
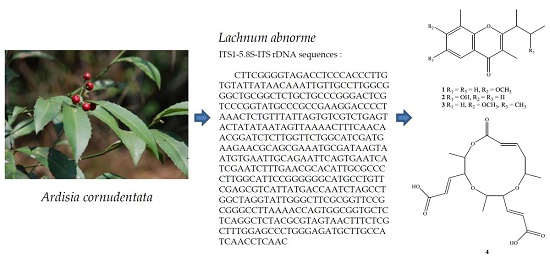
Secondary Metabolites of the Endophytic Fungus Lachnum abnorme from Ardisia cornudentata

Abstract
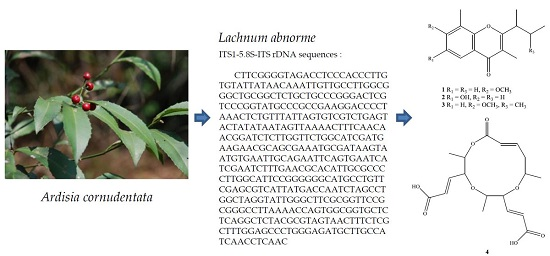
:
1. Introduction
2. Results
2.1. Purification and Characterization
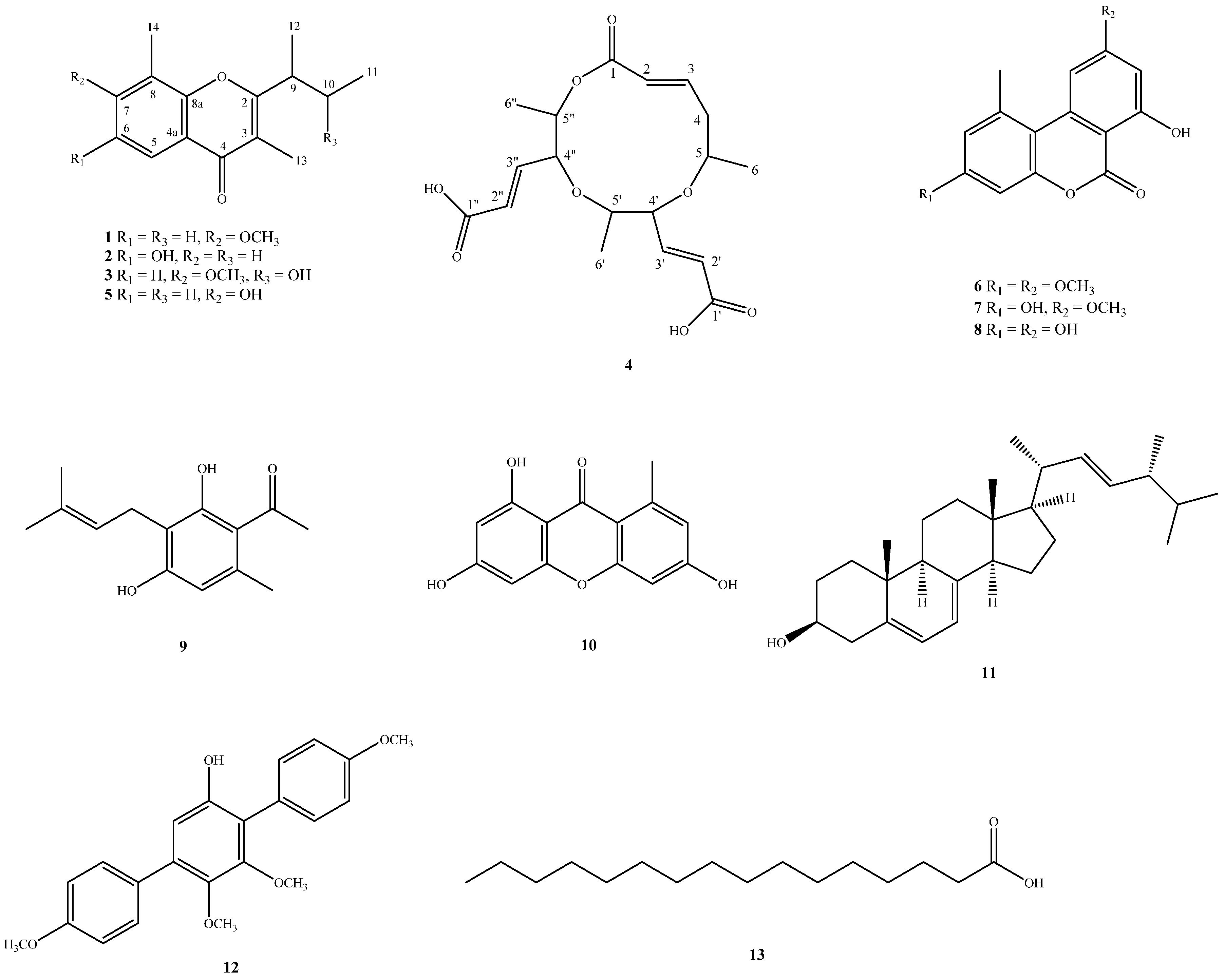
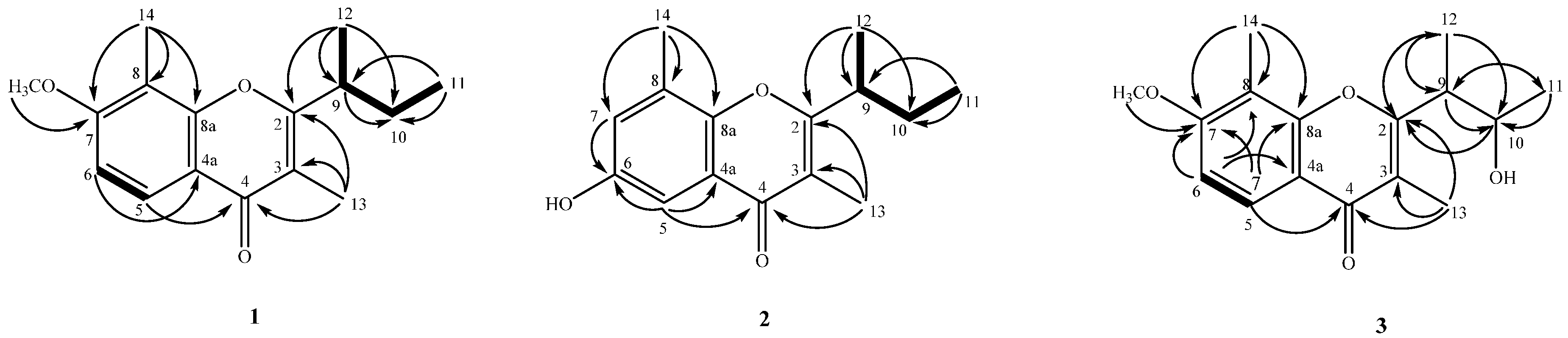
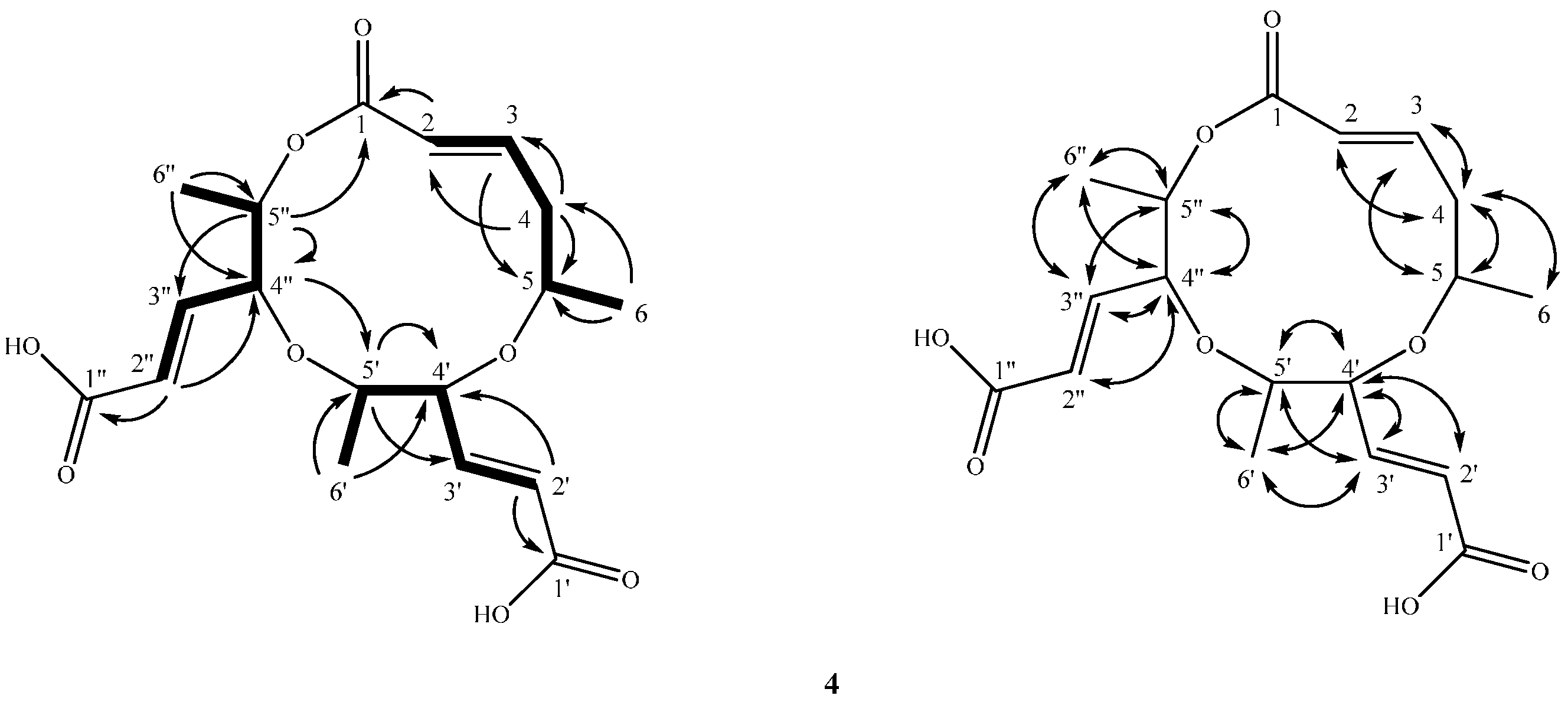
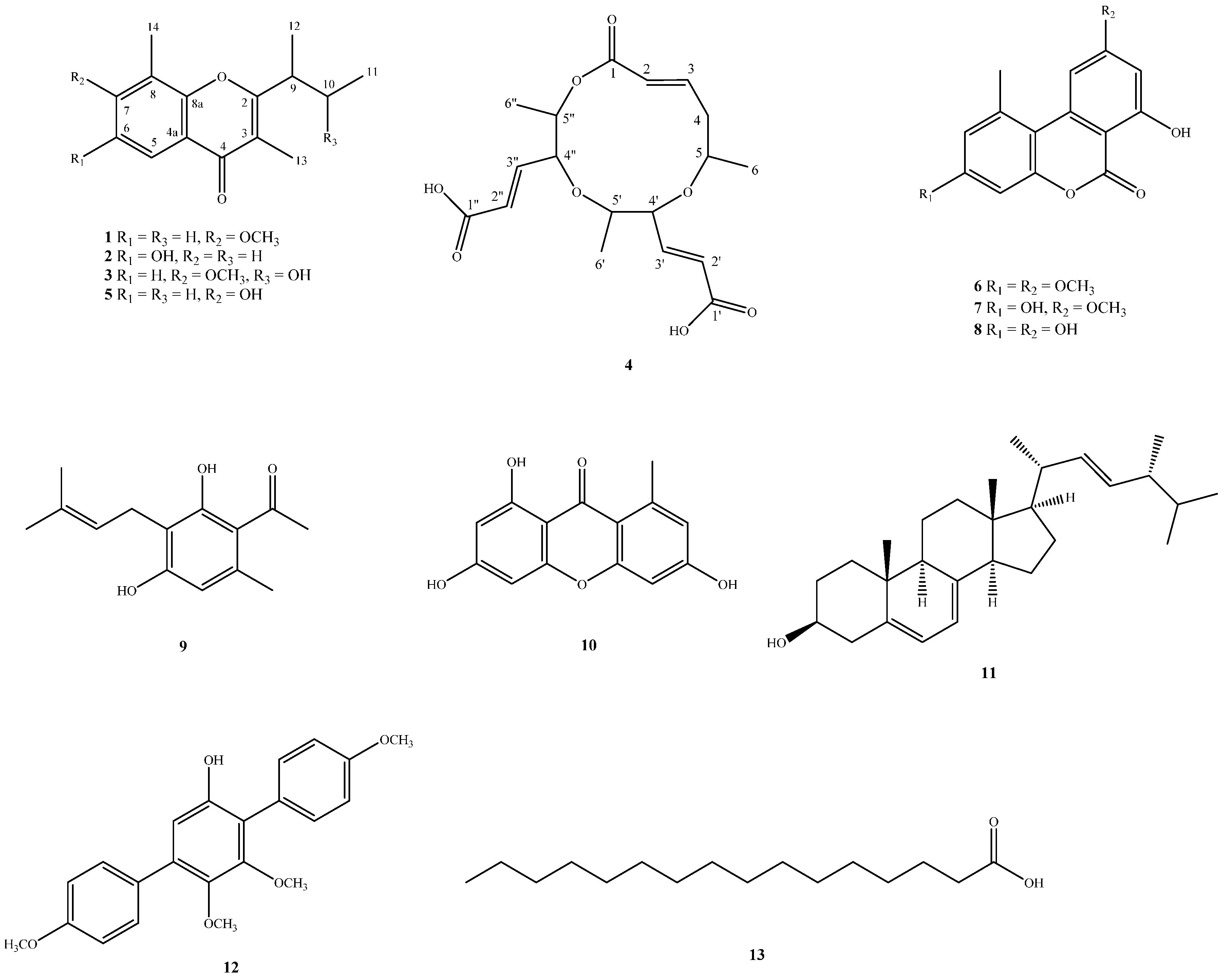
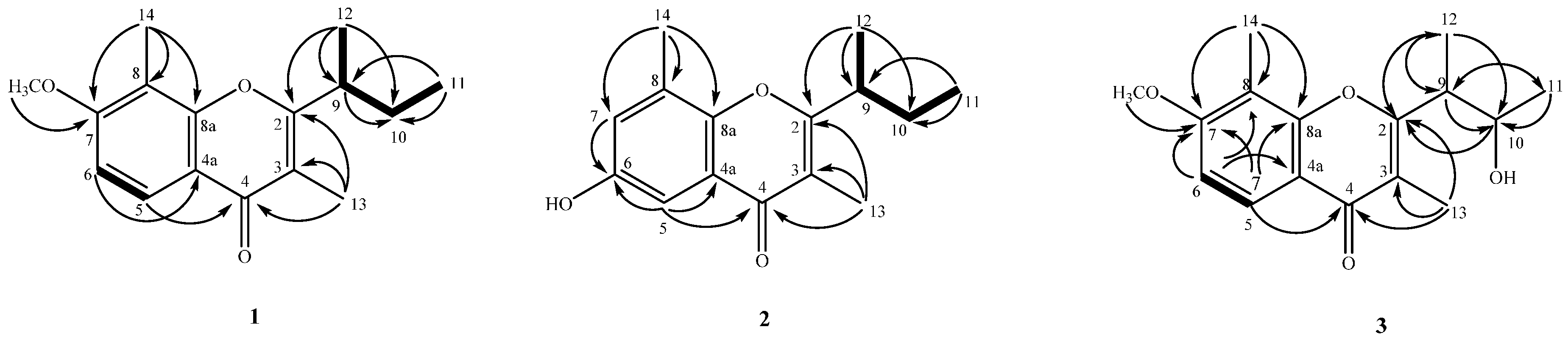
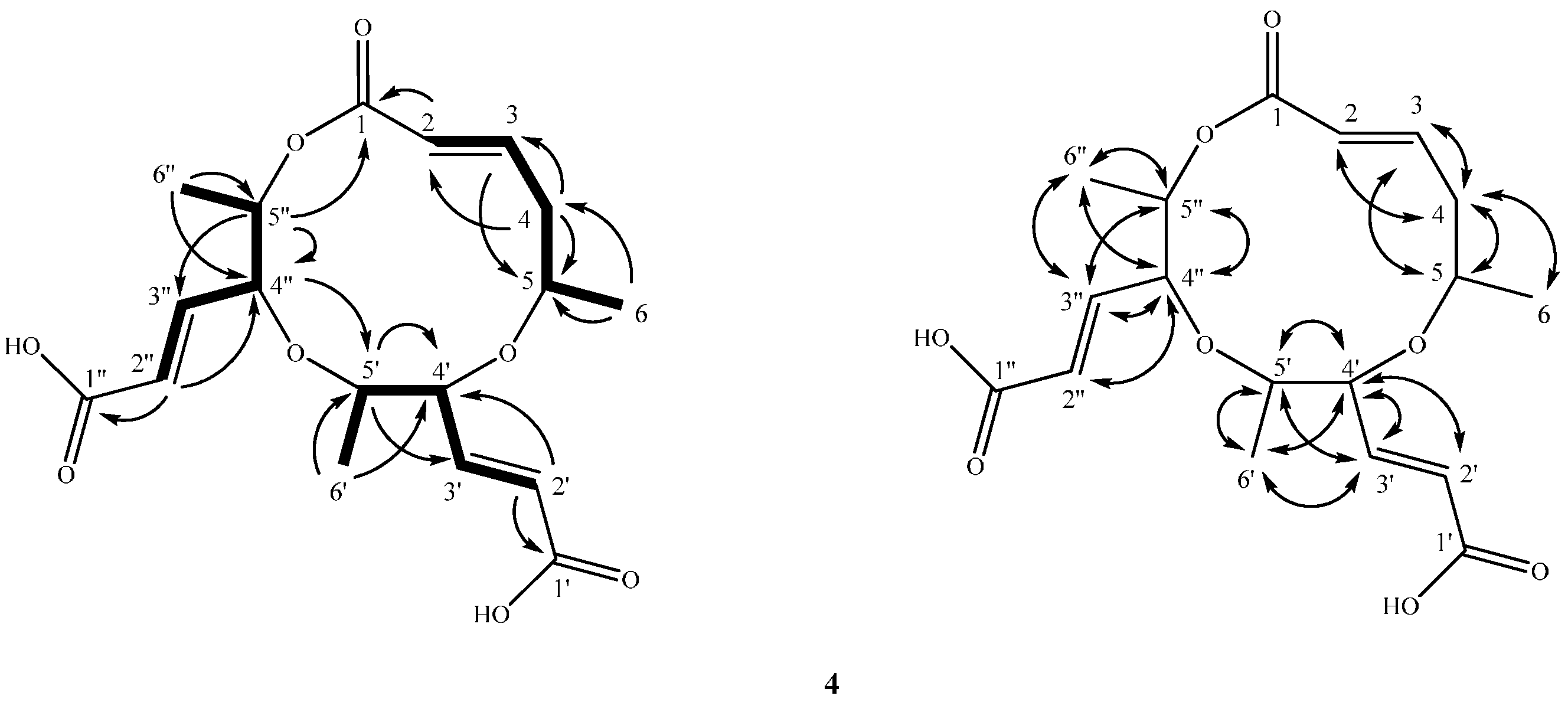
2.2. Structure Elucidation of Compounds 1–4
3. Discussion
4. Materials and Methods
4.1. General
4.2. Fungus Material
4.3. Extraction and Isolation
4.3.1. Lachnochromonin D (1)
4.3.2. Lachnochromonin E (2)
4.3.3. Lachnochromonin F (3)
4.3.4. Lachabnormic Acid (4)
4.3.5. Lachnochromonin C (5)
4.3.6. Alternariol-3,9-dimethyl Ether (6)
4.3.7. Alternariol-9-methyl Ether (7)
4.3.8. Alternariol (8)
4.3.9. Pestalotionol (9)
4.3.10. 1,3,6-Trihydroxy-8-methylxanthone (10)
4.3.11. Ergosterol (11)
4.3.12. 6′-Hydroxy-4,2′,3′,4′′-tetramethoxy-p-terphenyl (12)
4.3.13. Palmitic Acid (13)
4.4. Determination of NO Cell Viability Assay
4.4.1. NO Activity Assay
4.4.2. Cell Viability Assay
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gunatilaka, A.A. Natural products from plant-associated microorganisms: Distribution, structural diversity, bioactivity, and implications of their occurrence. J. Nat. Prod. 2006, 69, 509–526. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.X.; Zou, W.X. Endophytes: A rich source of functional metabolites. Nat. Prod. Rep. 2001, 18, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Stierle, A.; Strobel, G.; Stierle, D. Taxol and taxane production by Taxomyces andreanae, an endophytic fungus of Pacific yew. Science 1993, 260, 214–216. [Google Scholar] [CrossRef] [PubMed]
- Strobel, G.A.; Miller, R.V.; Martinez-Miller, C.; Condron, M.M.; Teplow, D.B.; Hess, W.M. Cryptocandin, a potent antimycotic from the endophytic fungus Cryptosporiopsis cf. quercina. Microbiology 1999, 145, 1919–1926. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Strobel, G.; Harper, J.; Lobkovsky, E.; Clardy, J. Cryptocin, a potent tetramic acid antimycotic from the endophytic fungus Cryptosporiopsis cf. quercina. Org. Lett. 2000, 2, 767–770. [Google Scholar] [CrossRef] [PubMed]
- Matumoto, T.; Hosoya, T.; Shigemori, H. Palmariols A and B, two new chlorinated dibenzo-α-pyrones from discomycete Lachnum palmae. Heterocycles 2010, 81, 1231–1237. [Google Scholar] [CrossRef]
- Shan, R.; Stabler, M.; Anke, H.; Sterner, O. Naphthalenone and phthalide metabolites from Lachnum papyraceum. J. Nat. Prod. 1997, 60, 804–805. [Google Scholar] [CrossRef]
- Shan, R.; Stadler, M.; Sterner, O.; Anke, H. New metabolites with nematicidal and antimicrobial activities from the ascomycete Lachnum papyraceum (Karst.) Karst. VIII. Isolation, structure determination and biological activities of minor metabolites structurally related to mycorrhizin A. J. Antibiot. 1996, 49, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Stadler, M.; Anke, H.; Sterner, O. Metabolites with nematicidal and antimicrobial activities from the ascomycete Lachnum papyraceum (Karst.) Karst. V. Production, isolation and biological activities of bromine-containing mycorrhizin and lachnumon derivatives and four additional new bioactive metabolites. J. Antibiot. 1995, 48, 149–153. [Google Scholar] [PubMed]
- Stadler, M.; Anke, H.; Sterner, O. Metabolites with nematicidal and antimicrobial activities from the ascomycete Lachnum papyraceum (Karst.) Karst. III. Production of novel isocoumarin derivatives, isolation, and biological activities. J. Antibiot. 1995, 48, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Ondeyka, J.; Harris, G.; Zink, D.; Basilio, A.; Vicente, F.; Bills, G.; Platas, G.; Collado, J.; Gonzalez, A.; de la Cruz, M.; et al. Isolation, structure elucidation, and biological activity of virgineone from Lachnum virgineum using the genome-wide Candida albicans fitness test. J. Nat. Prod. 2009, 72, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, B.K.; Bagchi, P. Studies on the ultraviolet absorption spectra of coumarins and chromones. Part I. J. Org. Chem. 1956, 21, 1415–1419. [Google Scholar] [CrossRef]
- Yano, K.; Tanaka, I.; Miyauchi, T. Novel Compound, Lachnochromonin Compound. WO 2009101959 A1, 20 August 2009. [Google Scholar]
- Tan, N.; Tao, Y.W.; Pan, J.H.; Wang, S.Y.; Xu, F.; She, Z.G.; Lin, Y.C.; Jones, E.B.G. Isolation, structure elucidation, and mutagenicity of four alternariol derivatives produced by the mangrove endophytic fungus No. 2240. Chem. Nat. Compd. 2008, 44, 296–300. [Google Scholar] [CrossRef]
- Yang, Z.J.; Yang, T.; Luo, M.Y.; Xia, X.; Chen, D.J.; Qian, X.P. A new sesquiterpenoid from fungus Colletotrichum sp. and its cytotoxicity. Acta Pharm. Sin. 2013, 48, 891–895. [Google Scholar]
- Liang, D.; Luo, H.; Liu, Y.F.; Hao, Z.Y.; Wang, Y.; Zhang, C.L.; Zhang, Q.J.; Chen, R.Y.; Yu, D.Q. Lysilactones A-C, three 6H-dibenzo[b,d]pyran-6-one glycosides from Lysimachia clethroides, total synthesis of lysilactone A. Tetrahedron 2013, 69, 2093–2097. [Google Scholar] [CrossRef]
- Freeman, G.G. Isolation of alternariol and alternariol monomethyl ether from Alternaria dauci (Kühn) Groves et Skolko. Phytochemistry 1965, 5, 719–725. [Google Scholar] [CrossRef]
- Aly, A.H.; Edrada-Ebel, R.; Indriani, I.D.; Wray, V.; Müller, W.E.G.; Totzke, F.; Zirrgibel, U.; Schächtele, C.; Kubbutat, M.H.G.; Lin, W.H.; et al. Cytotoxic metabolites from the fungal endophyte Alternaria sp. and their subsequent detection in its host plant Polygonum senegalense. J. Nat. Prod. 2008, 71, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Arunpanichlert, J.; Rukachaisirikul, V.; Phongpaichit, S.; Supaphon, O.; Sakayaroj, J. Meroterpenoid, isocoumarin, and phenol derivatives from the seagrass-derived fungus Pestalotiopsis sp. PSU-ES194. Tetrahedron 2015, 71, 882–888. [Google Scholar] [CrossRef]
- Mutanyatta, J.; Matapa, B.G.; Shushu, D.D.; Abegaz, B.M. Homoisoflavonoids and xanthones from the tubers of wild and in vitro regenerated Ledebouria graminifolia and cytotoxic activities of some of the homoisoflavonoids. Phytochemistry 2003, 62, 797–804. [Google Scholar] [CrossRef]
- Kanokmedhakul, S.; Kanokmedhakul, K.; Phonkerd, N.; Soytong, K.; Kongsaeree, P.; Suksamrarn, A. Antimycobacterial anthraquinone-chromanone compound and diketopiperazine alkaloid from the fungus Chaetomium globosum KMITL-N0802. Planta Med. 2002, 68, 834–836. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.Z.; Pu, X.; Luo, G.; Zhao, L.X.; Xu, L.H.; Li, W.J.; Luo, Y. Isolation and characterization of new p-terphenyls with antifungal, antibacterial, and antioxidant activities from halophilic actinomycete Nocardiopsis gilva YIM 90087. J. Agric. Food Chem. 2013, 61, 3006–3012. [Google Scholar] [CrossRef] [PubMed]
- Walter, E.D. Isolation of α-spinasteryl-d-glucoside and α-spinasterol from Alfalfa. J. Pharm. Sci. 1964, 24, 1917–1919. [Google Scholar] [CrossRef]
- Rukachaisirikul, V.; Chantaruk, S.; Pongcharoen, W.; Isaka, M.; Lapanun, S. Chromone derivatives from the filamentous fungus Lachnum sp. BCC 2424. J. Nat. Prod. 2006, 69, 980–982. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.; Chang, H.S.; Liu, T.W.; Hsieh, S.Y.; Yuan, G.F.; Cheng, M.J.; Chen, I.S. Secondary metabolites and bioactivity of the endophytic fungus Phomopsis theicola from Taiwanese endemic plant. Rec. Nat. Prod. 2016, 10, 189–194. [Google Scholar]
- Ting, Y.C.; Ko, H.H.; Wang, H.C.; Peng, C.F.; Chang, H.S.; Hsieh, P.C.; Chen, I.S. Biological evaluation of secondary metabolites from the roots of Myrica adenophora. Phytochemistry 2014, 103, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Wu, C.C.; Cheng, T.S.; Kuo, C.Y.; Tsai, Y.C.; Chiang, S.Y.; Wong, T.S.; Wu, Y.C.; Chang, F.R. Active constituents from Liriope platyphylla root against cancer growth in vitro. J. Evid. Based Complement. Altern. Med. 2013, 2013. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | 5 | ||||
|---|---|---|---|---|---|---|---|---|
| δ (J in Hz) | δC | δ (J in Hz) | δC | δ (J in Hz) | δC | δ (J in Hz) | δC | |
| 2 | 167.2 | 167.7 | 165.0 | 168.2 | ||||
| 3 | 115.1 | 115.6 | 116.8 | 115.1 | ||||
| 4 | 178.4 | 178.3 | 178.2 | 179.2 | ||||
| 4a | 116.5 | 115.9 | 116.5 | 115.6 | ||||
| 5 | 8.03 d (8.8) | 124.1 | 6.90 s | 101.8 | 8.03 d (8.9) | 124.4 | 7.90 d (8.8) | 123.8 |
| 6 | 6.92 d (8.8) | 108.0 | 156.2 | 6.94 d (8.9) | 108.2 | 114.2 | ||
| 7 | 160.6 | 7.96 s | 127.1 | 160.8 | 7.03 d (8.8) | 159.5 | ||
| 8 | 113.3 | 123.4 | 113.2 | 111.4 | ||||
| 8a | 156.0 | 159.4 | 154.8 | 155.9 | ||||
| 9 | 3.00 m | 37.6 | 3.00 m | 37.5 | 3.11 dq (7.2, 6.6) | 44.9 | 3.05 m | 37.8 |
| 10 | 1.82 m | 27.7 | 1.77 m | 27.5 | 4.15 dq (6.6, 6.0) | 70.2 | 1.85 m | 27.8 |
| 1.68 m | 1.64 m | 1.68 m | ||||||
| 11 | 0.89 t (7.4) | 11.9 | 0.89 t (7.6) | 12.0 | 1.34 d (6.0) | 21.1 | 0.93 dd (7.6, 7.2) | 12.0 |
| 12 | 1.29 d (6.8) | 18.0 | 1.27 d (6.8) | 17.8 | 1.32 d (7.2) | 14.6 | 1.33 d (6.8) | 18.0 |
| 13 | 2.05 s | 9.4 | 2.09 s | 9.7 | 2.09 s | 9.7 | 2.11 s | 9.6 |
| 14 | 2.27 s | 7.8 | 2.33 s | 15.6 | 2.29 s | 8.0 | 2.35 s | 7.9 |
| OH-6 | 7.35 br s | |||||||
| OH-7 | 8.91 br s | |||||||
| OCH3-7 | 3.91 s | 55.9 | 3.94 s | 56.0 | ||||
| Compounds | Nitrite Production (%) | Emax (%) b | Cell Viability (%) |
|---|---|---|---|
| lachnochromonin F (3) | 88.48 ± 6.45 | 11.52 ± 6.45 | 98.17 ± 13.65 |
| lachabnormic acid (4) | 82.46 ± 1.67 | 17.54 ± 1.67 | 73.62 ± 7.68 |
| alternariol-9-methyl ether (7) | 101.83 ± 0.69 | −1.83 ± 0.69 | 83.00 ± 4.67 |
| alternariol (8) | 86.65 ± 2.22 | 13.35 ± 2.22 | 106.41 ± 8.19 |
| pestalorionol (9) | 91.62 ± 2.24 | 8.38 ± 2.24 | 107.33 ± 5.87 |
| 1,3,6-trihydroxy-8-methyl-xanthone (10) | 83.77 ± 0.53 | 16.23 ± 0.53 | 96.40 ± 11.07 |
| palmitic acid (13) | 77.75 ± 2.89 | 22.25 ± 2.89 | 92.83 ± 0.13 |
| aminoguanidine | 35.59 ± 0.2 | 78.74 ± 0.64 | 88.87 ± 2.98 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, H.-S.; Lin, C.-H.; Chen, Y.-S.; Wang, H.-C.; Chan, H.-Y.; Hsieh, S.-Y.; Wu, H.-C.; Cheng, M.-J.; Yuan, G.-F.; Lin, S.-Y.; et al. Secondary Metabolites of the Endophytic Fungus Lachnum abnorme from Ardisia cornudentata. Int. J. Mol. Sci. 2016, 17, 1512. https://doi.org/10.3390/ijms17091512
Chang H-S, Lin C-H, Chen Y-S, Wang H-C, Chan H-Y, Hsieh S-Y, Wu H-C, Cheng M-J, Yuan G-F, Lin S-Y, et al. Secondary Metabolites of the Endophytic Fungus Lachnum abnorme from Ardisia cornudentata. International Journal of Molecular Sciences. 2016; 17(9):1512. https://doi.org/10.3390/ijms17091512
Chicago/Turabian StyleChang, Hsun-Shuo, Chu-Hung Lin, Yi-Shuan Chen, Hui-Chun Wang, Hing-Yuen Chan, Sung-Yuan Hsieh, Ho-Cheng Wu, Ming-Jen Cheng, Gwo-Fang Yuan, Shan-Yu Lin, and et al. 2016. "Secondary Metabolites of the Endophytic Fungus Lachnum abnorme from Ardisia cornudentata" International Journal of Molecular Sciences 17, no. 9: 1512. https://doi.org/10.3390/ijms17091512