
Focal Adhesion Kinase: Insight into Molecular Roles and Functions in Hepatocellular Carcinoma



Abstract
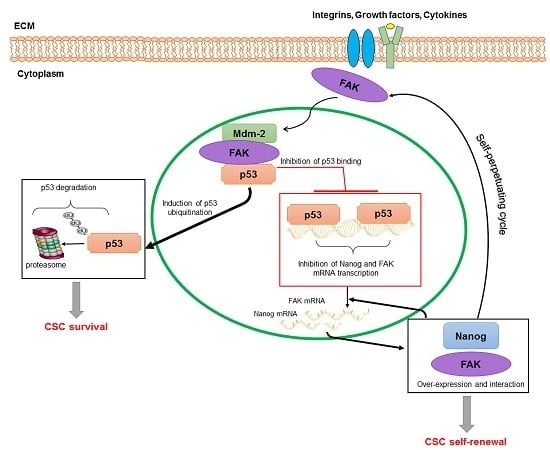
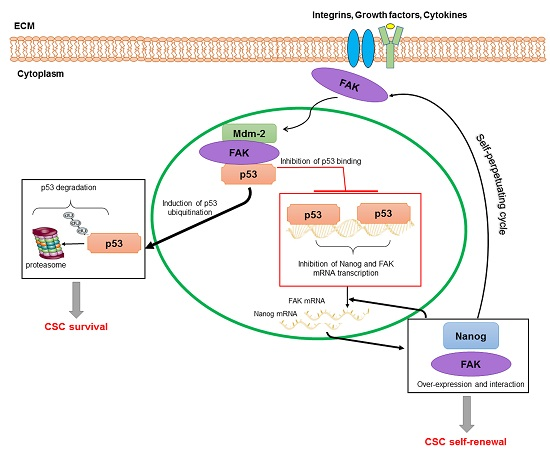
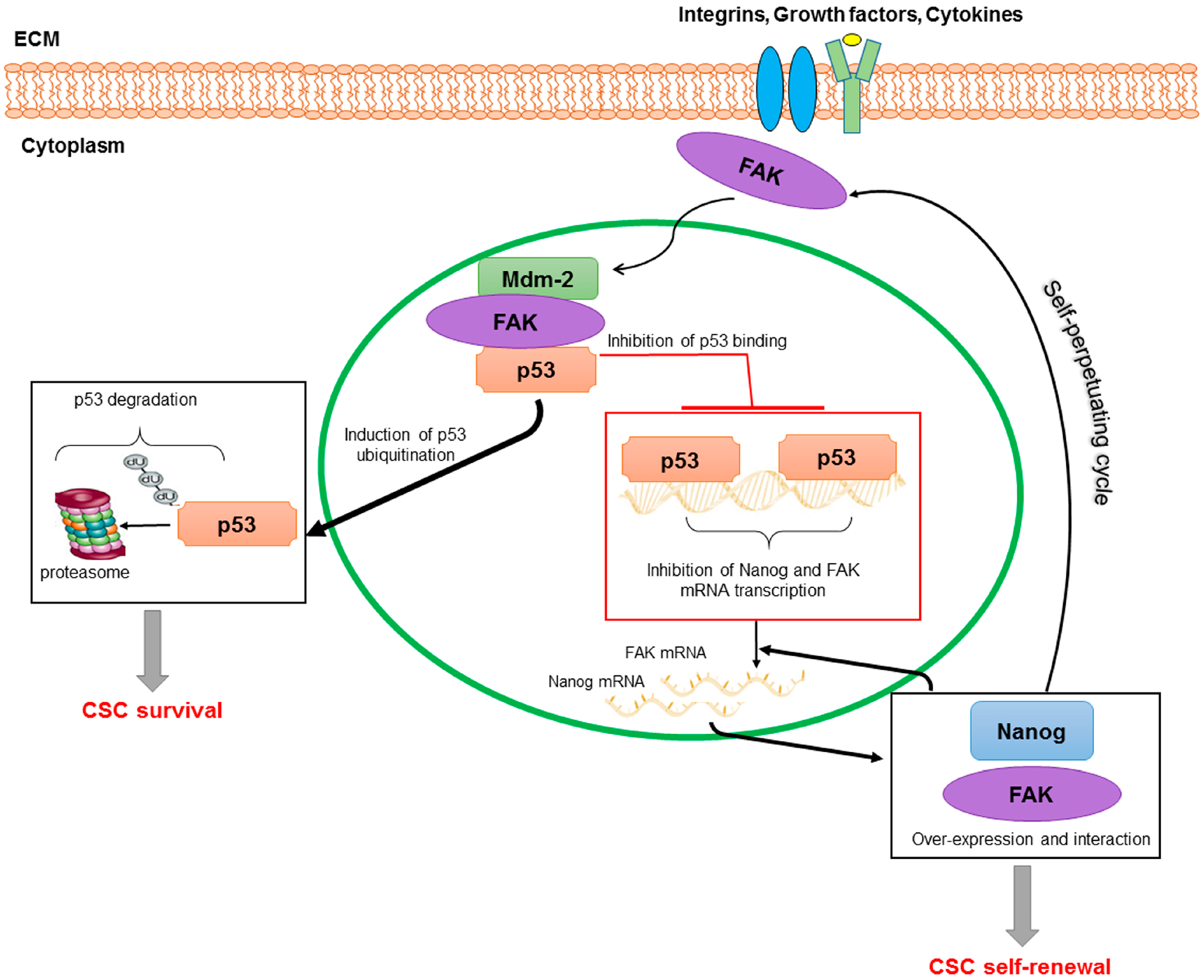
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
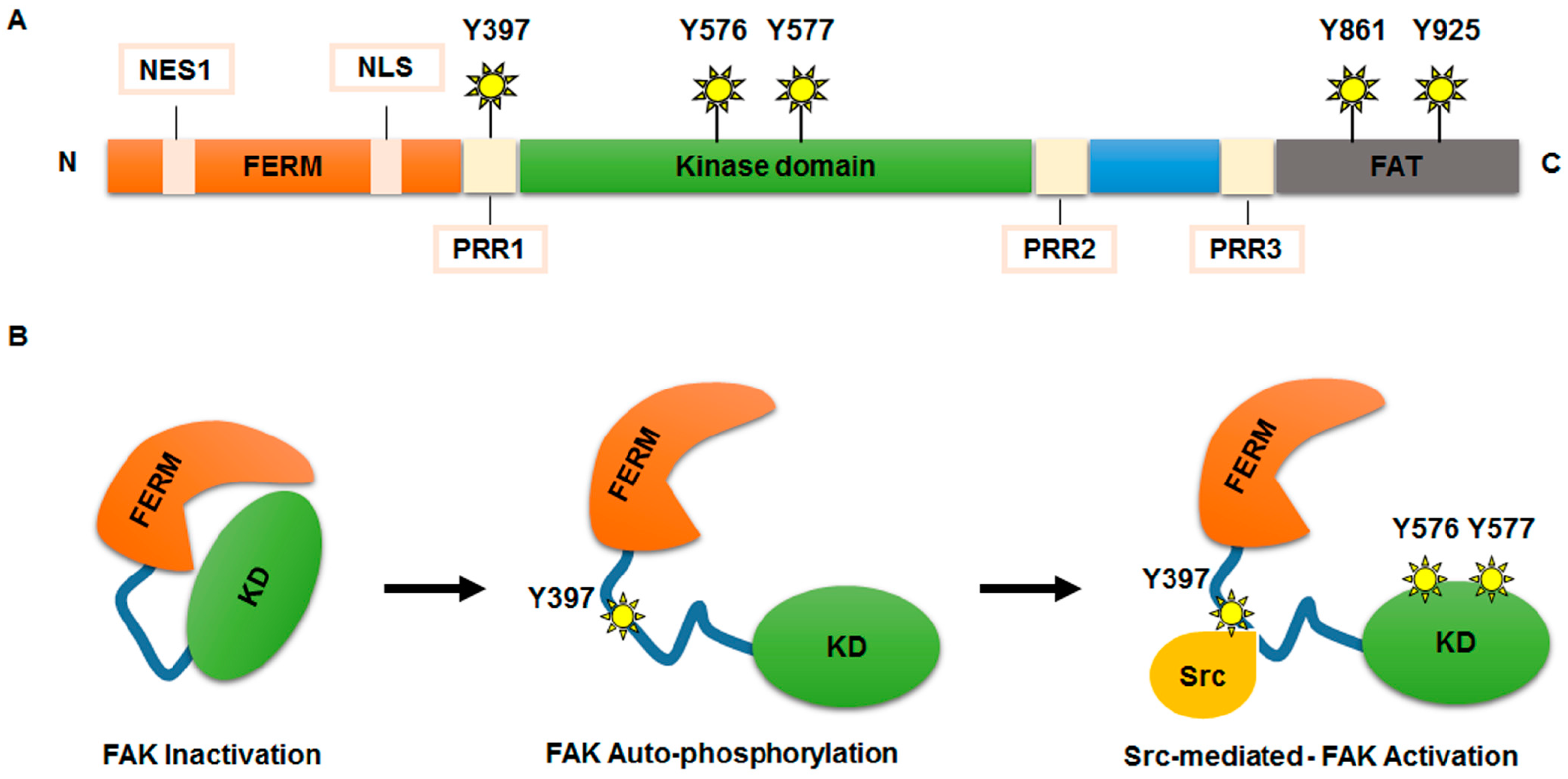
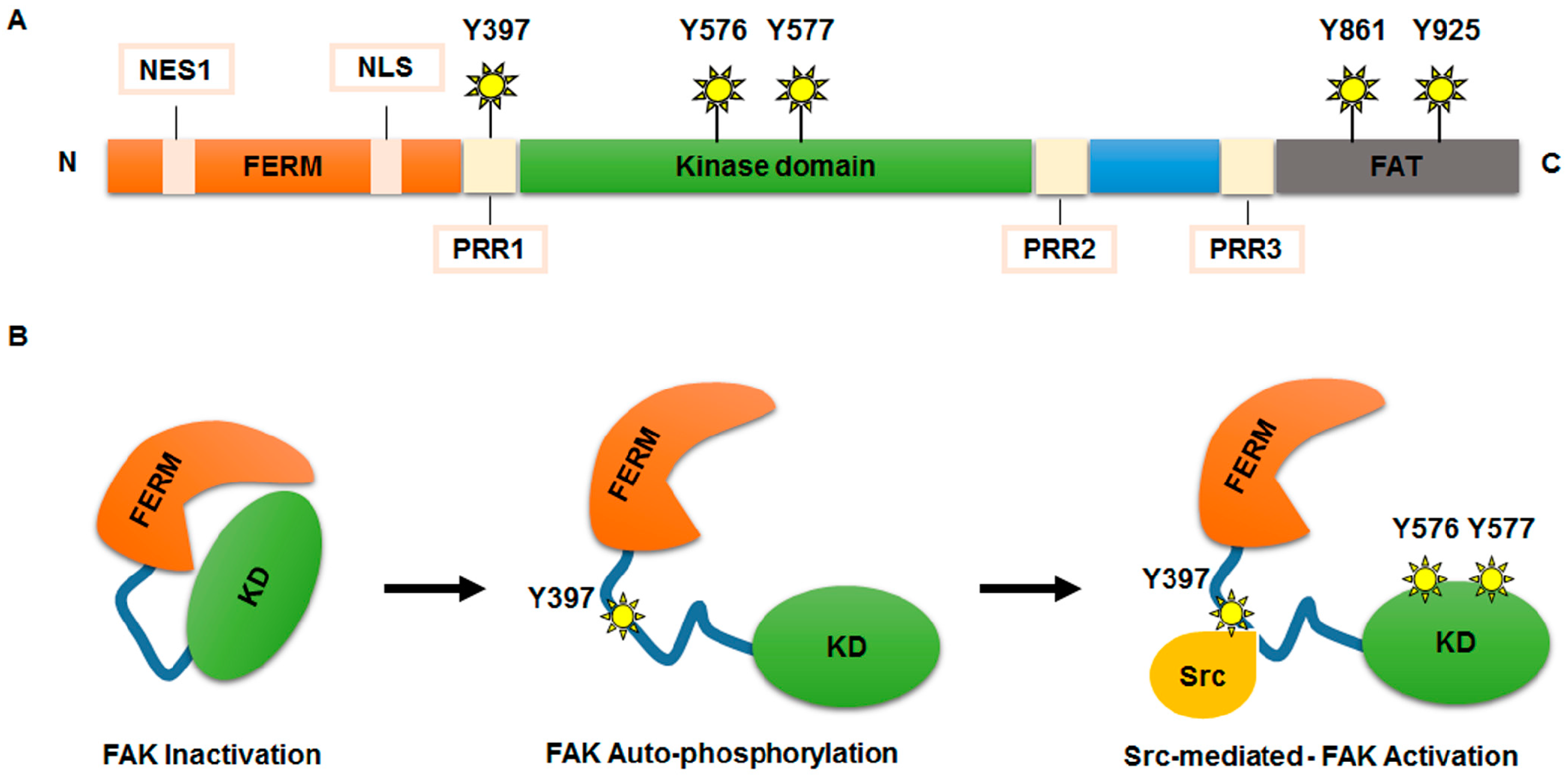
2. Structure and Functions of FAK
2.1. FAK Functional Domains
2.2. Functions of FAK in Cancer
3. Role of FAK in HCC
3.1. FAK in Human HCC Samples
3.2. FAK in HCC Cells
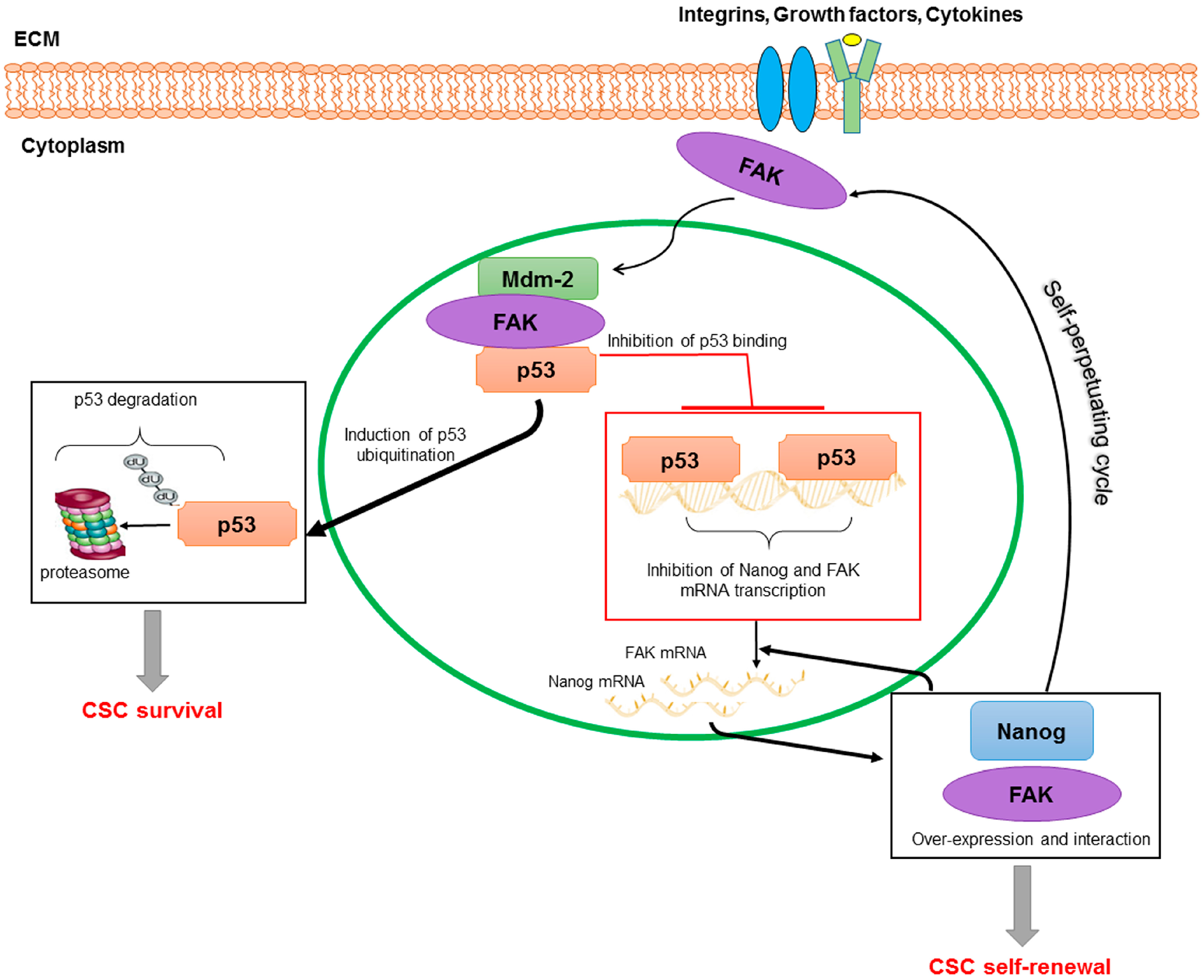
4. FAK in Cancer Stem Cells: A Look at HCC
4.1. Cancer Stem Cells in HCC Pathogenesis
4.2. FAK and Stemness
4.3. FAK in HCC CSCs
5. FAK Inhibitors in Clinical Applications
5.1. FAK Inhibitors
5.2. FAK Inhibitors in HCC Models
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Njei, B.; Rotman, Y.; Ditah, I.; Lim, J.K. Emerging trends in hepatocellular carcinoma incidence and mortality. Hepatology 2015, 1, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Schwartz, M.; Mazzaferro, V. Resection and liver transplantation for hepatocellular carcinoma. Semin. Liver Dis. 2005, 25, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Hoffmann, K.; Schemmer, P. Treatment of hepatocellular carcinoma: A systematic review. Liver Cancer 2012, 1, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Kishi, Y.; Hasegawa, K.; Sugawara, Y.; Kokudo, N. Hepatocellular carcinoma: Current management and future development-improved outcomes with surgical resection. Int. J. Hepatol. 2011, 2011, 728103–728113. [Google Scholar] [CrossRef] [PubMed]
- Waly, R.S.; Yangde, Z.; Yuxiang, C. Hepatocellular carcinoma: Focus on different aspects of management. ISRN Oncol. 2012, 2012, 421673–421685. [Google Scholar]
- Kocabayoglu, P.; Friedman, S.L. Cellular basis of hepatic fibrosis and its role in inflammation and cancer. Front. Biosci. 2013, 5, 217–230. [Google Scholar] [CrossRef]
- Gomaa, A.I.; Khan, S.A.; Toledano, M.B.; Waked, I.; Taylor-Robinson, S.D. Hepatocellular carcinoma: Epidemiology, risk factors and pathogenesis. World J. Gastroenterol. 2008, 14, 4300–4308. [Google Scholar] [CrossRef] [PubMed]
- Wörns, M.A.; Bosslet, T.; Victor, A.; Koch, S.; Hoppe-Lotichius, M.; Heise, M.; Hansen, T.; Pitton, M.B.; Niederle, I.M.; Schuchmann, M.; et al. Prognostic factors and outcomes of patients with hepatocellular carcinoma in non-cirrhotic liver. Scand. J. Gastroenterol. 2012, 47, 718–728. [Google Scholar]
- Oishi, N.; Yamashita, T.; Kaneko, S. Molecular biology of liver cancer stem cells. Liver Cancer 2014, 3, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Kanda, M.; Sugimoto, H.; Kodera, Y. Genetic and epigenetic aspects of initiation and progression of hepatocellular carcinoma. World J. Gastroenterol. 2015, 21, 10584–10597. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Jiang, L.; Guan, X.Y. The genetic and epigenetic alterations in human hepatocellular carcinoma: A recent update. Protein Cell 2014, 5, 673–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S. The mutational landscape of hepatocellular carcinoma. Clin. Mol. Hepatol. 2015, 21, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Mao, M. Next generation sequencing reveals genetic landscape of hepatocellular carcinomas. Cancer Lett. 2013, 340, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Kan, Z.; Zheng, H.; Liu, X.; Li, S.; Barber, T.D.; Gong, Z.; Gao, H.; Hao, K.; Willard, M.D.; Xu, J.; et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013, 23, 1422–1433. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Guan, J.L. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009, 28, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M. Targeting FAK in human cancer: From finding to first clinical trials. Front. Biosci. 2014, 19, 687–706. [Google Scholar] [CrossRef]
- Brami-Cherrier, K.; Gervasi, N.; Arsenieva, D.; Walkiewicz, K.; Boutterin, M.C.; Ortega, A.; Leonard, P.G.; Seantier, B.; Gasmi, L.; Bouceba, T.; et al. FAK dimerization controls its kinase-dependent functions at focal adhesions. EMBO J. 2014, 33, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Geiger, B.; Spatz, J.P.; Bershadsky, A.D. Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol. 2009, 10, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.W.; Roca-Cusachs, P.; Sheetz, M.P. Stretchy proteins on stretchy substrates: The important elements of integrin-mediated rigidity sensing. Dev. Cell 2010, 19, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.D. Cellular functions of FAK kinases: Insight into molecular mechanisms and novel functions. J. Cell Sci. 2010, 123, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, D.F.; Song, H.K.; Poy, F.; Schaller, M.D.; Eck, M.J. Crystal structure of the FERM domain of focal adhesion kinase. J. Biol. Chem. 2006, 281, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Frame, M.C.; Patel, H.; Serrels, B.; Lietha, D.; Eck, M.J. The FERM domain: Organizing the structure and function of FAK. Nat. Rev. Mol. Cell Biol. 2010, 11, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Brunton, V.G.; Avizienyte, E.; Fincham, V.J.; Serrels, B.; Metcalf, C.A.; Sawyer, T.K.; Frame, M.C. Identification of Src-specific phosphorylation site on focal adhesion kinase: Dissection of the role of Src SH2 and catalytic functions and their consequences for tumor cell behavior. Cancer Res. 2005, 15, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; Lu, S.; Szeto, K.W.; Sun, J.; Wang, Y.; Lasheras, J.C.; Chien, S. FAK and paxillin dynamics at focal adhesions in the protrusions of migrating cells. Sci. Rep. 2014, 4, 6024–6031. [Google Scholar] [CrossRef] [PubMed]
- Tomar, A.; Schlaepfer, D.D. Focal adhesion kinase: Switching between GAPs and GEFs in the regulation of cell motility. Curr. Opin. Cell Biol. 2009, 21, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.; Lim, S.T.; Uryu, S.; Chen, X.L.; Calderwood, D.A.; Schlaepfer, D.D. FAK promotes recruitment of talin to nascent adhesions to control cell motility. J. Cell Biol. 2012, 196, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Serrels, B.; Serrels, A.; Brunton, V.G.; Holt, M.; McLean, G.W.; Gray, C.H.; Jones, G.E.; Frame, M.C. Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat. Cell Biol. 2007, 9, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.T. Nuclear FAK: A new mode of gene regulation from cellular adhesions. Mol. Cells 2013, 36, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.T.; Chen, X.L.; Lim, Y.; Hanson, D.A.; Vo, T.T.; Howerton, K.; Larocque, N.; Fisher, S.J.; Schlaepfer, D.D.; Ilic, D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell 2008, 29, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Cance, W.G. FAK and p53 protein interactions. Anticancer Agents Med. Chem. 2011, 11, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.T.; Mikolon, D.; Stupack, D.G.; Schlaepfer, D.D. FERM control of FAK function: Implications for cancer therapy. Cell Cycle 2008, 7, 2306–2314. [Google Scholar] [CrossRef] [PubMed]
- Serrels, A.; Lund, T.; Serrels, B.; Byron, A.; McPherson, R.C.; von Kriegsheim, A.; Gómez-Cuadrado, L.; Canel, M.; Muir, M.; Ring, J.E.; et al. Nuclear FAK controls chemokine transcription, tregs, and evasion of anti-tumor immunity. Cell 2015, 163, 160–173. [Google Scholar] [CrossRef] [PubMed]
- McLean, G.W.; Carragher, N.O.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M.C. The role of focal-adhesion kinase in cancer—A new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.Y.; Timpson, P.; Horvath, L.G.; Daly, R.J. FAK signaling in human cancer as a target for therapeutics. Pharmacol. Ther. 2015, 146, 132–149. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.L.; Chen, L.C.; Shen, T.L. Emerging roles of focal adhesion kinase in cancer. BioMed Res. Int. 2015, 2015, 690690–690703. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, V.; Demers, M.J.; Thibodeau, S.; Laquerre, V.; Fujita, N.; Tsuruo, T.; Beaulieu, J.F.; Gauthier, R.; Vézina, A.; Villeneuve, L.; et al. FAK/Src signaling in human intestinal epithelial cell survival and anoikis: Differentiation state-specific uncoupling with the PI3-K/Akt-1 and MEK/Erk pathways. J. Cell. Physiol. 2007, 212, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, J.C.; Rogers, D.S.; Sharma, V.; Vittal, R.; White, E.S.; Cui, Z.; Thannickal, V.J. Combinatorial activation of FAK and AKT by transforming growth factor-β-1 confers an anoikis-resistant phenotype to myofibroblasts. Cell Signal. 2007, 19, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Pestell, R.; Guan, J.L. Transcriptional activation of cyclin D1 promoter by FAK contributes to cell cycle progression. Mol. Biol. Cell 2001, 12, 4066–4077. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Grammer, J.R.; Nelson, M.A.; Guan, J.L.; Stewart, J.E., Jr.; Gladson, C.L. p27Kip1 and cyclin D1 are necessary for focal adhesion kinase regulation of cell cycle progression in glioblastoma cells propagated in vitro and in vivo in the SCID mouse brain. J. Biol. Chem. 2005, 280, 6802–6815. [Google Scholar] [CrossRef] [PubMed]
- Avizienyte, E.; Frame, M.C. Src and FAK signalling controls adhesion fate and the epithelial-to-mesenchymal transition. Curr. Opin. Cell Biol. 2005, 17, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Kikkawa, F.; Nawa, A.; Thant, A.A.; Naruse, K.; Mizutani, S.; Hamaguchi, M. Both focal adhesion kinase and c-RAS are required for the enhanced matrix metalloproteinase 9 secretion by fibronectin in ovarian cancer cells. Cancer Res. 1998, 58, 900–903. [Google Scholar] [PubMed]
- Sein, T.T.; Thant, A.A.; Hiraiwa, Y.; Amin, A.R.; Sohara, Y.; Liu, Y.; Matsuda, S.; Yamamoto, T.; Hamaguchi, M. A role for FAK in the concanavalin A-dependent secretion of matrix metalloproteinase-2 and -9. Oncogene 2000, 19, 5539–5542. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Nam, J.O.; Jean, C.; Lawson, C.; Walsh, C.T.; Goka, E.; Lim, S.T.; Tomar, A.; Tancioni, I.; Uryu, S.; et al. VEGF-induced vascular permeability is mediated by FAK. Dev. Cell 2012, 22, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, Y.; Enomoto, N.; Nagayama, K.; Izumi, N.; Marumo, F.; Watanabe, M.; Sato, C. Analysis of differentially expressed genes in human hepatocellular carcinoma using suppression subtractive hybridization. Br. J. Cancer 2001, 85, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; Maeda, T.; Shimada, M.; Aishima, S.; Shirabe, K.; Tanaka, S.; Maehara, Y. Role of expression of focal adhesion kinase in progression of hepatocellular carcinoma. Clin. Cancer Res. 2004, 10, 2812–2817. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Koshikawa, K.; Nomoto, S.; Okochi, O.; Kaneko, T.; Inoue, S.; Yatabe, Y.; Takeda, S.; Nakao, A. Focal adhesion kinase is overexpressed in hepatocellular carcinoma and can be served as an independent prognostic factor. J. Hepatol. 2004, 4, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Huang, X.H.; Wang, Q.; Chen, X.L.; Fu, X.H.; Tan, H.X.; Zhang, L.J.; Li, W.; Bi, J. FAK is involved in invasion and metastasis of hepatocellular carcinoma. Clin. Exp. Metastasis 2010, 27, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Yasui, K.; Zhao, C.; Arii, S.; Inazawa, J. PTK2 and EIF3S3 genes may be amplification targets at 8q23–q24 and are associated with large hepatocellular carcinomas. Hepatology 2003, 38, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Mori, N.; Tamesa, T.; Okada, T.; Kawauchi, S.; Oga, A.; Furuya, T.; Tangokum, A.; Oka, M.; Sasaki, K. Analysis of DNA copy number aberrations in hepatitis C virus-associated hepatocellular carcinomas by conventional CGH and array CGH. Mod. Pathol. 2004, 17, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.M.; Jang, S.J.; Shim, J.H.; Kim, D.; Hong, S.M.; Sung, C.O.; Baek, D.; Haq, F.; Ansari, A.A.; Lee, S.Y.; et al. Genomic portrait of resectable hepatocellular carcinomas: Implications of RB1 and FGF19 aberrations for patient stratification. Hepatology 2014, 60, 1972–1982. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, 269–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillory, L.A.; Stewart, J.E.; Megison, M.L.; Nabers, H.C.; Mroczek-Musulman, E.; Beierle, E.A. FAK inhibition decreases hepatoblastoma survival both in vitro and in vivo. Transl. Oncol. 2013, 6, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Von Sengbusch, A.; Gassmann, P.; Fisch, K.M.; Enns, A.; Nicolson, G.L.; Haier, J. Focal adhesion kinase regulates metastatic adhesion of carcinoma cells within liver sinusoids. Am. J. Pathol. 2005, 166, 585–596. [Google Scholar] [CrossRef]
- Cheng, N.; Li, Y.; Han, Z.G. Argonaute2 promotes tumor metastasis by way of up-regulating focal adhesion kinase expression in hepatocellular carcinoma. Hepatology 2013, 57, 1906–1918. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.J.; Li, H.Y.; Qiu, K.J.; Li, D.C.; Zhou, J.H.; Hu, Y.H.; Zhang, F.M. Downregulation of Notch1 inhibits the invasion of human hepatocellular carcinoma HepG2 and MHCC97H cells through the regulation of PTEN and FAK. Int. J. Mol. Med. 2014, 34, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef] [PubMed]
- Chetram, M.A.; Hinton, C.V. PTEN regulation of ERK1/2 signaling in cancer. J. Recept. Signal. Transduct. Res. 2012, 32, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Gu, J.; Danen, E.H.; Takino, T.; Miyamoto, S.; Yamada, K.M. PTEN interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-kinase/Akt cell survival pathway. J. Biol. Chem. 1999, 274, 20693–20703. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Kim, M.J.; Park, S.Y.; Kim, J.S.; Lim, W.; Nam, J.S.; Yhong Sheen, Y. TIMP-1 mediates TGF-β-dependent crosstalk between hepatic stellate and cancer cells via FAK signaling. Sci. Rep. 2015, 5, 16492–16506. [Google Scholar] [CrossRef] [PubMed]
- Shang, N.; Arteaga, M.; Zaidi, A.; Stauffer, J.; Cotler, S.J.; Zeleznik-Le, N.J.; Zhang, J.; Qiu, W. FAK is required for c-Met/β-catenin-driven hepatocarcinogenesis. Hepatology 2015, 61, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Breslin, P.; Kuo, P.; Ding, X.; Cotler, S.; Nishimura, M.; Zhang, J.; Qiu, W.; Arteaga, M.; Zaidi, A.; Shang, N. FAK kinase activity is required for the progression of c-Met/β-catenin-driven HCC. Gene Expr. 2016, 17, 79–88. [Google Scholar]
- Chung, I.H.; Chen, C.Y.; Lin, Y.H.; Chi, H.C.; Huang, Y.H.; Tai, P.J.; Liao, C.J.; Tsai, C.Y.; Lin, S.L.; Wu, M.H.; et al. Thyroid hormone-mediated regulation of lipocalin 2 through the Met/FAK pathway in liver cancer. Oncotarget 2015, 6, 15050–15064. [Google Scholar] [CrossRef] [PubMed]
- Geng, D.; Zhao, W.; Feng, Y.; Liu, J. Overexpression of Rab5a promotes hepatocellular carcinoma cell proliferation and invasion via FAK signaling pathway. Tumour Biol. 2016, 37, 3341–3347. [Google Scholar] [CrossRef] [PubMed]
- Rovida, E.; di Maira, G.; Tusa, I.; Cannito, S.; Paternostro, C.; Navari, N.; Vivoli, E.; Deng, X.; Gray, N.S.; Esparís-Ogando, A.; et al. The mitogen-activated protein kinase ERK5 regulates the development and growth of hepatocellular carcinoma. Gut 2015, 64, 1454–1465. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.F.; Li, K.S.; Shen, Y.H.; Gao, P.T.; Dong, Z.R.; Cai, J.B.; Zhang, C.; Huang, X.Y.; Tian, M.X.; Hu, Z.Q.; et al. Galectin-1 induces hepatocellular carcinoma EMT and sorafenib resistance by activating FAK/PI3K/AKT signaling. Cell Death Dis. 2016, 7, e2201. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Columbano, A. MicroRNAs: New tools for diagnosis, prognosis, and therapy in hepatocellular carcinoma? Hepatology 2013, 57, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Borel, F.; Konstantinova, P.; Jansen, P.L. Diagnostic and therapeutic potential of miRNA signatures in patients with hepatocellular carcinoma. J. Hepatol. 2012, 56, 1371–1383. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Li, H.S.; Huang, J.Q.; Dong, S.H.; Huang, Z.J.; Yi, W.; Zhan, G.F.; Feng, J.T.; Sun, J.C.; Huang, X.H. MicroRNA-379-5p inhibits tumor invasion and metastasis by targeting FAK/AKT signaling in hepatocellular carcinoma. Cancer Lett. 2016, 375, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Mishra, L.; Banker, T.; Murray, J.; Byers, S.; Thenappan, A.; He, A.R.; Shetty, K.; Johnson, L.; Reddy, E.P. Liver stem cells and hepatocellular carcinoma. Hepatology 2009, 49, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Z.; Chen, D. Origin of hepatocellular carcinoma: Role of stem cells. J. Gastroenterol. Hepatol. 2006, 21, 1093–1098. [Google Scholar] [PubMed]
- Li, Y.; Laterra, J. Cancer stem cells: Distinct entities or dynamically regulated phenotypes? Cancer Res. 2012, 72, 576–580. [Google Scholar] [PubMed]
- Yamashita, T.; Wang, X.W. Cancer stem cells in the development of liver cancer. J. Clin. Investig. 2013, 123, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Sun, L.; Jiang, K.; Gao, D.M.; Kang, X.N.; Wang, C.; Zhang, S.; Huang, S.; Qin, X.; Li, Y.; et al. NANOG promotes liver cancer cell invasion by inducing epithelial-mesenchymal transition through NODAL/SMAD3 signaling pathway. Int. J. Biochem. Cell Biol. 2013, 45, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Zhang, B.H.; Zheng, S.S.; Gao, D.M.; Qiu, S.J.; Wu, W.Z.; Ren, Z.G. Coexpression of gene Oct4 and Nanog initiates stem cell characteristics in hepatocellular carcinoma and promotes epithelial-mesenchymal transition through activation of STAT3/Snail signaling. J. Hematol. Oncol. 2015, 11, 8–23. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Zheng, Q.; Dong, Y.; Xie, X.; Wang, Y.; Wu, S.; Zhang, L.; Wang, Y.; Xue, T.; Wang, Z.; et al. Matrix stiffness-mediated effects on stemness characteristics occurring in HCC cells. Oncotarget 2016, 7, 32221–32231. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Baffet, G. Self-renewal of tumor-initiating cells: What’s new about hepatocellular carcinoma? Gastroenterology 2012, 142, 1414–1416. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Fan, H.; Nagy, T.; Wei, H.; Wang, C.; Liu, S.; Wicha, M.S.; Guan, J.L. Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res. 2009, 69, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.E.; Bundred, N.J.; Landberg, G.; Clarke, R.B.; Farnie, G. Focal adhesion kinase and Wnt signaling regulate human ductal carcinoma in situ stem cell activity and response to radiotherapy. Stem Cells 2015, 33, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, I.M.; Kolev, V.N.; Vidal, C.M.; Kadariya, Y.; Ring, J.E.; Wright, Q.; Weaver, D.T.; Menges, C.; Padval, M.; McClatchey, A.I.; et al. Merlin deficiency predicts FAK inhibitor sensitivity: A synthetic lethal relationship. Sci. Transl. Med. 2014, 6, 237–268. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, R.A.; Serrels, B.; Mason, S.; Kinnaird, A.; Muir, M.; Patel, H.; Muller, W.J.; Sansom, O.J.; Brunton, V.G. Focal adhesion kinase is required for β-catenin-induced mobilization of epidermal stem cells. Carcinogenesis 2012, 33, 2369–2376. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.; Olson, G.; Figel, S.; Gelman, I.; Cance, W.G.; Golubovskaya, V.M. Nanog increases focal adhesion kinase (FAK) promoter activity and expression and directly binds to FAK protein to be phosphorylated. J. Biol. Chem. 2012, 25, 18656–18673. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Kaur, A.; Cance, W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: Nuclear factor κB and p53 binding sites. Biochim. Biophys. Acta 2004, 25, 111–112. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Chao, C.; Saito, S.; Mazur, S.J.; Murphy, M.E.; Appella, E.; Xu, Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat. Cell Biol. 2005, 7, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M. FAK and Nanog cross talk with p53 in cancer stem cells. Anticancer Agents Med. Chem. 2013, 13, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Finch, R.; Cance, W.G. Direct interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53. J. Biol. Chem. 2005, 280, 25008–25021. [Google Scholar] [CrossRef] [PubMed]
- Cance, W.G.; Golubovskaya, V.M. Focal adhesion kinase versus p53: Apoptosis or survival? Sci. Signal. 2008, 1, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Tong, X.; Zhang, S.; Yin, F.; Li, X.; Wei, H.; Li, C.; Guo, Y.; Zhao, J. ASPP2 suppresses stem cell-like characteristics and chemoresistance by inhibiting the Src/FAK/Snail axis in hepatocellular carcinoma. Tumour Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Dehart, J.P.; Murphy, J.M.; Lim, S.T. Understanding the roles of FAK in cancer: Inhibitors, genetic models, and new insights. J. Histochem. Cytochem. 2015, 63, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Schultze, A.; Fiedler, W. Therapeutic potential and limitations of new FAK inhibitors in the treatment of cancer. Expert Opin. Investig. Drugs 2010, 19, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Hjelmeland, A.B.; Keir, S.T.; Song, L.; Wickman, S.; Jackson, D.; Ohmori, O.; Bigner, D.D.; Friedman, H.S.; Rich, J.N. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Mol. Carcinog. 2007, 46, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Roberts, W.G.; Ung, E.; Whalen, P.; Cooper, B.; Hulford, C.; Autry, C.; Richter, D.; Emerson, E.; Lin, J.; Kath, J.; et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008, 68, 1935–1944. [Google Scholar] [CrossRef] [PubMed]
- Tanjoni, I.; Walsh, C.; Uryu, S.; Tomar, A.; Nam, J.O.; Mielgo, A.; Lim, S.T.; Liang, C.; Koenig, M.; Sun, C.; et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol. Ther. 2010, 9, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Figel, S.; Ho, B.T.; Johnson, C.P.; Yemma, M.; Huang, G.; Zheng, M.; Nyberg, C.; Magis, A.; Ostrov, D.A.; et al. A small molecule focal adhesion kinase (FAK) inhibitor, targeting Y397 site: 1-(2-hydroxyethyl)-3, 5, 7-triaza-1-azoniatricyclo (3.3.1.1(3,7))decane; bromide effectively inhibits FAK autophosphorylation activity and decreases cancer cell viability, clonogenicity and tumor growth in vivo. Carcinogenesis 2012, 33, 1004–1013. [Google Scholar] [PubMed]
- Cance, W.G.; Kurenova, E.; Marlowe, T.; Golubovskaya, V. Disrupting the scaffold to improve focal adhesion kinase-targeted cancer therapeutics. Sci. Signal. 2013. [Google Scholar] [CrossRef] [PubMed]
- Balogh, J.; Victor, D.; Asham, E.H.; Burroughs, S.G.; Boktour, M.; Saharia, A.; Li, X.; Ghobrial, R.M.; Monsour, H.P., Jr. Hepatocellular carcinoma: A review. J. Hepatocell Carcinoma 2016, 3, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Von Felden, J.; Schulze, K.; Gil-Ibanez, I.; Werner, T.; Wege, H. First- and second-line targeted systemic therapy in hepatocellular carcinoma—An update on patient selection and response evaluation. Diagnostics (Basel) 2016. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia–Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Zhai, B.; Sun, X.Y. Mechanisms of resistance to sorafenib and the corresponding strategies in hepatocellular carcinoma. World J. Hepatol. 2013, 5, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Azzariti, A.; Mancarella, S.; Porcelli, L.; Quatrale, E.A.; Caligiuri, A.; Lupo, L.; Dituri, F.; Giannelli, G. Hepatic stellate cells induce HCC cell resistance to sorafenib through the laminin-332/α3 integrin axis recovery of FAK ubiquitination. Hepatology 2016. [Google Scholar] [CrossRef] [PubMed]
- Bagi, C.M.; Christensen, J.; Cohen, D.P.; Roberts, W.G.; Wilkie, D.; Swanson, T.; Tuthill, T.; Andresen, C.J. Sunitinib and PF-562,271 (FAK/Pyk2 inhibitor) effectively block growth and recovery of human hepatocellular carcinoma in a rat xenograft model. Cancer Biol. Ther. 2009, 8, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Li, W.X.; Chen, L.P.; Sun, M.Y.; Li, J.T.; Liu, H.Z.; Zhu, W. 3’3-Diindolylmethane inhibits migration, invasion and metastasis of hepatocellular carcinoma by suppressing FAK signaling. Oncotarget 2015, 6, 23776–23792. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Chen, Y.; Chen, Z.B.; Yan, F.J.; Dai, X.Y.; Ying, M.D.; Cao, J.; Ma, J.; Luo, P.; Han, Y.X.; et al. CT-707, a novel FAK inhibitor, synergizes with Cabozantinib to suppress hepatocellular carcinoma by blocking Cabozantinib-induced FAK activation. Mol. Cancer Ther. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.J.; Neoh, C.A.; Tsao, C.Y.; Su, J.H.; Li, H.H. Sinulariolide suppresses human hepatocellular carcinoma cell migration and invasion by inhibiting matrix metalloproteinase-2/-9 through MAPKs and PI3K/Akt signaling pathways. Int. J. Mol. Sci. 2015, 16, 16469–16482. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.Y.; Wang, Y.R.; Lin, H.Y.; Lu, S.C.; Lin, J.Y. Corosolic acid inhibits hepatocellular carcinoma cell migration by targeting the VEGFR2/Src/FAK pathway. PLoS ONE 2015, 10, e0126725. [Google Scholar] [CrossRef] [PubMed]
- Broekman, F.; Giovannetti, E.; Peters, G.J. Tyrosine kinase inhibitors: Multi-targeted or single-targeted? World J. Clin. Oncol. 2011, 2, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panera, N.; Crudele, A.; Romito, I.; Gnani, D.; Alisi, A. Focal Adhesion Kinase: Insight into Molecular Roles and Functions in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2017, 18, 99. https://doi.org/10.3390/ijms18010099
Panera N, Crudele A, Romito I, Gnani D, Alisi A. Focal Adhesion Kinase: Insight into Molecular Roles and Functions in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2017; 18(1):99. https://doi.org/10.3390/ijms18010099
Chicago/Turabian StylePanera, Nadia, Annalisa Crudele, Ilaria Romito, Daniela Gnani, and Anna Alisi. 2017. "Focal Adhesion Kinase: Insight into Molecular Roles and Functions in Hepatocellular Carcinoma" International Journal of Molecular Sciences 18, no. 1: 99. https://doi.org/10.3390/ijms18010099
APA StylePanera, N., Crudele, A., Romito, I., Gnani, D., & Alisi, A. (2017). Focal Adhesion Kinase: Insight into Molecular Roles and Functions in Hepatocellular Carcinoma. International Journal of Molecular Sciences, 18(1), 99. https://doi.org/10.3390/ijms18010099