
Genome-Wide Discriminatory Information Patterns of Cytosine DNA Methylation


Abstract
:1. Introduction
2. Results
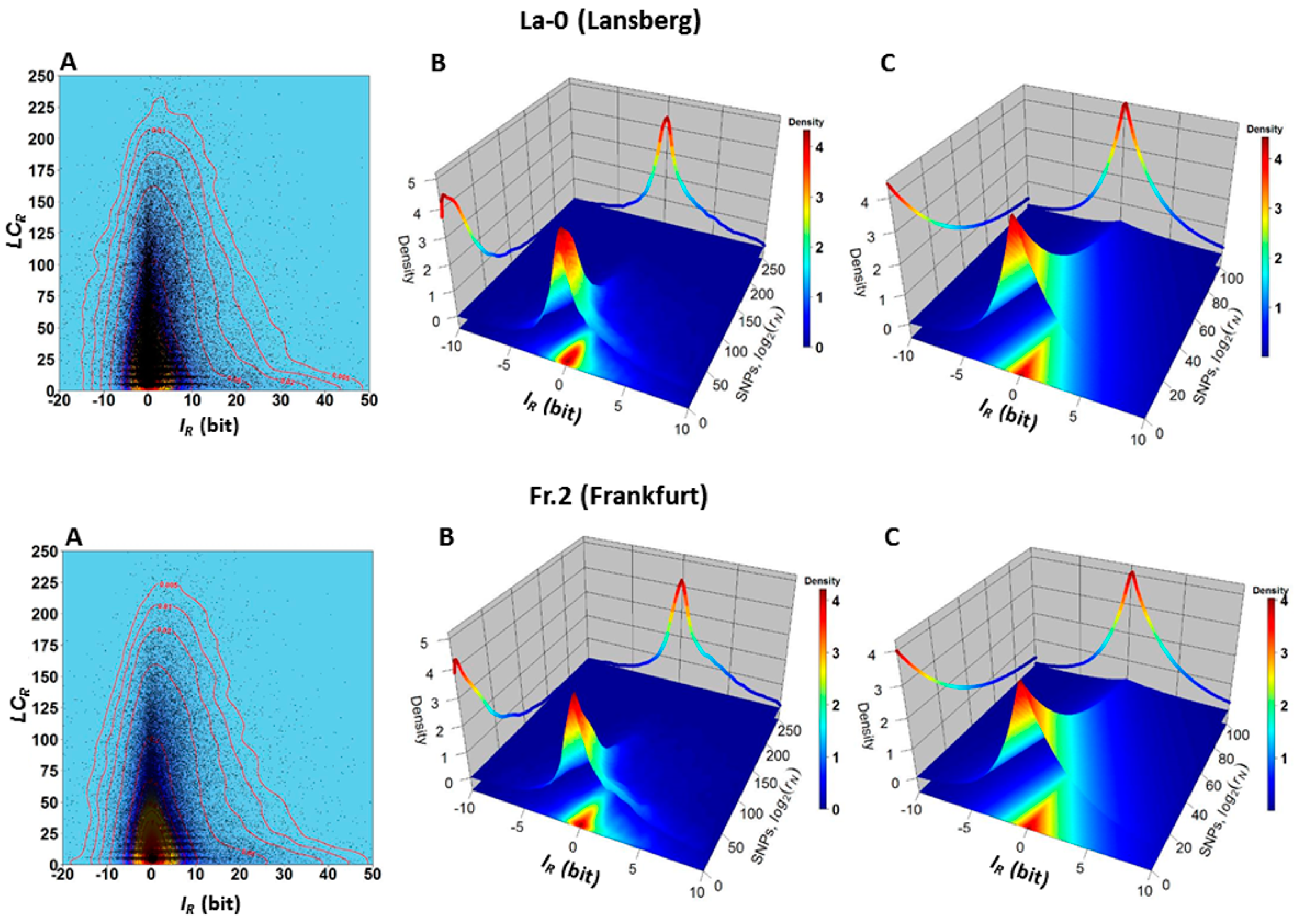
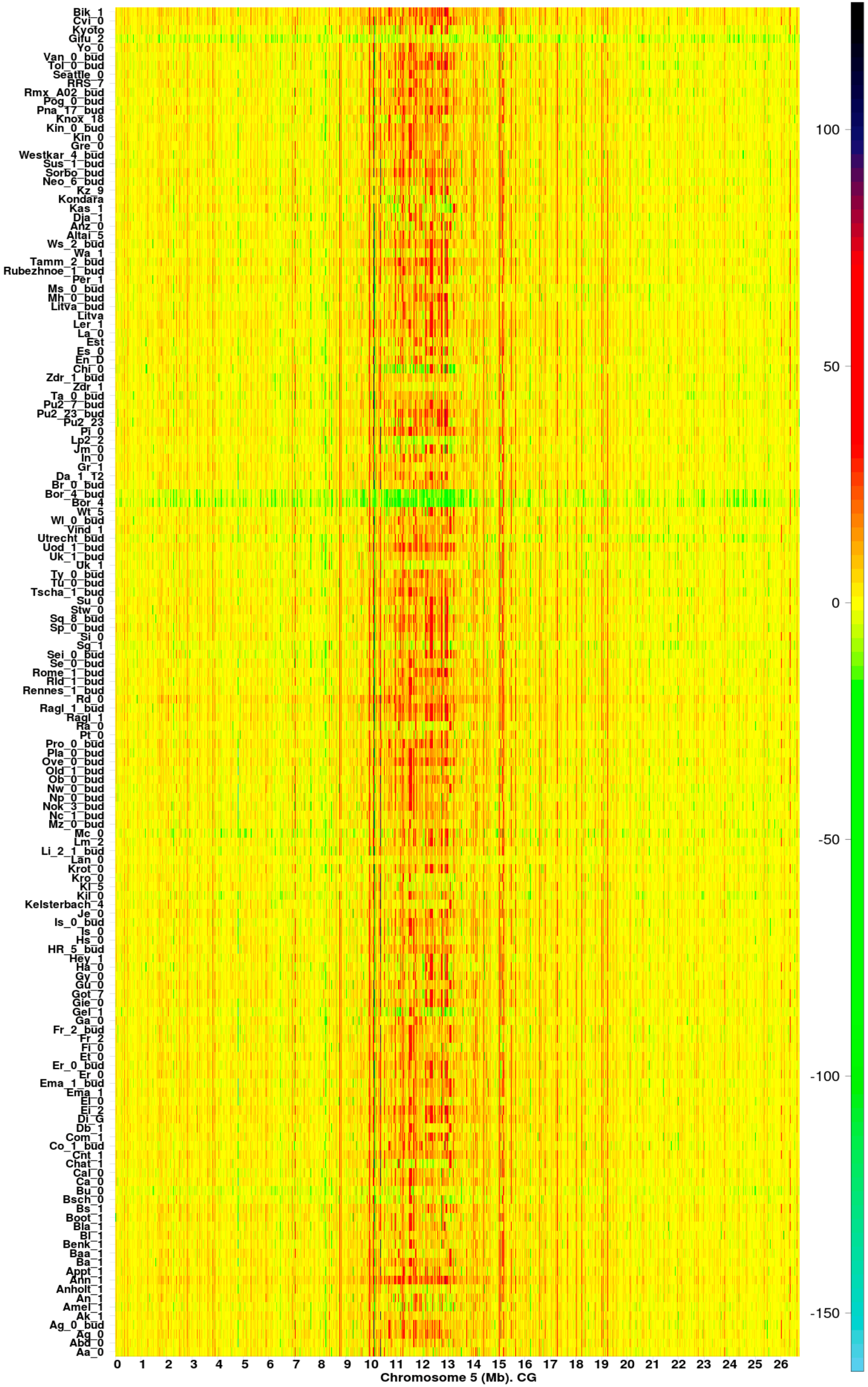
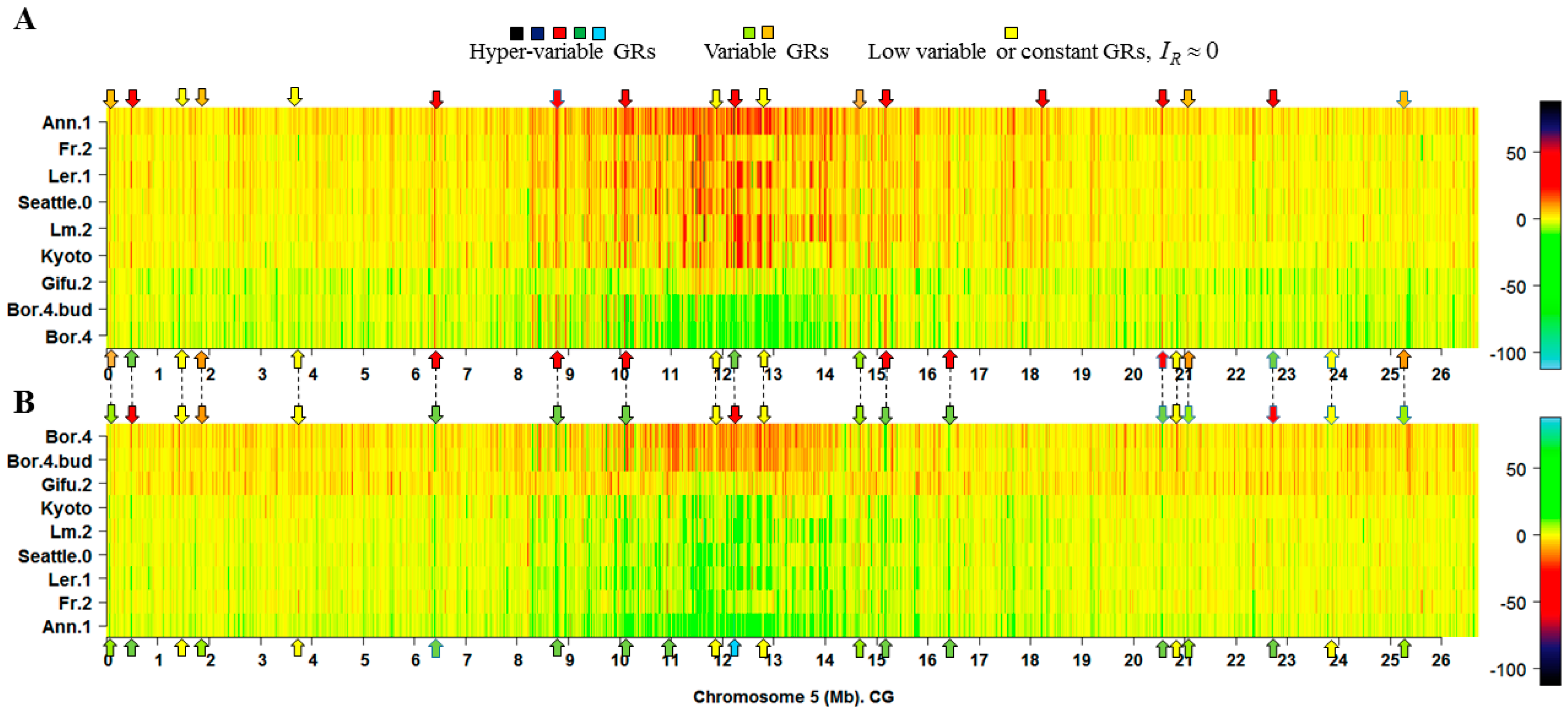
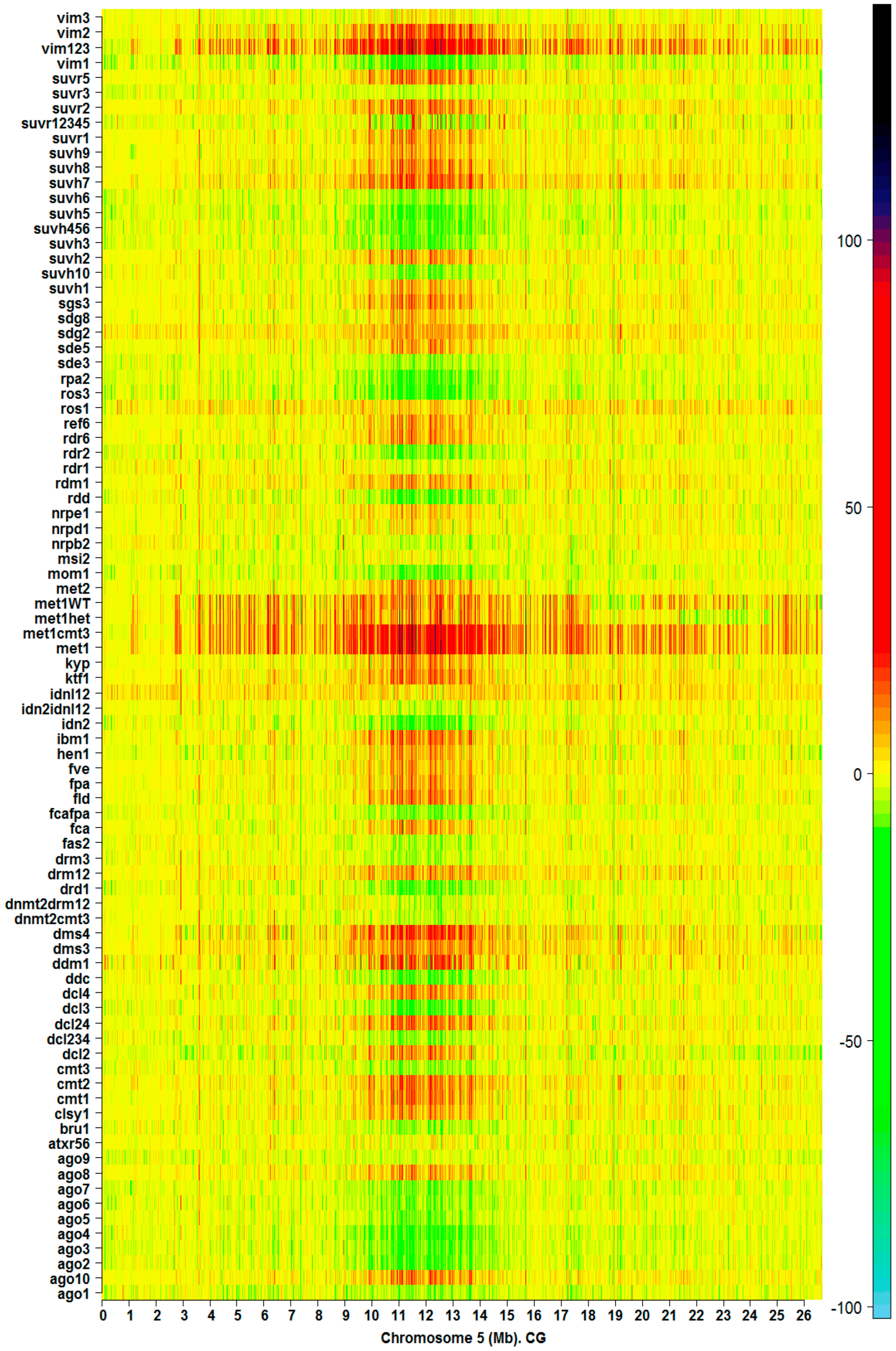
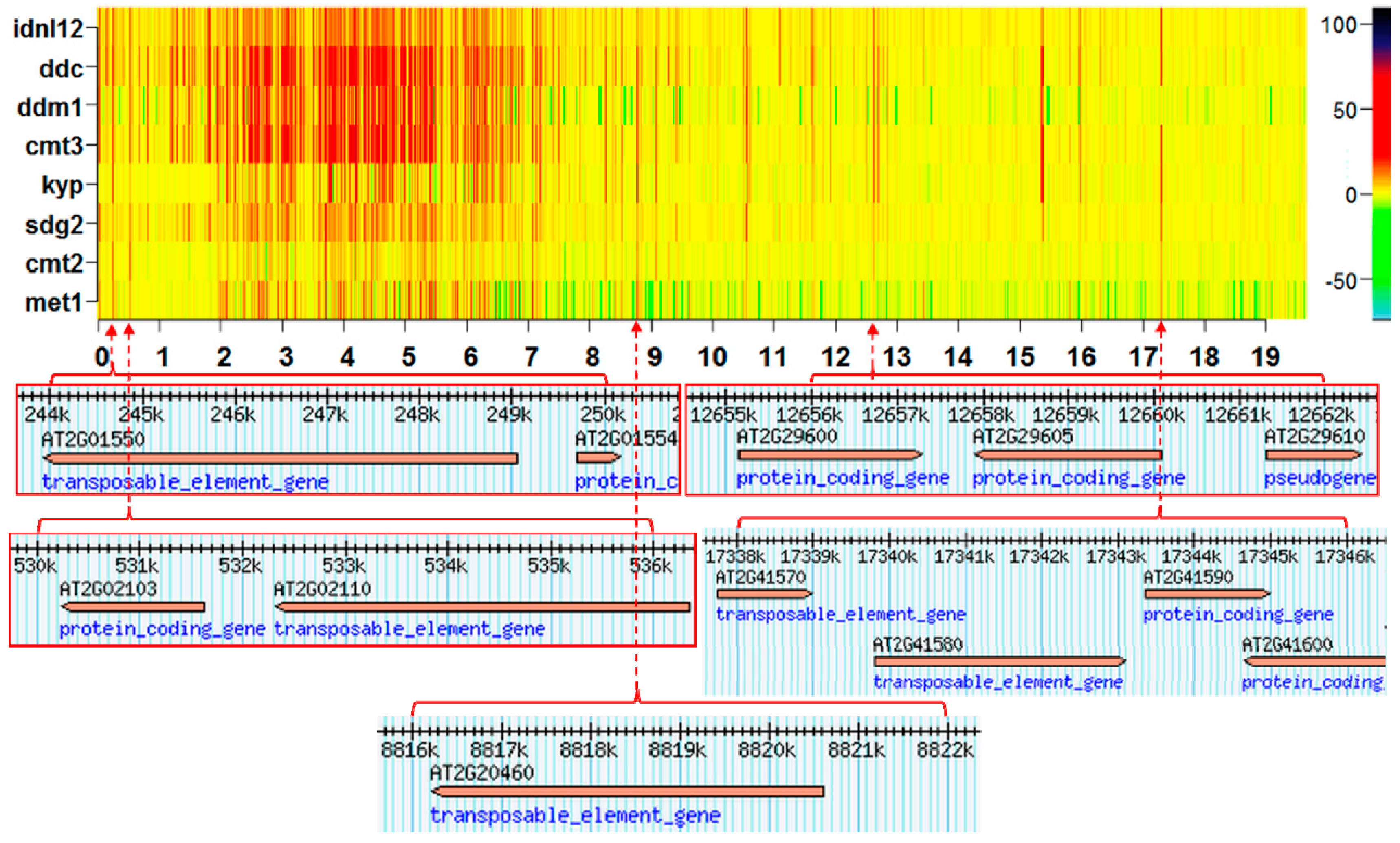
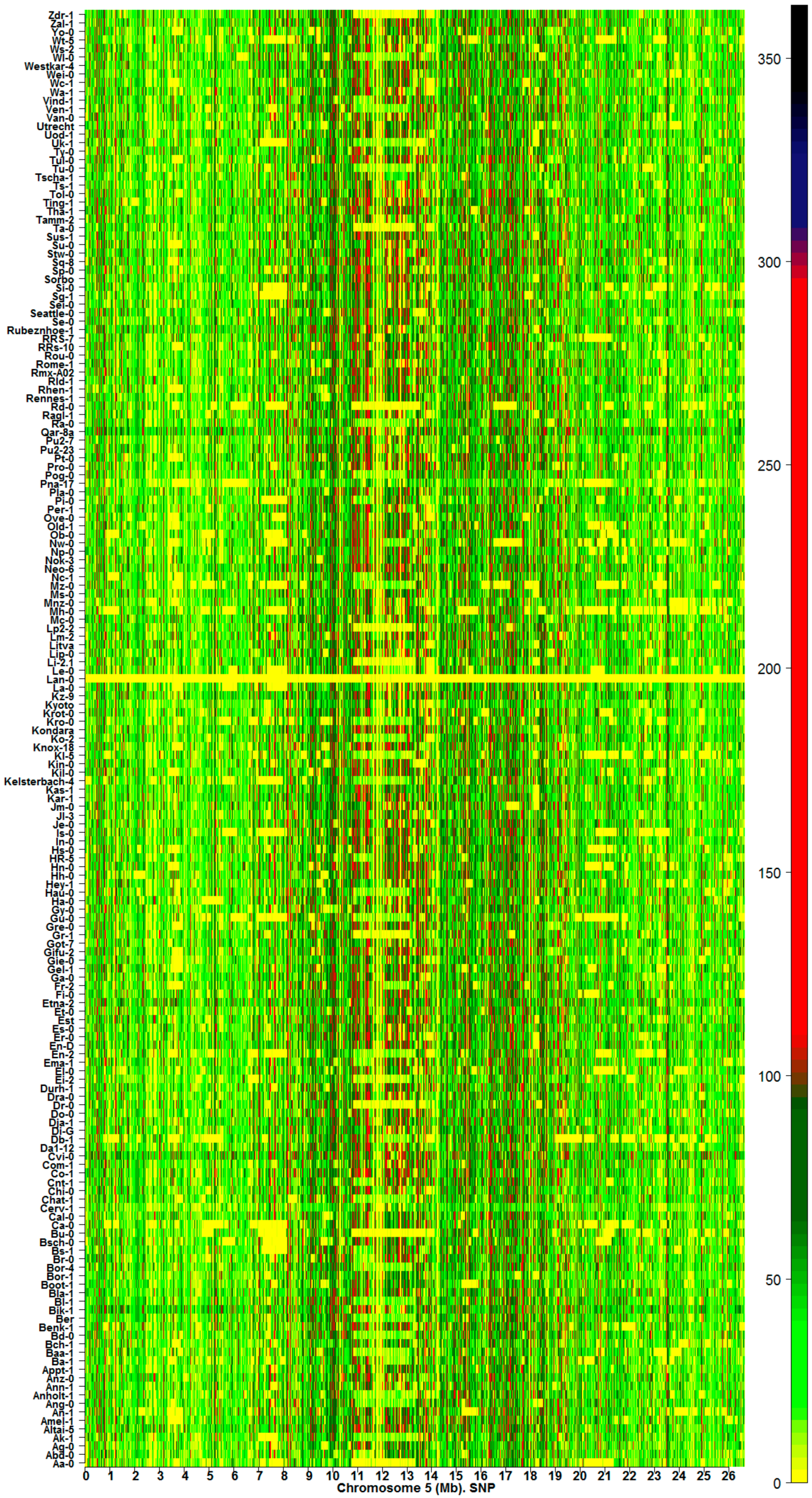
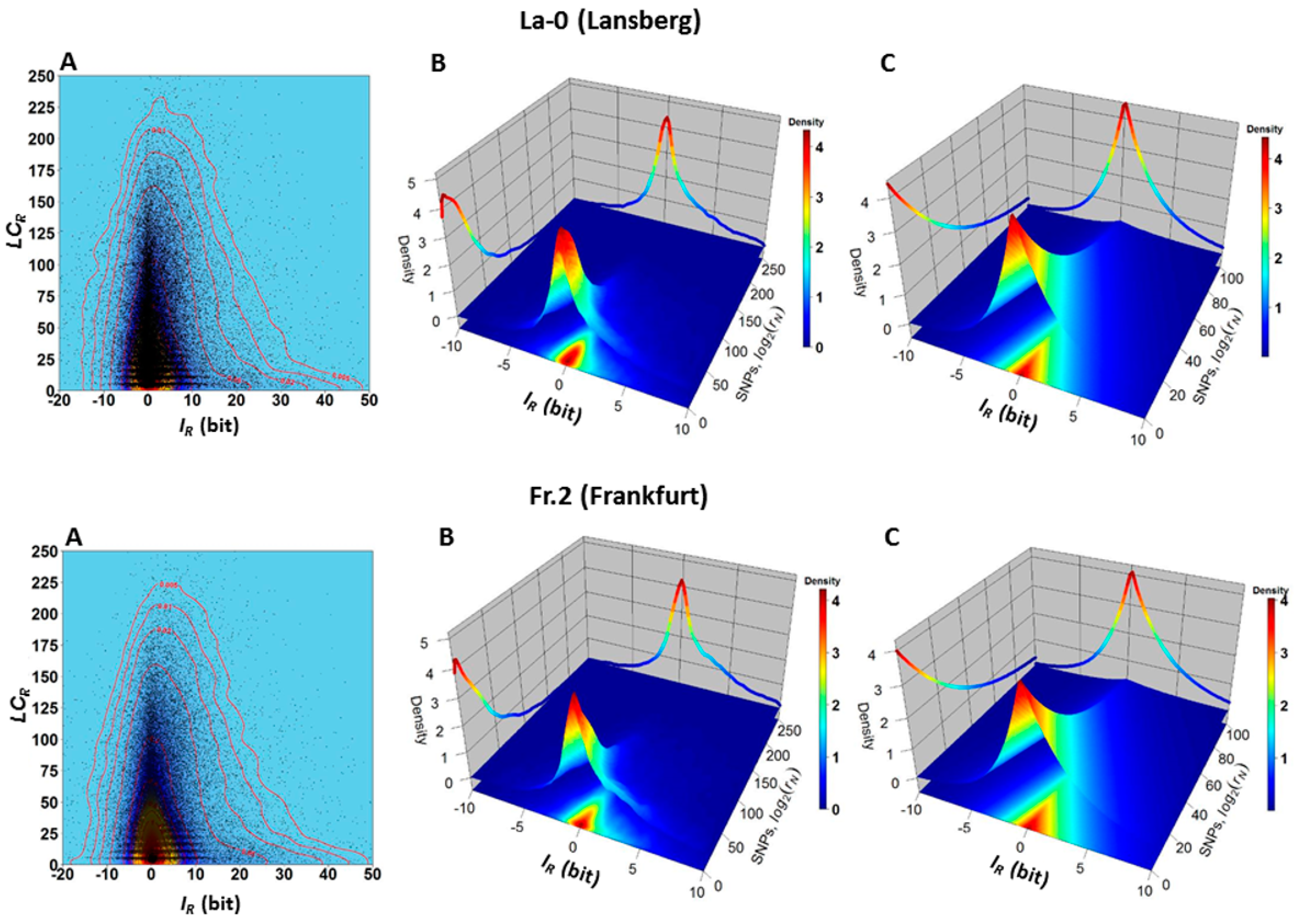
2.1. The Hotspots of Methylation and SNP Landmarks
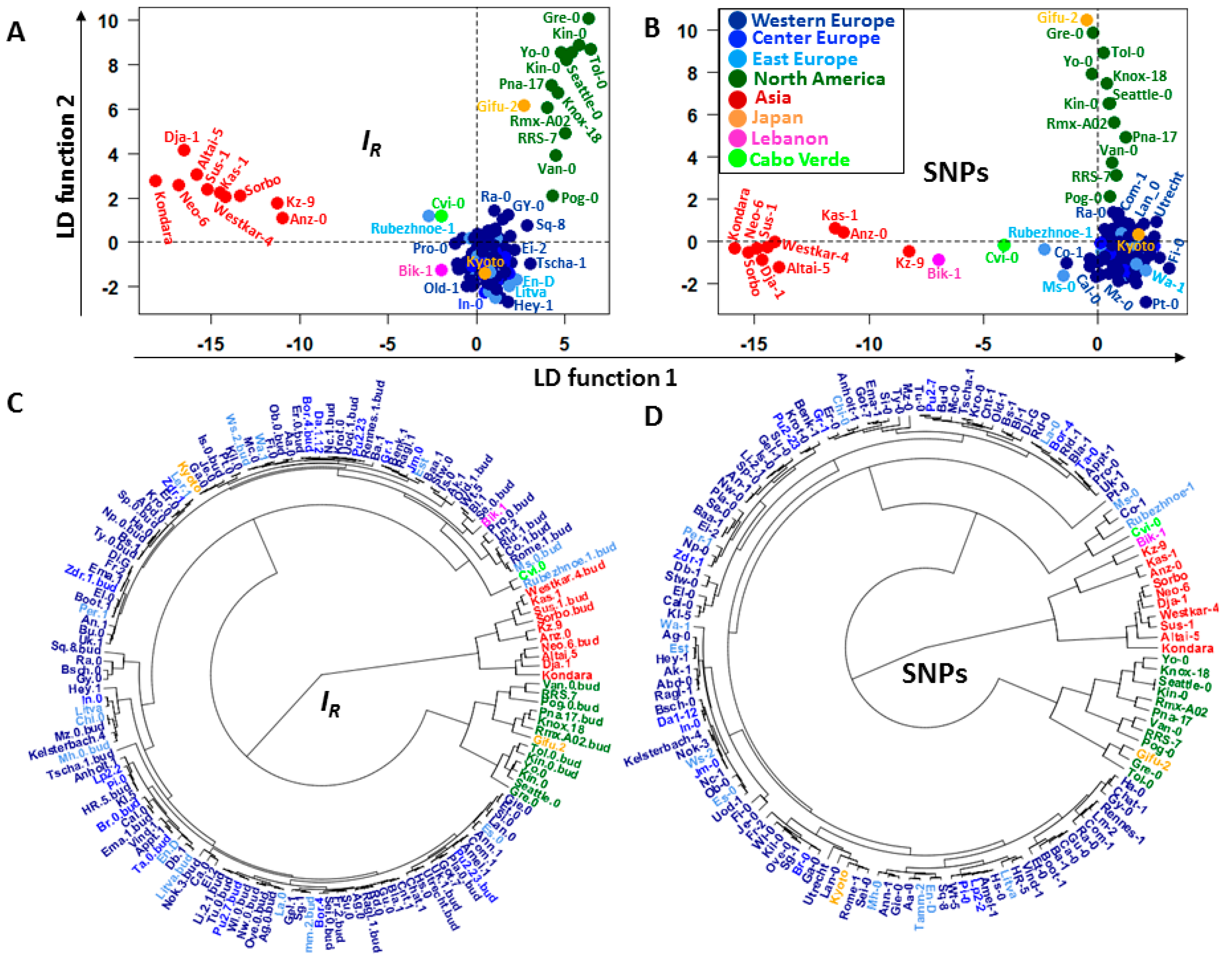
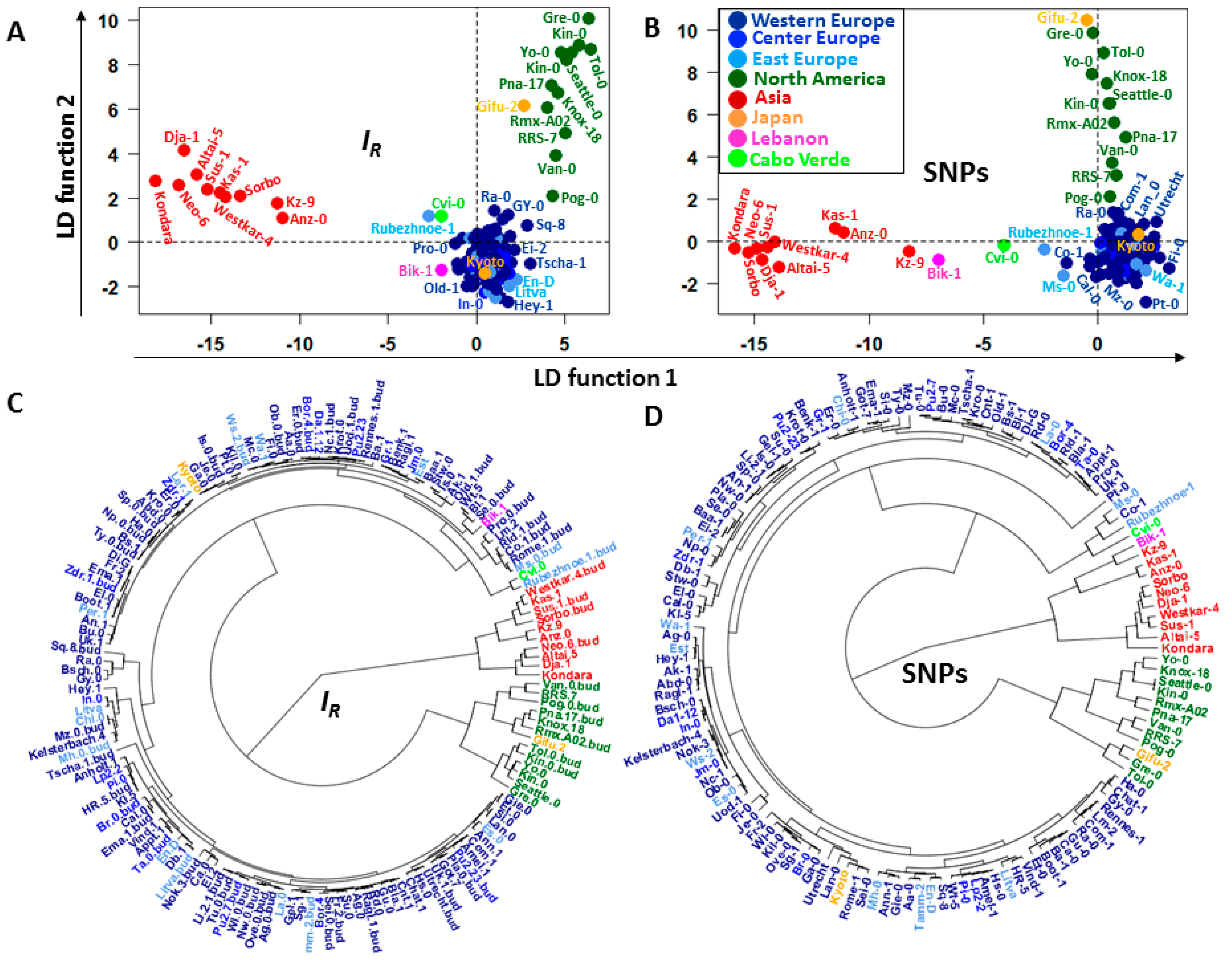
2.2. Discriminatory Information Patterns in Natural Arabidopsis Ecotypes
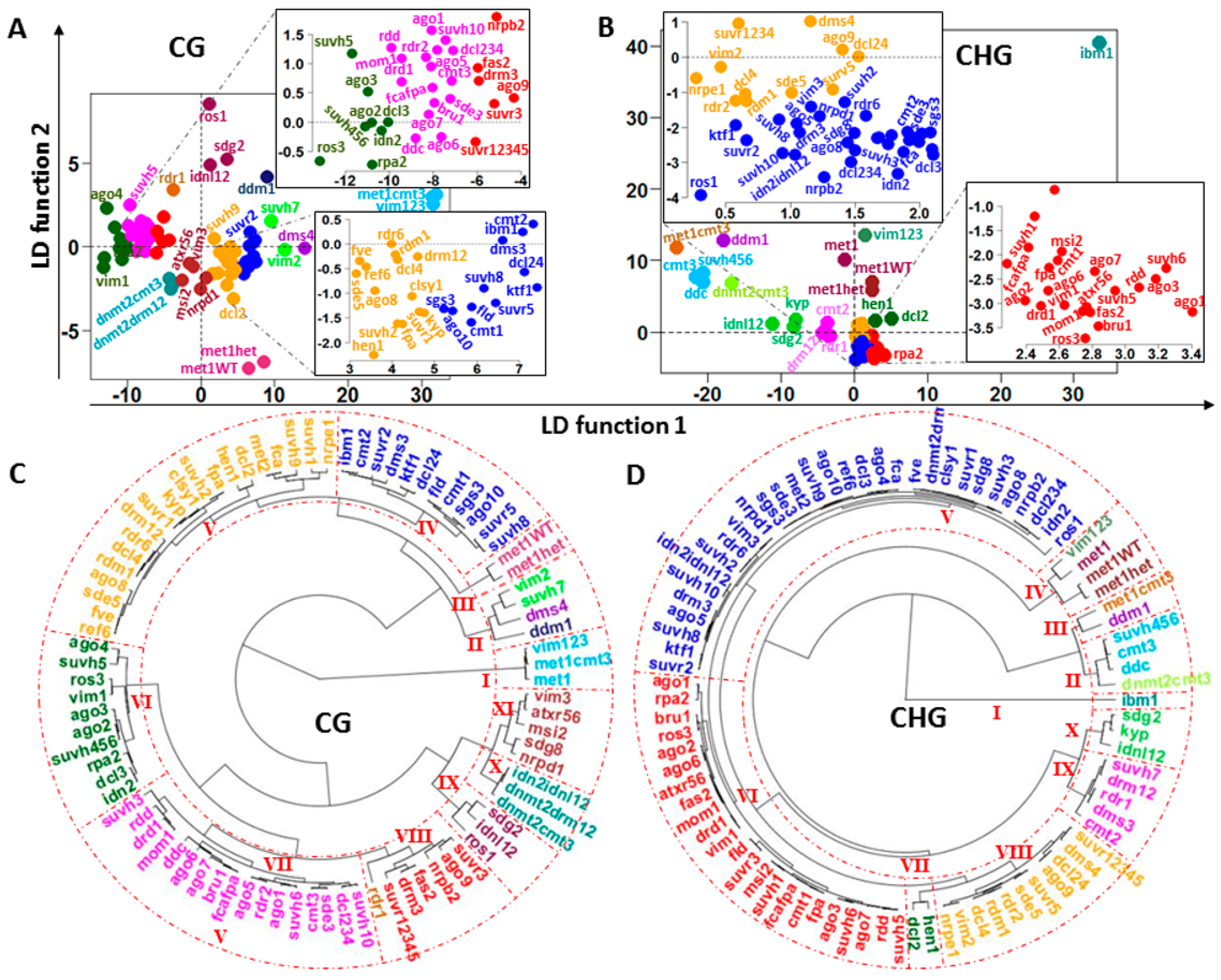
2.3. Discriminatory Information Patterns in Silencing Mutants
2.4. GF Selection and the Topology of the Dendrograms of Structured Populations
3. Discussion
3.1. Simlarities of Discriminatory Information Patterns in Silencing Mutants May Reflect Biological Relationships between Them
3.2. Links between the Discriminatory Informational and Mutational Patterns in Natural Arabidopsis Ecotypes
3.3. Consistent Topologies of the Population Structure Based on and
3.4. Potential Role of Methylations on the Fixation of New Mutations
4. Materials and Methods
4.1. Information Gain of a DNA Sequence Region R
4.2. Arabidopsis thaliana Methylation and SNP Data
4.3. Machine Learning Approach
4.4. Logarithm of the Normalized Reads Counts
4.5. Heatmaps
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AUC | Area under ROC curve |
| CDF | Cumulative distribution function |
| CDM | Cytosine DNA methylation |
| DI | Discriminatory information |
| DMR | differentially methylated region |
| FGM | Farlie–Gumbel–Morgenstern |
| LDA | Linear discriminant analysis |
| LD | Linear discriminant |
| GF | genomic feature |
| GR | genomic region |
| PC | Principal component |
| PCA | PC analysis |
| ROC | receiver operating characteristic |
| SVM | Support vector machine |
| SNP | Single nucleotide polymorphism |
| CMT3 | CHROMOMETHYLASE 3, a chromomethylase involved in methylating cytosine residues at non-CG sites |
| DDM1 | decreased dna methylation 1 |
| DRM(n) | domains rearranged methyltransferase n, e.g., DRM1 |
| DCL(n) | DICER-LIKE n, e.g., DICER-LIKE 4 (DCL4) |
| HEN1 | a methyltransferase that methylates miRNAs and siRNAs |
| IDN2 | involved in de novo 2 |
| IDNL(n) | involved in de novo 2 (IDN2)—LIKE n, e.g., IDNL1 |
| MET1 | methyltransferase 1 |
| sdg2 | mutant of the SET DOMAIN PROTEIN 2, a histone-lysine N-methyltransferase (H3-K4 specific) |
| RDR(n) | RNA-dependent RNA polymerases, e.g., RDR1 |
| SUVH(n) | histone 3 lysine 9 (H3K9) specific methyltransferase involved in the maintenance of DNA methylation |
| SUVH4 is also called KRYPTONITE (KYP); suvh456 denote the triple mutant suvh4, suvh5, and suvh6 | |
| UPGMA | Stands for Unweighted Pair Group Method with Arithmetic Mean |
| VIM(n) | variant in methylation family protein, e.g., VIM1 |
| ddc | triple mutant drm1 drm2 and cmt3 |
References
- Belanger, A.S.; Tojcic, J.; Harvey, M.; Guillemette, C. Regulation of UGT1A1 and HNF1 transcription factor gene expression by DNA methylation in colon cancer cells. BMC Mol. Biol. 2010, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Dantas Machado, A.C.; Zhou, T.; Rao, S.; Goel, P.; Rastogi, C.; Lazarovici, A.; Bussemaker, H.J.; Rohs, R. Evolving insights on how cytosine methylation affects protein-DNA binding. Brief. Funct. Genom. 2015, 14, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Draper, D. Bayesian Model Specification: Toward a Theory of Applied Statistics; ICSA 2010: Guangzhou, China, 2010. [Google Scholar]
- Lindley, D.V. The philosophy of statistics. J. R. Stat. Soc. Ser. D 2000, 49, 293–337. [Google Scholar] [CrossRef]
- Rényi, A. On measures of entropy and information. In Proceedings of the Fourth Berkeley Symposium on Mathematical Statistics and Probability; University of California Press: Berkeley, CA, USA, 1961; Volume 1, pp. 547–561. [Google Scholar]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Schneider, T.D. Theory of molecular machines. II. Energy dissipation from molecular machines. J. Theor. Biol. 1991, 148, 125–137. [Google Scholar] [CrossRef]
- Bérut, A.; Arakelyan, A.; Petrosyan, A.; Ciliberto, S.; Dillenschneider, R.; Lutz, E. Experimental verification of Landauer’s principle linking information and thermodynamics. Nature 2012, 483, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Toyabe, S.; Sagawa, T.; Ueda, M.; Muneyuki, E.; Sano, M. Experimental demonstration of information-to-energy conversion and validation of the generalized Jarzynski equality. Nat. Phys. 2010, 6, 988–992. [Google Scholar] [CrossRef]
- Sanchez, R.; Mackenzie, S.A. Information thermodynamics of cytosine DNA methylation. PLoS ONE 2016, 11, e0150427. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Plochberger, B.; Costa, P.; Cope, S.M.; Vaiana, S.M.; Lindsay, S. Hydrophobicity of methylated DNA as a possible mechanism for gene silencing. Phys. Biol. 2012, 9, 065001. [Google Scholar] [CrossRef] [PubMed]
- Choy, J.S.; Wei, S.; Lee, J.Y.; Tan, S.; Chu, S.; Lee, T.H. DNA methylation increases nucleosome compaction and rigidity. J. Am. Chem. Soc. 2010, 132, 1782–1783. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.J.; Schultz, M.D.; Urich, M.A.; Nery, J.R.; Pelizzola, M.; Libiger, O.; Alix, A.; McCosh, R.B.; Chen, H.; Schork, N.J.; et al. Patterns of population epigenomic diversity. Nature 2013, 495, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Stroud, H.; Greenberg, M.V.C.; Feng, S.; Bernatavichute, Y.V.; Jacobsen, S.E. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 2013, 152, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Stroud, H.; Hale, C.J.; Feng, S.; Caro, E.; Jacob, Y.; Michaels, S.D.; Jacobsen, S.E. DNA Methyltransferases are required to induce heterochromatic re-replication in Arabidopsis. PLoS Genet. 2012, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Christian, G.; Favre, A. Everything You always wanted to know about copula modeling but were afraid to ask. J. Hydrol. Eng. 2007, 12, 347–368. [Google Scholar]
- Schucany, W.R.; Parr, W.C.; Boyer, J.E. Correlation structure in Farlie-Gumbel-Morgenstern distributions. Biometrika 1978, 65, 650–653. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment analysis for Gene Ontology. R package version 2.22.0. 2015. Available online: http://bioconductor.org/packages/release/bioc/vignettes/topGO/inst/doc/topGO.pdf (accessed on 1 February 2016).
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014, 21, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Ebbs, M.L.; Bender, J. Locus-specific control of DNA methylation by the arabidopsis SUVH5 histone methyltransferase. Plant Cell 2006, 18, 1166–1176. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.R.; Dittmer, T.A.; Richards, E.J. Three SRA-domain methylcytosine-binding proteins cooperate to maintain global CpG methylation and epigenetic silencing in Arabidopsis. PLoS Genet 2008, 4, e1000156. [Google Scholar] [CrossRef] [PubMed]
- Ausin, I.; Greenberg, M.V.C.; Simanshu, D.K.; Hale, C.J.; Vashisht, A.A.; Simon, S.A.; Lee, T.; Feng, S.; Española, S.D.; Meyers, B.C.; et al. Involved in De novo 2-containing complex involved in RNA-directed DNA methylation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2012, 109, 8374–8381. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P. Transgenes and their contributions to epigenetic research. Int. J. Dev. Biol. 2013, 57, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Johansen, L.K.; Gustafson, A.M.; Kasschau, K.D.; Lellis, A.D.; Zilberman, D.; Jacobsen, S.E.; Carrington, J.C. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol. 2004, 2, E104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasciolli, V.; Mallory, A.C.; Bartel, D.P.; Vaucheret, H. Partially redundant functions of Arabidopsis DICER-like enzymes and a role for DCL4 in producing trans-acting siRNAs. Curr. Biol. 2005, 15, 1494–1500. [Google Scholar] [CrossRef] [PubMed]
- Mlotshwa, S.; Pruss, G.J.; Peragine, A.; Endres, M.W.; Li, J.; Chen, X.; Poethig, R.S.; Bowman, L.H.; Vance, V. DICER-LIKE2 plays a primary role in transitive silencing of transgenes in Arabidopsis. PLoS ONE 2008, 3, e1755. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, X.; Singh, J.; Li, D.; Qu, F. Temperature-dependent survival of Turnip crinkle virus-infected Arabidopsis plants relies on an RNA silencing-based defense that requires DCL2, AGO2, and HEN1. J. Virol. 2012, 86, 6847–6854. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, A.T.; Ream, T.S.; Haag, J.R.; Pikaard, C.S. RNA polymerase V transcription guides ARGONAUTE4 to chromatin. Nat. Genet. 2009, 41, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Pooggin, M.M. How can plant DNA viruses evade siRNA-directed DNA methylation and silencing? Int. J. Mol. Sci. 2013, 14, 15233–15259. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, A.; Fahlgren, N.; Garcia-Ruiz, H.; Gilbert, K.B.; Montgomery, T.A.; Nguyen, T.; Cuperus, J.T.; Carrington, J.C. Functional analysis of three Arabidopsis ARGONAUTES using slicer-defective mutants. Plant Cell 2012, 24, 3613–3629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severin, P.M.D.; Zou, X.; Gaub, H.E.; Schulten, K. Cytosine methylation alters DNA mechanical properties. Nucleic Acids Res. 2011, 39, 8740–8751. [Google Scholar] [CrossRef] [PubMed]
- Severin, P.M.D.; Zou, X.; Schulten, K.; Gaub, H.E. Effects of cytosine hydroxymethylation on DNA strand separation. Biophys. J. 2013, 104, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Osakabe, A.; Adachi, F.; Arimura, Y.; Maehara, K.; Ohkawa, Y.; Kurumizaka, H. Influence of DNA methylation on positioning and DNA flexibility of nucleosomes with pericentric satellite DNA. Open Biol. 2015, 5, 150128. [Google Scholar] [CrossRef] [PubMed]
- Yusufaly, T.I.; Li, Y.; Olson, W.K. 5-Methylation of cytosine in CG:CG base-pair steps: A physicochemical mechanism for the epigenetic control of DNA nanomechanics. J. Phys. Chem. B 2013, 117, 16436–16442. [Google Scholar] [CrossRef] [PubMed]
- Washburn, J.D.; Bird, K.A.; Conant, G.C.; Pires, J.C. Convergent evolution and the origin of complex phenotypes in the age of systems biology. Int. J. Plant Sci. 2016, 177, 305–318. [Google Scholar] [CrossRef]
- Stewart, C.-B.; Schilling, J.W.; Wilson, A.C. Adaptive evolution in the stomach lysozymes of foregut fermenters. Nature 1987, 330, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Foote, A.D.; Liu, Y.; Thomas, G.W.C.; Vinař, T.; Alföldi, J.; Deng, J.; Dugan, S.; van Elk, C.E.; Hunter, M.E.; Joshi, V.; et al. Convergent evolution of the genomes of marine mammals. Nat. Genet. 2015, 47, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA Data Mining Software: An Update. SIGKDD Explor. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Al-Zyoud, W.A.; Hynson, R.M.; Ganuelas, L.A.; Coster, A.C.; Duff, A.P.; Baker, M.A.; Stewart, A.G.; Giannoulatou, E.; Ho, J.W.; Gaus, K.; et al. Binding of transcription factor GabR to DNA requires recognition of DNA shape at a location distinct from its cognate binding site. Nucleic Acids Res. 2016, 44, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Blake, R.D.; Delcourt, S.G. Thermal stability of DNA. Nucleic Acids Res. 1998, 26, 3323–3332. [Google Scholar] [CrossRef] [PubMed]
- Nardo, L.; Lamperti, M.; Salerno, D.; Cassina, V.; Missana, N.; Bondani, M.; Tempestini, A.; Mantegazza, F. Effects of non-CpG site methylation on DNA thermal stability: a fluorescence study. Nucleic Acids Res. 2015, 43, 10722–10733. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar-Jog, Y.P.; van Dornshuld, E.; Brooks, T.A.; Tschumper, G.S.; Wadkins, R.M. Epigenetic modification, dehydration, and molecular crowding effects on the thermodynamics of i-motif structure formation from C-Rich DNA. Biochemistry 2014, 53, 1586–1594. [Google Scholar] [CrossRef] [PubMed]
- Římal, V.; Socha, O.; Štěpánek, J.; Štěpánková, H. Spectroscopic study of cytosine methylation effect on thermodynamics of DNA duplex containing CpG motif. J. Spectrosc. 2015, 2015, 1–8. [Google Scholar] [CrossRef]
- Portella, G.; Battistini, F.; Orozco, M. Understanding the connection between epigenetic DNA methylation and nucleosome positioning from computer simulations. PLoS Comput. Biol. 2013, 9, e1003354. [Google Scholar] [CrossRef] [PubMed]
- Flores, K.B.; Wolschin, F.; Amdam, G.V. The role of methylation of DNA in environmental adaptation. Integr. Comp. Biol. 2013, 53, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, Z.; Chen, H.; Su, Z.; Yang, J.; Lin, F.; Shi, S.; He, X. Nucleosomes suppress spontaneous mutations base-specifically in eukaryotes. Science 2012, 335, 1235–1238. [Google Scholar] [CrossRef] [PubMed]
- Felle, M.; Hoffmeister, H.; Rothammer, J.; Fuchs, A.; Exler, J.H.; Langst, G. Nucleosomes protect DNA from DNA methylation in vivo and in vitro. Nucleic Acids Res. 2011, 39, 6956–6969. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.E.; Mazrimas, J.A.; Bishop, C.C. Physical studies on synthetic DNAs containing 5-methylcytosine. Biochim. Biophys. Acta Nucleic Acids Protein Synth. 1974, 335, 330–348. [Google Scholar] [CrossRef]
- Ehrlich, M.; Ehrlich, K.; Mayo, J.A. Unusual properties of the DNA from Xanthomonas phage XP-12 in which 5-methylcytosine completely replaces cytosine. Biochim. Biophys. Acta Nucleic Acids Protein Synth. 1975, 395, 109–119. [Google Scholar] [CrossRef]
- Laprise, S.L.; Gray, M.R. Covalent genomic DNA modification patterns revealed by denaturing gradient gel blots. Gene 2007, 391, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Schultz, M.; Lewsey, M. Transgenerational epigenetic instability is a source of novel methylation variants. Science 2011, 334, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Hagmann, J.; Müller, J.; Koenig, D.; Stegle, O.; Borgwardt, K.; Weigel, D. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 2011, 480, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Wiley, R.H. A receiver–signaler equilibrium in the evolution of communication in noise. Behaviour 2013, 150, 1–37. [Google Scholar] [CrossRef]
- Stanislaw, H.; Todorov, N. Calculation of signal detection theory measures. Behav. Res. Methods Instrum. Comput. 1999, 31, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Wiley, R.H. Signal Detection, Noise, and the Evolution of Communication. In Animal Communication and Noise; Brumm, H., Ed.; Springer-Verlag: Berlin, Germany; Heidelberg, Germany, 2013; Volume 2, pp. 7–31. [Google Scholar]
- Wiley, R.H. Signal Detection and Animal Communication. Adv. Study Behav. 2006, 36, 217–247. [Google Scholar]
- Tribus, M.; McIrvine, E.C. Energy and Information. Sci. Am. 1971, 225, 179–188. [Google Scholar] [CrossRef]
- R Core Team. A Language and Environment for Statistical Computing. 2014. Available online: http://www.r-project.org/ (accessed on 1 February 2016).
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Miki, D.; Zhang, H.; Liu, Y.; Zhang, X.; Tang, K.; Kan, Y.; La, H.; Li, X.; Li, S.; et al. A histone acetyltransferase regulates active DNA demethylation in arabidopsis. Science 2012, 336, 1445–1448. [Google Scholar] [CrossRef] [PubMed]
- Hand, D.J.; Till, R.J. A Simple generalisation of the area under the ROC curve for multiple class classification problems. Mach. Learn. 2001, 45, 171–186. [Google Scholar] [CrossRef]
- Romanski, P.; Kotthoff, L. FSelector: Selecting Attributes. R Package Version 0.20. 2014. Available online: http://cran.r-project.org/package=FSelector (accessed on 1 February 2016).
- Fayyad, U.M.; Irani, K.B. Multi-Interval Discretization of Continuos-Valued Attributes for Classification Learning. In Proceedings of the International Joint Conference on Uncertainty in AI; Morgan Kaufmann Publishers: Burlington, MA, USA, 1993; pp. 1022–1027. [Google Scholar]
- Jombart, T. ADEGENET: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.; Dimitriadou, E.; Hornik, K.; Weingessel, A.; Leisch, F. e1071: Misc Functions of the Department of Statistics, Probability Theory Group, TU Wien. Available online: https://cran.r-project.org/web/packages/e1071/ (accessed on 1 February 2016).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample 1 | Classifier | Accuracy Mean | 2.5% Quantile | 97.5% Quantile |
|---|---|---|---|---|
| CG ecotypes (2482 DIRs) | AUC + PCA + LDA | 93.08352 | 88.05678 | 97.4359 |
| AUC + PCA + SVM | 93.52517 | 91.83673 | 95.2381 | |
| AUC + SVM | 96.42381 | 95.91837 | 96.59864 | |
| SNP ecotypes (2590 DIRs) | AUC + LDA | 90.85758 | 85.42125 | 95.89744 |
| AUC + PCA + SVM | 95.01642 | 94.02985 | 96.26866 | |
| AUC + SVM | 95.77007 | 95.23810 | 95.91837 |
| Sample a | Classifier | Accuracy Mean | 2.5% Quantile | 97.5% Quantile |
|---|---|---|---|---|
| CG mutants (all regions) | PCA + LDA | 93.81872 | 92.76714 | 94.85746 |
| PCA + SVM | 94.37882 | 90.58824 | 96.47059 | |
| CG mutants (9428 DIRs) | PCA + LDA | 83.46161 | 81.47742 | 85.34079 |
| SVM | 87.83059 | 84.70588 | 89.44118 | |
| PCA + SVM | 92.77412 | 89.41176 | 95.29412 | |
| CHG mutants (all regions) | PCA + LDA | 95.38752 | 94.16575 | 96.57823 |
| PCA + SVM | 97.10741 | 93.82716 | 98.76543 | |
| CHG mutants (9428 DIRs) | PCA + LDA | 93.77919 | 91.94159 | 95.3213 |
| SVM | 89.62963 | 85.18519 | 92.59259 | |
| PCA + SVM | 96.31970 | 94.94312 | 97.60015 |
| GO.ID | Term (Short Description) | Classic KS Ties a | elimF a | Fisher a |
|---|---|---|---|---|
| GO:0008150 | biological process | 0.007 | < 0.001 | – |
| GO:0071704 | organic substance metabolic process | 0.348 | 0.0015 | – |
| GO:0044238 | primary metabolic process | 0.164 | 0.0027 | – |
| GO:0044237 | cellular metabolic process | 0.305 | 0.0029 | – |
| GO:0044763 | single-organism cellular process | 0.246 | 0.0059 | – |
| GO:0009058 | biosynthetic process | 0.497 | 0.0091 | – |
| GO:1901576 | organic substance biosynthetic process | 0.442 | 0.0116 | – |
| GO:0050896 | response to stimulus | 0.068 | 0.0139 | – |
| GO:0043170 | macromolecule metabolic process | 0.083 | 0.0139 | – |
| GO:0044249 | cellular biosynthetic process | 0.54 | 0.0198 | – |
| GO:0044260 | cellular macromolecule metabolic process | 0.129 | 0.0209 | – |
| GO:0065007 | biological regulation | 0.811 | 0.0296 | – |
| GO:0006807 | nitrogen compound metabolic process | 0.333 | 0.0332 | – |
| GO:0044710 | single-organism metabolic process | 0.748 | 0.0371 | – |
| GO:0034641 | cellular nitrogen compound metabolic process | 0.629 | 0.0415 | <0.001 |
| GO:1901360 | organic cyclic compound metabolic process | 0.233 | 0.0439 | – |
| GO:0050789 | regulation of biological process | 0.913 | 0.0439 | – |
| GO:0006725 | cellular aromatic compound metabolic process | 0.327 | 0.0548 | <0.001 |
| GO:0046483 | heterocycle metabolic process | 0.294 | 0.0611 | <0.001 |
| GO:0050794 | regulation of cellular process | 0.889 | 0.0682 | <0.001 |
| GO:0009059 | macromolecule biosynthetic process | 0.369 | 0.0759 | <0.001 |
| GO:0006139 | nucleobase-containing compound metabolic | 0.202 | 0.0845 | <0.001 |
| GO:0034645 | cellular macromolecule biosynthetic process | 0.569 | 0.0892 | <0.001 |
| GO:0032502 | developmental process | 0.046 | 0.094 | – |
| GO:0006950 | response to stress | 0.094 | 0.094 | <0.001 |
| GO:0044767 | single-organism developmental process | 0.029 | 0.0991 | <0.001 |
| Gene.ID | Name (Short Description) | a |
|---|---|---|
| AT1G16540 | ABA3; K15631 molybdenum cofactor sulfurtransferase (C:2.8.1.9) | 1.11 |
| AT1G19180 | JAZ1; K13464 jasmonate ZIM domain-containing protein | 0.98 |
| AT1G19480 | K01247 DNA-3-methyladenine glycosylase II (EC:3.2.2.21) | 1.02 |
| AT1G28480 | GRX480; glutaredoxin-GRX480 | 1.54 |
| AT1G31812 | ACBP6; acyl-CoA-binding protein 6 | 0.98 |
| AT1G54610 | K08819 cyclin-dependent kinase 12/13 (EC:2.7.11.22 2.7.11.23) | 0.81 |
| AT1G69940 | PPME1; pectinesterase PPME1; K01051 pectinesterase (EC:3.1.1.11) | 0.55 |
| AT1G76500 | SOB3; AT-hook motif nuclear localized protein 29 | 0.42 |
| AT2G05990 | MOD1; K00208 enoyl-[acyl-carrier protein] reductase I (EC:1.3.1.9 1.3.1.10) | 0.77 |
| AT2G11000 | MAK10; MAK10-like protein | 1.07 |
| AT2G28660 | chloroplast-targeted copper chaperone protein | 1.43 |
| AT2G30750 | CYP71A12; cytochrome P450 71A12; K00517 (EC:1.14.-.-) | 1.13 |
| AT2G32660 | RLP22; receptor like protein 22 | 1.26 |
| AT2G34390 | NIP2;1; aquaporin NIP2-1; K09874 aquaporin NIP | 1.66 |
| AT2G43620 | chitinase family protein; K01183 chitinase (EC:3.2.1.14) | 0.76 |
| AT2G47000 | ABCB4; auxin efflux MDR4; K05658 (EC:3.6.3.44) | 1.17 |
| AT3G21860 | SK10; SKP1-like protein 10; K03094 S-phase kinase-associated protein 1 | 0.58 |
| AT3G28360 | PGP16; ABC transporter B family member 16; K05658 (EC:3.6.3.44) | 0.81 |
| AT3G32920 | DNA repair protein recA homolog 4; K03553 recombination protein RecA | 1.01 |
| AT3G44110 | ATJ3; chaperone protein dnaJ 3; K09503 | 1.1 |
| AT3G44480 | RPP1; TIR-NBS-LRR class disease resistance protein | 1.01 |
| AT3G45140 | LOX2; lipoxygenase 2; K00454 lipoxygenase (EC:1.13.11.12) | 1.21 |
| AT3G45260 | C2H2-like zinc finger protein | 0.96 |
| AT3G46970 | PHS2; K00688 starch phosphorylase (EC:2.4.1.1) | 1.08 |
| AT3G61220 | K15095 (+)-neomenthol dehydrogenase (EC:1.1.1.208) | 1.57 |
| AT4G04770 | ABC1; ATP binding cassette protein 1; K07033 uncharacterized protein | 1.15 |
| AT4G13920 | RLP50; receptor like protein 50 | 0.93 |
| AT4G19840 | PP2-A1; protein PHLOEM protein 2-LIKE A1 | 0.8 |
| AT4G24670 | TAR2; K16903 L-tryptophan—pyruvate aminotransferase (EC:2.6.1.99) | 1.28 |
| AT4G27410 | RD26; NAC transcription factor RD26 | 0.62 |
| AT5G24360 | IRE1-1; protein inositol requiring 1-1 | 1.52 |
| AT5G38340 | TIR-NBS-LRR class disease resistance protein | 1.16 |
| AT5G42020 | BIP2; Luminal-binding protein 2; K09490 heat shock 70kDa protein 5 | 1.03 |
| AT5G42540 | XRN2; 5’-3’ exoribonuclease 2; K12619 5’-3’ exoribonuclease 2 (EC:3.1.13.-) | 1.17 |
| AT5G43810 | ZLL; eIF2C Argonaute10; K11593 eukaryotic translation initiation factor 2C | 0.94 |
| AT5G44910 | Toll-Interleukin-Resistance domain-containing protein | 0.9 |
| AT5G51630 | TIR-NBS-LRR class disease resistance protein | 0.9 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanchez, R.; Mackenzie, S.A. Genome-Wide Discriminatory Information Patterns of Cytosine DNA Methylation. Int. J. Mol. Sci. 2016, 17, 938. https://doi.org/10.3390/ijms17060938
Sanchez R, Mackenzie SA. Genome-Wide Discriminatory Information Patterns of Cytosine DNA Methylation. International Journal of Molecular Sciences. 2016; 17(6):938. https://doi.org/10.3390/ijms17060938
Chicago/Turabian StyleSanchez, Robersy, and Sally A. Mackenzie. 2016. "Genome-Wide Discriminatory Information Patterns of Cytosine DNA Methylation" International Journal of Molecular Sciences 17, no. 6: 938. https://doi.org/10.3390/ijms17060938