
In Vitro Co-Culture Models of Breast Cancer Metastatic Progression towards Bone


Abstract
:1. Introduction
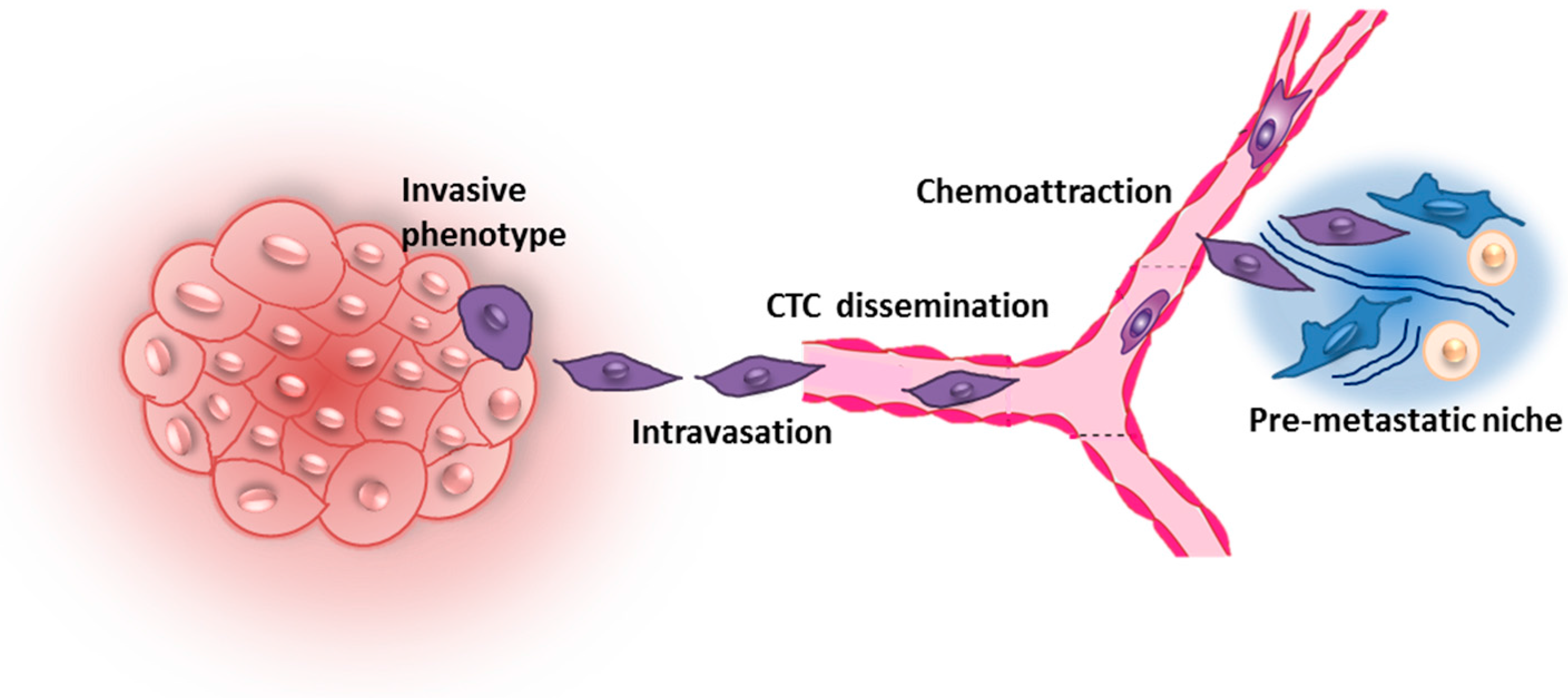
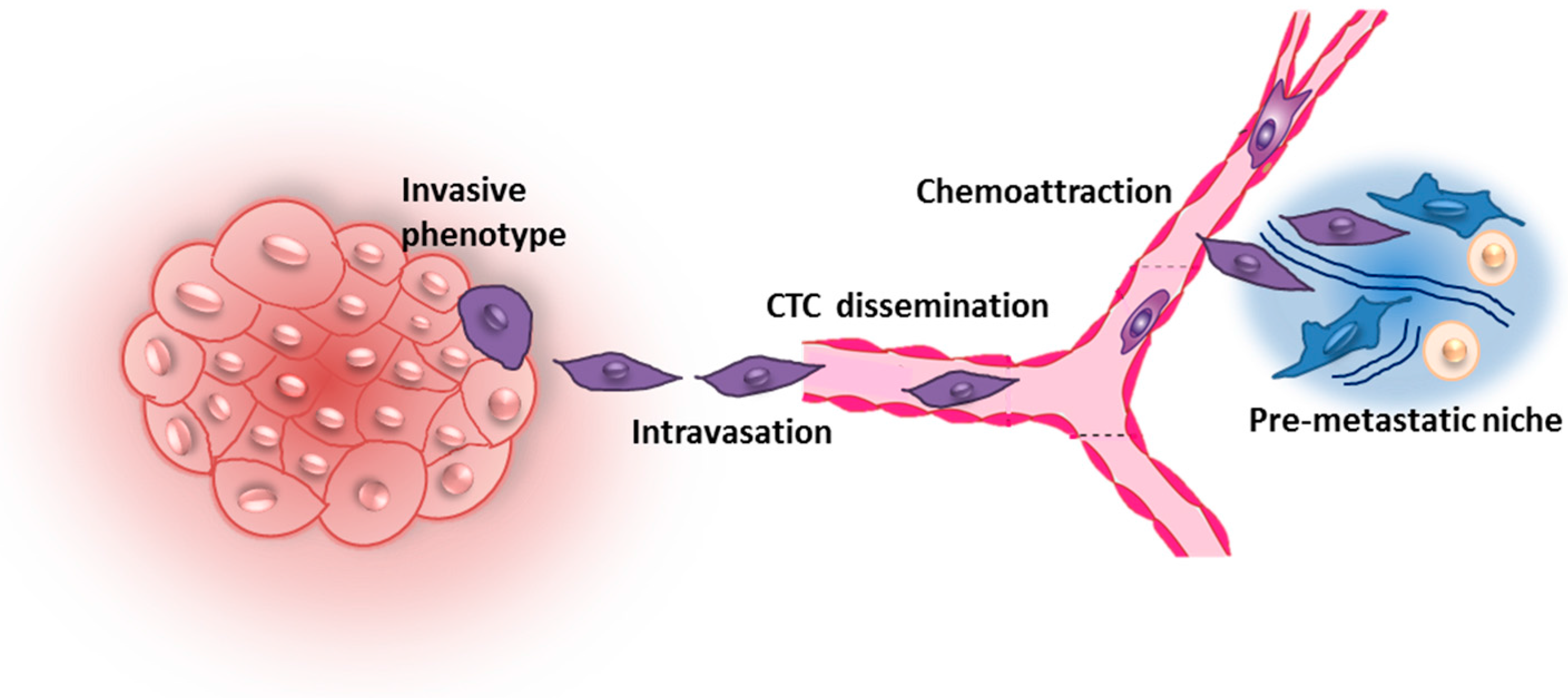
2. Early Steps of Metastatic Dissemination
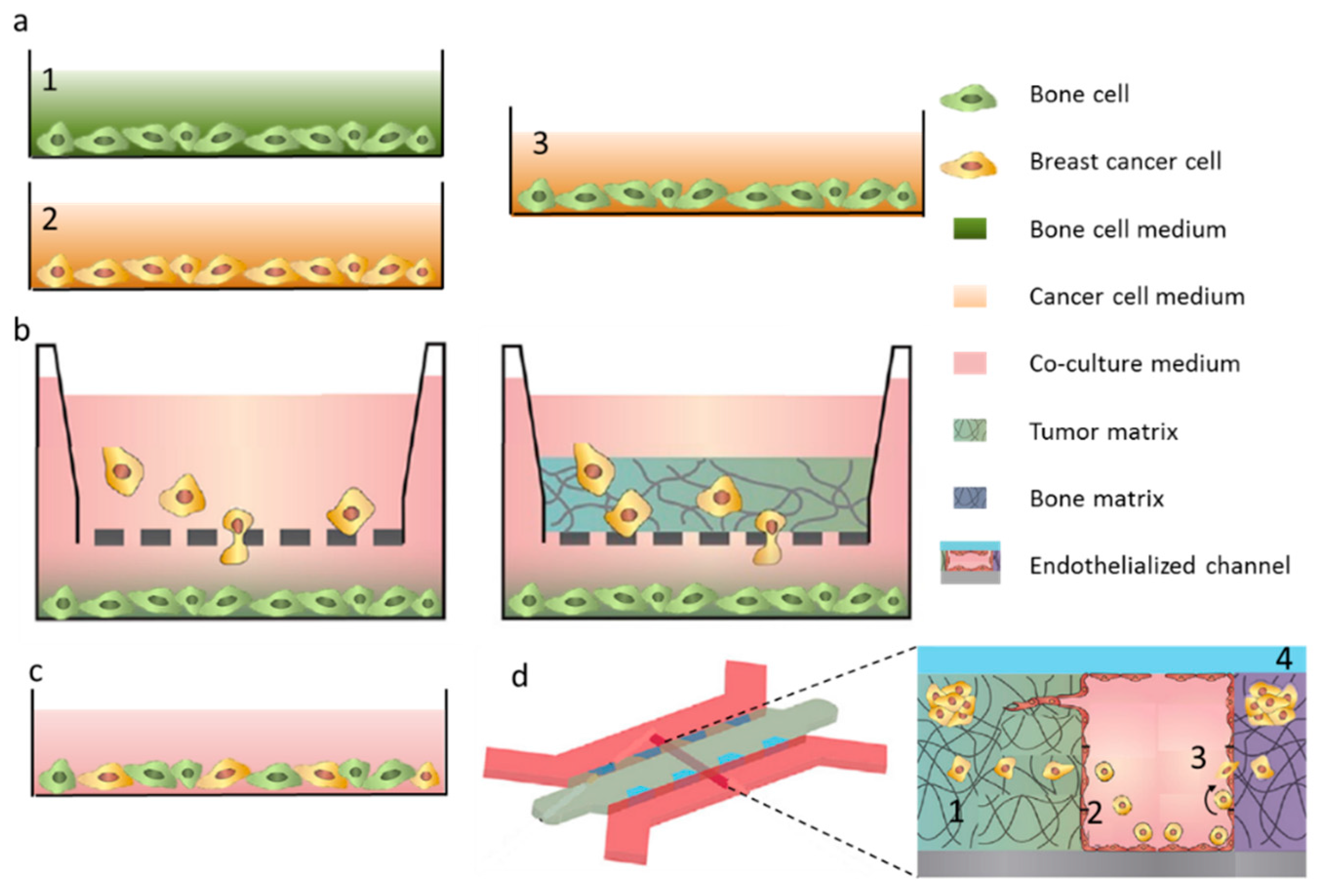
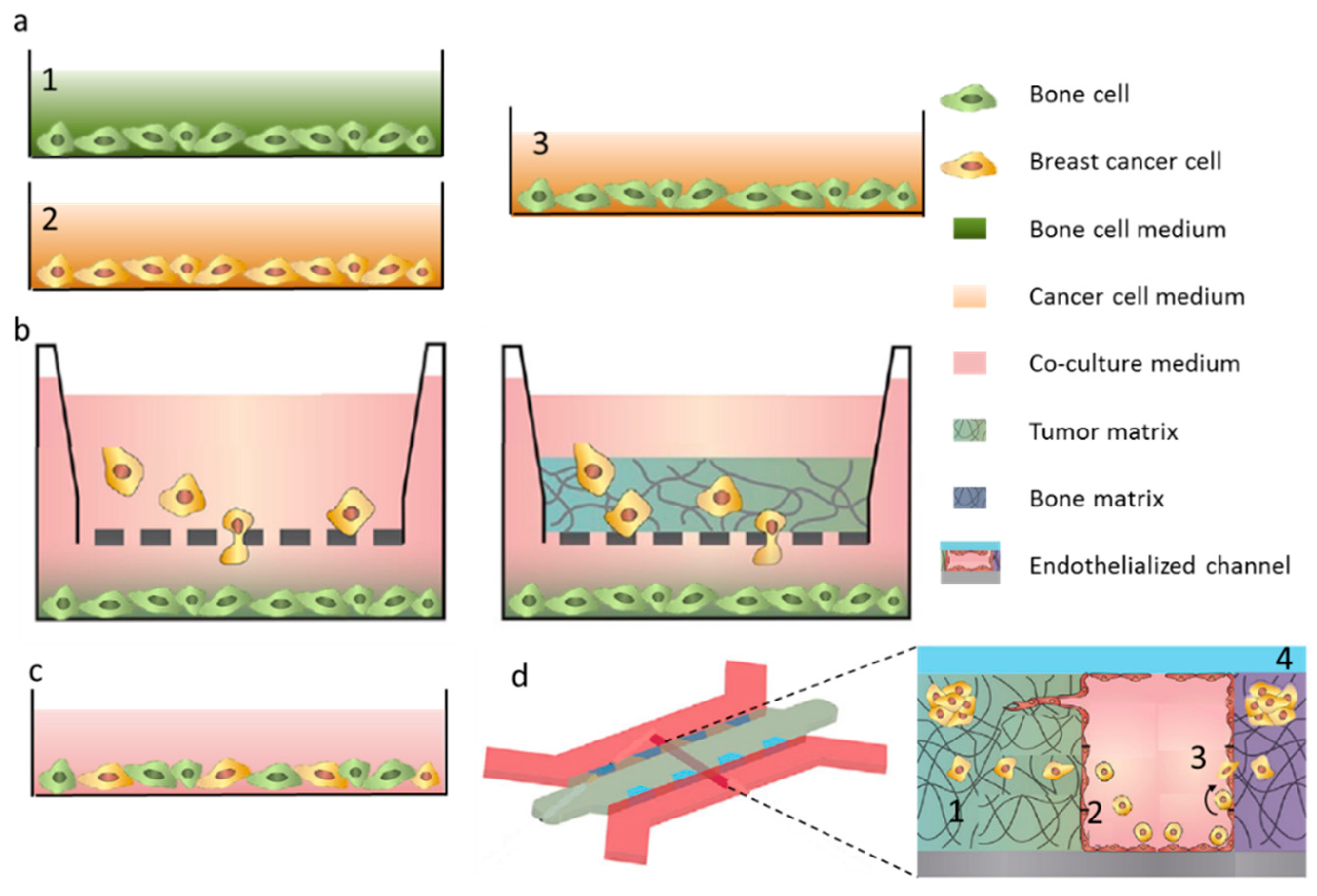
3. Breast Cancer Cell Extravasation to Bone—In Vitro Modeling of Key Mechanisms
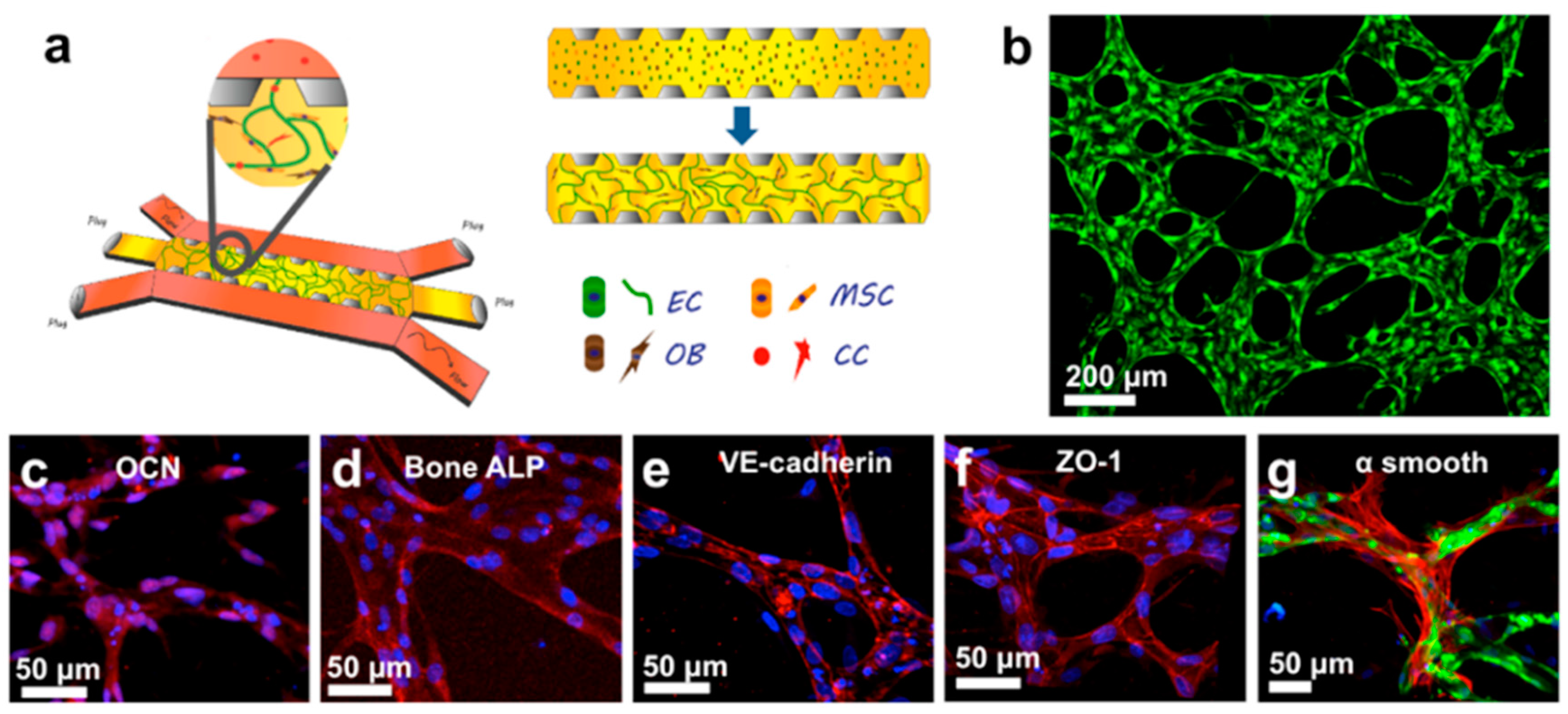
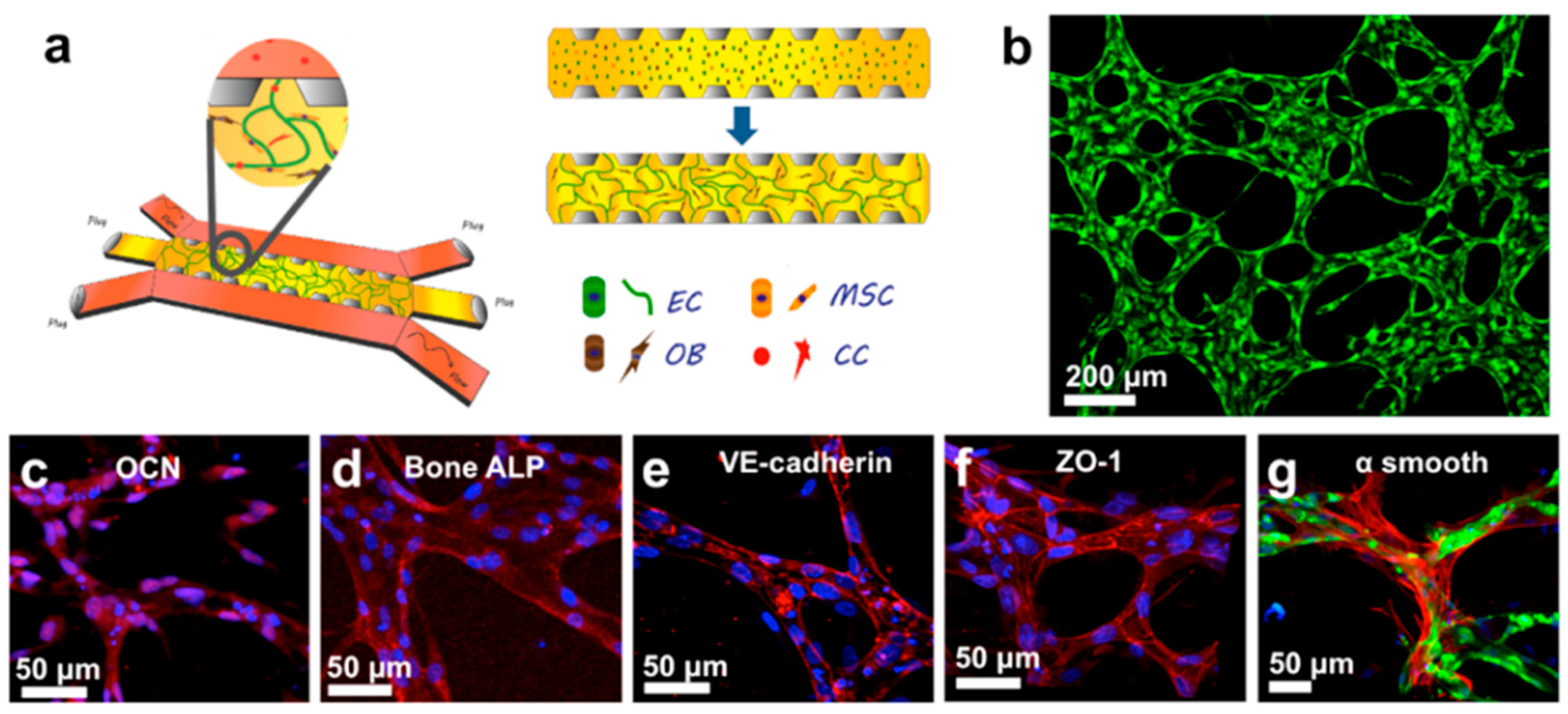
4. Bone Tissue Colonization
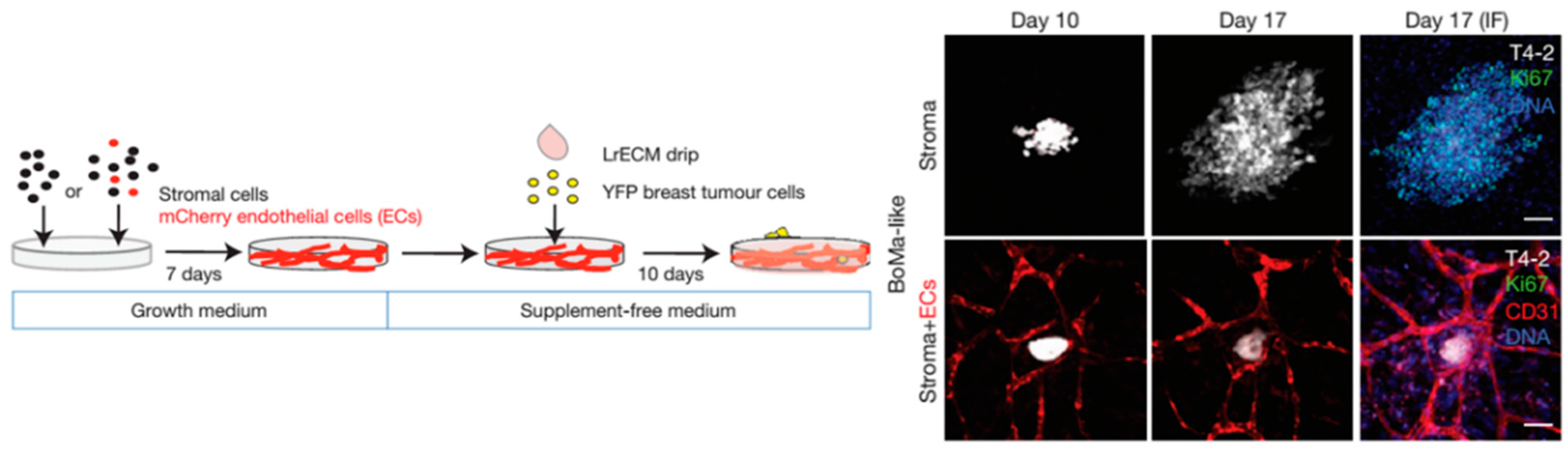
4.1. From Early Invasion to Cancer Cell Dormancy
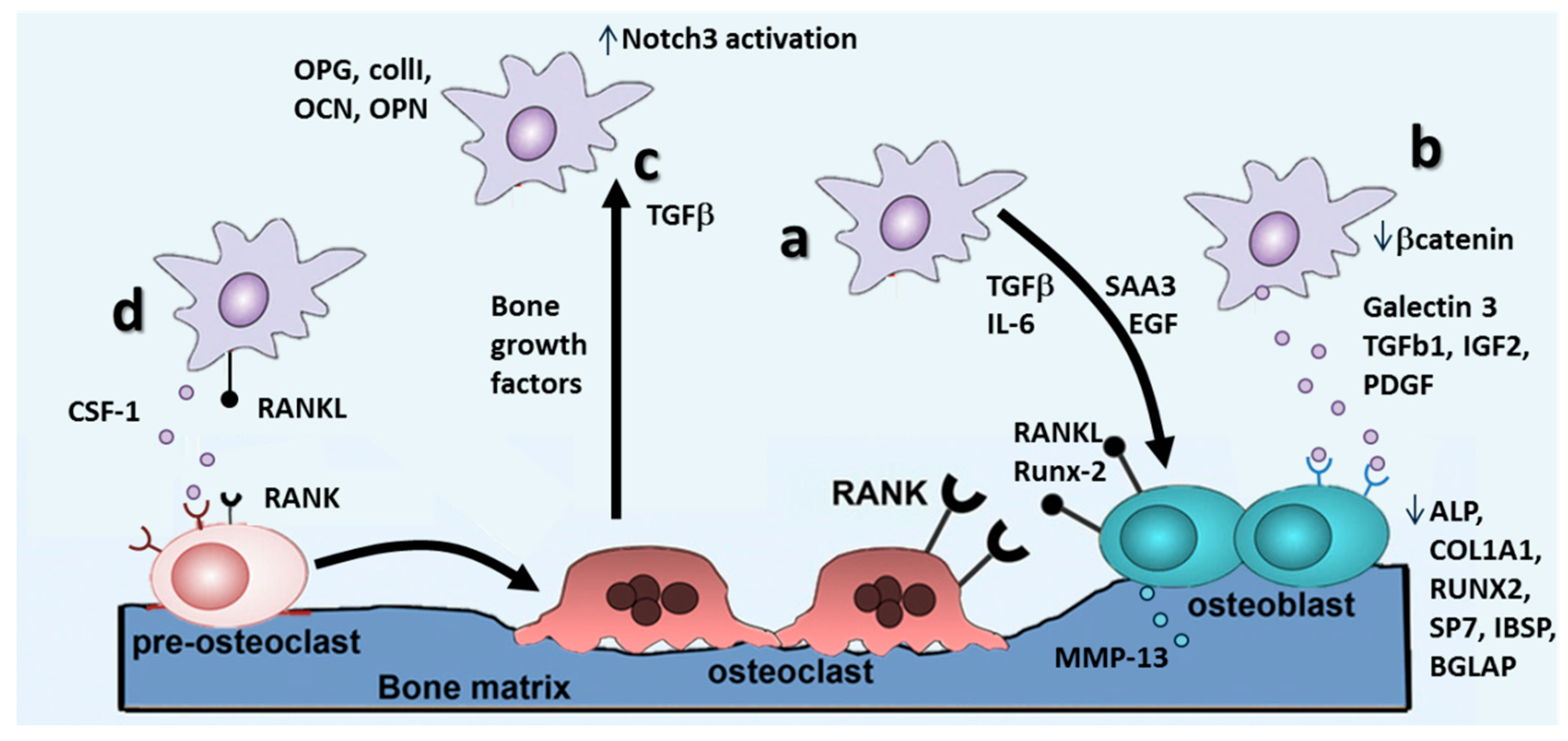
4.2. Metastasis Growth and Formation of Osteolytic Lesions
The RANKL/RANK/OPG Pathway
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Coleman, R.E. Skeletal complications of malignancy. Cancer 1997, 80, 1588–1594. [Google Scholar] [CrossRef]
- Kroep, J.R.; Charehbili, A.; Coleman, R.E.; Aft, R.L.; Hasegawa, Y.; Winter, M.C.; Weilbaecher, K.; Akazawa, K.; Hinsley, S.; Putter, H.; et al. Effects of neoadjuvant chemotherapy with or without zoledronic acid on pathological response: A meta-analysis of randomised trials. Eur. J. Cancer 2016, 54, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Reymond, N.; d’Agua, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Fazilaty, H.; Mehdipour, P. Genetics of breast cancer bone metastasis: A sequential multistep pattern. Clin. Exp. Metastasis 2014, 31, 595–612. [Google Scholar] [CrossRef] [PubMed]
- Futakuchi, M.; Fukamachi, K.; Suzui, M. Heterogeneity of tumor cells in the bone microenvironment: Mechanisms and therapeutic targets for bone metastasis of prostate or breast cancer. Adv. Drug Deliv. Rev. 2016, 99, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Simmons, J.K.; Hildreth, B.E.; Supsavhad, W.; Elshafae, S.M.; Hassan, B.B.; Dirksen, W.P.; Toribio, R.E.; Rosol, T.J. Animal models of bone metastasis. Vet. Pathol. 2015, 52, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Fan, Y.; Zhang, H.; Zhao, Z.; Hao, Y.; Li, J.; Sun, C.; Yang, J.; Yang, Z.; Yang, X.; et al. Differential TGF-β pathway targeting by miR-122 in humans and mice affects liver cancer metastasis. Nat. Commun. 2016, 7, 11012–11025. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.P.; Sutton, P.A.; Winiarski, B.K.; Fenwick, S.W.; Malik, H.Z.; Vimalachandran, D.; Tweedle, E.M.; Costello, E.; Palmer, D.H.; Park, B.K.; et al. From mice to men: Murine models of colorectal cancer for use in translational research. Crit. Rev. Oncol. Hematol. 2016, 98, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Shologu, N.; Szegezdi, E.; Lowery, A.; Kerin, M.; Pandit, A.; Zeugolis, D.I. Recreating complex pathophysiologies in vitro with extracellular matrix surrogates for anticancer therapeutics screening. Drug Discov. Today 2016. [Google Scholar] [CrossRef] [PubMed]
- Sanford, K.K.; Barker, B.E.; Woods, M.W.; Parshad, R.; Law, L.W. Search for “indicators” of neoplastic conversion in vitro. J. Natl. Cancer Inst. 1967, 39, 705–733. [Google Scholar]
- Haskell, C.M.; Sullivan, A. Comparative survival in tissue culture of normal and neoplastic human cells exposed to adriamycin. Cancer Res. 1974, 34, 2991–2994. [Google Scholar] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Katt, M.E.; Placone, A.L.; Wong, A.D.; Xu, Z.S.; Searson, P.C. In vitro tumor models: Advantages, disadvantages, variables, and selecting the right platform. Front. Bioeng. Biotechnol. 2016, 4, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Bersini, S.; Jeon, J.S.; Moretti, M.; Kamm, R.D. In vitro models of the metastatic cascade: From local invasion to extravasation. Drug Discov. Today 2014, 19, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Vogler, E.A.; Sosnoski, D.M.; Mastro, A.M. In vitro mimics of bone remodeling and the vicious cycle of cancer in bone. J. Cell. Physiol. 2014, 229, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Holen, I.; Nutter, F.; Wilkinson, J.M.; Evans, C.A.; Avgoustou, P.; Ottewell, P.D. Human breast cancer bone metastasis in vitro and in vivo: A novel 3D model system for studies of tumour cell-bone cell interactions. Clin. Exp. Metastasis 2015, 32, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Horas, K.; Zheng, Y.; Zhou, H.; Seibel, M.J. Animal models for breast cancer metastasis to bone: Opportunities and limitations. Cancer Investig. 2015, 33, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, A.V. In vitro microenvironments to study breast cancer bone colonisation. Adv. Drug Deliv. Rev. 2014, 79, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Hess, K.R.; Pusztai, L.; Buzdar, A.U.; Hortobagyi, G.N. Estrogen receptors and distinct patterns of breast cancer relapse. Breast Cancer Res. Treat. 2003, 78, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Sasser, A.K.; Sullivan, N.J.; Studebaker, A.W.; Hendey, L.F.; Axel, A.E.; Hall, B.M. Interleukin-6 is a potent growth factor for ER-α-positive human breast cancer. FASEB J. 2007, 21, 3763–3770. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jarrett, J.; Huang, C.C.; Satcher, R.L.; Levenson, A.S. Identification of estrogen-responsive genes involved in breast cancer metastases to the bone. Clin. Exp. Metastasis 2007, 24, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Koenders, P.G.; Beex, L.V.; Langens, R.; Kloppenborg, P.W.; Smals, A.G.; Benraad, T.J. Steroid hormone receptor activity of primary human breast cancer and pattern of first metastasis. The breast cancer study group. Breast Cancer Res. Treat. 1991, 18, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, J.; Van Der Valk, S.W.; Hilkens, J. A mechanism for inhibition of E-cadherin-mediated cell-cell adhesion by the membrane-associated mucin episialin/MUC1. Mol. Biol. Cell 1996, 7, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Bao, Z.; Li, J. Expression of MacMARCKS restores cell adhesion to ICAM-1-coated surface. Cell Adhes. Commun. 2000, 7, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.R.; Sikes, R.A.; Nicholson, B.E.; Sun, Y.X.; Pienta, K.J.; Taichman, R.S. Cancer cells homing to bone: The significance of chemotaxis and cell adhesion. Cancer Treat. Res. 2004, 118, 291–309. [Google Scholar] [PubMed]
- Pohorelic, B.; Singh, R.; Parkin, S.; Koro, K.; Yang, A.D.; Egan, C.; Magliocco, A. Role of SRC in breast cancer cell migration and invasion in a breast cell/bone-derived cell microenvironment. Breast Cancer Res. Treat. 2012, 133, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Kipps, T.J. CXCR4: A key receptor in the crosstalk between tumor cells and their microenvironment. Blood 2006, 107, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Uygur, B.; Wu, W.S. Slug promotes prostate cancer cell migration and invasion via CXCR4/CXCL12 axis. Mol. Cancer 2011, 10, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, T. CXCL12/SDF-1 and CXCR4. Front. Immunol. 2015, 6, 301–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, D.; Huang, J.; Gong, J. Bone morphogenetic protein 4 (BMP4) is required for migration and invasion of breast cancer. Mol. Cell. Biochem. 2012, 363, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Olbrich, T.; Ziegler, E.; Turk, G.; Schubert, A.; Emons, G.; Grundker, C. Kisspeptin-10 inhibits bone-directed migration of GPR54-positive breast cancer cells: Evidence for a dose-window effect. Gynecol. Oncol. 2010, 119, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Makri, A.; Pissimissis, N.; Lembessis, P.; Polychronakos, C.; Koutsilieris, M. The kisspeptin (KISS-1)/GPR54 system in cancer biology. Cancer Treat. Rev. 2008, 34, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Hinsche, O.; Girgert, R.; Emons, G.; Grundker, C. Estrogen receptor β selective agonists reduce invasiveness of triple-negative breast cancer cells. Int. J. Oncol. 2015, 46, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Marriott, I.; Gray, D.L.; Rati, D.M.; Fowler, V.G.; Stryjewski, M.E.; Levin, L.S.; Hudson, M.C.; Bost, K.L. Osteoblasts produce monocyte chemoattractant protein-1 in a murine model of staphylococcus aureus osteomyelitis and infected human bone tissue. Bone 2005, 37, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Molloy, A.P.; Martin, F.T.; Dwyer, R.M.; Griffin, T.P.; Murphy, M.; Barry, F.P.; O’Brien, T.; Kerin, M.J. Mesenchymal stem cell secretion of chemokines during differentiation into osteoblasts, and their potential role in mediating interactions with breast cancer cells. Int. J. Cancer 2009, 124, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Bussard, K.M.; Venzon, D.J.; Mastro, A.M. Osteoblasts are a major source of inflammatory cytokines in the tumor microenvironment of bone metastatic breast cancer. J. Cell. Biochem. 2010, 111, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Sceneay, J.; Smyth, M.J.; Moller, A. The pre-metastatic niche: Finding common ground. Cancer Metastasis Rev. 2013, 32, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Chiovaro, F.; Martina, E.; Bottos, A.; Scherberich, A.; Hynes, N.E.; Chiquet-Ehrismann, R. Transcriptional regulation of tenascin-W by TGF-β signaling in the bone metastatic niche of breast cancer cells. Int. J. Cancer 2015, 137, 1842–1854. [Google Scholar] [CrossRef] [PubMed]
- Wobus, M.; List, C.; Dittrich, T.; Dhawan, A.; Duryagina, R.; Arabanian, L.S.; Kast, K.; Wimberger, P.; Stiehler, M.; Hofbauer, L.C.; et al. Breast carcinoma cells modulate the chemoattractive activity of human bone marrow-derived mesenchymal stromal cells by interfering with CXCL12. Int. J. Cancer 2015, 136, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Massague, J. Extracellular matrix players in metastatic niches. EMBO J. 2012, 31, 254–256. [Google Scholar] [CrossRef] [PubMed]
- Scherberich, A.; Tucker, R.P.; Samandari, E.; Brown-Luedi, M.; Martin, D.; Chiquet-Ehrismann, R. Murine tenascin-W: A novel mammalian tenascin expressed in kidney and at sites of bone and smooth muscle development. J. Cell Sci. 2004, 117, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, K.E.; Trzaska, K.A.; Fernandes, H.; Bryan, M.; Taborga, M.; Srinivas, V.; Packman, K.; Patel, P.S.; Rameshwar, P. Mesenchymal stem cells in early entry of breast cancer into bone marrow. PLoS ONE 2008, 3, e2563. [Google Scholar] [CrossRef] [PubMed]
- Bersini, S.; Jeon, J.S.; Dubini, G.; Arrigoni, C.; Chung, S.; Charest, J.L.; Moretti, M.; Kamm, R.D. A microfluidic 3D in vitro model for specificity of breast cancer metastasis to bone. Biomaterials 2014, 35, 2454–2461. [Google Scholar] [CrossRef] [PubMed]
- Varani, K.; Vincenzi, F.; Targa, M.; Paradiso, B.; Parrilli, A.; Fini, M.; Lanza, G.; Borea, P.A. The stimulation of A3 adenosine receptors reduces bone-residing breast cancer in a rat preclinical model. Eur. J. Cancer 2013, 49, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.S.; Bersini, S.; Gilardi, M.; Dubini, G.; Charest, J.L.; Moretti, M.; Kamm, R.D. Human 3D vascularized organotypic microfluidic assays to study breast cancer cell extravasation. Proc. Natl. Acad. Sci. USA 2015, 112, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, L.A.; Fournier, P.G.; Chirgwin, J.M.; Guise, T.A. Molecular biology of bone metastasis. Mol. Cancer Ther. 2007, 6, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Rajski, M.; Vogel, B.; Baty, F.; Rochlitz, C.; Buess, M. Global gene expression analysis of the interaction between cancer cells and osteoblasts to predict bone metastasis in breast cancer. PLoS ONE 2012, 7, e29743. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, J.; Fatatis, A. Osteoblasts modulate Ca2+ signaling in bone-metastatic prostate and breast cancer cells. Clin. Exp. Metastasis 2009, 26, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Marlow, R.; Honeth, G.; Lombardi, S.; Cariati, M.; Hessey, S.; Pipili, A.; Mariotti, V.; Buchupalli, B.; Foster, K.; Bonnet, D.; et al. A novel model of dormancy for bone metastatic breast cancer cells. Cancer Res. 2013, 73, 6886–6899. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zuo, D.; Wang, M.; Zhang, Y.; Yu, M.; Yang, J.; Yao, Z. Effect of truncated neurokinin-1 receptor expression changes on the interaction between human breast cancer and bone marrow-derived mesenchymal stem cells. Genes Cells 2014, 19, 676–691. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Kosaka, N.; Tominaga, N.; Yoshioka, Y.; Takeshita, F.; Takahashi, R.U.; Yoshida, M.; Tsuda, H.; Tamura, K.; Ochiya, T. Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci. Signal. 2014. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.K.; Bliss, S.A.; Patel, S.A.; Taborga, M.; Dave, M.A.; Gregory, L.A.; Greco, S.J.; Bryan, M.; Patel, P.S.; Rameshwar, P. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011, 71, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Sosnoski, D.M.; Norgard, R.J.; Grove, C.D.; Foster, S.J.; Mastro, A.M. Dormancy and growth of metastatic breast cancer cells in a bone-like microenvironment. Clin. Exp. Metastasis 2015, 32, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Sosnoski, D.M.; Mastro, A.M. Breast cancer metastasis to the bone: Mechanisms of bone loss. Breast Cancer Res. 2010, 12, 215. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Bryant, R.; Roudier, M.; Branstetter, D.G.; Dougall, W.C. Rankl inhibition combined with tamoxifen treatment increases anti-tumor efficacy and prevents tumor-induced bone destruction in an estrogen receptor-positive breast cancer bone metastasis model. Breast Cancer Res. Treat. 2012, 135, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.; Mancini, S.; Cipollone, J.; Kappelhoff, R.; Roskelley, C.; Overall, C. Microarray and proteomic analysis of breast cancer cell and osteoblast co-cultures: Role of osteoblast matrix metalloproteinase (MMP)-13 in bone metastasis. J. Biol. Chem. 2011, 286, 34271–34285. [Google Scholar] [CrossRef] [PubMed]
- David, L.W.; Theresa, A.G. Cancer-associated muscle weakness: What’s bone got to do with it. Bonedey Rep. 2015. [Google Scholar] [CrossRef]
- Chen, Y.; Shi, H.Y.; Stock, S.R.; Stern, P.H.; Zhang, M. Regulation of breast cancer-induced bone lesions by β-catenin protein signaling. J. Biol. Chem. 2011, 286, 42575–42584. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Kho, D.H.; Yanagawa, T.; Harazono, Y.; Gao, X.; Hogan, V.; Raz, A. Galectin-3 inhibits osteoblast differentiation through Notch signaling. Neoplasia 2014, 16, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, H.; Ikeda, S.; Fahey, F.; Bielenberg, D.; Smits, P.; Hauschka, P.V. Notch3 in human breast cancer cell lines regulates osteoblast-cancer cell interactions and osteolytic bone metastasis. Am. J. Pathol. 2010, 177, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Du, X.; Li, D.M.; Kong, P.Z.; Sun, Y.; Liu, P.F.; Wang, Q.S.; Feng, Y.M. ITGBL1 is a RUNX2 transcriptional target and promotes breast cancer bone metastasis by activating the TGF-β signaling pathway. Cancer Res. 2015, 75, 3302–3313. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Maeda, K.; Ishihara, A.; Uehara, S.; Kobayashi, Y. Regulatory mechanism of osteoclastogenesis by RANKl and Wnt signals. Front. Biosci. 2011, 16, 21–30. [Google Scholar] [CrossRef]
- Yao, G.Q.; Sun, B.H.; Weir, E.C.; Insogna, K.L. A role for cell-surface CSF-1 in osteoblast-mediated osteoclastogenesis. Calcif. Tissue Int. 2002, 70, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Day, C.J.; Morrison, N.A. MCP-1 is induced by receptor activator of nuclear factor-κB ligand, promotes human osteoclast fusion, and rescues granulocyte macrophage colony-stimulating factor suppression of osteoclast formation. J. Biol. Chem. 2005, 280, 16163–16169. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKl. Proc. Natl. Acad. Sci. USA 1998, 95, 3597–3602. [Google Scholar] [CrossRef] [PubMed]
- Shalhoub, V.; Faust, J.; Boyle, W.J.; Dunstan, C.R.; Kelley, M.; Kaufman, S.; Scully, S.; Van, G.; Lacey, D.L. Osteoprotegerin and osteoprotegerin ligand effects on osteoclast formation from human peripheral blood mononuclear cell precursors. J. Cell. Biochem. 1999, 72, 251–261. [Google Scholar] [CrossRef]
- Liverani, C.; Mercatali, L.; Spadazzi, C.; La Manna, F.; De Vita, A.; Riva, N.; Calpona, S.; Ricci, M.; Bongiovanni, A.; Gunelli, E.; et al. CSF-1 blockade impairs breast cancer osteoclastogenic potential in co-culture systems. Bone 2014, 66, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Nicolin, V.; Bortul, R.; Bareggi, R.; Baldini, G.; Martinelli, B.; Narducci, P. Breast adenocarcinoma MCF-7 cell line induces spontaneous osteoclastogenesis via a RANK-ligand-dependent pathway. Acta Histochem. 2008, 110, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni, C.; De Luca, P.; Gilardi, M.; Previdi, S.; Broggini, M.; Moretti, M. Direct but not indirect co-culture with osteogenically differentiated human bone marrow stromal cells increases RANKl/OPG ratio in human breast cancer cells generating bone metastases. Mol. Cancer 2014, 13, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, C.; Gao, X.; Welte, T.; Muscarella, A.M.; Tian, L.; Zhao, H.; Zhao, Z.; Du, S.; Tao, J.; et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell 2015, 27, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ning, L.L.; Wang, Z.Y.; Li, H.T.; Qiao, D.; Yao, Y.; Qin, H.L. Calcitonin gene-related peptide inhibits osteolytic factors induced by osteoblast in co-culture system with breast cancer. Cell Biochem. Biophys. 2014, 70, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Chow, S.O.; Boernert, K.; Basel, D.; Mikuscheva, A.; Kim, S.; Fong-Yee, C.; Trivedi, T.; Buttgereit, F.; Sutherland, R.L.; et al. Direct crosstalk between cancer and osteoblast lineage cells fuels metastatic growth in bone via auto-amplification of IL-6 and RANKl signaling pathways. J. Bone Miner. Res. 2014, 29, 1938–1949. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Jia, X.; Xiao, G.; Kang, Y.; Partridge, N.C.; Qin, L. EGF-like ligands stimulate osteoclastogenesis by regulating expression of osteoclast regulatory factors by osteoblasts: Implications for osteolytic bone metastases. J. Biol. Chem. 2007, 282, 26656–26664. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, P.; Suva, L.J.; Welch, D.R.; Donahue, H.J. Osteoprotegrin and the bone homing and colonization potential of breast cancer cells. J. Cell. Biochem. 2008, 103, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.A.; Chen, C.S. Cell biology. Deconstructing dimensionality. Science 2013, 339, 402–404. [Google Scholar] [CrossRef] [PubMed]
- Bersini, S.; Gilardi, M.; Arrigoni, C.; Talo, G.; Zamai, M.; Zagra, L.; Caiolfa, V.; Moretti, M. Human in vitro 3D co-culture model to engineer vascularized bone-mimicking tissues combining computational tools and statistical experimental approach. Biomaterials 2016, 76, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Bongio, M.; Lopa, S.; Gilardi, M.; Bersini, S.; Moretti, M. A 3D vascularized bone remodeling model combining osteoblasts and osteoclasts in a CaP nanoparticle-enriched matrix. Nanomedicine 2016, 11, 1073–1091. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Platelets guide the formation of early metastatic niches. Proc. Natl. Acad. Sci. USA 2014, 111, 3053–3061. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, A.; Brooks, M.W.; Houshyar, S.; Reinhardt, F.; Ardolino, M.; Fessler, E.; Chen, M.B.; Krall, J.A.; DeCock, J.; Zervantonakis, I.K.; et al. Neutrophils suppress intraluminal NK cell-mediated tumor cell clearance and enhance extravasation of disseminated carcinoma cells. Cancer Discov. 2016, 6, 630–649. [Google Scholar] [CrossRef] [PubMed]
- Whittle, J.R.; Lewis, M.T.; Lindeman, G.J.; Visvader, J.E. Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Res. 2015, 17, 17–30. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step of Metastatic Dissemination | Mechanism Studied | Type of In Vitro Model Used | Molecules Involved | References |
|---|---|---|---|---|
| Early steps of bone metastasis | Acquisition of bone-aggressive phenotype | 2D direct co-culture | IL-6 | [21] |
| ER-α, ER-β | [22] | |||
| Chemotactic migration | Transwell | Src-kinase | [26] | |
| CXCR-4/CXCL-12 | [31,32,34] | |||
| CCL-2 | [36,37] | |||
| Pre-metastatic niche | Transwell, conditioned medium | Tenascin W | [39] | |
| TGF-β1, CXCL-12 | [40] | |||
| Extravasation | Transedothelial migration | Transwell | SDF-1α, CXCR-4, Tac-1 | [44] |
| Microfluidic | CXCR-2/CXCL-5, adenosine | [45,47] | ||
| Bone colonization | Early invasion | 2D direct co-culture | IL-6 | [50] |
| Ca2+ | [51] | |||
| Dormancy | 3D direct co-culture | TSP-1, TGF-β, p38 | [50,53] | |
| 2D direct co-culture | Tac-1, NRK-1 | [54] | ||
| miRNAs from BMCs | [55,56] | |||
| Cancer cell growth in the bone | 2D and 3D direct co-culture | ILα, TNF-β | [57] | |
| E-cadh, N-cadh | [73] | |||
| Interaction with osteoblasts | 2D direct co-culture | MMP-13 | [60] | |
| β-catenin | [61] | |||
| Transwell, 2D direct co-culture | Galectin-3, Notch, TGF-β | [62,63] | ||
| Osteoclast maturation | Conditioned medium | Integrin β1, TGF-β | [64] | |
| CSF-1 | [70] | |||
| 2D and 3D direct co-culture | RANKL/RANK, CGRP, IL-6 | [71,72,74,75] | ||
| EGF, MCP-1 | [76] | |||
| OPG | [77] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arrigoni, C.; Bersini, S.; Gilardi, M.; Moretti, M. In Vitro Co-Culture Models of Breast Cancer Metastatic Progression towards Bone. Int. J. Mol. Sci. 2016, 17, 1405. https://doi.org/10.3390/ijms17091405
Arrigoni C, Bersini S, Gilardi M, Moretti M. In Vitro Co-Culture Models of Breast Cancer Metastatic Progression towards Bone. International Journal of Molecular Sciences. 2016; 17(9):1405. https://doi.org/10.3390/ijms17091405
Chicago/Turabian StyleArrigoni, Chiara, Simone Bersini, Mara Gilardi, and Matteo Moretti. 2016. "In Vitro Co-Culture Models of Breast Cancer Metastatic Progression towards Bone" International Journal of Molecular Sciences 17, no. 9: 1405. https://doi.org/10.3390/ijms17091405