Combined Virtual and Experimental Screening for CK1 Inhibitors Identifies a Modulator of p53 and Reveals Important Aspects of in Silico Screening Performance

 , ,
, , 

Abstract
1. Introduction
2. Results and Discussion
2.1. The National Cancer Institute Developmental Therapeutics Program (NCI/DTP) Diversity Set-II Compound Collection
2.2. Complete In Vitro Evaluation
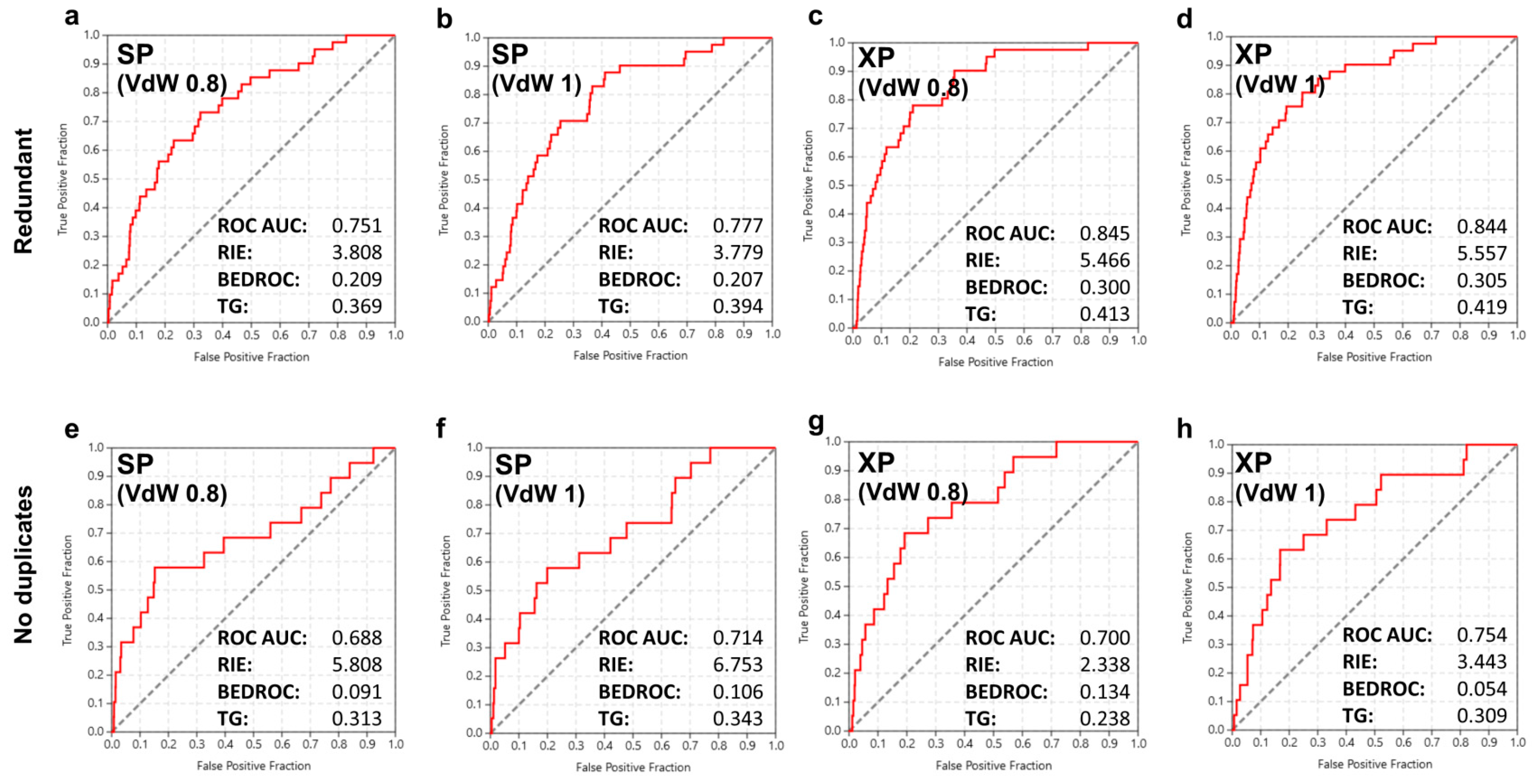
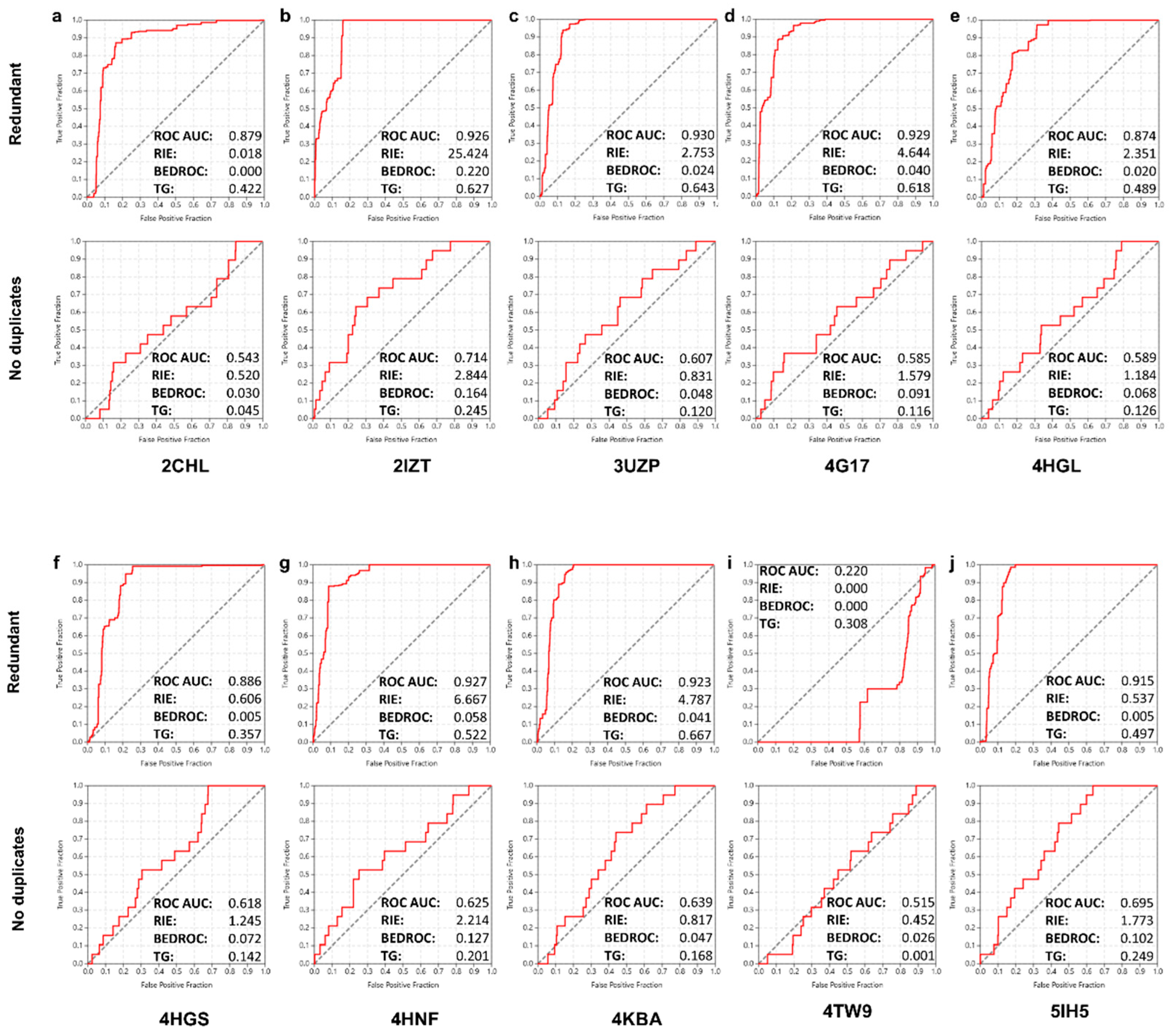
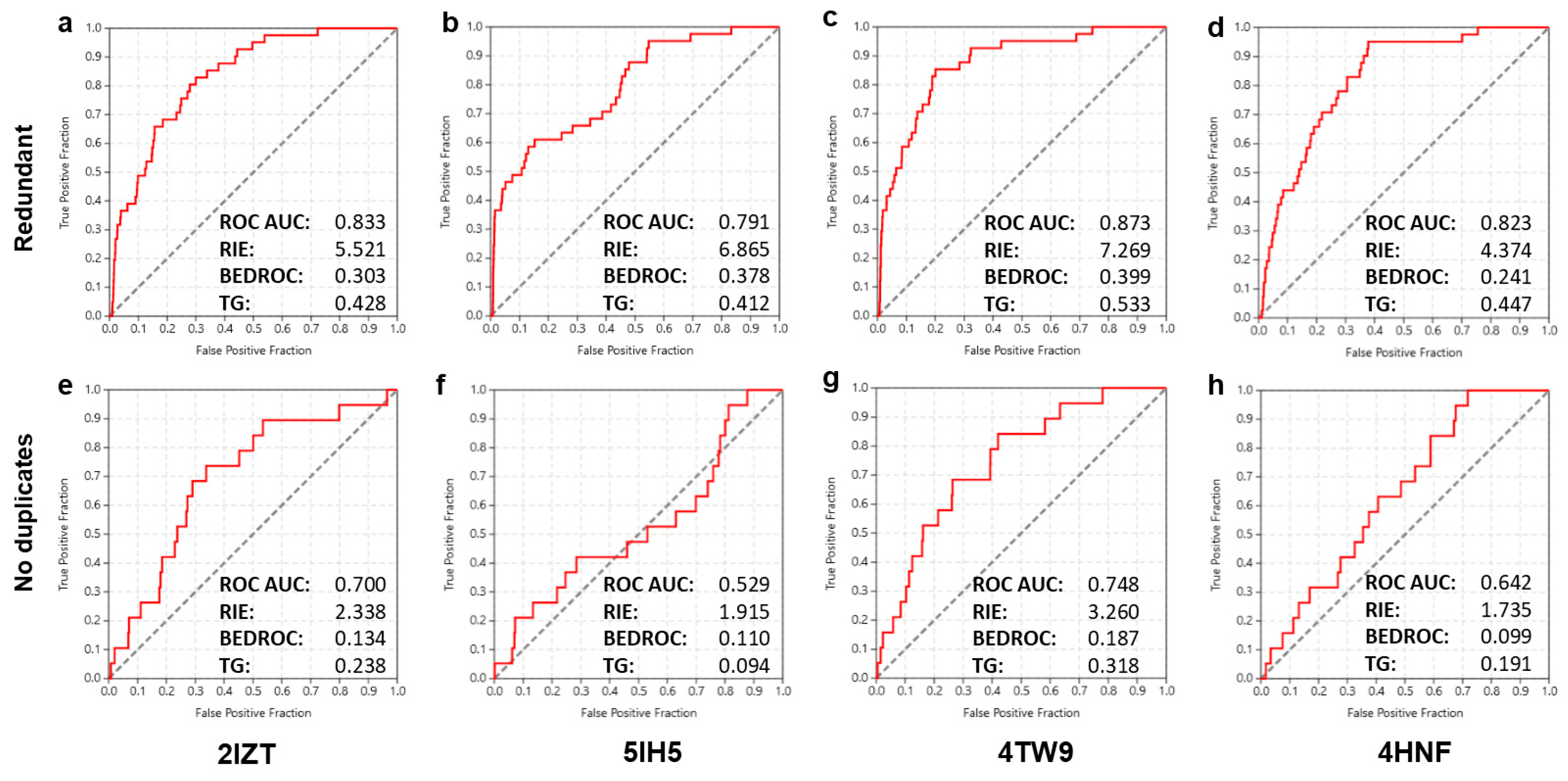
2.3. Virtual Screening Performance and Enrichment Metrics
2.4. Structure-Based Screening
2.5. Ligand-Based Screening
2.6. Combined Screening Approach
2.7. Overview of Ranking Success and Practical Considerations
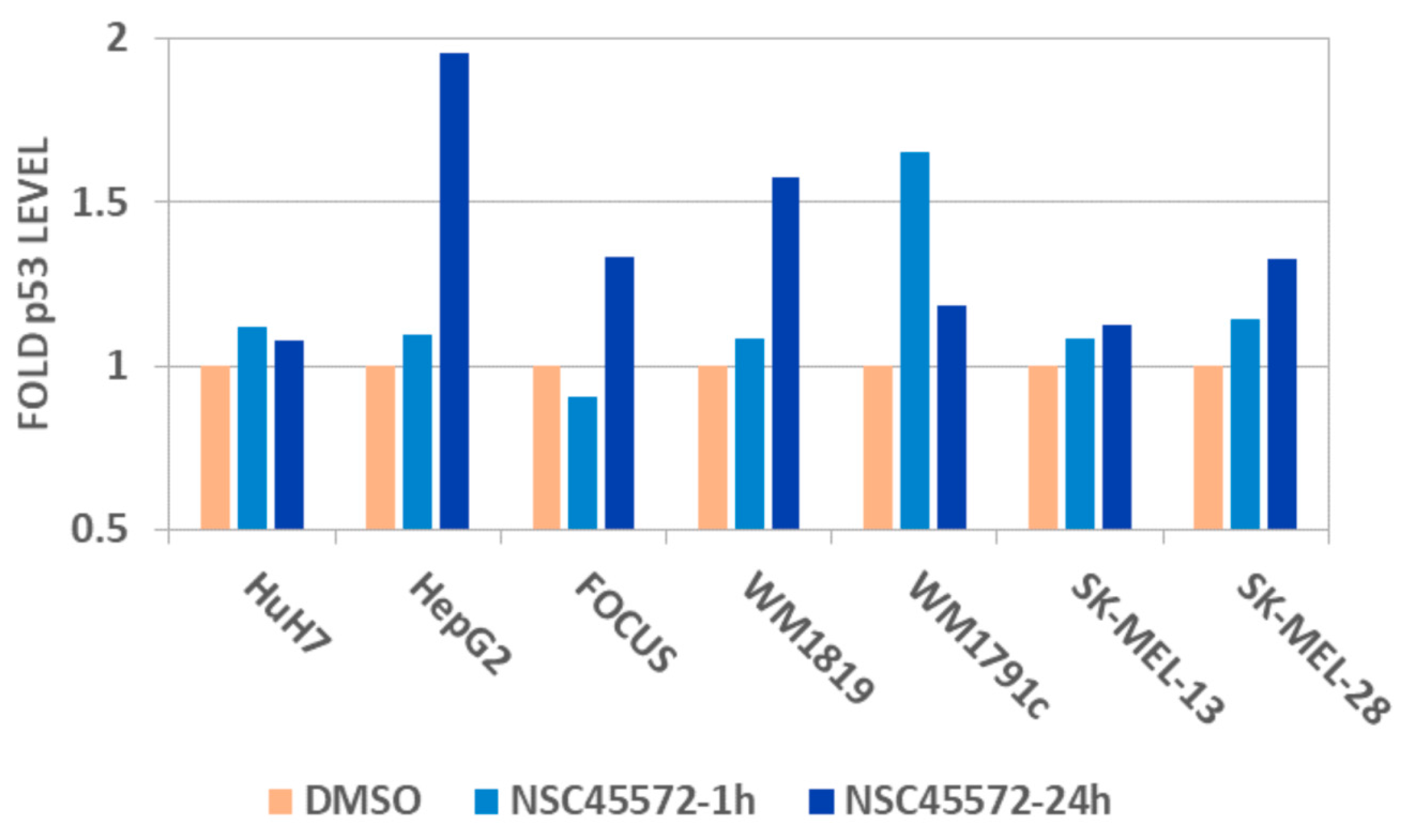
2.8. Cell-Based Assessment of p53-Modulating Capacity of NSC45572
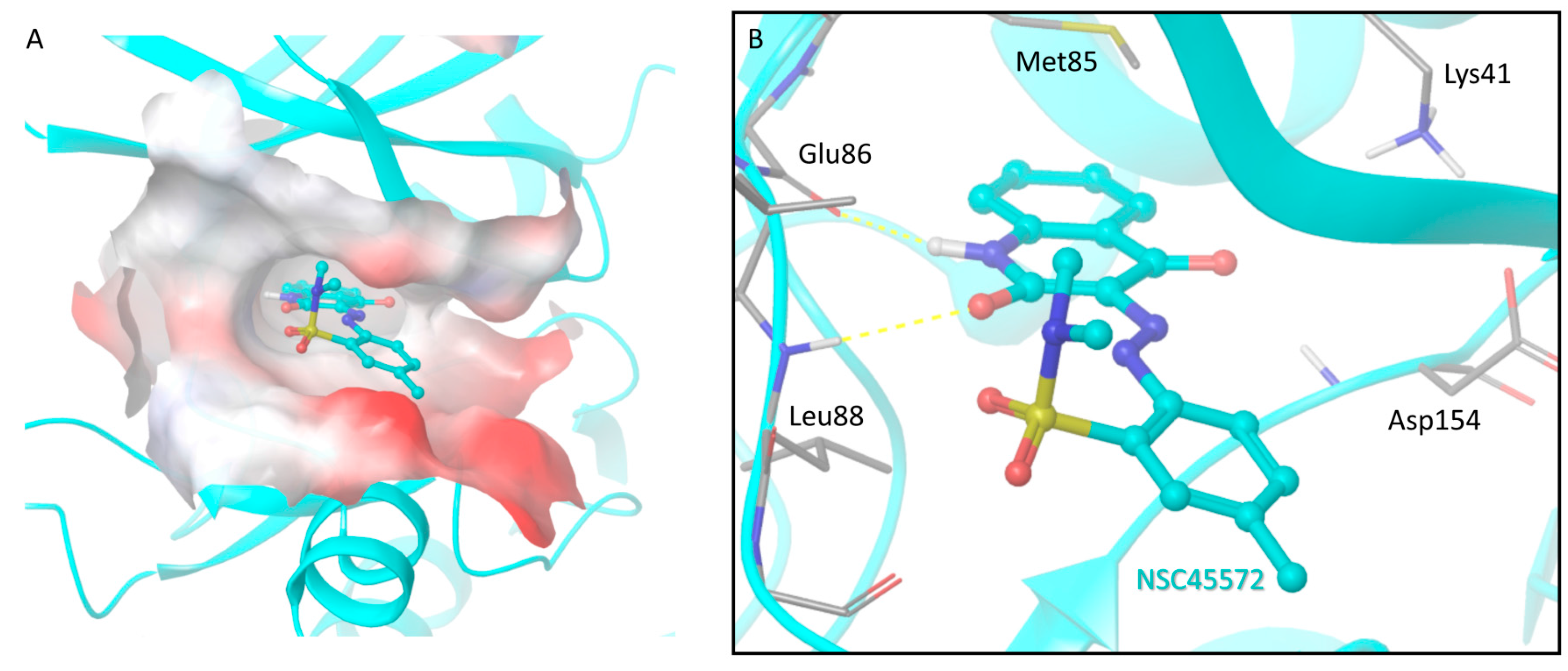
2.9. Docking of NSC45572 in the CK1 Active Site
3. Materials and Methods
3.1. Protein Kinase Assays
3.2. Cell Cultures
3.3. Cell Viability Assay
3.4. xMAP Assays
3.5. Preparation of the Compound Collection, Virtual Screening and Induced-Fit Docking
3.6. Enrichment Quantification
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Hevener, K.E.; White, S.W.; Lee, R.E.; Boyett, J.M. A statistical framework to evaluate virtual screening. BMC Bioinform. 2009, 10, 225. [Google Scholar] [CrossRef] [PubMed]
- Chaput, L.; Martinez-Sanz, J.; Saettel, N.; Mouawad, L. Benchmark of four popular virtual screening programs: Construction of the active/decoy dataset remains a major determinant of measured performance. J. Cheminform. 2016, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Kirchmair, J.; Markt, P.; Distinto, S.; Wolber, G.; Langer, T. Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection—What can we learn from earlier mistakes? J. Comput. Aided Mol. Des. 2008, 22, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Tang, Y.; Wells, J.A. Small-molecule inhibitors of protein–protein interactions: Progressing toward the reality. Chem. Biol. 2014, 21, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Laraia, L.; McKenzie, G.; Spring, D.R.; Venkitaraman, A.R.; Huggins, D.J. Overcoming chemical, biological, and computational challenges in the development of inhibitors targeting protein–protein interactions. Chem. Biol. 2015, 22, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M.G. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Petrakis, T.G.; Komseli, E.-S.; Papaioannou, M.; Vougas, K.; Polyzos, A.; Myrianthopoulos, V.; Mikros, E.; Trougakos, I.P.; Thanos, D.; Branzei, D.; et al. Exploring and exploiting the systemic effects of deregulated replication licensing. Semin. Cancer Biol. 2016, 37, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, H.-B.; Brinkman, K.L.; Han, S.-X.; Xu, B. Strategies for targeting the DNA damage response for cancer therapeutics. Chin. J. Cancer 2012, 31, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Knippschild, U.; Gocht, A.; Wolff, S.; Huber, N.; Löhler, J.; Stöter, M. The casein kinase 1 family: Participation in multiple cellular processes in eukaryotes. Cell. Signal. 2005, 17, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Knippschild, U.; Krüger, M.; Richter, J.; Xu, P.; García-Reyes, B.; Peifer, C.; Halekotte, J.; Bakulev, V.; Bischof, J. The CK1 family: Contribution to cellular stress response and its role in carcinogenesis. Front. Oncol. 2014, 4, 96. [Google Scholar] [CrossRef] [PubMed]
- Huart, A.-S.; MacLaine, N.J.; Meek, D.W.; Hupp, T.R. CK1α plays a central role in mediating MDM2 control of p53 and E2F-1 protein stability. J. Biol. Chem. 2009, 284, 32384–32394. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W. Regulation of the p53 response and its relationship to cancer. Biochem. J. 2015, 469, 325. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844. [Google Scholar] [CrossRef] [PubMed]
- Greer, Y.E.; Gao, B.; Yang, Y.; Nussenzweig, A.; Rubin, J.S. Lack of casein kinase 1 delta promotes genomic instability—The accumulation of DNA damage and down-regulation of checkpoint kinase 1. PLoS ONE 2017, 12, e0170903. [Google Scholar] [CrossRef] [PubMed]
- Peifer, C.; Abadleh, M.; Bischof, J.; Hauser, D.; Schattel, V.; Hirner, H.; Knippschild, U.; Laufer, S. 3,4-Diaryl-isoxazoles and -imidazoles as potent dual inhibitors of p38α mitogen activated protein kinase and casein kinase 1δ. J. Med. Chem. 2009, 52, 7618–7630. [Google Scholar] [CrossRef] [PubMed]
- Oumata, N.; Bettayeb, K.; Ferandin, Y.; Demange, L.; Lopez-Giral, A.; Goddard, M.-L.; Myrianthopoulos, V.; Mikros, E.; Flajolet, M.; Greengard, P.; et al. Roscovitine-derived, dual-specificity inhibitors of cyclin-dependent kinases and casein kinases 1. J. Med. Chem. 2008, 51, 5229–5242. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Bain, J.; Elliott, M.; Cohen, P. D4476, a cell-permeant inhibitor of CK1, suppresses the site-specific phosphorylation and nuclear exclusion of FOXO1a. EMBO Rep. 2004, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Laco, F.; Low, J.-L.; Seow, J.; Woo, T.L.; Zhong, Q.; Seayad, J.; Liu, Z.; Wei, H.; Reuveny, S.; Elliott, D.A.; et al. Cardiomyocyte differentiation of pluripotent stem cells with SB203580 analogues correlates with Wnt pathway CK1 inhibition independent of p38 MAPK signaling. J. Mol. Cell. Cardiol. 2015, 80, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Hirota, T.; Peters, E.C.; Garcia, M.; Gonzalez, R.; Cho, C.Y.; Wu, X.; Schultz, P.G.; Kay, S.A. A small molecule modulates circadian rhythms through phosphorylation of the period protein. Angew. Chem. Int. Ed. 2011, 50, 10608–10611. [Google Scholar] [CrossRef] [PubMed]
- Walton, K.M.; Fisher, K.; Rubitski, D.; Marconi, M.; Meng, Q.-J.; Sládek, M.; Adams, J.; Bass, M.; Chandrasekaran, R.; Butler, T.; et al. Selective inhibition of casein kinase 1ε minimally alters circadian clock period. J. Pharmacol. Exp. Ther. 2009, 330, 430. [Google Scholar] [CrossRef] [PubMed]
- Badura, L.; Swanson, T.; Adamowicz, W.; Adams, J.; Cianfrogna, J.; Fisher, K.; Holland, J.; Kleiman, R.; Nelson, F.; Reynolds, L.; et al. An inhibitor of casein kinase Iε induces phase delays in circadian rhythms under free-running and entrained conditions. J. Pharmacol. Exp. Ther. 2007, 322, 730. [Google Scholar] [CrossRef] [PubMed]
- Myrianthopoulos, V.; Gaboriaud-Kolar, N.; Tallant, C.; Hall, M.-L.; Grigoriou, S.; Brownlee, P.M.; Fedorov, O.; Rogers, C.; Heidenreich, D.; Wanior, M.; et al. Discovery and optimization of a selective ligand for the switch/sucrose nonfermenting-related bromodomains of polybromo protein-1 by the use of virtual screening and hydration analysis. J. Med. Chem. 2016, 59, 8787–8803. [Google Scholar] [CrossRef] [PubMed]
- Myrianthopoulos, V.; Cartron, P.F.; Liutkevičiūtė, Z.; Klimašauskas, S.; Matulis, D.; Bronner, C.; Martinet, N.; Mikros, E. Tandem virtual screening targeting the {SRA} domain of {UHRF1} identifies a novel chemical tool modulating {DNA} methylation. Eur. J. Med. Chem. 2016, 114, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.D.; Skillman, A.G.; Nicholls, A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Empereur-mot, C.; Guillemain, H.; Latouche, A.; Zagury, J.-F.; Viallon, V.; Montes, M. Predictiveness curves in virtual screening. J. Cheminform. 2015, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Feher, M. Consensus scoring for protein–ligand interactions. Drug Discov. Today 2006, 11, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Triballeau, N.; Acher, F.; Brabet, I.; Pin, J.-P.; Bertrand, H.-O. Virtual screening workflow development guided by the “receiver operating characteristic” curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J. Med. Chem. 2005, 48, 2534–2547. [Google Scholar] [CrossRef] [PubMed]
- Truchon, J.-F.; Bayly, C.I. Evaluating virtual screening methods: Good and bad metrics for the “early recognition” problem. J. Chem. Inf. Model. 2007, 47, 488–508. [Google Scholar] [CrossRef] [PubMed]
- Empereur-Mot, C.; Zagury, J.-F.; Montes, M. Screening explorer–An interactive tool for the analysis of screening results. J. Chem. Inf. Model. 2016, 56, 2281–2286. [Google Scholar] [CrossRef] [PubMed]
- Polychronopoulos, P.; Magiatis, P.; Skaltsounis, A.-L.; Myrianthopoulos, V.; Mikros, E.; Tarricone, A.; Musacchio, A.; Roe, S.M.; Pearl, L.; Leost, M.; et al. Structural basis for the synthesis of indirubins as potent and selective inhibitors of glycogen synthase kinase-3 and cyclin-dependent kinases. J. Med. Chem. 2004, 47, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Huse, M.; Kuriyan, J. The conformational plasticity of protein kinases. Cell 2002, 109, 275–282. [Google Scholar] [CrossRef]
- Chen, H.; Lyne, P.D.; Giordanetto, F.; Lovell, T.; Li, J. On evaluating molecular-docking methods for pose prediction and enrichment factors. J. Chem. Inf. Model. 2006, 46, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, K.; Saito, S.; Higashimoto, Y.; Roy, S.; Anderson, C.W.; Appella, E. Damage-mediated phosphorylation of human p53 threonine 18 through a cascade mediated by a casein 1-like kinase: Effect on MDM2 binding. J. Biol. Chem. 2000, 275, 9278–9283. [Google Scholar] [CrossRef] [PubMed]
- MacLaine, N.J.; Øster, B.; Bundgaard, B.; Fraser, J.A.; Buckner, C.; Lazo, P.A.; Meek, D.W.; Höllsberg, P.; Hupp, T.R. A central role for CK1 in catalyzing phosphorylation of the p53 transactivation domain at serine 20 after HHV-6B viral infection. J. Biol. Chem. 2008, 283, 28563–28573. [Google Scholar] [CrossRef] [PubMed]
- Inuzuka, H.; Tseng, A.; Gao, D.; Zhai, B.; Zhang, Q.; Shaik, S.; Wan, L.; Ang, X.L.; Mock, C.; Yin, H.; et al. Phosphorylation by casein kinase I promotes the turnover of the MDM2 oncoprotein via the SCFβ-TRCP ubiquitin ligase. Cancer Cell 2010, 18, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, L.G.; Saez-Rodriguez, J.; Cosgrove, B.D.; Lauffenburger, D.A.; Sorger, P.K. networks inferred from biochemical data reveal profound differences in toll-like receptor and inflammatory signaling between normal and transformed hepatocytes. Mol. Cell. Proteom. MCP 2010, 9, 1849–1865. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug Des. 2006, 67, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef] [PubMed]
- Bettayeb, K.; Oumata, N.; Echalier, A.; Ferandin, Y.; Endicott, J.A.; Galons, H.; Meijer, L. CR8, a potent and selective, roscovitine-derived inhibitor of cyclin-dependent kinases. Oncogene 2008, 27, 5797–5807. [Google Scholar] [CrossRef] [PubMed]
- Bach, S.; Knockaert, M.; Reinhardt, J.; Lozach, O.; Schmitt, S.; Baratte, B.; Koken, M.; Coburn, S.P.; Tang, L.; Jiang, T.; et al. Roscovitine targets, protein kinases and pyridoxal kinase. J. Biol. Chem. 2005, 280, 31208–31219. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, J.; Ferandin, Y.; Meijer, L. Purification of CK1 by affinity chromatography on immobilised axin. Protein Expr. Purif. 2007, 54, 101–109. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Code | Structure | Residual Kinase Activity | |||
|---|---|---|---|---|---|
| CK1δ/ε | GSK3α/β | CDK5/p25 | DYRK1α | ||
| NSC45572 (1) |  | 7% | 82% | 77% | 94% |
| NSC252777 (2) |  | 18% | 96% | 98% | 72% |
| NSC71866 (3) |  | 14% | 74% | 25% | 72% |
| NSC105827 (4) |  | 20% | 33% | 7% | 12% |
| Active Compound | Structure-Based Screening | Ligand-Based Screening | Combined Screening |
|---|---|---|---|
| NSC45572 | 82.52% | 45.82% | 78.41% |
| NSC252777 | 7.48% | 9.80% | 10.79% |
| NSC71866 | 1.76% | 1.21% | 40.01% |
| NSC105827 | 0.81% | 19.67% | 0.66% |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Myrianthopoulos, V.; Lozach, O.; Zareifi, D.; Alexopoulos, L.; Meijer, L.; Gorgoulis, V.G.; Mikros, E. Combined Virtual and Experimental Screening for CK1 Inhibitors Identifies a Modulator of p53 and Reveals Important Aspects of in Silico Screening Performance. Int. J. Mol. Sci. 2017, 18, 2102. https://doi.org/10.3390/ijms18102102
Myrianthopoulos V, Lozach O, Zareifi D, Alexopoulos L, Meijer L, Gorgoulis VG, Mikros E. Combined Virtual and Experimental Screening for CK1 Inhibitors Identifies a Modulator of p53 and Reveals Important Aspects of in Silico Screening Performance. International Journal of Molecular Sciences. 2017; 18(10):2102. https://doi.org/10.3390/ijms18102102
Chicago/Turabian StyleMyrianthopoulos, Vassilios, Olivier Lozach, Danae Zareifi, Leonidas Alexopoulos, Laurent Meijer, Vassilis G. Gorgoulis, and Emmanuel Mikros. 2017. "Combined Virtual and Experimental Screening for CK1 Inhibitors Identifies a Modulator of p53 and Reveals Important Aspects of in Silico Screening Performance" International Journal of Molecular Sciences 18, no. 10: 2102. https://doi.org/10.3390/ijms18102102
APA StyleMyrianthopoulos, V., Lozach, O., Zareifi, D., Alexopoulos, L., Meijer, L., Gorgoulis, V. G., & Mikros, E. (2017). Combined Virtual and Experimental Screening for CK1 Inhibitors Identifies a Modulator of p53 and Reveals Important Aspects of in Silico Screening Performance. International Journal of Molecular Sciences, 18(10), 2102. https://doi.org/10.3390/ijms18102102