
Molecular Biomarkers for Prediction of Targeted Therapy Response in Metastatic Breast Cancer: Trick or Treat?

 , ,
, ,
Abstract
:
1. Introduction
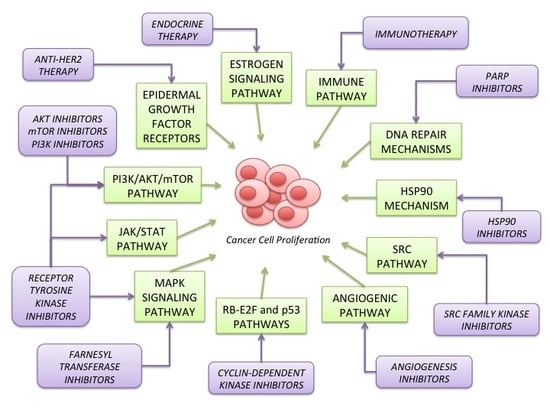
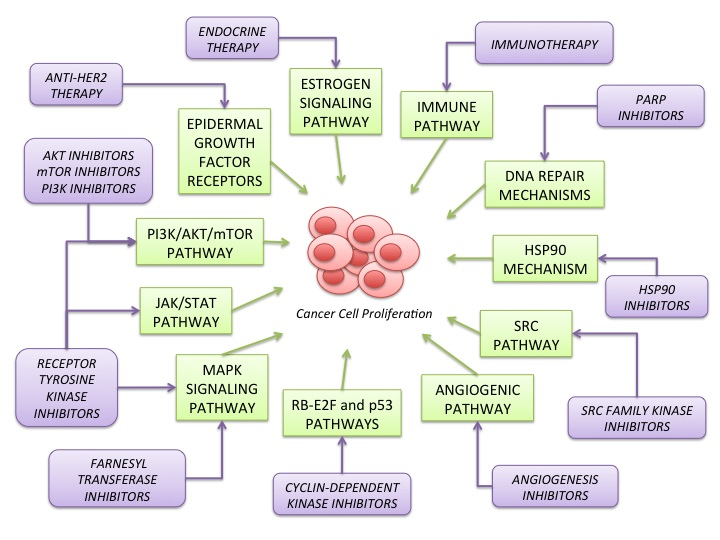
2. Signaling Pathways Involved in Breast Cancer Carcinogenesis
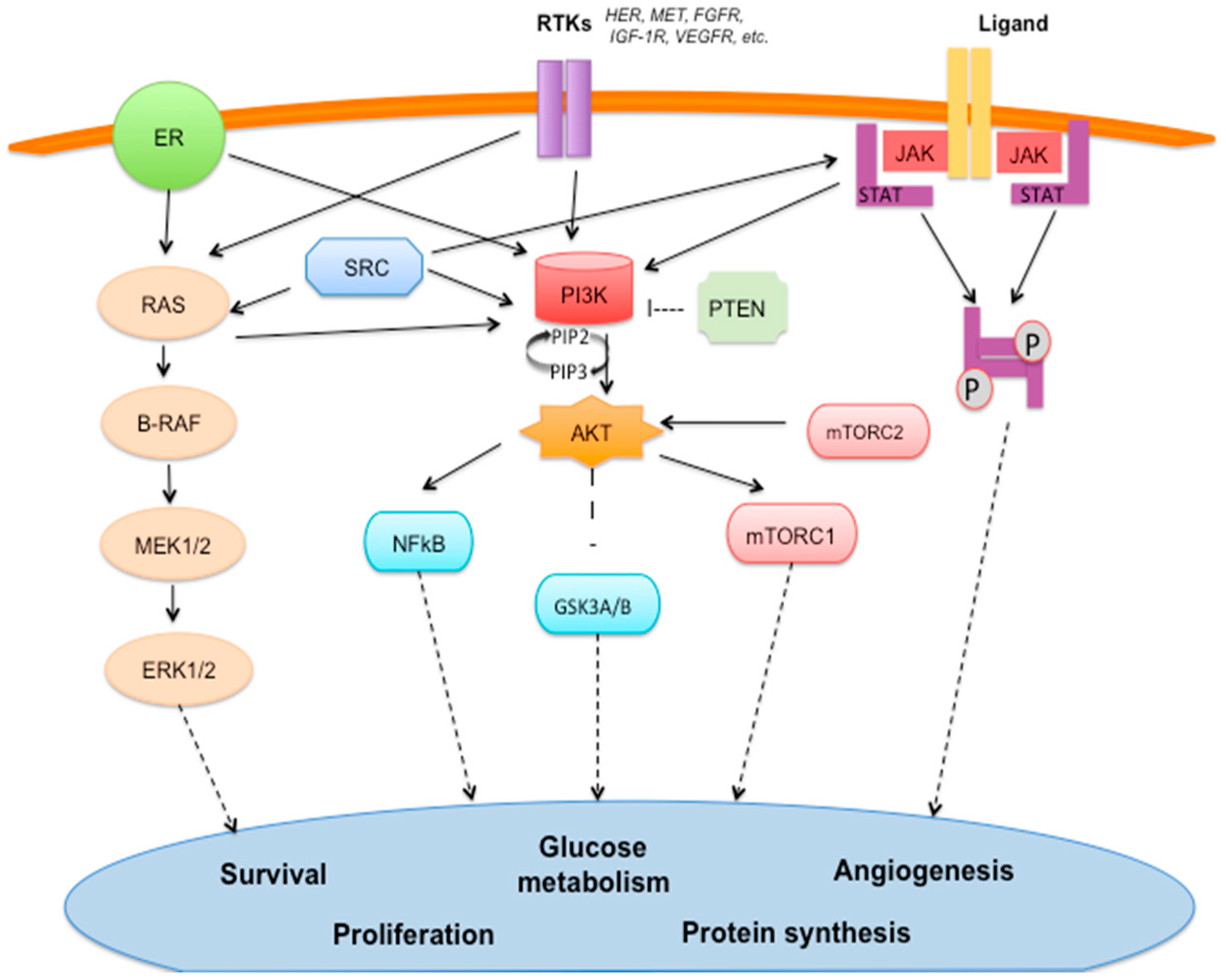
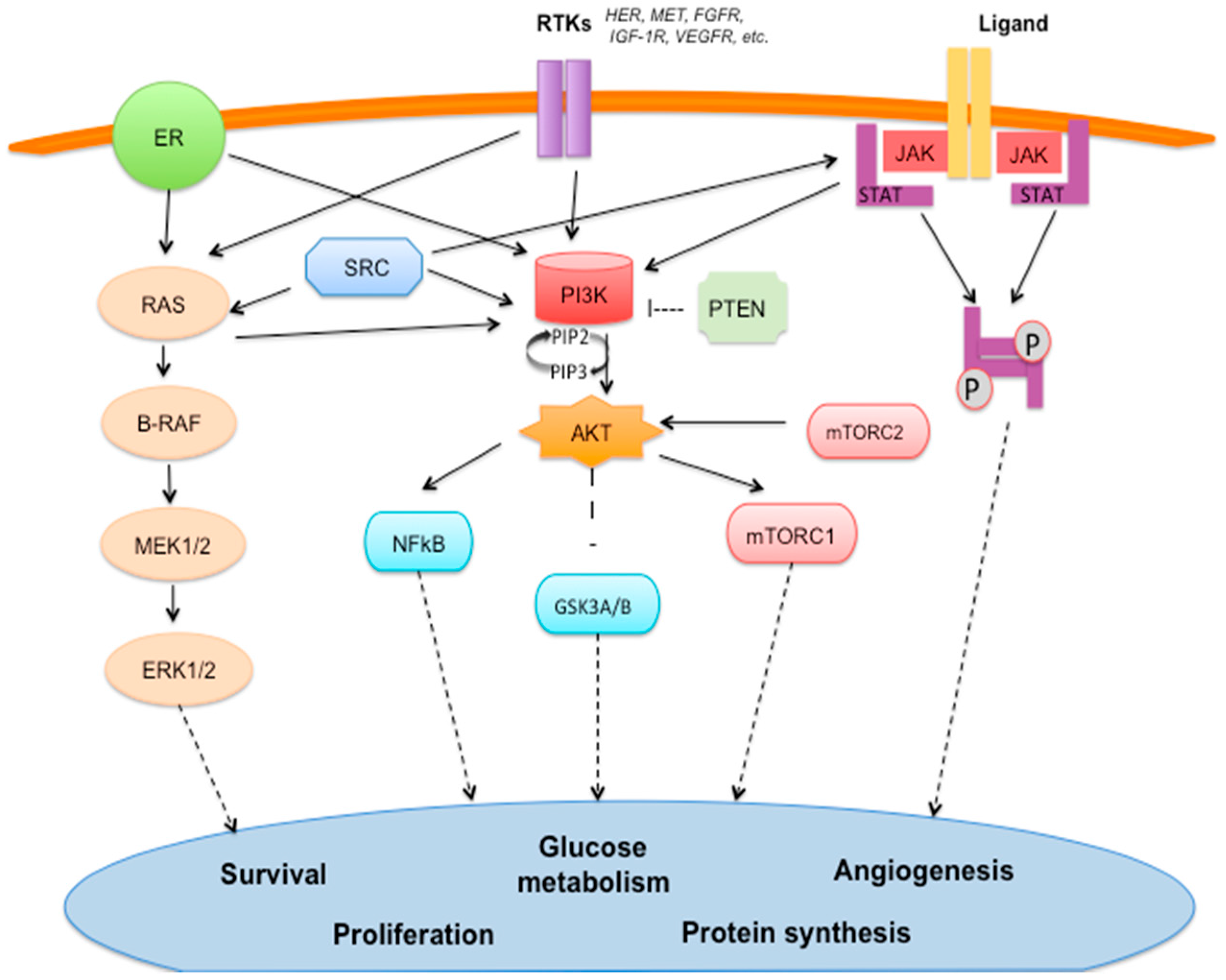
2.1. Estrogen Signaling Pathway (Figure 1)
2.2. Epidermal Growth Factor Receptors (HER) Family (Figure 1)
2.3. PI3K/AKT/mTOR Pathway (Figure 1)
2.4. MAPK Signaling Pathway (Figure 1)
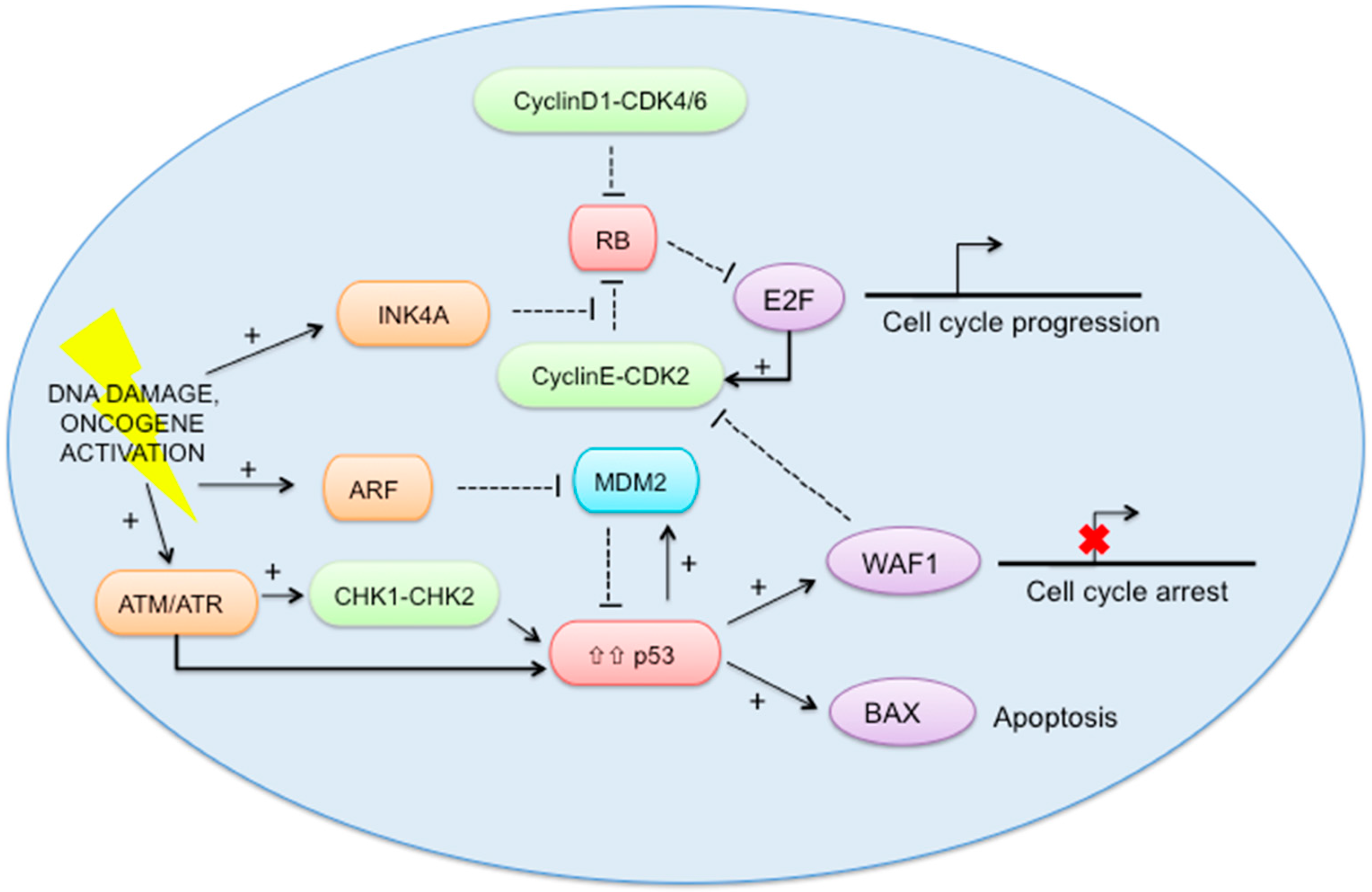
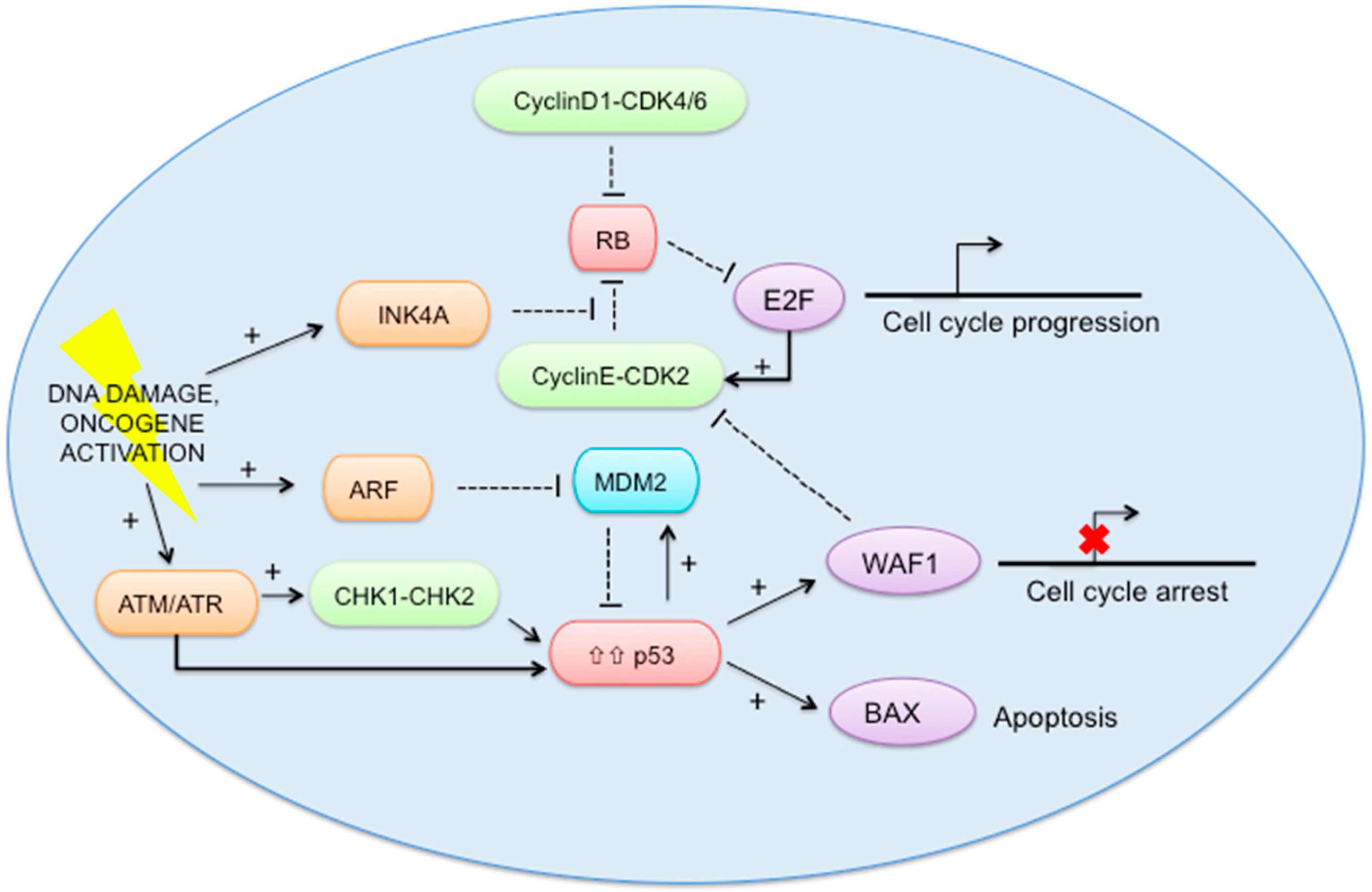
2.5. RB-E2F and p53 Pathways (Figure 2)
2.6. Angiogenic Pathway (Figure 1)
2.7. SRC Pathway (Figure 1)
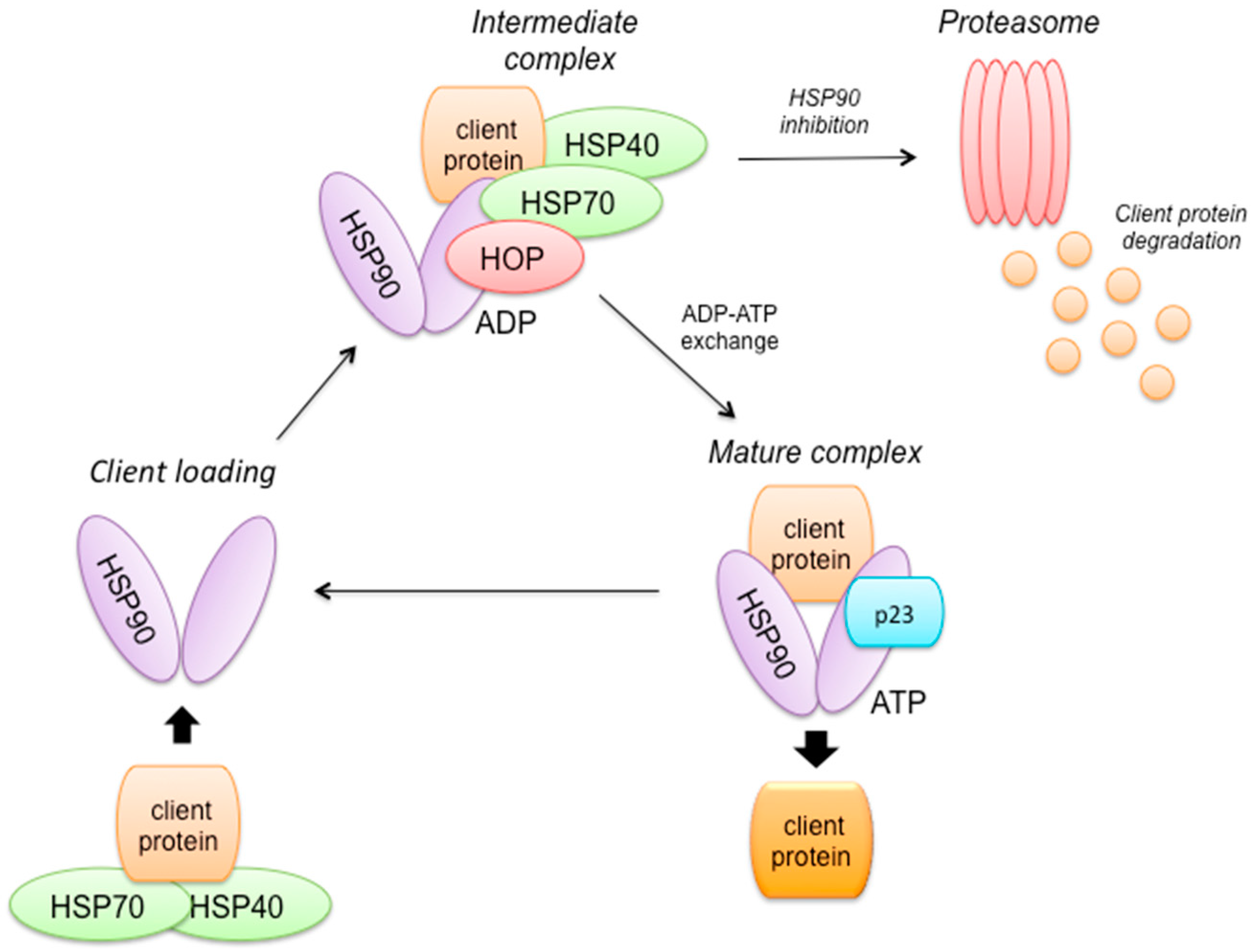
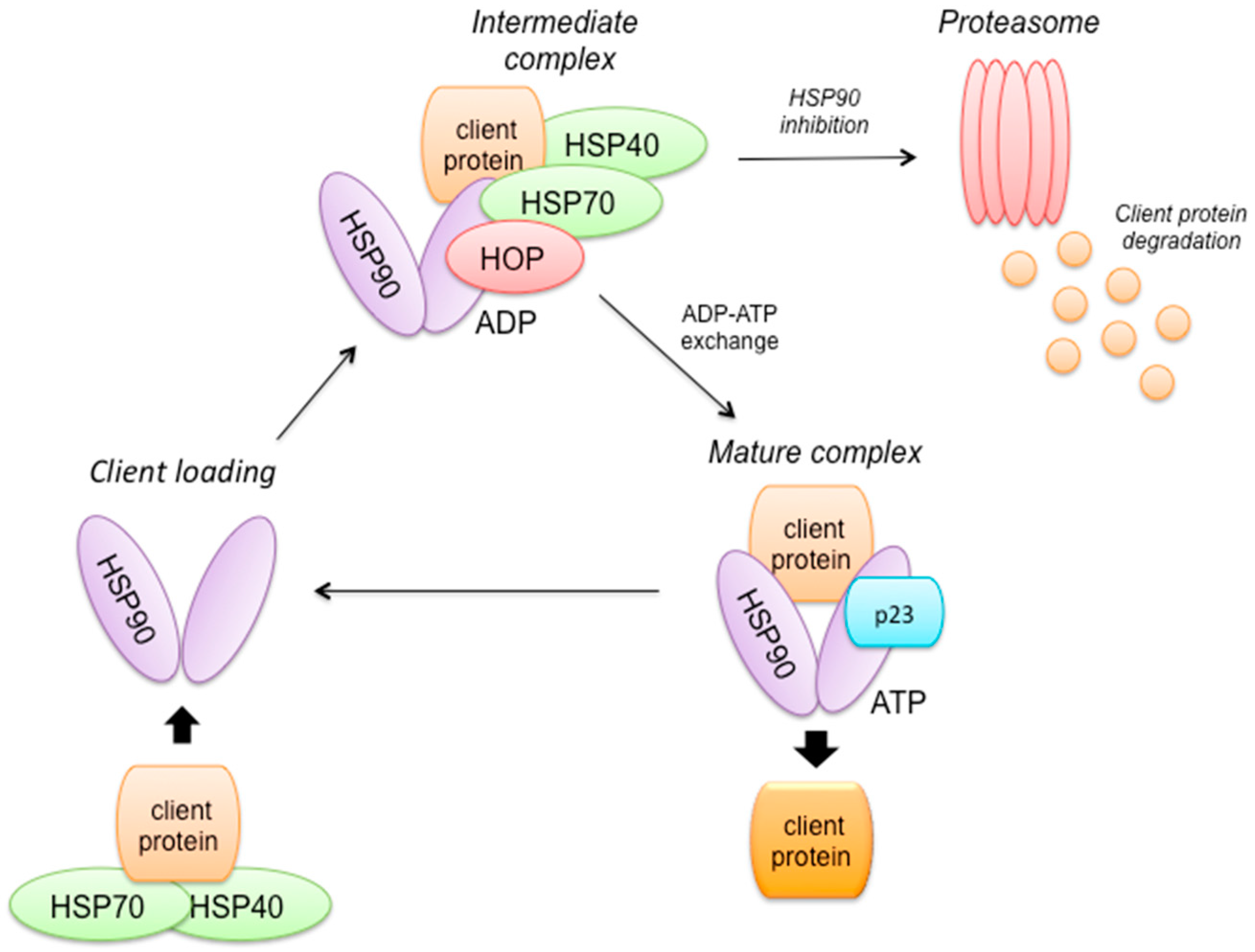
2.8. HSP90 Mechanism of Action (Figure 3)
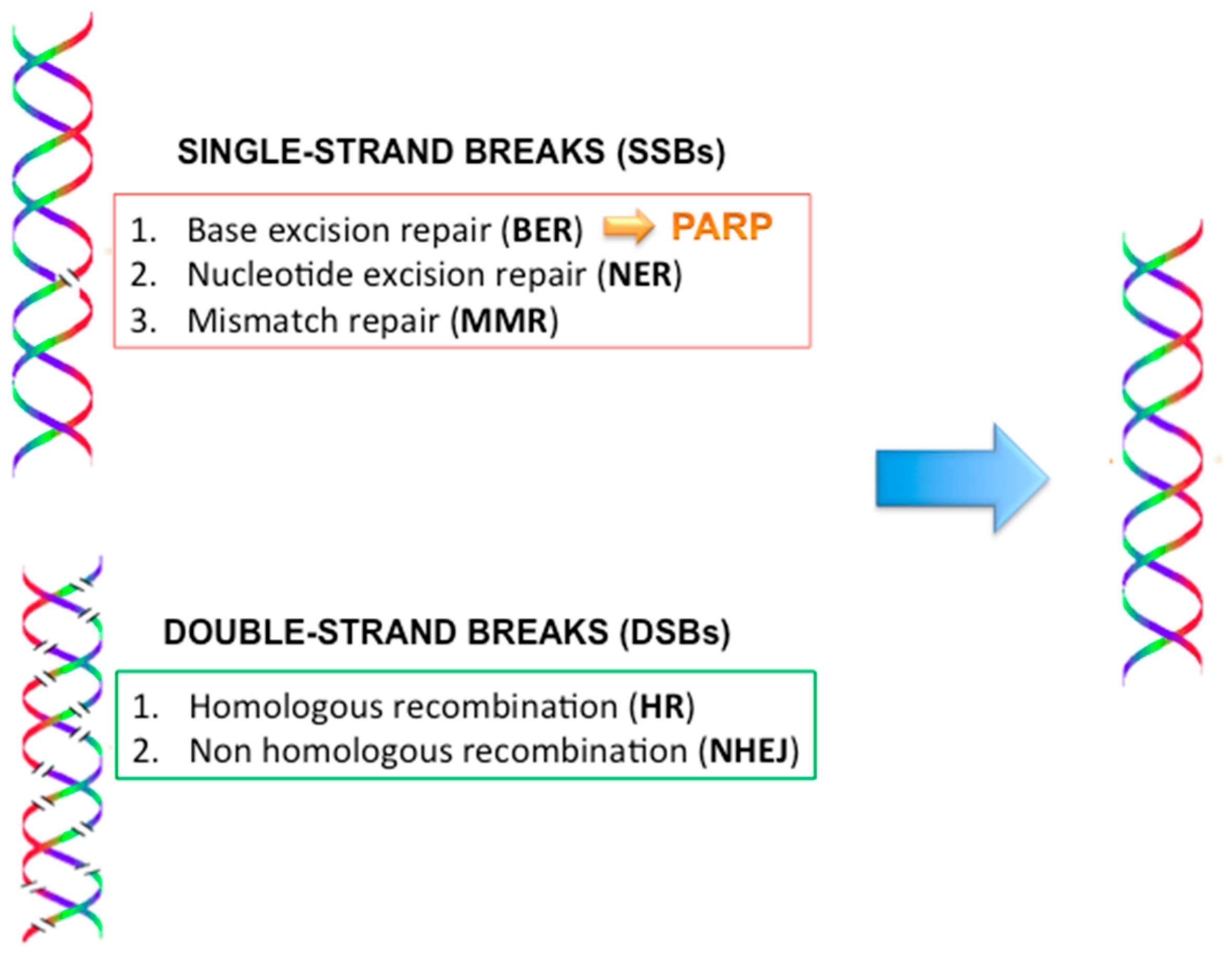
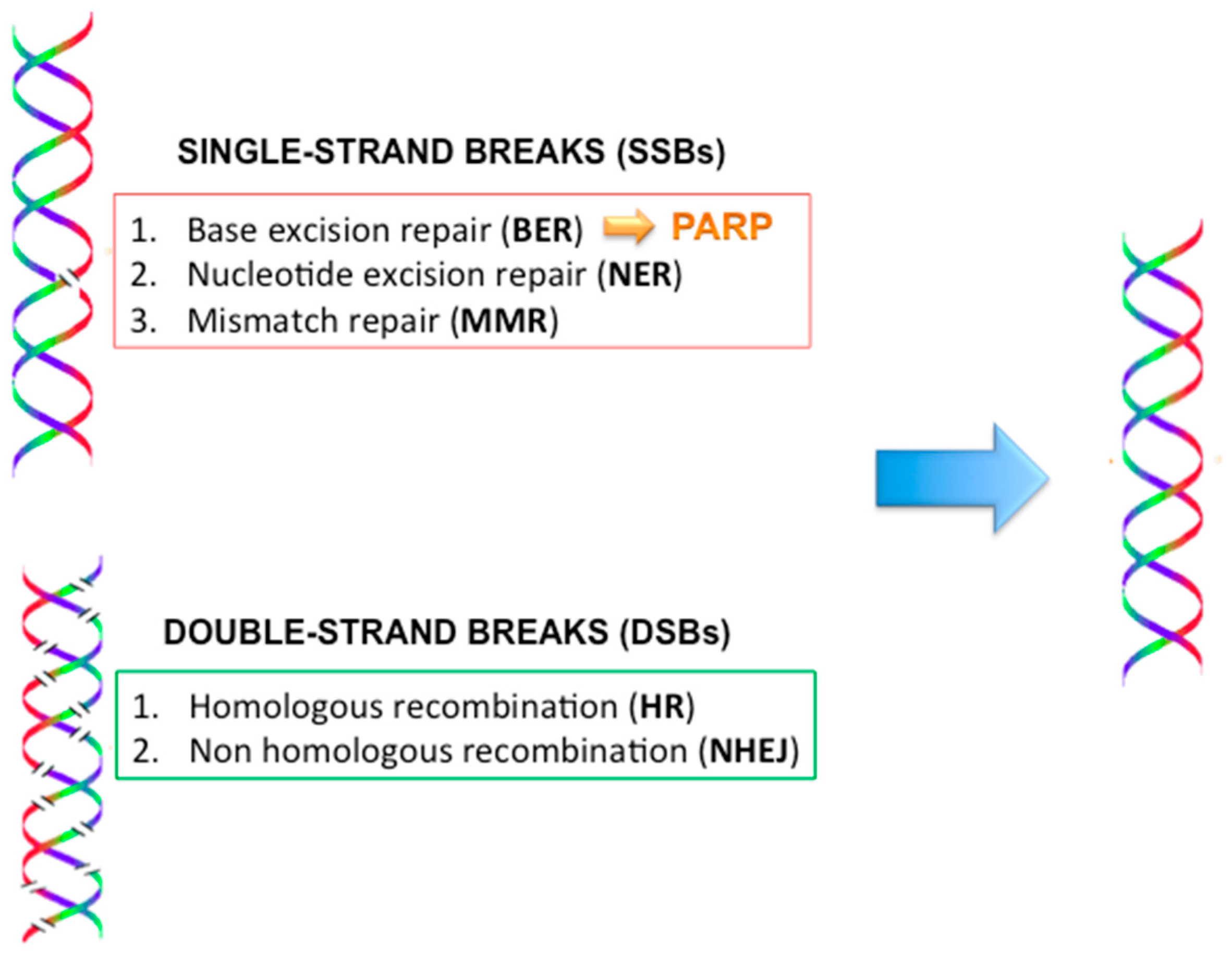
2.9. DNA Repair Mechanisms (Figure 4)
2.10. JAK/STAT Pathway (Figure 1)
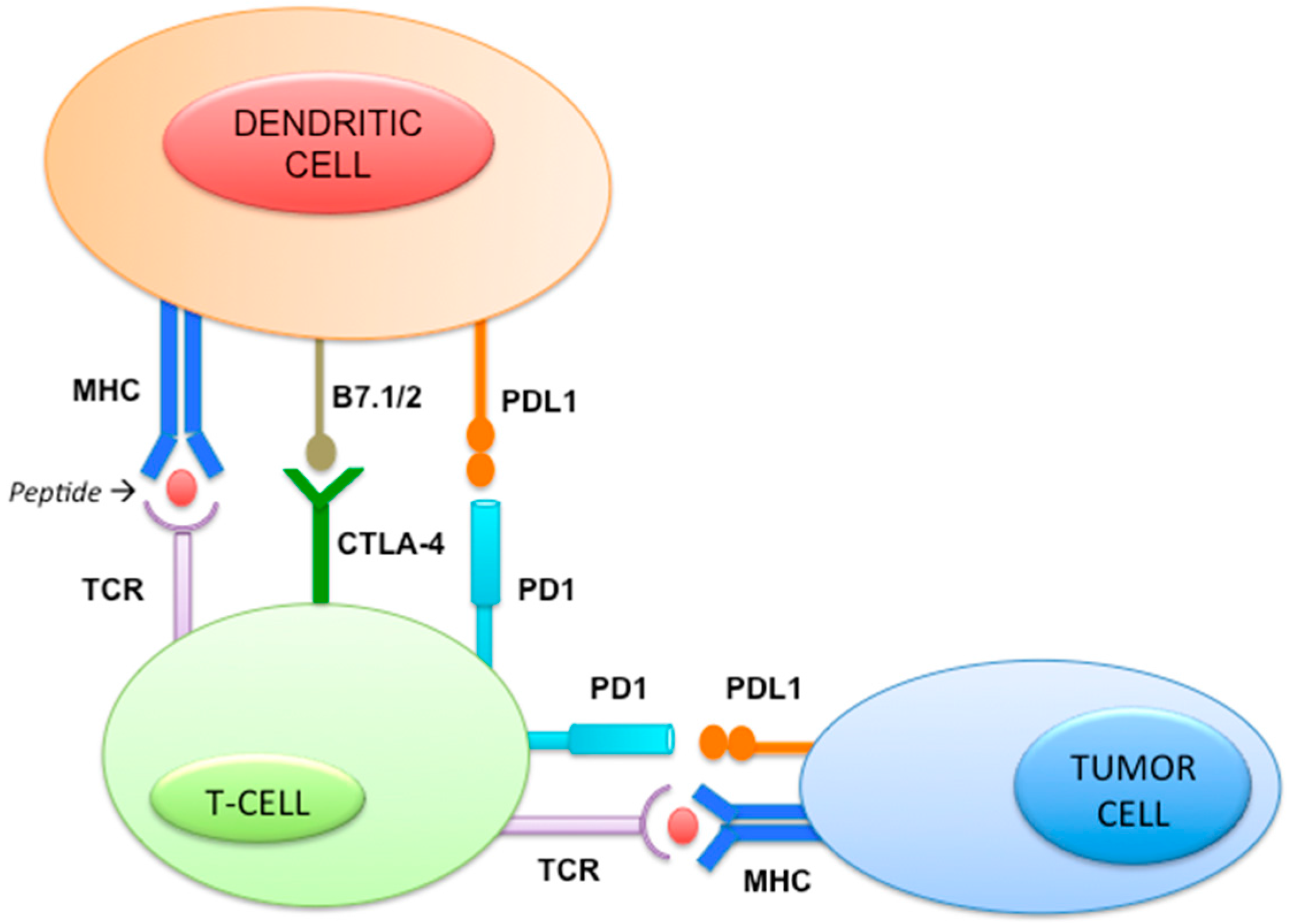
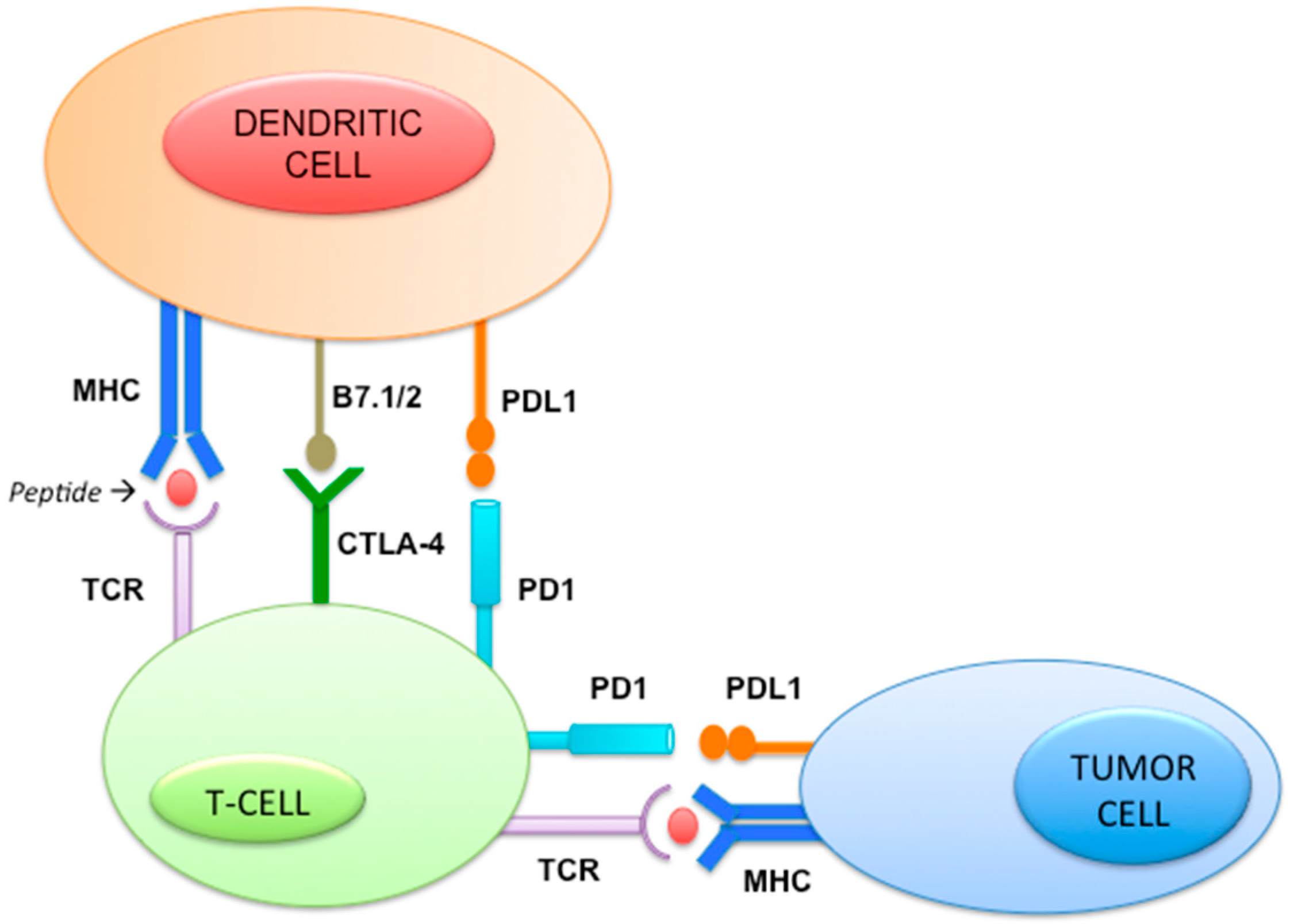
2.11. Immune Pathway (Figure 5)
3. Targeted Therapies for the Treatment of Advanced Breast Cancer
3.1. Endocrine Therapy
- SERMs (selective estrogen receptor modulators): they are competitive partial agonists of the estrogen receptor. Particularly, tamoxifen is the oldest and the most well-known drug of this category [53]. Subsequently, toremifene citrate was developed with the goal of achieving efficacy similar to that of tamoxifen and with an improved safety profile. To date, although studies have not confirmed a better safety profile, clinical data have supported the efficacy and safety of toremifene for the treatment of BC in postmenopausal patients [54].
- Aromatase Inhibitors: they stop the production of estrogen in postmenopausal women by inhibiting the activity of aromatase. The third-generation aromatase inhibitors have largely replaced tamoxifen in the treatment of postmenopausal HR positive BC patients. They are classified into irreversible steroidal inhibitors, such as exemestane, that form a permanent and deactivating bond with the aromatase enzyme, and non-steroidal inhibitors, such as anastrozole and letrozole, that act via reversible competition for the aromatase enzyme [55,56,57].
3.2. Anti-HER Agents
- Recombinant humanized monoclonal antibodies (trastuzumab and pertuzumab): binding the extracellular domain of HER2, trastuzumab blocks the dimerization of HER2 while pertuzumab inhibits the heterodimerization of HER2 with other HER receptors, inhibiting the downstream signaling pathways (PI3K and MAPK) with a cytostatic mechanism; they also have a cytotoxic mechanism through the activation of the antibody dependent cell-mediated cytotoxicity (ADCC) [62,63,64].
- Receptor tyrosine kinase inhibitors (RTKIs) (lapatinib): they inhibit enzyme function of HER family intracellularly, binding competitively to the intracellular kinase domain ATP-binding site of EGFR and/or HER2 [67].
- Other anti-HER2 compounds are still under evaluation in clinical trials such as HER2 vaccines, other monoclonal antibodies (such as ertumaxomab and margetuximab), and defucosylated trastuzumab [11].
3.3. Compounds Targeting PI3K/AKT/mTOR Pathway
3.4. Farnesyl Transferase Inhibitors
3.5. Anti-RTKs (FGFR, MET, and IGF-1R)
3.6. Cyclin-Dependent Kinase (CDK) Inhibitors
3.7. Angiogenesis Inhibitors
3.8. SFK Inhibitors
3.9. HSP90 Function Inhibitors
3.10. PARP Inhibitors
3.11. Immunotherapy
4. Resistance Mechanisms to Targeted Therapies
5. Predictive Molecular Biomarkers
5.1. Fulvestrant
5.2. Everolimus
5.3. Buparlisib
5.4. Pictilisib
5.5. Alpelisib and Taselisib
5.6. Dovitinib
5.7. Trastuzumab
5.8. Pertuzumab
5.9. LAPATINIB and TDM-1
5.10. Neratinib
5.11. Palbociclib
5.12. Bevacizumab
5.13. Entinostat
5.14. Parp-Inhibitors
5.15. Immunotherapy
6. Conclusions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Cortesi, L.; Toss, A.; Cirilli, C.; Marcheselli, L.; Braghiroli, B.; Sebastiani, F.; Federico, M. Twenty-years experience with de novo metastatic breast cancer. Int. J. Cancer 2015, 137, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Zeichner, S.B.; Herna, S.; Mani, A.; Ambros, T.; Montero, A.J.; Mahtani, R.L.; Ahn, E.R.; Vogel, C.L. Survival of patients with de-novo metastatic breast cancer: Analysis of data from a large breast cancer-specific private practice, a university-based cancer center and review of the literature. Breast Cancer Res. Treat. 2015, 153, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Mauri, D.; Polyzos, N.P.; Salanti, G.; Pavlidis, N.; Ioannidis, J.P.A. Multiple-Treatments Meta-analysis of Chemotherapy and Targeted Therapies in Advanced Breast Cancer. J. Natl. Cancer Inst. 2008, 100, 1780–1791. [Google Scholar] [CrossRef] [PubMed]
- Cnossen, J.A.; Heinemann, V.; Laessig, D.; Stemmler, H.J. Long term survival with metastatic breast cancer (MBC): Results of a retrospective, single-centre analysis from 2000–2005. Anticancer Drugs 2011, 22, 933–939. [Google Scholar]
- Minicozzi, P.; Bella, F.; Toss, A.; Giacomin, A.; Fusco, M.; Zarcone, M.; Tumino, R.; Falcini, F.; Cesaraccio, R.; Candela, G.; et al. Relative and disease-free survival for breast cancer in relation to subtype: A population-based study. J. Cancer Res. Clin. Oncol. 2013, 139, 1569–1577. [Google Scholar] [CrossRef] [PubMed]
- Sledge, G.W., Jr.; Miller, K.D. Exploiting the hallmarks of cancer: The future conquest of breast cancer. Eur. J. Cancer 2003, 39, 1668–1675. [Google Scholar] [CrossRef]
- Ingvarsson, S. Genetics of breast cancer. Drugs Today 2004, 40, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Harbeck, N.; Fallowfield, L.; Kyriakides, S.; Senkus, E. ESMO Guidelines Working Group. Locally recurrent or metastatic breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012, 23, vii11–vii19. [Google Scholar] [CrossRef] [PubMed]
- Sonnenblick, A.; Pondé, N.; Piccart, M. Metastatic breast cancer: The Odyssey of personalization. Mol. Oncol. 2016, 10, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Toss, A.; Cristofanilli, M. Molecular characterization and targeted therapeutic approaches in breast cancer. Breast Cancer Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Edler, L.; Kopp-Schneider, A. Origins of the mutational origin of cancer. Int. J. Epidemiol. 2005, 34, 1168–1170. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, V. Early references to the mutational origin of cancer. Int. J. Epidemiol. 2007, 36, 246–247. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Becerra, R.; Santos, N.; Diaz, L.; Camacho, J. Mechanisms of Resistance to Endocrine Therapy in Breast Cancer: Focus on Signaling Pathways, miRNAs and Genetically Based Resistance. Int. J. Mol. Sci. 2013, 14, 108–145. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.; Galluzzo, P.; Ascenzi, P. Estrogen Signaling Multiple Pathways to Impact Gene Transcription. Curr. Genom. 2006, 7, 497–508. [Google Scholar] [CrossRef]
- Lin, N.U.; Winer, E.P. New targets for therapy in breast cancer: Small molecule tyrosine kinase inhibitors. Breast Cancer Res. 2004, 6, 204–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ménard, S.; Fortis, S.; Castiglioni, F.; Agresti, R.; Balsari, A. HER2 as a prognostic factor in breast cancer. Oncology 2001, 61, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Atalay, G.; Cardoso, F.; Awada, A.; Piccart, M.J. Novel therapeutic strategies targeting the epidermal growth factor receptor (EGFR) family and its downstream effectors in breast cancer. Ann. Oncol. 2003, 14, 1346–1363. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J. Common and distinct elements in cellular signaling via EGF and FGF receptors. Science 2004, 306, 1506–1507. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Franke, T.F.; Kaplan, D.R.; Cantley, L.C.; Toker, A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science 1997, 275, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.; Jin, J.; Cardiff, R.D.; Woodgett, J.R.; Muller, W. Activation of Akt (protein kinase B) in mammary epithelium provides a critical cell survival signal required for tumor progression. Mol. Cell. Biol. 2001, 21, 2203–2212. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Gu, J.; Matsumoto, K.; Aota, S.; Parsons, R.; Yamada, K.M. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 1998, 280, 1614–1617. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Lin, Y.Z.; LaPushin, R.; Cuevas, B.; Fang, X.; Yu, S.X.; Davies, M.A.; Khan, H.; Furui, T.; Mao, M.; et al. The PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and induces apoptosis and anoikis in breast cancer cells. Oncogene 1999, 18, 7034–7045. [Google Scholar] [CrossRef] [PubMed]
- Tamguney, T.; Stokoe, D. New insights into PTEN. J. Cell Sci. 2007, 120, 4071–4079. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.; Cheng, S.; Sun, S.P.; An, C.; Liu, Z.Y.; Feng, X.; Liu, G.J. Prognostic role of PIK3CA mutations and their association with hormone receptor expression in breast cancer: A meta-analysis. Sci. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Kalinsky, K.; Jacks, L.M.; Heguy, A.; Patil, S.; Drobnjak, M.; Bhanot, U.K.; Hedvat, C.V.; Traina, T.A.; Solit, D.; Gerald, W.; et al. PIK3CA mutation associates with improved outcome in breast cancer. Clin. Cancer Res. 2009, 15, 5049–5059. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Liang, F.; Jia, Z.L.; Song, S.T.; Jiang, Z.F. PTEN mutation, methylation and expression in breast cancer patients. Oncol. Lett. 2013, 6, 161–168. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed]
- Kolch, W. Meaningful relationships: The regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J. 2000, 351, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef] [PubMed]
- Sandal, T. Molecular aspects of the mammalian cell cycle and cancer. Oncologist 2002, 7, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Park, M.T.; Lee, S.J. Cell cycle and cancer. J. Biochem. Mol. Biol. 2003, 36, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; Thijssen, B.; Mc Dermott, U.; Garnett, M.; Wessels, L.F.; Bernards, R. Targeting the RB-E2F pathway in breast cancer. Oncogene 2016, 35, 4829–4835. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Cancer and ageing: Rival demons? Nat. Rev. Cancer 2003, 3, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002, 29, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; de Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, B.S.; Vroom, T.M.; Adriaansen-Slot, S.S.; Ottenhoff-Kalff, A.E.; Geertzema, J.G.; Hennipman, A.; Rijksen, G. c-Src protein expression is increased in human breast cancer. An immunohistochemical and biochemical analysis. J. Pathol. 1996, 180, 383–388. [Google Scholar] [CrossRef]
- Elsberger, B. Translational evidence on the role of Src kinase and activated Src kinase in invasive breast cancer. Crit. Rev. Oncol. Hematol. 2014, 89, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Burrows, F. Targeting multiple signal transduction pathways through inhibition of Hsp90. J. Mol. Med. 2004, 82, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. A growing network of cancer-susceptibility genes. N. Engl. J. Med. 2003, 348, 1917–1919. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.N.; Kachnic, L.A. Roles of BRCA1 and BRCA2 in homologous recombination. DNA replication fidelity and the cellular response to ionizing radiation. Oncogene 2003, 22, 5784–5791. [Google Scholar] [CrossRef] [PubMed]
- Weil, M.K.; Chen, A.P. PARP inhibitor treatment in ovarian and breast cancer. Curr. Probl. Cancer 2011, 35, 7–50. [Google Scholar] [CrossRef] [PubMed]
- Toss, A.; Cortesi, L. Molecular Mechanisms of PARP Inhibitors in BRCA-related Ovarian Cancer. J. Cancer Sci. Ther. 2013, 5, 409–416. [Google Scholar]
- Thomas, S.J.; Snowden, J.A.; Zeidler, M.P.; Danson, S.J. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Criscitiello, C.; Curigliano, G. Immunotherapy of Breast Cancer. Prog. Tumor Res. 2015, 42, 30–43. [Google Scholar] [PubMed]
- Bedognetti, D.; Maccalli, C.; Bader, S.B.; Marincola, F.M.; Seliger, B. Checkpoint Inhibitors and Their Application in Breast Cancer. Breast Care 2016, 11, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Beatson, G.T. On the treatment of inoperable cases of carcinoma of the mamma: Suggestions for a new method of treament, with illustrative cases. Lancet 1896, 148, 162–165. [Google Scholar] [CrossRef]
- Osborne, C.K. Tamoxifen in the treatment of breast cancer. N. Engl. J. Med. 1998, 339, 1609–1618. [Google Scholar] [PubMed]
- Vogel, C.L.; Johnston, M.A.; Capers, C.; Braccia, D. Toremifene for breast cancer: A review of 20 years of data. Clin. Breast Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Nabholtz, J.M.; Buzdar, A.; Pollak, M.; Harwin, W.; Burton, G.; Mangalik, A.; Steinberg, M.; Webster, A.; von Euler, M. Anastrozole is superior to tamoxifen as first-line therapy for advanced breast cancer in postmenopausal women: Results of a North American multicenter randomized trial. Arimidex Study Group. J. Clin. Oncol. 2000, 18, 3758–3767. [Google Scholar] [PubMed]
- Mouridsen, H.T. Letrozole in advanced breast cancer: The PO25 trial. Breast Cancer Res. Treat. 2007, 105, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Paridaens, R.J.; Dirix, L.Y.; Beex, L.V.; Nooij, M.; Cameron, D.A.; Cufer, T.; Piccart, M.J.; Bogaerts, J.; Therasse, P. Phase III study comparing exemestane with tamoxifen as first-line hormonal treatment of metastatic breast cancer in postmenopausal women: The European Organisation for Research and Treatment of Cancer Breast Cancer Cooperative Group. J. Clin. Oncol. 2008, 26, 4883–4890. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Pippen, J.; Jones, S.E.; Parker, L.M.; Ellis, M.; Come, S.; Gertler, S.Z.; May, J.T.; Burton, G.; Dimery, I.; et al. Double-blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: Results of a North American trial. J. Clin. Oncol. 2002, 20, 3386–3395. [Google Scholar] [CrossRef] [PubMed]
- Howell, A.; Robertson, J.F.; Quaresma Albano, J.; Aschermannova, A.; Mauriac, L.; Kleeberg, U.R.; Vergote, I.; Erikstein, B.; Webster, A.; Morris, C. Fulvestrant, formerly ICI 182,780, is as effective as anastrozole in postmenopausal women with advanced breast cancer progressing after prior endocrine treatment. J. Clin. Oncol. 2002, 20, 3396–3403. [Google Scholar] [CrossRef] [PubMed]
- Dawood, S.; Broglio, K.; Buzdar, A.U.; Hortobagyi, G.N.; Giordano, S.H. Prognosis of women with metastatic breast cancer by HER-2 status and trastuzumab treatment: An institutional-based review. J. Clin. Oncol. 2010, 28, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Cortesi, L.; de Matteis, E.; Cirilli, C.; Marcheselli, L.; Proietto, M.; Federico, M. Outcome evaluation in pre-trastuzumab era between different breast cancer phenotypes: A population-based study on Italian women. Tumori 2012, 98, 743–750. [Google Scholar] [PubMed]
- Baselga, J.; Albanell, J.; Molina, M.A.; Arribas, J. Mechanism of action of trastuzumab and scientific update. Semin. Oncol. 2001, 28, 4–11. [Google Scholar] [CrossRef]
- Gennari, R.; Menard, S.; Fagnoni, F.; Ponchio, L.; Scelsi, M.; Tagliabue, E.; Castiglioni, F.; Villani, L.; Magalotti, C.; Gibelli, N.; et al. Pilot study of the mechanism of action of preoperative trastuzumab in patients with primary operable breast tumors overexpressing HER2. Clin. Cancer Res. 2004, 10, 5650–5655. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Cortés, J.; Im, S.A.; Clark, E.; Ross, G.; Kiermaier, A.; Swain, S.M. Biomarker Analyses in CLEOPATRA: A Phase III, Placebo-Controlled Study of Pertuzumab in Human Epidermal Growth Factor Receptor 2-Positive, First-Line Metastatic Breast Cancer. J. Clin. Oncol. 2014, 32, 3753–3761. [Google Scholar] [CrossRef] [PubMed]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Weiss, D.; Guardino, E.; Girish, S.; Sliwkowski, M.X. Trastuzumab emtansine: A unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin. Cancer Res. 2011, 17, 6437–6447. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L.S.; Ashurst, H.L.; Chap, L. Targeting signal transduction pathways in metastatic breast cancer: A comprehensive review. Oncologist 2010, 5, 216–235. [Google Scholar] [CrossRef] [PubMed]
- Paplomata, E.; O’Regan, R. The PI3K/AKT/mTOR pathway in breast cancer: Targets, trials and biomarkers. Ther. Adv. Med. Oncol. 2014, 6, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Kondapaka, S.B.; Singh, S.S.; Dasmahapatra, G.P.; Sausville, E.A.; Roy, K.K. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol. Cancer Ther. 2003, 2, 1093–1103. [Google Scholar] [PubMed]
- Ma, C.X.; Sanchez, C.; Gao, F.; Crowder, R.; Naughton, M.; Pluard, T.; Creekmore, A.; Guo, Z.; Hoog, J.; Lockhart, A.C.; et al. A Phase I Study of the AKT Inhibitor MK-2206 in Combination with Hormonal Therapy in Postmenopausal Women with Estrogen Receptor-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2016, 22, 2650–2658. [Google Scholar] [CrossRef] [PubMed]
- Beaver, J.A.; Park, B.H. The BOLERO-2 trial: The addition of everolimus to exemestane in the treatment of postmenopausal hormone receptor-positive advanced breast cancer. Future Oncol. 2012, 8, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, S.A.; Andre, F.; Jiang, Z.; Shao, Z.; Mano, M.S.; Neciosup, S.P.; Tseng, L.M.; Zhang, Q.; Shen, K.; Liu, D.; et al. Combination of everolimus with trastuzumab plus paclitaxel as first-line treatment for patients with HER2-positive advanced breast cancer (BOLERO-1): A phase 3, randomised, double-blind, multicentre trial. Lancet Oncol. 2015, 16, 816–829. [Google Scholar] [CrossRef]
- André, F.; O’Regan, R.; Ozguroglu, M.; Toi, M.; Xu, B.; Jerusalem, G.; Masuda, N.; Wilks, S.; Arena, F.; Isaacs, C.; et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014, 15, 580–591. [Google Scholar] [CrossRef]
- Baselga, J.; Im, S.A. PIK3CA status in circulating tumor DNA predicts efficacy of buparlisib plus fulvestrant in postmenopausal women with endocrine-resistant HR+/Her2− advanced breast cancer: First results from the randomized, phase III BELLE-2. In Proceedings of the 38th Annual San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 8–12 December 2015.
- Krop, I.E.; Mayer, I.A.; Ganju, V.; Dickler, M.; Johnston, S.; Morales, S.; Yardley, D.A.; Melichar, B.; Forero-Torres, A.; Lee, S.C.; et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): A randomized, double-blind, placebo-controlloed, phase 2 trial. Lancet Oncol. 2016, 17, 811–821. [Google Scholar] [CrossRef]
- Vuylsteke, P.; Huizing, M.; Petrakova, K.; Roylance, R.; Laing, R.; Chan, S.; Abell, F.; Gendreau, S.; Rooney, I.; Apt, D.; et al. Pictilisib plus paclitaxel for the treatment of hormone receptor-positive, HER2-negative, locally recurrent, or metastatic breast cancer: Interim analysis of the multicentre, placebo-controlled, phase II randomised PEGGY study. Ann. Oncol. 2016, 27, 2059–2066. [Google Scholar] [CrossRef] [PubMed]
- Dickler, M.N.; Saura, C.; Richards, D.A.; Krop, I.E.; Cervantes, A.; Bedard, P.L.; Patel, M.R.; Pusztai, L.; Oliveira, M.; Ware, J.A.; et al. A phase II study of the PI3K inhibitor taselisib (GDC-0032) combined with fulvestrant (F) in patients (pts) with HER2-negative (HER2−), hormone receptor-positive (HR+) advanced breast cancer. In Proceedings of the 2016 ASCO Annual Meeting, Chicago, IL, USA, 3–7 June 2016.
- Johnston, S.R.; Semiglazov, V.F.; Manikhas, G.M.; Spaeth, D.; Romieu, G.; Dodwell, D.J.; Wardley, A.M.; Neven, P.; Bessems, A.; Park, Y.C.; et al. A phase II, randomized, blinded study of the farnesyltransferase inhibitor tipifarnib combined with letrozole in the treatment of advanced breast cancer after antiestrogen therapy. Breast Cancer Res. Treat. 2008, 110, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Sparano, J.A.; Moulder, S.; Kazi, A.; Vahdat, L.; Li, T.; Pellegrino, C.; Munster, P.; Malafa, M.; Lee, D.; Hoschander, S.; et al. Targeted inhibition of farnesyltransferase in locally advanced breast cancer: A phase I and II trial of tipifarnib plus dose-dense doxorubicin and cyclophosphamide. J. Clin. Oncol. 2006, 24, 3013–3018. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Guo, M.; Gradishar, W.J.; Sparano, J.A.; Perez, E.A.; Wang, M.; Sledge, G.W. A phase II trial of capecitabine in combination with the farnesyl transferase inhibitor tipifarnib in patients with anthracycline-treated and taxane-resistant metastatic breast cancer: An Eastern Cooperative Oncology Group Study (E1103). Breast Cancer Res. Treat. 2012, 134, 345–352. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Bachelot, T.; Campone, M.; Dalenc, F.; Perez-Garcia, J.M.; Hurvitz, S.A.; Turner, N.; Rugo, H.; Smith, J.W.; Deudon, S.; et al. Targeting FGFR with dovitinib (TKI258): Preclinical and clinical data in breast cancer. Clin. Cancer Res. 2013, 19, 3693–3702. [Google Scholar] [CrossRef] [PubMed]
- Tolaney, S.M.; Tan, S.; Guo, H.; Barry, W.; van Allen, E.; Wagle, N.; Brock, J.; Larrabee, K.; Paweletz, C.; Ivanova, E.; et al. Phase II study of tivantinib (ARQ 197) in patients with metastatic triple-negative breast cancer. Investig. New Drugs 2015, 33, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Tolaney, S.M.; Nechushtan, H.; Ron, I.G.; Schöffski, P.; Awada, A.; Yasenchak, C.A.; Laird, A.D.; O’Keeffe, B.; Shapiro, G.I.; Winer, E.P. Cabozantinib for metastatic breast carcinoma: Results of a phase II placebo-controlled randomized discontinuation study. Breast Cancer Res. Treat. 2016, 160, 305–312. [Google Scholar] [CrossRef] [PubMed]
- O’Flanagan, C.H.; O’Shea, S.; Lyons, A.; Fogarty, F.M.; McCabe, N.; Kennedy, R.D.; O’Connor, R. IGF-1R inhibition sensitizes breast cancer cells to ATM-Related Kinase (ATR) inhibitor and cisplatin. Oncotarget 2016. [Google Scholar] [CrossRef]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar] [PubMed]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.L.; Blackwell, K.L.; André, F.; Winer, E.P.; et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- Dickler, M.N.; Tolaney, S.M.; Rugo, H.S.; Cortes, J.; Dieras, V.; Patt, D.A.; Wildiers, H.; Frenzel, M.; Koustenis, A.; Baselga, J. MONARCH1: Results from a phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as monotherapy, in patients with HR+/HER2− breast cancer, after chemotherapy for advanced disease. In Proceedings of the 2016 ASCO Annual Meeting, Chicago, IL, USA, 3–7 June 2016.
- Miller, K.; Wang, M.; Gralow, J.; Dickler, M.; Cobleigh, M.; Perez, E.A.; Shenkier, T.; Cella, D.; Davidson, N.E. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N. Engl. J. Med. 2007, 357, 2666–2676. [Google Scholar] [CrossRef] [PubMed]
- Brufsky, A.M.; Hurvitz, S.; Perez, E.; Swamy, R.; Valero, V.; O’Neill, V.; Rugo, H.S. RIBBON-2: A randomized, double-blind, placebo-controlled, phase III trial evaluating the efficacy and safety of bevacizumab in combination with chemotherapy for second-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J. Clin. Oncol. 2011, 29, 4286–4293. [Google Scholar] [PubMed]
- Miles, D.W.; Chan, A.; Dirix, L.Y.; Cortés, J.; Pivot, X.; Tomczak, P.; Delozier, T.; Sohn, J.H.; Provencher, L.; Puglisi, F.; et al. Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J. Clin. Oncol. 2010, 28, 3239–3247. [Google Scholar] [CrossRef] [PubMed]
- Robert, N.J.; Diéras, V.; Glaspy, J.; Brufsky, A.M.; Bondarenko, I.; Lipatov, O.N.; Perez, E.A.; Yardley, D.A.; Chan, S.Y.; Zhou, X.; et al. RIBBON-1: Randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J. Clin. Oncol. 2011, 29, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Robert, N.J.; Saleh, M.N.; Paul, D.; Generali, D.; Gressot, L.; Copur, M.S.; Brufsky, A.M.; Minton, S.E.; Giguere, J.K.; Smith, J.W.; et al. Sunitinib plus paclitaxel versus bevacizumab plus paclitaxel for first-line treatment of patients with advanced breast cancer: A phase III, randomized, open-label trial. Clin. Breast Cancer 2011, 11, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Reeves, K.; Han, X.; Fairchild, C.; Platero, S.; Wong, T.W.; Lee, F.; Shaw, P.; Clark, E. Identification of candidate molecular markers predicting sensitivity in solid tumors to dasatinib: Rationale for patient selection. Cancer Res. 2007, 67, 2226–2238. [Google Scholar] [CrossRef] [PubMed]
- Nautiyal, J.; Majumder, P.; Patel, B.B.; Lee, F.Y.; Majumdar, A.P. Src inhibitor dasatinib inhibits growth of breast cancer cells by modulating EGFR signaling. Cancer Lett. 2009, 283, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Sanjay, A.; Neff, L.; Tanaka, S.; Horne, W.C.; Baron, R. Src kinase activity is essential for osteoclast function. J. Biol. Chem. 2004, 279, 17660–17666. [Google Scholar] [CrossRef] [PubMed]
- Schott, A.F.; Barlow, W.E.; van Poznak, C.H.; Hayes, D.F.; Moinpour, C.M.; Lew, D.L.; Dy, P.A.; Keller, E.T.; Keller, J.M.; Hortobagyi, G.P. Phase II studies of two different schedules of dasatinib in bone metastasis predominant metastatic breast cancer: SWOG S0622. Breast Cancer Res. Treat. 2016, 159, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Carbonero, R.; Carnero, A.; Paz-Ares, L. Inhibition of HSP90 molecular chaperones: Moving into the clinic. Lancet Oncol. 2013, 14, e358–e369. [Google Scholar] [CrossRef]
- Modi, S.; Stopeck, A.; Linden, H.; Solit, D.; Chandarlapaty, S.; Rosen, N.; D’Andrea, G.; Dickler, M.; Moynahan, M.E.; Sugarman, S.; et al. HSP90 inhibition is effective in breast cancer: A phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin. Cancer Res. 2011, 17, 5132–5139. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, K.; Chandarlapaty, S.; Lake, D.; Gilewski, T.; Robson, M.; Goldfarb, S.; Drullinsky, P.; Sugarman, S.; Wasserheit-Leiblich, C.; Fasano, J.; et al. A phase II open-label study of ganetespib, a novel heat shock protein 90 inhibitor for patients with metastatic breast cancer. Clin. Breast Cancer 2014, 14, 154–160. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J.; Schwartzberg, L.; Danso, M.A.; Miller, K.D.; Rugo, H.S.; Neubauer, M.; Robert, N.; Hellerstedt, B.; Saleh, M.; Richards, P.; et al. Phase III study of iniparib plus gemcitabine and carboplatin versus gemcitabine and carboplatin in patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2014, 32, 3840–3847. [Google Scholar] [CrossRef] [PubMed]
- Sonnenblick, A.; de Azambuja, E.; Azim, H.A., Jr.; Piccart, M. An update on PARP inhibitors—Moving to the adjuvant setting. Nat. Rev. Clin. Oncol. 2015, 12, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Dizdar, O.; Arslan, C.; Altundag, K. Advances in PARP inhibitors for the treatment of breast cancer. Expert Opin. Pharmacother. 2015, 16, 2751–2758. [Google Scholar] [CrossRef] [PubMed]
- Livraghi, L.; Garber, J.E. PARP inhibitors in the management of breast cancer: Current data and future prospects. BMC Med. 2015, 13, 188. [Google Scholar] [CrossRef] [PubMed]
- Maj, T.; Wei, S.; Welling, T.; Zou, W. T cells and costimulation in cancer. Cancer J. 2013, 19, 473–482. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Safety Study Of Chemotherapy Combined With Dendritic Cell Vaccine to Treat Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02018458 (accessed on 18 December 2016).
- Disis, M.L.; Wallace, D.R.; Gooley, T.A.; Dang, Y.; Slota, M.; Lu, H.; Coveler, A.L.; Childs, J.S.; Higgins, D.M.; Fintak, P.A.; et al. Concurrent trastuzumab and HER2/neu-specific vaccination in patients with metastatic breast cancer. J. Clin. Oncol. 2009, 27, 4685–4692. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Vaccine Therapy With or Without Sirolimus in Treating Patients with NY-ESO-1 Expressing Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT01522820 (accessed on 18 December 2016).
- Nanda, R.; Chow, L.Q.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktank, G.; Cheng, J.D.; et al. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M.; Hicklin, D.J. Resistance to Targeted Therapies: Refining Anticancer Therapy in the Era of Molecular Oncology. Clin. Cancer Res. 2009, 15, 7471–7478. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ellis, M.; Li, S.; Larson, D.E.; Chen, K.; Wallis, J.W.; Harris, C.C.; McLellan, M.D.; Fulton, R.S.; Fulton, L.L.; et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 2010, 464, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.E. The first five years of single-cell cancer genomics and beyond. Genome Res. 2015, 25, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Amir, E.; Clemons, M.; Purdie, C.A.; Miller, N.; Quinlan, P.; Geddie, W.; Coleman, R.E.; Freedman, O.C.; Jordan, L.B.; Thompson, A.M. Tissue confirmation of disease recurrence in breast cancer patients: Pooled analysis of multi-centre, multi-disciplinary prospective studies. Cancer Treat. Rev. 2012, 38, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Guarneri, V.; Giovannelli, S.; Ficarra, G.; Bettelli, S.; Maiorana, A.; Piacentini, F.; Barbieri, E.; Dieci, M.V.; D’Amico, R.; Jovic, G.; et al. Comparison of HER-2 and hormone receptor expression in primary breast cancers and asynchronous paired metastases: Impact on patient management. Oncologist 2008, 13, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Jeselsohn, R.; Buchwalter, G.; de Angelis, C.; Brown, M.; Schiff, R. ESR1 mutations—A mechanism for acquired endocrine resistance in breast cancer. Nat. Rev. Clin. Oncol. 2015, 12, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Balko, J.M.; Arteaga, C.L. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J. Clin. Oncol. 2011, 29, 4452–4461. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Yardley, D.A.; Ismail-Khan, R.R.; Melichar, B.; Lichinitser, M.; Munster, P.N.; Klein, P.M.; Cruickshank, S.; Miller, K.D.; Lee, M.J.; Trepel, J.B. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J. Clin. Oncol. 2013, 31, 2128–2135. [Google Scholar] [PubMed]
- Tortora, G. Mechanisms of resistance to HER2 target therapy. J. Natl. Cancer Inst. Monogr. 2011, 2011, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Fribbens, C.; O’Leary, B.; Kilburn, L.; Hrebien, S.; Garcia-Murillas, I.; Beaney, M.; Cristofanilli, M.; Andre, F.; Loi, S.; Loibl, S.; et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor–Positive Advanced Breast Cancer. J. Clin. Oncol. 2016, 34, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Chandarlapaty, S.; Chen, D.; He, W.; Sung, P.; Samoila, A.; You, D.; Bhatt, T.; Patel, P.; Voi, M.; Gnant, M.; et al. Prevalence of ESR1 Mutations in Cell-Free DNA and Outcomes in Metastatic Breast Cancer: A Secondary Analysis of the BOLERO-2 Clinical Trial. JAMA Oncol. 2016, 2, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Hurvitz, S.; Fasolo, A.; Tseng, L.M.; Jerusalem, G.; Wilks, S.; O’Regan, R.; Isaacs, C.; Toi, M.; Burris, H.; et al. Molecular Alterations and Everolimus Efficacy in Human Epidermal Growth Factor Receptor 2–Overexpressing Metastatic Breast Cancers: Combined Exploratory Biomarker Analysis from BOLERO-1 and BOLERO-3. J. Clin. Oncol. 2016, 34, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Lewis Phillips, G.D.; Verma, S.; Ro, J.; Huober, J.; Guardino, A.E.; Samant, M.K.; Olsen, S.; de Haas, S.L.; Pegram, M.D. Relationship Between Tumor Biomarkers and Efficacy in EMILIA, a Phase III Study of Trastuzumab Emtansine in HER2-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2016, 22, 3755–3763. [Google Scholar] [CrossRef] [PubMed]
- Treilleux, I.; Arnedos, M.; Cropet, C.; Wang, Q.; Ferrero, J.M.; Abadie-Lacourtoisie, S.; Levy, C.; Legouffe, E.; Lortholary, A.; Pujade-Lauraine, E.; et al. Translational studies within the TAMRAD randomized GINECO trial: Evidence for mTORC1 activation marker as a predictive factor for everolimus efficacy in advanced breast cancer. Ann. Oncol. 2015, 26, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.W.; de Haas, S.L.; Dirix, L.Y.; Romieu, G.; Chan, A.; Pivot, X.; Tomczak, P.; Provencher, L.; Cortés, J.; Delmar, P.R.; et al. Biomarker results from the AVADO phase 3 trial of first-line bevacizumab plus docetaxel for HER2-negative metastatic breast cancer. Br. J. Cancer 2013, 108, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Jerusalem, G.; Andre, F.; Chen, D.; Robinson, D.; Ozgguroglu, M.; Lang, I.; White, M.; Toi, M.; Taran, T.; Gianni, L. Evaluation of everolimus (EVE) in HER2+ advanced breast cancer (BC) with activated PI3K/mTOR pathway: Exploratory biomarker observations from the BOLERO-3 trial. In Proceedings of the European Cancer Congress 2013 (ECCO, ESMO, ESTRO), Amsterdam, The Netherlands, 27 September–1 October 2013.
- Hortobagyi, G.N.; Chen, D.; Piccart, M.; Rugo, H.S.; Burris, H.A.; Pritchard, K.I.; Campone, M.; Noguchi, S.; Perez, A.T.; Deleu, I.; et al. Correlative Analysis of Genetic Alterations and Everolimus Benefit in Hormone Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Advanced Breast Cancer: Results from BOLERO-2. J. Clin. Oncol. 2015, 34, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Sung, P.; Chen, D.; He, W.; Samoila, A.; You, D.; Bhatt, T.; Patel, P.; Ringeisen, F.P.; Hortobagyi, G.N.; et al. Correlation of PIK3CA mutations in cell-free DNA (cfDNA) and efficacy of everolimus (EVE) in metastatic breast cancer: Results from BOLERO-2. In Proceedings of the 2016 ASCO Annual Meeting, Chicago, IL, USA, 3–7 June 2016.
- Di Leo, A.; Ciruelos, E.; Janni, W.; Lonning, P.E.; O’Regan, R.; Hubalek, M.; Csõszi, T.; Decker, T.; Tjan-Heijnen, V.C.; Weber, D.; et al. BELLE-3: A Phase III study of the pan-phosphatidylinositol 3-kinase (PI3K) inhibitor buparlisib (BKM120) with fulvestrant in postmenopausal women with HR+/HER2− locally advanced/metastatic breast cancer (BC) pretreated with aromatase inhibitors (AIs) and refractory to mTOR inhibitor (mTORi)-based treatment. In Proceedings of the 2015 ASCO Annual Meeting, Chicago, IL, USA, 29 May–2 June 2015.
- Andrè, F.; Campone, M.; Ciruelos, E.M.; Iwata, H.; Loibl, S.; Rugo, H.S.; Wilke, C.; Mills, D.; Chol, M.; Longin, A.S.; et al. SOLAR-1: A phase III study of alpelisib + fulvestrant in men and postmenopausal women with HR+/HER2− advanced breast cancer (BC) progressing on or after prior aromatase inhibitor therapy. In Proceedings of the 2016 ASCO Annual Meeting, Chicago, IL, USA, 3–7 June 2016.
- Massacesi, C.; Di Tomaso, E.; Urban, P.; Germa, C.; Quadt, C.; Trandafir, L.; Aimone, P.; Fretault, N.; Dharan, B.; Tavorath, R.; et al. PI3K inhibitors as new cancer therapeutics: Implications for clinical trial design. Oncol. Targets Ther. 2016, 9, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Cortes, J.; de Laurentiis, M.; Dieras, V.; Harbeck, N.; Hsu, J.Y.; Ng, V.; Schimmoller, F.; Wilson, T.R.; Im, Y.M.; et al. SANDPIPER: Phase III study of the PI3-kinase (PI3K) inhibitor taselisib (GDC-0032) plus fulvestrant in patients (pts) with estrogen receptor (ER)-positive, HER2-negative locally advanced or metastatic breast cancer (BC) enriched for pts with PIK3CA-mutant tumors. In Proceedings of the 2016 ASCO Annual Meeting, Chicago, IL, USA, 3–7 June 2016.
- ClinicalTrials.gov. Trial Evaluating Dovitinib Combined With Fulvestrant, in Postmenopausal Patients With HER2− and HR+ Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT01528345 (accessed on 18 December 2016).
- Carter, P.; Presta, L.; Gorman, C.M.; Ridgway, J.B.; Henner, D.; Wong, W.L.; Rowland, A.M.; Kotts, C.; Carver, M.E.; Shepard, H.M. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. USA 1992, 89, 4285–4289. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Rugo, H.S.; Vukelja, S.J.; Vogel, C.L.; Borson, R.A.; Limentani, S.; Tan-Chiu, E.; Krop, I.E.; Michaelson, R.A.; Girish, S.; et al. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. J. Clin. Oncol. 2011, 29, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.E.; LoRusso, P.; Miller, K.D.; Modi, S.; Yardley, D.; Rodriguez, G.; Guardino, E.; Lu, M.; Zheng, M.; Girish, S.; et al. A phase II study of trastuzumab emtansine in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer who were previously treated with trastuzumab, lapatinib, an anthracycline, a taxane, and capecitabine. J. Clin. Oncol. 2012, 30, 3234–3241. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A.; Hurvitz, S.A.; Amler, L.C.; Mundt, K.E.; Ng, V.; Guardino, E.; Gianni, L. Relationship between HER2 expression and efficacy with first-line trastuzumab emtansine compared with trastuzumab plus docetaxel in TDM4450g: A randomized phase II study of patients with previously untreated HER2-positive metastatic breast cancer. Breast Cancer Res. 2014, 16. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, P.J.; Gili, M.; Scaltriti, M.; Serra, V.; Guzman, M.; Nijkamp, W.; Beijersbergen, R.L.; Valero, V.; Seoane, J.; Bernards, R.; et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008, 68, 9221–9230. [Google Scholar] [CrossRef] [PubMed]
- Bose, R.; Kavuri, S.M.; Searleman, A.C.; Shen, W.; Shen, D.; Koboldt, D.C.; Monsey, J.; Goel, N.; Aronson, A.B.; Li, S.; et al. Activating HER2 Mutations in HER2 Gene Amplification Negative Breast Cancer. Cancer Discov. 2013, 3, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Feldinger, K.; Kong, A. Profile of neratinib and its potential in the treatment of breast cancer. Breast Cancer 2015, 7, 147–162. [Google Scholar] [PubMed]
- Hyman, D.; Piha-Paul, S.A.; Rodón, J.; Saura, C.; Puzanov, I.; Shapiro, G.I.; Loi, S.; Joensuu, H.; Hanrahan, A.J.; Modi, S.; et al. Neratinib for ERBB2 Mutant, HER2 Nonamplified, Metastatic Breast Cancer: Preliminary Analysis from a Multicenter, Open-Label, Multi-Histology Phase II Basket Trial. In Proceedings of the San Antonio Breast Cancer Symposium 2015, San Antonio, TX, USA, 8–12 December 2015.
- Murphy, C.G.; Dickler, M.N. Endocrine resistance in hormone-responsive breast cancer: Mechanisms and therapeutic strategies. Endocr. Relat. Cancer 2016, 23, R337–R352. [Google Scholar] [CrossRef] [PubMed]
- Connolly, R.M.; Zhao, F.; Miller, K.; Tevaarwerk, A.; Wagner, L.I.; Lee, M.J.; Murray, J.; Gray, R.J.; Piekarz, R.; Zujewski, J.A.; et al. E2112: Randomized phase III trial of endocrine therapy plus entinostat/placebo in patients with hormone receptor-positive advanced breast cancer. In Proceedings of the 2015 ASCO Annual Meeting, Chicago, IL, USA, 29 May–2 June 2015.
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Huang, Z.; Teng, F.; Xing, L.; Yu, J. Predictive biomarkers in PD-1/PD-L1 checkpoint blockade immunotherapy. Cancer Treat. Rev. 2015, 41, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.M.; Wen, S.; Hong, D.S.; Wheler, J.J.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; Naing, A.; Janku, F.; Aldape, K.; et al. Personalized medicine for patients with advanced cancer in the phase I program at MD Anderson: Validation and landmark analyses. Clin. Cancer Res. 2014, 20, 4827–4836. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Stephenson, J.J., Jr.; Rosen, P.; Loesch, D.M.; Borad, M.J.; Anthony, S.; Jameson, G.; Brown, S.; Cantafio, N.; Richards, D.A.; et al. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J. Clin. Oncol. 2010, 28, 4877–4883. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.M.; Iskander, N.G.; Hong, D.S.; Wheler, J.J.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; Naing, A.; Janku, F.; Luthra, R.; et al. Personalized medicine in a phase I clinical trials program: The MD Anderson Cancer Center initiative. Clin. Cancer Res. 2012, 18, 6373–6383. [Google Scholar] [CrossRef] [PubMed]
- Le Tourneau, C.; Delord, J.P.; Gonçalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Trédan, O.; Massiani, M.A.; Mauborgne, C.; et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef]
- Iorio, M.V.; Ferracin, M.; Liu, C.G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Rothé, F.; Ignatiadis, M.; Chaboteaux, C.; Haibe-Kains, B.; Kheddoumi, N.; Majjaj, S.; Badran, B.; Fayyad-Kazan, H.; Desmedt, C.; Harris, A.L.; et al. Global microRNA expression profiling identifies MiR-210 associated with tumor proliferation, invasion and poor clinical outcome in breast cancer. PLoS ONE 2011, 6, e20980. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.J.; Santarpia, L.; Kim, J.; Esteva, F.J.; Moretti, E.; Buzdar, A.U.; Di Leo, A.; Le, X.F.; Bast, R.C., Jr.; Park, S.T.; et al. Plasma microRNA 210 levels correlate with sensitivity to trastuzumab and tumor presence in breast cancer patients. Cancer 2012, 118, 2603–2614. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Biomarkers | Method of Analysis | Targeted Therapy | References |
|---|---|---|---|
| ESR1 mutations | ctDNA | sensitivity to FULVESTRANT | Fribbens 2016; [121] |
| ctDNA | resistance to EXEMESTANE | Fribbens 2016; [121] | |
| Y537S mutation in ctDNA | resistance to EVEROLIMUS | Chandarlapaty 2016; [122] | |
| PIK3CA mutations | Tumor tissue | sensitivity to EVEROLIMUS | André 2016; [123] |
| ctDNA | sensitivity to BUPARLISIB | Baselga 2015; [74] | |
| Tumor tissue | sensitivity to TASELISIB | Dickler 2016; [89] | |
| Tumor tissue | resistance to LAPATINIB | Baselga 2016; [124] | |
| AKT1 mutations | Tumor tissue | sensitivity to EVEROLIMUS | André 2016; [123] |
| mTORC1 activation (high p4EBP1, low 4EBP1, low liver kinase B1, low pAkt, and low PI3K) | Tumor tissue | sensitivity to EVEROLIMUS | Treilleux 2015; [125] |
| FGF pathway amplified | Tumor tissue | sensitivity to DOVITINIB | André 2013; [81] |
| HER2 amplification | Tumor tissue | sensitivity to TRASTUZUMAB | Dawood 2010; [60] |
| Serum samples and tumor tissue | sensitivity to PERTUZUMAB | Baselga 2014; [64] | |
| Tumor tissue | sensitivity to LAPATINIB | Baselga 2016; [124] | |
| Tumor tissue | sensitivity to TDM1 | Baselga 2016; [124] | |
| EGFR down expression | Tumor tissue | sensitivity to TDM1 | Baselga 2016; [124] |
| HER3 down expression | Tumor tissue | sensitivity to TDM1 | Baselga 2016; [124] |
| VEGF-A and VEGFR-2 high concentration | Serum samples | sensitivity to BEVACIZUMAB | Miles 2013; [126] |
| Low PTEN concentration | Tumor tissue | sensitivity to EVEROLIMUS | Jerusalem 2013; [127] André 2016; [123] |
| sensitivity to TDM1 | Baselga 2016; [124] | ||
| High pS6 concentration | Tumor tissue | sensitivity to EVEROLIMUS | Jerusalem 2013; [127] |
| Hyperacetylation of lysines | Serum samples | sensitivity to ENTINOSTAT | Yardley 2013; [119] |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toss, A.; Venturelli, M.; Peterle, C.; Piacentini, F.; Cascinu, S.; Cortesi, L. Molecular Biomarkers for Prediction of Targeted Therapy Response in Metastatic Breast Cancer: Trick or Treat? Int. J. Mol. Sci. 2017, 18, 85. https://doi.org/10.3390/ijms18010085
Toss A, Venturelli M, Peterle C, Piacentini F, Cascinu S, Cortesi L. Molecular Biomarkers for Prediction of Targeted Therapy Response in Metastatic Breast Cancer: Trick or Treat? International Journal of Molecular Sciences. 2017; 18(1):85. https://doi.org/10.3390/ijms18010085
Chicago/Turabian StyleToss, Angela, Marta Venturelli, Chiara Peterle, Federico Piacentini, Stefano Cascinu, and Laura Cortesi. 2017. "Molecular Biomarkers for Prediction of Targeted Therapy Response in Metastatic Breast Cancer: Trick or Treat?" International Journal of Molecular Sciences 18, no. 1: 85. https://doi.org/10.3390/ijms18010085
APA StyleToss, A., Venturelli, M., Peterle, C., Piacentini, F., Cascinu, S., & Cortesi, L. (2017). Molecular Biomarkers for Prediction of Targeted Therapy Response in Metastatic Breast Cancer: Trick or Treat? International Journal of Molecular Sciences, 18(1), 85. https://doi.org/10.3390/ijms18010085