google Scholar] [CrossRef] [PubMed]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Fanburg, B.L. Activation of an H2O2-generating nadh oxidase in human lung fibroblasts by transforming growth factor beta 1. J. Biol. Chem. 1995, 270, 30334–30338. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Andre-Levigne, D.; Modarressi, A.; Pignel, R.; Bochaton-Piallat, M.L.; Pittet-Cuenod, B. Hyperbaric oxygen therapy promotes wound repair in ischemic and hyperglycemic conditions, increasing tissue perfusion and collagen deposition. Wound Repair Regen. 2016, 24, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Tomasek, J.J.; Haaksma, C.J.; Schwartz, R.J.; Howard, E.W. Whole animal knockout of smooth muscle alpha-actin does not alter excisional wound healing or the fibroblast-to-myofibroblast transition. Wound Repair Regen. 2013, 21, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Au, K.; Ehrlich, H.P. When the smad signaling pathway is impaired, fibroblasts advance open wound contraction. Exp. Mol. Pathol. 2010, 89, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. The myofibroblast: Paradigm for a mechanically active cell. J. Biomech. 2010, 43, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Goffin, J.M.; Pittet, P.; Csucs, G.; Lussi, J.W.; Meister, J.J.; Hinz, B. Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibers. J. Cell Biol. 2006, 172, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, H.P.; Rajaratnam, J.B. Cell locomotion forces versus cell contraction forces for collagen lattice contraction: An in vitro model of wound contraction. Tissue Cell 1990, 22, 407–417. [Google Scholar] [CrossRef]
- Martin, P. Wound healing—Aiming for perfect skin regeneration. Science 1997, 276, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Jarman, E.R.; Khambata, V.S.; Cope, C.; Jones, P.; Roger, J.; Ye, L.Y.; Duggan, N.; Head, D.; Pearce, A.; Press, N.J.; et al. An inhibitor of NADPH oxidase-4 attenuates established pulmonary fibrosis in a rodent disease model. Am. J. Respir. Cell Mol. Biol. 2014, 50, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Hopf, H.W.; Humphrey, L.M.; Puzziferri, N.; West, J.M.; Attinger, C.E.; Hunt, T.K. Adjuncts to preparing wounds for closure: Hyperbaric oxygen, growth factors, skin substitutes, negative pressure wound therapy (vacuum-assisted closure). Foot Ankle Clin. 2001, 6, 661–682. [Google Scholar] [CrossRef]
- Myllyla, R.; Tuderman, L.; Kivirikko, K.I. Mechanism of the prolyl hydroxylase reaction. 2. Kinetic analysis of the reaction sequence. Eur. J. Biochem. FEBS 1977, 80, 349–357. [Google Scholar] [CrossRef]
- Jonsson, K.; Jensen, J.A.; Goodson, W.H., 3rd; Scheuenstuhl, H.; West, J.; Hopf, H.W.; Hunt, T.K. Tissue oxygenation, anemia, and perfusion in relation to wound healing in surgical patients. Ann. Surg. 1991, 214, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Frost, H.M. A 2003 update of bone physiology and wolff's law for clinicians. Angle Orthod. 2004, 74, 3–15. [Google Scholar] [PubMed]
- Garcia-Cardena, G.; Comander, J.; Anderson, K.R.; Blackman, B.R.; Gimbrone, M.A., Jr. Biomechanical activation of vascular endothelium as a determinant of its functional phenotype. Proc. Natl. Acad. Sci. USA 2001, 98, 4478–4485. [Google Scholar] [CrossRef] [PubMed]
- Larios, J.M.; Budhiraja, R.; Fanburg, B.L.; Thannickal, V.J. Oxidative protein cross-linking reactions involving l-tyrosine in transforming growth factor-beta1-stimulated fibroblasts. J. Biol. Chem. 2001, 276, 17437–17441. [Google Scholar] [CrossRef] [PubMed]
- Lardinois, O.M.; Medzihradszky, K.F.; Ortiz de Montellano, P.R. Spin trapping and protein cross-linking of the lactoperoxidase protein radical. J. Biol. Chem. 1999, 274, 35441–35448. [Google Scholar] [CrossRef] [PubMed]
- Edens, W.A.; Sharling, L.; Cheng, G.; Shapira, R.; Kinkade, J.M.; Lee, T.; Edens, H.A.; Tang, X.; Sullards, C.; Flaherty, D.B.; et al. Tyrosine cross-linking of extracellular matrix is catalyzed by duox, a multidomain oxidase/peroxidase with homology to the phagocyte oxidase subunit gp91phox. J. Cell. Biol. 2001, 154, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Heinecke, J.W.; Shapiro, B.M. The respiratory burst oxidase of fertilization. A physiological target for regulation by protein kinase c. J. Biol. Chem. 1992, 267, 7959–7962. [Google Scholar] [PubMed]
- Nelson, R.E.; Fessler, L.I.; Takagi, Y.; Blumberg, B.; Keene, D.R.; Olson, P.F.; Parker, C.G.; Fessler, J.H. Peroxidasin: A novel enzyme-matrix protein of drosophila development. EMBO J. 1994, 13, 3438–3447. [Google Scholar] [PubMed]
- Peterfi, Z.; Donko, A.; Orient, A.; Sum, A.; Prokai, A.; Molnar, B.; Vereb, Z.; Rajnavolgyi, E.; Kovacs, K.J.; Muller, V.; et al. Peroxidasin is secreted and incorporated into the extracellular matrix of myofibroblasts and fibrotic kidney. Am. J. Pathol. 2009, 175, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Lazar, E.; Peterfi, Z.; Sirokmany, G.; Kovacs, H.A.; Klement, E.; Medzihradszky, K.F.; Geiszt, M. Structure-function analysis of peroxidasin provides insight into the mechanism of collagen iv crosslinking. Free Radic. Biol. Med. 2015, 83, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, G.; Li, H.; Cao, Z.; Qiu, X.; McCormick, S.; Thannickal, V.J.; Nauseef, W.M. Vascular peroxidase-1 is rapidly secreted, circulates in plasma, and supports dityrosine cross-linking reactions. Free Radic. Biol. Med. 2011, 51, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Luo, X.J.; Yang, Z.B.; Zhang, J.J.; Li, T.B.; Zhang, X.J.; Ma, Q.L.; Zhang, G.G.; Hu, C.P.; Peng, J. Inhibition of NOX/vpo1 pathway and inflammatory reaction by trimethoxystilbene in prevention of cardiovascular remodeling in hypoxia-induced pulmonary hypertensive rats. J. Cardiovasc. Pharmacol. 2014, 63, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A.; Szot, S.; Williams, M.A.; Kagan, H.M. Oxidation of lysine side-chains of elastin by the myeloperoxidase system and by stimulated human neutrophils. Biochem. Biophys. Res. Commun. 1986, 135, 451–457. [Google Scholar] [CrossRef]
- Hazen, S.L.; Gaut, J.P.; Hsu, F.F.; Crowley, J.R.; d’Avignon, A.; Heinecke, J.W. P-hydroxyphenylacetaldehyde, the major product of l-tyrosine oxidation by the myeloperoxidase-H2O2-chloride system of phagocytes, covalently modifies epsilon-amino groups of protein lysine residues. J. Biol. Chem. 1997, 272, 16990–16998. [Google Scholar] [CrossRef] [PubMed]
- Georges, P.C.; Hui, J.J.; Gombos, Z.; McCormick, M.E.; Wang, A.Y.; Uemura, M.; Mick, R.; Janmey, P.A.; Furth, E.E.; Wells, R.G. Increased stiffness of the rat liver precedes matrix deposition: Implications for fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G1147–G1154. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, M.R.; Efron, P.A.; Thornton, F.J.; Klingel, K.; Gross, S.S.; Barbul, A. Nitric oxide, an autocrine regulator of wound fibroblast synthetic function. J. Immunol. 1997, 158, 2375–2381. [Google Scholar] [PubMed]
- Amadeu, T.P.; Costa, A.M. Nitric oxide synthesis inhibition alters rat cutaneous wound healing. J. Cutan. Pathol. 2006, 33, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Desmouliere, A.; Redard, M.; Darby, I.; Gabbiani, G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am. J. Pathol. 1995, 146, 56–66. [Google Scholar] [PubMed]
- Mignatti, P. Extracellular matrix remodeling by metalloproteinases and plasminogen activators. Kidney Int. Suppl. 1995, 49, S12–S14. [Google Scholar] [PubMed]
- Niland, S.; Cremer, A.; Fluck, J.; Eble, J.A.; Krieg, T.; Sollberg, S. Contraction-dependent apoptosis of normal dermal fibroblasts. J. Investig. Dermatol. 2001, 116, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Fluck, J.; Querfeld, C.; Cremer, A.; Niland, S.; Krieg, T.; Sollberg, S. Normal human primary fibroblasts undergo apoptosis in three-dimensional contractile collagen gels. J. Investig. Dermatol. 1998, 110, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Sarrazy, V.; Billet, F.; Micallef, L.; Coulomb, B.; Desmouliere, A. Mechanisms of pathological scarring: Role of myofibroblasts and current developments. Wound Repair Regen. 2011, 19 (Suppl. 1), s10–s15. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jiao, H.; Stewart, T.L.; Shankowsky, H.A.; Scott, P.G.; Tredget, E.E. Increased TGF-beta-producing cd4+ t lymphocytes in postburn patients and their potential interaction with dermal fibroblasts in hypertrophic scarring. Wound Repair Regen. 2007, 15, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Meitzler, J.L.; Antony, S.; Wu, Y.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Roy, K.; Doroshow, J.H. NADPH oxidases: A perspective on reactive oxygen species production in tumor biology. Antioxid. Redox Signal. 2014, 20, 2873–2889. [Google Scholar] [CrossRef] [PubMed]
- Assi, M. The differential role of reactive oxygen species in early and late stages of cancer. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH oxidase-mediated redox signaling: Roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; He, Q.; Cheng, Y.; Zhao, B.; Zhang, Y.; Zhang, S.; Miao, J. A benzoxazine derivative induces vascular endothelial cell apoptosis in the presence of fibroblast growth factor-2 by elevating NADPH oxidase activity and reactive oxygen species levels. Toxicol. In Vitro 2009, 23, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, S.; Akasaka, Y.; Kiguchi, H.; Suzuki, T.; Imaizumi, R.; Ishikawa, Y.; Ito, K.; Ishii, T. Basic fibroblast growth factor induces down-regulation of alpha-smooth muscle actin and reduction of myofibroblast areas in open skin wounds. Wound Repair Regen. 2009, 17, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Akasaka, Y.; Ono, I.; Tominaga, A.; Ishikawa, Y.; Ito, K.; Suzuki, T.; Imaizumi, R.; Ishiguro, S.; Jimbow, K.; Ishii, T. Basic fibroblast growth factor in an artificial dermis promotes apoptosis and inhibits expression of alpha-smooth muscle actin, leading to reduction of wound contraction. Wound Repair Regen. 2007, 15, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Akasaka, Y.; Ono, I.; Yamashita, T.; Jimbow, K.; Ishii, T. Basic fibroblast growth factor promotes apoptosis and suppresses granulation tissue formation in acute incisional wounds. J. Pathol. 2004, 203, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Yokoyama, Y.; Ishikawa, O. A possible mechanism of basic fibroblast growth factor-promoted scarless wound healing: The induction of myofibroblast apoptosis. Eur. J. Dermatol. 2012, 22, 46–53. [Google Scholar] [PubMed]
- Xue, M.; Jackson, C.J. Extracellular matrix reorganization during wound healing and its impact on abnormal scarring. Adv. Wound Care 2015, 4, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.H.; Moon, Y.J.; Seo, J.E.; Lee, Y.; Kim, K.H.; Chung, J.H. Reactive oxygen species produced by NADPH oxidase, xanthine oxidase, and mitochondrial electron transport system mediate heat shock-induced mmp-1 and mmp-9 expression. Free Radic. Biol. Med. 2008, 44, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Rosner, K.; Ross, C.; Karlsmark, T.; Petersen, A.A.; Gottrup, F.; Vejlsgaard, G.L. Immunohistochemical characterization of the cutaneous cellular infiltrate in different areas of chronic leg ulcers. APMIS 1995, 103, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Newton, P.M.; Watson, J.A.; Wolowacz, R.G.; Wood, E.J. Macrophages restrain contraction of an in vitro wound healing model. Inflammation 2004, 28, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Wlaschek, M.; Scharffetter-Kochanek, K. Oxidative stress in chronic venous leg ulcers. Wound Repair Regen. 2005, 13, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Wall, I.B.; Moseley, R.; Baird, D.M.; Kipling, D.; Giles, P.; Laffafian, I.; Price, P.E.; Thomas, D.W.; Stephens, P. Fibroblast dysfunction is a key factor in the non-healing of chronic venous leg ulcers. J. Investig. Dermatol. 2008, 128, 2526–2540. [Google Scholar] [CrossRef] [PubMed]
- James, T.J.; Hughes, M.A.; Cherry, G.W.; Taylor, R.P. Evidence of oxidative stress in chronic venous ulcers. Wound Repair Regen. 2003, 11, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Kumin, A.; Schafer, M.; Epp, N.; Bugnon, P.; Born-Berclaz, C.; Oxenius, A.; Klippel, A.; Bloch, W.; Werner, S. Peroxiredoxin 6 is required for blood vessel integrity in wounded skin. J. Cell Biol. 2007, 179, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A. Oxidative stress and “senescent” fibroblasts in non-healing wounds as potential therapeutic targets. J. Investig. Dermatol. 2008, 128, 2361–2364. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.S.; Lee, B.Y.; Hwang, E.S. Dinstinct ros and biochemical profiles in cells undergoing DNA damage-induced senescence and apoptosis. Mech. Ageing Dev. 2005, 126, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Mendez, M.V.; Stanley, A.; Phillips, T.; Murphy, M.; Menzoian, J.O.; Park, H.Y. Fibroblasts cultured from distal lower extremities in patients with venous reflux display cellular characteristics of senescence. J. Vasc. Surg. 1998, 28, 1040–1050. [Google Scholar] [CrossRef]
- Jun, J.I.; Lau, L.F. The matricellular protein ccn1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Niinikoski, J.; Hunt, T.K.; Dunphy, J.E. Oxygen supply in healing tissue. Am. J. Surg. 1972, 123, 247–252. [Google Scholar] [CrossRef]
- Scheid, A.; Wenger, R.H.; Christina, H.; Camenisch, I.; Ferenc, A.; Stauffer, U.G.; Gassmann, M.; Meuli, M. Hypoxia-regulated gene expression in fetal wound regeneration and adult wound repair. Pediatr. Surg. Int. 2000, 16, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Peschen, M.; Lahaye, T.; Hennig, B.; Weyl, A.; Simon, J.C.; Vanscheidt, W. Expression of the adhesion molecules icam-1, vcam-1, lfa-1 and vla-4 in the skin is modulated in progressing stages of chronic venous insufficiency. Acta Derm.-Venereol. 1999, 79, 27–32. [Google Scholar] [PubMed]
- Xia, Y.P.; Zhao, Y.; Tyrone, J.W.; Chen, A.; Mustoe, T.A. Differential activation of migration by hypoxia in keratinocytes isolated from donors of increasing age: Implication for chronic wounds in the elderly. J. Investig. Dermatol. 2001, 116, 50–56. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, E.A.; Marinkovich, M.P.; Peavey, C.L.; Amieva, M.R.; Furthmayr, H.; Mustoe, T.A.; Woodley, D.T. Hypoxia increases human keratinocyte motility on connective tissue. J. Clin. Investig. 1997, 100, 2881–2891. [Google Scholar] [CrossRef] [PubMed]
- Falanga, V.; Qian, S.W.; Danielpour, D.; Katz, M.H.; Roberts, A.B.; Sporn, M.B. Hypoxia upregulates the synthesis of TGF-β 1 by human dermal fibroblasts. J. Investig. Dermatol. 1991, 97, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.; Galiano, R.D.; Connors, D.; Gruskin, E.; Wu, L.; Mustoe, T.A. Differential effects of oxygen on human dermal fibroblasts: Acute versus chronic hypoxia. Wound Repair Regen. 1996, 4, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Steinbrech, D.S.; Longaker, M.T.; Mehrara, B.J.; Saadeh, P.B.; Chin, G.S.; Gerrets, R.P.; Chau, D.C.; Rowe, N.M.; Gittes, G.K. Fibroblast response to hypoxia: The relationship between angiogenesis and matrix regulation. J. Surg. Res. 1999, 84, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Modarressi, A.; Pietramaggiori, G.; Godbout, C.; Vigato, E.; Pittet, B.; Hinz, B. Hypoxia impairs skin myofibroblast differentiation and function. J. Investig. Dermatol. 2010, 130, 2818–2827. [Google Scholar] [CrossRef] [PubMed]
- Lokmic, Z.; Musyoka, J.; Hewitson, T.D.; Darby, I.A. Hypoxia and hypoxia signaling in tissue repair and fibrosis. Int. Rev. Cell Mol. Biol. 2012, 296, 139–185. [Google Scholar] [PubMed]
- Alizadeh, N.; Pepper, M.S.; Modarressi, A.; Alfo, K.; Schlaudraff, K.; Montandon, D.; Gabbiani, G.; Bochaton-Piallat, M.L.; Pittet, B. Persistent ischemia impairs myofibroblast development in wound granulation tissue: A new model of delayed wound healing. Wound Repair Regen. 2007, 15, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.T.; Mustoe, T.A. Effects of ischemia on ulcer wound healing: A new model in the rabbit ear. Ann. Plast. Surg. 1990, 24, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Xia, Y.P.; Roth, S.I.; Gruskin, E.; Mustoe, T.A. Transforming growth factor-beta1 fails to stimulate wound healing and impairs its signal transduction in an aged ischemic ulcer model: Importance of oxygen and age. Am. J. Pathol. 1999, 154, 301–309. [Google Scholar] [CrossRef]
- Kim, B.C.; Kim, H.T.; Park, S.H.; Cha, J.S.; Yufit, T.; Kim, S.J.; Falanga, V. Fibroblasts from chronic wounds show altered TGF-β-signaling and decreased TGF-β type ii receptor expression. J. Cell. Physiol. 2003, 195, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Schmid, T.; Zhou, J.; Kohl, R.; Brune, B. P300 relieves p53-evoked transcriptional repression of hypoxia-inducible factor-1 (HIF-1). Biochem. J. 2004, 380, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ros production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Mustoe, T. Understanding chronic wounds: A unifying hypothesis on their pathogenesis and implications for therapy. Am. J. Surg. 2004, 187, 65S–70S. [Google Scholar] [CrossRef]
- Anaya-Prado, R.; Toledo-Pereyra, L.H. The molecular events underlying ischemia/reperfusion injury. Transpl. Proc. 2002, 34, 2518–2519. [Google Scholar] [CrossRef]
- Toledo-Pereyra, L.H.; Toledo, A.H.; Walsh, J.; Lopez-Neblina, F. Molecular signaling pathways in ischemia/reperfusion. Exp. Clin. Transpl. 2004, 2, 174–177. [Google Scholar]
- Peirce, S.M.; Skalak, T.C.; Rodeheaver, G.T. Ischemia-reperfusion injury in chronic pressure ulcer formation: A skin model in the rat. Wound Repair Regen. 2000, 8, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Reid, R.R.; Sull, A.C.; Mogford, J.E.; Roy, N.; Mustoe, T.A. A novel murine model of cyclical cutaneous ischemia-reperfusion injury. J. Surg. Res. 2004, 116, 172–180. [Google Scholar] [CrossRef]
- Braunersreuther, V.; Montecucco, F.; Asrih, M.; Pelli, G.; Galan, K.; Frias, M.; Burger, F.; Quindere, A.L.; Montessuit, C.; Krause, K.H.; et al. Role of NADPH oxidase isoforms NOX1, NOX2 and NOX4 in myocardial ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 2013, 64, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed]
- Kota, S.K.; Meher, L.K.; Jammula, S.; Kota, S.K.; Krishna, S.V.; Modi, K.D. Aberrant angiogenesis: The gateway to diabetic complications. Indian J. Endocrinol. Metab. 2012, 16, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Kolluru, G.K.; Bir, S.C.; Kevil, C.G. Endothelial dysfunction and diabetes: Effects on angiogenesis, vascular remodeling, and wound healing. Int. J. Vasc. Med. 2012, 2012, 918267. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.Z.; Soares, R. Neovascularization in diabetes and its complications. Unraveling the angiogenic paradox. Life Sci. 2013, 92, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Katakami, N.; Matsuhisa, M.; Matsuoka, T.A. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediat. Inflamm. 2010, 2010, 453892. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, M.S.; van Golde, J.M.; Schaper, N.C.; Stehouwer, C.D.; Huijberts, M.S. Diabetes impairs arteriogenesis in the peripheral circulation: Review of molecular mechanisms. Clin. Sci. 2010, 119, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, M.R.; Tantry, U.; Efron, P.A.; Ahrendt, G.M.; Thornton, F.J.; Barbul, A. Diabetes-impaired healing and reduced wound nitric oxide synthesis: A possible pathophysiologic correlation. Surgery 1997, 121, 513–519. [Google Scholar] [CrossRef]
- Witte, M.B.; Thornton, F.J.; Tantry, U.; Barbul, A. L-arginine supplementation enhances diabetic wound healing: Involvement of the nitric oxide synthase and arginase pathways. Metab. Clin. Exp. 2002, 51, 1269–1273. [Google Scholar] [CrossRef] [PubMed]
- Masters, K.S.; Leibovich, S.J.; Belem, P.; West, J.L.; Poole-Warren, L.A. Effects of nitric oxide releasing poly(vinyl alcohol) hydrogel dressings on dermal wound healing in diabetic mice. Wound Repair Regen. 2002, 10, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Witte, M.B.; Kiyama, T.; Barbul, A. Nitric oxide enhances experimental wound healing in diabetes. Br. J. Surg. 2002, 89, 1594–1601. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Shin, B.S.; Ma, H.; Van Hoecke, M.; Brennan, A.M.; Yenari, M.A.; Swanson, R.A. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann. Neurol. 2008, 64, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Olmez, I.; Ozyurt, H. Reactive oxygen species and ischemic cerebrovascular disease. Neurochem. Int. 2012, 60, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Blokhina, O.; Virolainen, E.; Fagerstedt, K.V. Antioxidants, oxidative damage and oxygen deprivation stress: A review. Ann. Bot. 2003, 91, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Gyurko, R.; Siqueira, C.C.; Caldon, N.; Gao, L.; Kantarci, A.; Van Dyke, T.E. Chronic hyperglycemia predisposes to exaggerated inflammatory response and leukocyte dysfunction in akita mice. J. Immunol. 2006, 177, 7250–7256. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Cao, X.; Song, F.; Xie, T.; Ji, X.; Miao, M.; Dong, J.; Tian, M.; Lin, Y.; Lu, S. Reduced dermis thickness and age accumulation in diabetic abdominal skin. Int. J. Lower Extrem. Wounds 2012, 11, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Xie, T.; Ge, K.; Lin, Y.; Lu, S. Effects of extracellular matrix glycosylation on proliferation and apoptosis of human dermal fibroblasts via the receptor for advanced glycosylated end products. Am. J. Dermatopathol. 2008, 30, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Ceolotto, G.; Bevilacqua, M.; Papparella, I.; Baritono, E.; Franco, L.; Corvaja, C.; Mazzoni, M.; Semplicini, A.; Avogaro, A. Insulin generates free radicals by an NAD(P)H, phosphatidylinositol 3′-kinase-dependent mechanism in human skin fibroblasts ex vivo. Diabetes 2004, 53, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, N.J.; Ikeyama, S. Age-related decline in cellular response to oxidative stress: Links to growth factor signaling pathways with common defects. Biochem. Pharmacol. 2002, 64, 999–1005. [Google Scholar] [CrossRef]
- Tandara, A.A.; Kloeters, O.; Kim, I.; Mogford, J.E.; Mustoe, T.A. Age effect on hsp70: Decreased resistance to ischemic and oxidative stress in hdf. J. Surg. Res. 2006, 132, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Mogford, J.E.; Tawil, N.; Chen, A.; Gies, D.; Xia, Y.; Mustoe, T.A. Effect of age and hypoxia on tgfbeta1 receptor expression and signal transduction in human dermal fibroblasts: Impact on cell migration. J. Cell. Physiol. 2002, 190, 259–265. [Google Scholar] [CrossRef] [PubMed]
- West, M.D.; Pereira-Smith, O.M.; Smith, J.R. Replicative senescence of human skin fibroblasts correlates with a loss of regulation and overexpression of collagenase activity. Exp. Cell Res. 1989, 184, 138–147. [Google Scholar] [CrossRef]
- Rivard, A.; Berthou-Soulie, L.; Principe, N.; Kearney, M.; Curry, C.; Branellec, D.; Semenza, G.L.; Isner, J.M. Age-dependent defect in vascular endothelial growth factor expression is associated with reduced hypoxia-inducible factor 1 activity. J. Biol. Chem. 2000, 275, 29643–29647. [Google Scholar] [CrossRef] [PubMed]
- Dumas, M.; Chaudagne, C.; Bonte, F.; Meybeck, A. Age-related response of human dermal fibroblasts to l-ascorbic acid: Study of type i and iii collagen synthesis. C. R. Acad. Sci. Ser. III Sci. 1996, 319, 1127–1132. [Google Scholar]
- Sahoo, S.; Meijles, D.N.; Pagano, P.J. NADPH oxidases: Key modulators in aging and age-related cardiovascular diseases? Clin. Sci. 2016, 130, 317–335. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.H. Aging: A revisited theory based on free radicals generated by NOX family NADPH oxidases. Exp. Gerontol. 2007, 42, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.A.; Asmis, R.; Evans, K.K.; Mustoe, T.A. Oxygen and wound care: A review of current therapeutic modalities and future direction. Wound Repair Regen. 2013, 21, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Knighton, D.R.; Halliday, B.; Hunt, T.K. Oxygen as an antibiotic. The effect of inspired oxygen on infection. Arch. Surg. 1984, 119, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Hirn, M. Hyperbaric oxygen in the treatment of gas gangrene and perineal necrotizing fasciitis. A clinical and experimental study. Eur. J. Surg. Suppl. 1993, 1–36. [Google Scholar]
- Bosco, M.C.; Delfino, S.; Ferlito, F.; Battaglia, F.; Puppo, M.; Gregorio, A.; Gambini, C.; Gattorno, M.; Martini, A.; Varesio, L. Hypoxic synovial environment and expression of macrophage inflammatory protein 3gamma/ccl20 in juvenile idiopathic arthritis. Arthr. Rheum. 2008, 58, 1833–1838. [Google Scholar] [CrossRef] [PubMed]
- Hohn, D.C.; MacKay, R.D.; Halliday, B.; Hunt, T.K. Effect of o2 tension on microbicidal function of leukocytes in wounds and in vitro. Surg. Forum 1976, 27, 18–20. [Google Scholar] [PubMed]
- Knighton, D.R.; Silver, I.A.; Hunt, T.K. Regulation of wound-healing angiogenesis-effect of oxygen gradients and inspired oxygen concentration. Surgery 1981, 90, 262–270. [Google Scholar] [PubMed]
- Hunt, T.K.; Pai, M.P. The effect of varying ambient oxygen tensions on wound metabolism and collagen synthesis. Surg. Gynecol. Obstet. 1972, 135, 561–567. [Google Scholar] [PubMed]
- Hehenberger, K.; Brismar, K.; Lind, F.; Kratz, G. Dose-dependent hyperbaric oxygen stimulation of human fibroblast proliferation. Wound Repair Regen. 1997, 5, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, D. Role of hyperbaric oxygen therapy in the management of lower extremity wounds. Int. J. Low Extrem. Wounds 2006, 5, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Goldman, R.J. Hyperbaric oxygen therapy for wound healing and limb salvage: A systematic review. PM R 2009, 1, 471–489. [Google Scholar] [CrossRef] [PubMed]
- Gordillo, G.M.; Sen, C.K. Evidence-based recommendations for the use of topical oxygen therapy in the treatment of lower extremity wounds. Int. J. Low Extrem. Wounds 2009, 8, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.; Davidson, J.D.; Mustoe, T.A. Ischemic tissue oxygen capacitance after hyperbaric oxygen therapy: A new physiologic concept. Plast. Reconstr. Surg. 1997, 99, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Kranke, P.; Bennett, M.H.; Martyn-St James, M.; Schnabel, A.; Debus, S.E. Hyperbaric oxygen therapy for chronic wounds. Cochrane Database Syst. Rev. 2012, 4, CD004123. [Google Scholar]
- Feldman-Idov, Y.; Melamed, Y.; Ore, L. Improvement of ischemic non-healing wounds following hyperoxygenation: The experience at rambam-elisha hyperbaric center in israel, 1998–2007. Isr. Med. Assoc. J. 2011, 13, 524–529. [Google Scholar] [PubMed]
- Zhao, L.L.; Davidson, J.D.; Wee, S.C.; Roth, S.I.; Mustoe, T.A. Effect of hyperbaric oxygen and growth factors on rabbit ear ischemic ulcers. Arch. Surg. 1994, 129, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Said, H.K.; Hijjawi, J.; Roy, N.; Mogford, J.; Mustoe, T. Transdermal sustained-delivery oxygen improves epithelial healing in a rabbit ear wound model. Arch. Surg. 2005, 140, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Londahl, M.; Landin-Olsson, M.; Katzman, P. Hyperbaric oxygen therapy improves health-related quality of life in patients with diabetes and chronic foot ulcer. Diabet. Med. 2011, 28, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K. Wound healing essentials: Let there be oxygen. Wound Repair Regen. 2009, 17, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.A.; Moon, R.E. Hyperbaric oxygen in the treatment of life-threatening soft-tissue infections. Respir. Care Clin. N. Am. 1999, 5, 203–219. [Google Scholar] [PubMed]
- Mader, J.T.; Brown, G.L.; Guckian, J.C.; Wells, C.H.; Reinarz, J.A. A mechanism for the amelioration by hyperbaric oxygen of experimental staphylococcal osteomyelitis in rabbits. J. Infect. Dis. 1980, 142, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Boykin, J.V., Jr. The nitric oxide connection: Hyperbaric oxygen therapy, becaplermin, and diabetic ulcer management. Adv. Skin Wound Care 2000, 13, 169–174. [Google Scholar] [PubMed]
- Hopf, H.W.; Gibson, J.J.; Angeles, A.P.; Constant, J.S.; Feng, J.J.; Rollins, M.D.; Zamirul Hussain, M.; Hunt, T.K. Hyperoxia and angiogenesis. Wound Repair Regen. 2005, 13, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Tra, W.M.; Spiegelberg, L.; Tuk, B.; Hovius, S.E.; Perez-Amodio, S. Hyperbaric oxygen treatment of tissue-engineered mucosa enhances secretion of angiogenic factors in vitro. Tissue Eng. Part A 2014, 20, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.Y.; Gibson, J.J.; Rollins, M.D.; Hopf, H.W.; Hussain, Z.; Hunt, T.K. Effect of hyperoxia on vascular endothelial growth factor levels in a wound model. Arch. Surg. 2000, 135, 1293–1297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chang, Q.; Cox, R.A.; Gong, X.; Gould, L.J. Hyperbaric oxygen attenuates apoptosis and decreases inflammation in an ischemic wound model. J. Investig. Dermatol. 2008, 128, 2102–2112. [Google Scholar] [CrossRef] [PubMed]
- Thom, S.R. Oxidative stress is fundamental to hyperbaric oxygen therapy. J. Appl. Physiol. 2009, 106, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Ema, M.; Hirota, K.; Mimura, J.; Abe, H.; Yodoi, J.; Sogawa, K.; Poellinger, L.; Fujii-Kuriyama, Y. Molecular mechanisms of transcription activation by hlf and hif1alpha in response to hypoxia: Their stabilization and redox signal-induced interaction with cbp/p300. EMBO J. 1999, 18, 1905–1914. [Google Scholar] [CrossRef] [PubMed]
- Kaga, S.; Zhan, L.; Matsumoto, M.; Maulik, N. Resveratrol enhances neovascularization in the infarcted rat myocardium through the induction of thioredoxin-1, heme oxygenase-1 and vascular endothelial growth factor. J. Mol. Cell. Cardiol. 2005, 39, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Milovanova, T.N.; Bhopale, V.M.; Sorokina, E.M.; Moore, J.S.; Hunt, T.K.; Hauer-Jensen, M.; Velazquez, O.C.; Thom, S.R. Lactate stimulates vasculogenic stem cells via the thioredoxin system and engages an autocrine activation loop involving hypoxia-inducible factor 1. Mol. Cell. Biol. 2008, 28, 6248–6261. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.S.; Gorti, G.K.; Quan, S.Y.; Ho, M.; Koch, R.J. Effect of hyperbaric oxygen on the growth factor profile of fibroblasts. Arch. Facial Plast. Surg. 2004, 6, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Miyanaga, Y.; Shimojo, H.; Ushida, T.; Tateishi, T. Effects of hyperbaric oxygen on procollagen messenger rna levels and collagen synthesis in the healing of rat tendon laceration. Tissue Eng. 1999, 5, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Motta, S.; Monti, M. Photodynamic therapy—A promising treatment option for autoimmune skin ulcers: A case report. Photochem. Photobiol. Sci. 2007, 6, 1150–1151. [Google Scholar] [CrossRef] [PubMed]
- Clichici, S.; Filip, A.; Daicoviciu, D.; Ion, R.M.; Mocan, T.; Tatomir, C.; Rogojan, L.; Olteanu, D.; Muresan, A. The dynamics of reactive oxygen species in photodynamic therapy with tetra sulfophenyl-porphyrin. Acta Physiol. Hung 2010, 97, 41–51. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
André-Lévigne, D.; Modarressi, A.; Pepper, M.S.; Pittet-Cuénod, B. Reactive Oxygen Species and NOX Enzymes Are Emerging as Key Players in Cutaneous Wound Repair. Int. J. Mol. Sci. 2017, 18, 2149. https://doi.org/10.3390/ijms18102149
André-Lévigne D, Modarressi A, Pepper MS, Pittet-Cuénod B. Reactive Oxygen Species and NOX Enzymes Are Emerging as Key Players in Cutaneous Wound Repair. International Journal of Molecular Sciences. 2017; 18(10):2149. https://doi.org/10.3390/ijms18102149
Chicago/Turabian StyleAndré-Lévigne, Dominik, Ali Modarressi, Michael S. Pepper, and Brigitte Pittet-Cuénod. 2017. "Reactive Oxygen Species and NOX Enzymes Are Emerging as Key Players in Cutaneous Wound Repair" International Journal of Molecular Sciences 18, no. 10: 2149. https://doi.org/10.3390/ijms18102149