
Review on Graph Clustering and Subgraph Similarity Based Analysis of Neurological Disorders


Abstract
:
1. Introduction
1.1. Characterizing Neurological Disorders with Graphs
1.2. Graph Clustering and Graph Similarity
2. Types of Bio-Networks and Applied Analysis on Neurological Disorders
2.1. Types of Bio-Networks
2.2. Bio-Network-Based Neurological Disorder Analysis
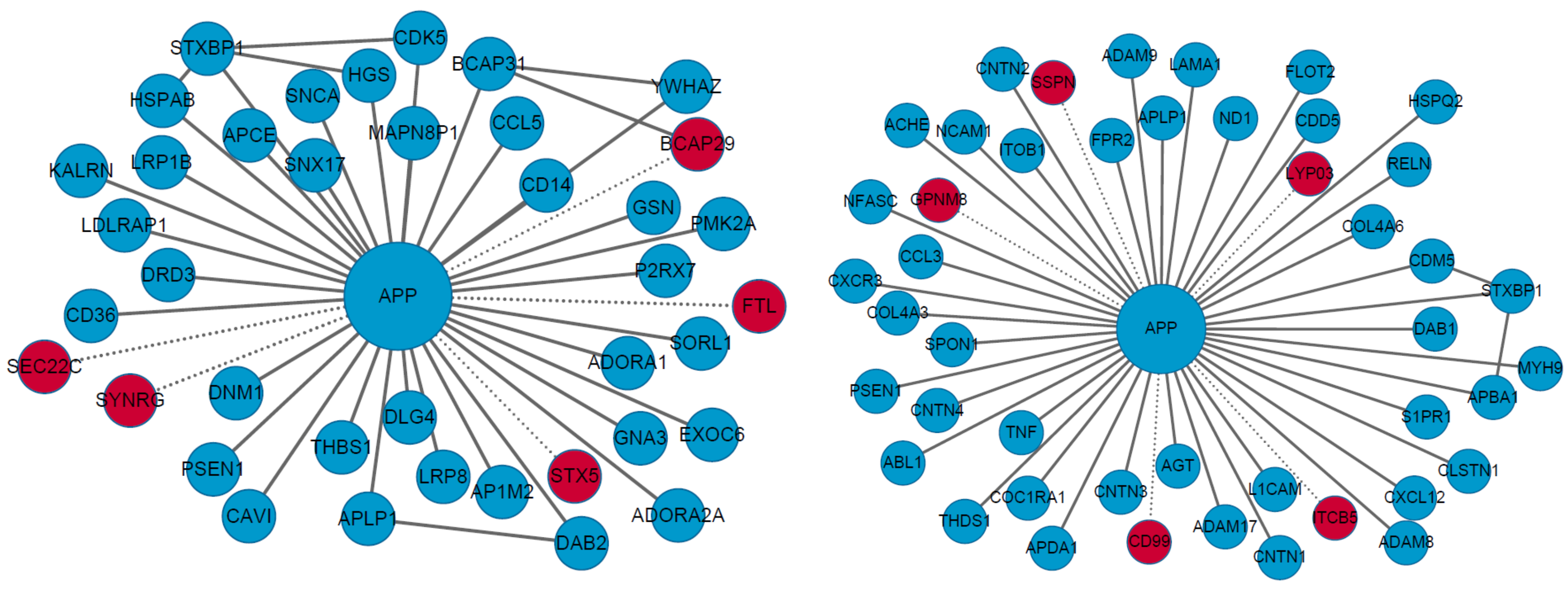
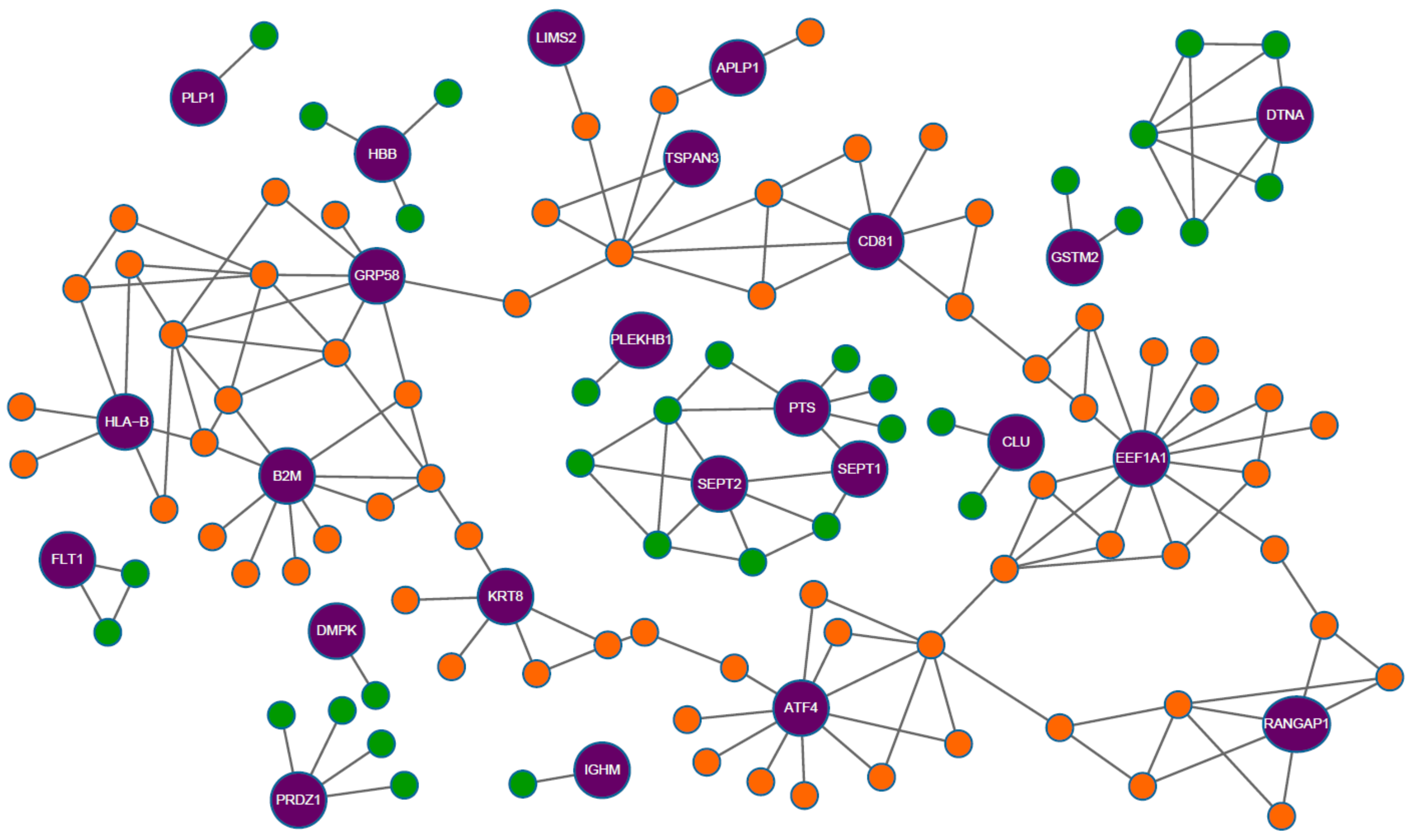
2.2.1. Causal and Susceptible Gene Finding
2.2.2. Disease Characterization
3. Types of Brain Networks Used in the Studies of Neurological Disorder
3.1. Types of Brain Networks
3.1.1. Functional Brain Networks
3.1.2. Structural Brain Networks
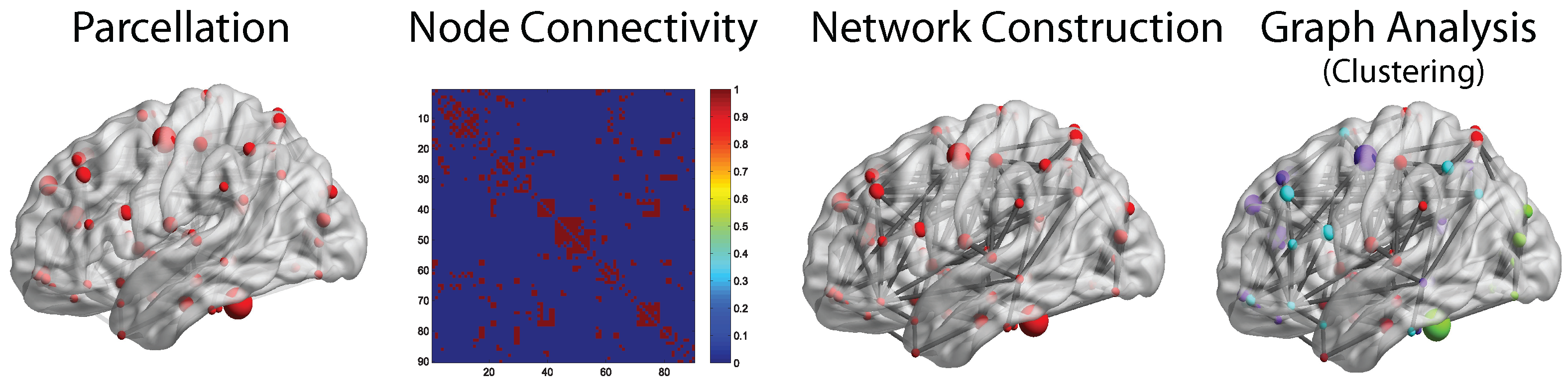
3.2. Graph Analysis Applications on Brain Networks
3.2.1. Analysis of Functional Brain Networks
3.2.2. Analysis of Structural Brain Networks
4. Need for Integrative Analysis on Large Graphs
4.1. Integrative Analysis for Single-Layered Graphs
4.2. Integrative Analysis of Multi-Layer Graphs
4.2.1. Multi-Layer Graphs
4.2.2. Existing Application of Multi-Layer Graph Analysis
5. Discussion and Conclusions
Acknowledgments
Conflicts of Interest
References
- Ridley, R.M.; Frith, C.D.; Crow, T.J.; Conneally, P.M. Anticipation in Huntington’s disease is inherited through the male line but may originate in the female. J. Med. Genet. 1988, 25, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Gatz, M.; Pedersen, N.L.; Berg, S.; Johansson, B.; Johansson, K.; Mortimer, J.A.; Posner, S.F.; Viitanen, M.; Winblad, B.; Ahlbom, A. Heritability for Alzheimers Disease: The study of dementia in Swedish twins. J. Gerontol. A Biol. Sci. Med. Sci. 1997, 52A, M117–M125. [Google Scholar] [CrossRef] [PubMed]
- Warner, T.T.; Schapira, A.H. Genetic and environmental factors in the cause of Parkinsons disease. Ann. Neurol. 2003, 53, S16–S23. [Google Scholar] [CrossRef] [PubMed]
- Stam, C.J.; Jones, B.F.; Nolte, G.; Breakspear, M.; Scheltens, P. Small-world networks and functional connectivity in Alzheimer’s disease. Cereb. Cortex 2007, 17, 92–99. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Evans, A.C. Graph theoretical modeling of brain connectivity. Curr. Opin. Neurol. 2010, 23, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, S. Graph Clustering by Flow Simulation. Ph.D. Thesis, University of Utrecht, Utrecht, The Netherlands, 1 May 2000. [Google Scholar]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Hartuv, E.; Shamir, R. A clustering algorithm based on graph connectivity. Inf. Process. Lett. 2000, 76, 175–181. [Google Scholar] [CrossRef]
- King, A.D.; Przulj, N.; Jurisica, I. Protein complex prediction via cost-based clustering. Bioinformatics 2004, 20, 3013–3020. [Google Scholar] [CrossRef] [PubMed]
- Brohée, S.; van Helden, J. Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinform. 2006, 7, 488. [Google Scholar] [CrossRef] [PubMed]
- Vlasblom, J.; Wodak, S.J. Markov clustering versus affinity propagation for the partitioning of protein interaction graphs. BMC Bioinform. 2009, 10, 99. [Google Scholar] [CrossRef] [PubMed]
- Asur, S.; Ucar, D.; Parthasarathy, S. An ensemble framework for clustering protein-protein interaction networks. Bioinformatics 2007, 23, i29–i40. [Google Scholar] [CrossRef] [PubMed]
- Hartuv, E.; Schmitt, A.O.; Langeb, J.; Meier-Ewert, S.; Lehrach, H.; Shamir, R. An algorithm for clustering cDNA fingerprints. Genomics 2000, 66, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Shasha, D.; Wang, J.T.L.; Rosalba, G. Algorithmics and applications of tree and graph searching. In Proceedings of the Twenty-First ACM SIGMOD-SIGACT-SIGART Symposium on Principles of Database Systems, Madison, WI, USA, 3–5 June 2002; pp. 39–52.
- Sharan, R.; Suthram, S.; Kelley, R.M.; Kuhn, T.; McCuine, S.; Uetz, P.; Sittler, T.; Karp, R.M.; Ideker, T. Conserved patterns of protein interaction in multiple species. Proc. Natl. Acad. Sci. USA 2005, 102, 1974–1979. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; McEachin, R.C.; Santos, C.; States, D.J.; Patel, J.M. SAGA: A subgraph matching tool for biological graphs. Bioinformatics 2007, 23, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Dubbelink, K.T.E.O.; Hillebrand, A.; Stoffers, D.; Deijen, J.B.; Twisk, J.W.R.; Stam, C.J.; Berendse, H.W. Disrupted brain network topology in Parkinson’s disease: A longitudinal magnetoencephalography study. Brain 2014, 137, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Watts, D.J.; Strogatz, S.H. Collective dynamics of ’small-world’ networks. Lett. Nat. 1997, 393, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Tewarie, P.; van Dellen, E.; Hillebrand, A.; Stam, C. The minimum spanning tree: An unbiased method for brain network analysis. NeuroImage 2015, 104, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Kruskal, J.B. On the shortest spanning subtree of a graph and the traveling salesman problem. Proc. Am. Math. Soc. 1956, 7, 48–50. [Google Scholar] [CrossRef]
- Andrei, A.; Kendziorski, C. An efficient method for identifying statistical interactors in gene association networks. Biostatistics 2009, 10, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.H.; Jung, J.Y.; DeLuca, T.F.; Hinebaugh, B.K.; Gabriel, K.C.S.; Wall, D.P. Autworks: A cross-disease network biology application for Autism and related disorders. BMC Med. Genom. 2012, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Gaiteri, C.; Bodea, L.G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Rivas, J.D.L.; Fontanillo, C. Protein-Protein interactions essentials: Key concepts to building and analyzing interactome networks. PLoS Comput. Biol. 2010, 6, e1000807. [Google Scholar]
- Liang, W.S.; Dunckley, T.; Beach, T.G.; Grover, A.; Mastroeni, D.; Ramsey, K.; Caselli, R.J.; Kukull, W.A.; McKeel, D.; Morris, J.C.; et al. Altered neuronal gene expression in brain regions differentially affected by Alzheimer’s disease: A reference data set. Physiol. Genom. 2008, 33, 240–256. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed]
- Chatr-aryamontri, A.; Ceol, A.; Palazzi, L.M.; Nardelli, G.; Schneider, M.V.; Castagnoli, L.; Cesareni, G. MINT: The Molecular INTeraction database. Nucleic Acids Res. 2007, 35, D572–D574. [Google Scholar] [CrossRef] [PubMed]
- Mering, C.V.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005, 33 (Suppl. S1), D433–D437. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M.; et al. STRING 8-a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, 37, D412–D416. [Google Scholar] [CrossRef] [PubMed]
- Xenarios, I.; Rice, D.W.; Salwinski, L.; Baron, L.; Marisa, K.A.; Marcotte, E.M.; Eisenberga, D. DIP: The database of interacting proteins. Nucleic Acids Res. 2000, 28, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Prasad, T.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human protein reference database-2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Milacic, M.; Haw, R.; Rothfels, K.; Wu, G.; Croft, D.; Hermjakob, H.; D’Eustachio, P.; Stein, L. Annotating cancer variants and anti-cancer therapeutics in reactome. Cancers 2012, 4, 1180–1211. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Iijima, R.; Ogishima, S.; Kikuchi, M.; Matsuoka, Y.; Ghosh, S.; Miyamoto, T.; Miyashita, A.; Kuwano, R.; Hiroshi, T. AlzPathway: A comprehensive map of signaling pathways of Alzheimer’s disease. BMC Syst. Biol. 2012, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.G.; Gross, B.E.; Demir, E.; Rodchenkov, I.; Babur, O.; Anwar, N.; Schultz, N.; Bader, G.D.; Sander, C. Pathway commons, a web resource for biological pathway data. Nucleic Acids Res. 2011, 39, D685–D690. [Google Scholar] [CrossRef] [PubMed]
- Bauer-Mehren, A.; Rautschka, M.; Sanz, F.; Furlong, L.I. Disgenet: A cytoscape plugin to visualize, integrate, search and analyze gene-disease networks. Bioinformatics 2010, 26, 2924–2926. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P.; Grondin, C.J.; Lennon-Hopkins, K.; Saraceni-Richards, C.; Sciaky, D.; King, B.L.; Wiegers, T.C.; Mattingly, C.J. The comparative toxicogenomics database’s 10th year anniversary: Update 2015. Nucleic Acids Res. 2015, 43, D914–D920. [Google Scholar] [CrossRef] [PubMed]
- Aranda, B.; Achuthan, P.; Alam-Faruque, Y.; Armean, I.; Bridge, A.; Derow, C.; Feuermann, M.; Ghanbarian, A.T.; Kerrien, S.; Khadake, J.; et al. The IntAct molecular interaction database in 2010. Nucleic Acids Res. 2010, 38, D525–D531. [Google Scholar] [CrossRef] [PubMed]
- Kerrien, S.; Aranda, B.; Breuza, L.; Bridge, A.; Broackes-Carter, F.; Chen, C.; Duesbury, M.; Dumousseau, M.; Feuermann, M.; Hinz, U.; et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012, 40, D841–D846. [Google Scholar] [CrossRef] [PubMed]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [PubMed]
- Kohler, S.; Bauer, S.; Horn, D.; Robinson, P.N. Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Genet. 2008, 82, 888–905. [Google Scholar] [CrossRef] [PubMed]
- Soler-López, M.; Zanzoni, A.; Lluis, R.; Stelzl, U.; Aloy, P. Interactome mapping suggests new mechanistic details underlying Alzheimer’s disease. Genome Res. 2011, 21, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Diao, B.; Liu, Y.; Zhang, Y.; Xu, G.Z. A graph-clustering approach to search important molecular markers and pathways of Parkinson’s disease. Afr. J. Biotechnol. 2011, 10, 15656–15661. [Google Scholar] [CrossRef]
- Altaf-Ul-Amin, M.; Shinbo, Y.; Mihara, K.; Kurokawa, K.; Kanaya, S. Development and implementation of an algorithm for detection of protein complexes in large interaction networks. BMC Bioinform. 2006, 7, 207. [Google Scholar] [CrossRef] [PubMed]
- Talwar, P.; Silla, Y.; Grover, S.; Gupta, M.; Agarwal, R.; Kushwaha, S.; Kukreti, R. Genomic convergence and network analysis approach to identify candidate genes in Alzheimer’s disease. BMC Genom. 2014, 15, 199. [Google Scholar] [CrossRef] [PubMed]
- Guney, E.; Oliva, B. Exploiting protein-protein interaction networks for genome-wide disease-gene prioritization. PLoS ONE 2012, 7, e43557. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.M.; Fox, H.S. Transcriptome meta-analysis reveals a central role for sex steroids in the degeneration of hippocampal neurons in Alzheimer’s disease. BMC Syst. Biol. 2013, 7, 51. [Google Scholar] [CrossRef] [PubMed]
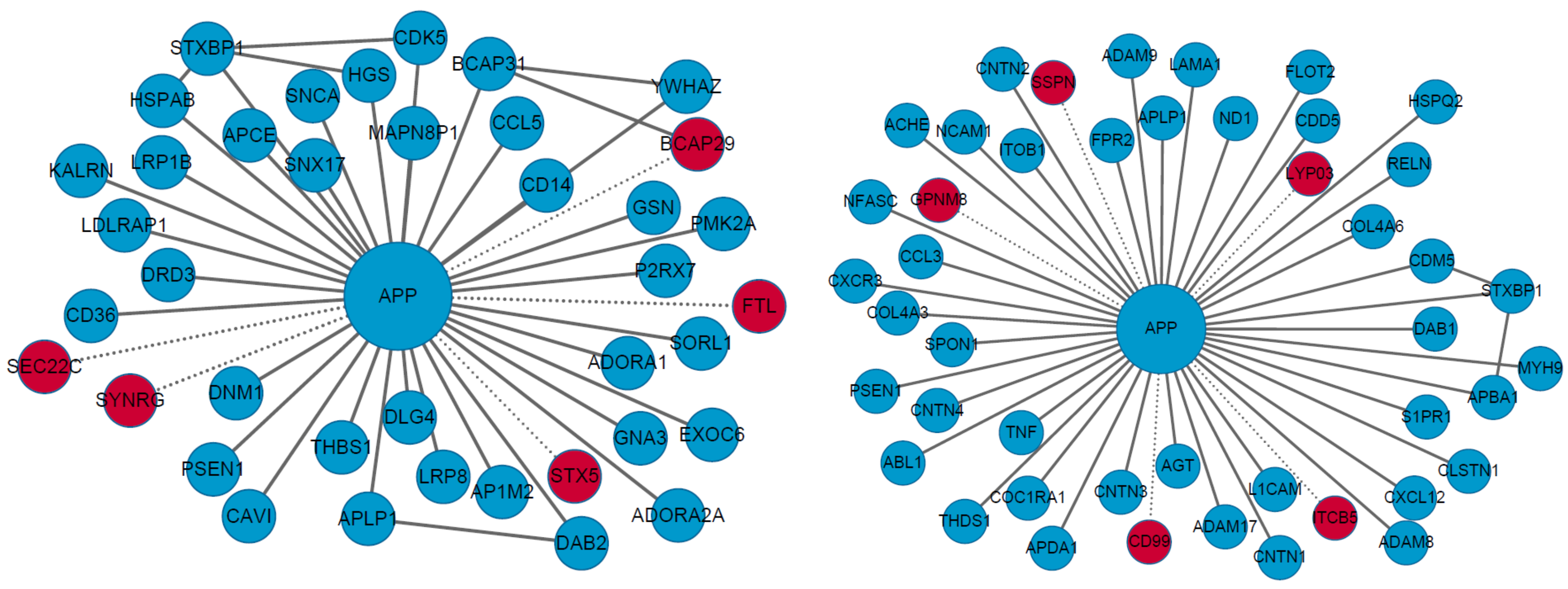
- Silva, J.V.; Yoon, S.; Domingues, S.; Guimarães, S.; Goltsev, A.V.; da Cruz e Silva, E.F.; Mendes, J.F.F.; da Cruz e Silva, O.B.A.; Fardilha, M. Amyloid precursor protein interaction network in human testis: Sentinel proteins for male reproduction. BMC Bioinform. 2015, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Seah, B.S.; Bhowmick, S.S.; Dewey, C.F.; Yu, H. FUSE: Towards multi-level functional summarization of protein interaction networks. In Proceedings of the 2nd ACM Conference on Bioinformatics, Computational Biology and Biomedicine, Chicago, IL, USA, 31 July–3 August 2011; ACM: New York, NY, USA, 2011; pp. 2–11. [Google Scholar]
- Khuller, S.; Mossb, A.; Naor, J. The budgeted maximum coverage problem. Inf. Process. Lett. 1999, 70, 39–45. [Google Scholar] [CrossRef]
- Rakshit, H.; Rathi, N.; Roy, D. Construction and analysis of the protein-protein interaction networks based on gene expression profiles of Parkinson’s disease. PLoS ONE 2014, 9, e103047. [Google Scholar]
- Mukherjeee, S.; Kaeberlein, M.; Kauwe, J.; Naj, A.C.; Crane, P. A systems-biology approach to identify candidate genes for Alzheimer’s disease by integrating protein-protein interaction network and subsequent in vivo validation of candidate genes using A C. Elegans model of AB toxicity. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2013, 10, 298–299. [Google Scholar] [CrossRef]
- Correia, C.; Oliveira, G.; Vicente, A.M. Protein interaction networks reveal novel autism risk genes within GWAS statistical noise. PLoS ONE 2014, 9, e112399. [Google Scholar] [CrossRef] [PubMed]
- Giorgini, F.; Muchowsk, P.J. Connecting the dots in huntington’s disease with protein interaction networks. Genome Biol. 2005, 6, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Shi, M.; Ma, Z.; Zhao, S.; Euskirchen, G.; Ziskin, J.; Urban, A.; Hallmayer, J.; Snyder, M. Integrated systems analysis reveals a molecular network underlying autism spectrum disorders. Mol. Syst. Biol. 2014, 10, 774. [Google Scholar] [CrossRef] [PubMed]
- Blondel, V.D.; Guillaume, J.L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [CrossRef]
- Goñi, J.; Esteban, F.J.; Mendizabal, N.V.D.; Sepulcre, J.; Ardanza-Trevijano, S.; Agirrezabal, I.; Villoslada, P. A computational analysis of protein-protein interaction networks in neurodegenerative diseases. BMC Syst. Biol. 2008, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M.L.; Moussa, M.N.; Paolini, B.M.; Lyday, R.G.; Burdette, J.H.; Laurienti, P.J. Defining nodes in complex brain networks. Front. Comput. Neurosci. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Crosson, B.; Ford, A.; McGregor, K.M.; Meinzer, M.; Cheshkov, S.; Li, X.; Walker-Batson, D.; Briggs, R.W. Functional imaging and related techniques: An introduction for rehabilitation researchers. J. Rehabil. Res. Dev. 2010, 47, 7–34. [Google Scholar] [CrossRef]
- Rombouts, S.A.; Barkhof, F.; Goekoop, R.; Stam, C.J.; Scheltens, P. Altered resting state networks in mild cognitive impairment and mild Alzheimer’s disease: An fMRI study. Hum. Brain Mapp. 2005, 26, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Supekar, K.; Menon, V.; Rubin, D.; Musen, M.; Greicius, M.D. Network analysis of intrinsic functional brain connectivity in Alzheimer’s disease. PLoS Comput. Biol. 2008, 4, e1000100. [Google Scholar] [CrossRef] [PubMed]
- Göttlich, M.; Münte, T.F.; Heldmann, M.; Kasten, M.; Hagenah, J.; Krämer, U.M. Altered resting state brain networks in Parkinson’s Disease. PLoS ONE 2013, 8, e77336. [Google Scholar]
- Bluhm, R.L.; Miller, J.; Lanius, R.A.; Osuch, E.A.; Boksman, K.; Neufeld, R.; Théberge, J.; Schaefer, B.; Williamson, P. Spontaneous low-frequency fluctuations in the BOLD signal in Schizophrenic patients: Anomalies in the default network. Schizophr. Bull. 2007, 33, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Heuvel, M.P.v.d.; Hulshoff Pol, H.E. Exploring the brain network: A review on resting-state fMRI functional connectivity. Eur. Neuropsychopharmacol. 2010, 20, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Bullmore, E.; Sporns, O. Complex brain networks: Graph theoretical analysis of structural and functional systems. Nat. Rev. Neurosci. 2009, 10, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Vissersa, M.E.; Cohena, M.X.; Geurtsa, H.M. Brain connectivity and high functioning Autism: A promising path of research that needs refined models, methodological convergence, and stronger behavioral links. Neurosci. Biobehav. Rev. 2012, 36, 604–625. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, D.; Galvána, A. The use of functional and effective connectivity techniques to understand the developing brain. Dev. Cogn. Neurosci. 2015, 12, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Bassett, D.S.; Yang, M.; Wymbs, N.F.; Grafton, S.T. Learning-Induced autonomy of sensorimotor systems. Nat. Neurosci. 2015, 18, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Fallani, F.D.V.; Richiardi, J.; Chavez, M.; Achard, S. Graph analysis of functional brain networks: Practical issues in translational neuroscience. Philos. Trans. R. Soc. B Biol. Sci. 2014. [Google Scholar] [CrossRef]
- Cheng, H.; Wang, Y.; Sheng, J.; Kronenberger, W.G.; Mathews, V.P.; Hummer, T.A.; Saykin, A.J. Characteristics and variability of structural networks derived from diffusion tensor imaging. Neuroimage 2012, 61, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.; Wagner, G.; Dahnke, R.; Schachtzabel, C.; Schultz, C.; Roebel, M.; Güllmar, D.; Reichenbach, J.R.; Sauer, H.; Schlösser, R.G.M. Disrupted white matter integrity of corticopontine-cerebellar circuitry in Schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2010, 260, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Clayden, J.D. Imaging connectivity: MRI and the structural networks of the brain. Funct. Neurol. 2013, 28, 197–203. [Google Scholar] [PubMed]
- Wang, Z.; Dai, Z.; Gong, G.; Zhou, C.; He, Y. Understanding structural-functional relationships in the human brain: A large-scale network perspective. Neuroscientist 2014, 21, 290–305. [Google Scholar] [CrossRef] [PubMed]
- Tewarie, P.; Hillebrand, A.; Schoonheimc, M.; Dijk, B.V.; Geurts, J.; Barkhof, F.; Polman, C.; Stamb, C. Functional brain network analysis using minimum spanning trees in multiple sclerosis: An meg source-space study. NeuroImage 2014, 88, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Sang, L.; Zhang, J.; Wang, L.; Zhang, J.; Zhang, Y.; Li, P.; Wang, J.; Qiu, M. Alteration of brain functional networks in early-stage Parkinson’s disease: A resting-state fmri study. PLoS ONE 2015, 10, e0141815. [Google Scholar] [CrossRef] [PubMed]
- Tzourio-Mazoyer, N.; Landeau, B.; Papathanassiou, D.; Crivello, F.; Etard, O.; Delcroix, N.; Mazoyer, B.; Joliot, M. Automated anatomical labeling of activations in spm using a macroscopic anatomical parcellation of the mni mri single-subject Brain. NeuroImage 2002, 15, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Dubbelink, K.T.E.O.; Stoffers, D.; Deijen, J.B.; Twisk, J.W.; Stam, C.J.; Hillebrand, A.; Berendse, H.W. Resting-state functional connectivity as a marker of disease progression in Parkinson’s disease: A longitudinal MEG study. NeuroImage Clin. 2013, 2, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Taquet, M.; Vega, C.; Jeste, S.S.; Fernández, I.S.; Tan, J.; Nelson, C.A.; Sahin, M.; Warfield, S.K. Brain functional networks in syndromic and non-syndromic autism: A graph theoretical study of EEG connectivity. BMC Med. 2013, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Ortega, G.J.; Sola, R.G.; Pastor, J. Complex network analysis of human ECoG data. Neurosci. Lett. 2008, 447, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Fakhri, G.E.; Li, Q. Evaluating structural symmetry of weighted brain networks via graph matching. In Medical Image Computing and Computer-Assisted Intervention-MICCAI 2014; Lecture Notes in Computer Science; Golland, P., Hata, N., Barillot, C., Hornegger, J., Howe, R., Eds.; Springer International Publishing: Berlin/Heidelberg, Germany, 2014; Volume 8674, pp. 733–740. [Google Scholar]
- Umeyama, S. An eigendecomposition approach to weighted graph matching problems. IEEE Trans. Pattern Anal. Mach. Intell. 1988, 10, 695–703. [Google Scholar] [CrossRef]
- He, Y.; Chen, Z.; Gong, G.; Evans, A. Structural insights into aberrant topological patterns of large-scale cortical networks in Alzheimer’s disease. J. Neurosci. 2008, 28, 4756–4766. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.Y.; Wang, P.N.; Chou, K.H.; Wang, J.; He, Y.; Lin, C.P. Diffusion tensor tractography reveals abnormal topological organization in structural cortical networks in Alzheimer’s disease. J. Neurosci. 2010, 30, 16876–16885. [Google Scholar] [CrossRef] [PubMed]
- Bassett, D.S.; Bullmore, E.; Verchinski, B.A.; Mattay, V.S.; Weinberger, D.R.; Meyer-Lindenberg, A. Hierarchical organization of human cortical networks in health and schizophrenia. J. Neurosci. 2008, 28, 9239–9248. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.; Atluri, G.; Xie, M.; Dey, S.; Hong, C.; Kumar, V.; Kuang, R. Co-clustering phenome-genome for phenotype classification and disease gene discovery. Nucleic Acids Res. 2012, 40, e146. [Google Scholar] [CrossRef] [PubMed]
- Rudie, J.D.; Brown, J.; Beck-Pancer, D.; Hernandez, L.; Dennis, E.; Thompson, P.; Bookheimer, S.; Daprettoa, M. Altered functional and structural brain network organization in Autism. NeuroImage Clin. 2013, 2, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Power, J.D.; Cohen, A.L.; Nelson, S.M.; Wig, G.S.; Barnes, K.A.; Church, J.A.; Vogel, A.C.; Laumann, T.O.; Miezin, F.M.; Schlaggar, B.L.; Petersen, S.E. Functional network organization of the human brain. Neuron 2011, 72, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Hagmann, P.; Sporns, O.; Madan, N.; Cammoun, L.; Pienaar, R.; Wedeen, V.J.; Meuli, R.; Thiran, J.P.; Grant, P.E. White matter maturation reshapes structural connectivity in the late developing human brain. Proc. Natl. Acad. Sci. USA 2010, 107, 19067–19072. [Google Scholar] [CrossRef] [PubMed]
- Kivelä, M.; Arenas, A.; Barthelemy, M.; Gleeson, J.P.; Moreno, Y.; Porter, M.A. Multilayer Networks. J. Complex Netw. 2014, 2, 203–271. [Google Scholar] [CrossRef]
- De Domenico, M.; Solé-Ribalta, A.; Cozzo, E.; Kivelä, M.; Moreno, Y.; Porter, M.A.; Gómez, S.; Arenas, A. Mathematical Formulation of Multilayer Networks. Phys. Rev. X 2013, 3, 041022. [Google Scholar] [CrossRef]
- Sael, L.; Jeon, I.; Kang, U. Scalable Tensor Mining. Big Data Res. 2015, 2, 82–86. [Google Scholar] [CrossRef]
- Jeon, B.S.; Jeon, L.S.I.; Kang, U. SCouT: Scalable coupled matrix-tensor factorization—Algorithm and discoveries. Int. Conf. Data Eng. 2016, in press. [Google Scholar]
- Gustafsson, M.; Nestor, C.E.; Zhang, H.; Barabási, A.L.; Baranzini, S.; Brunak, S.; Chung, K.F.; Federoff, H.J.; Gavin, A.C.; Meehan, R.R.; et al. Modules, networks and systems medicine for understanding disease and aiding diagnosis. Genome Med. 2014, 6, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, S.; Ozcaglar, C. Hybrid coexpression link similarity graph clustering for mining biological modules from multiple gene expression datasets. Bio. Data Mining 2014, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Didier, G.; Brun, C.; Baudot, A. Identifying communities from multiplex biological networks. Peer J. 2015, 3, e1525. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.E.J.; Girvan, M. Finding and evaluating community structure in networks. Phys. Rev. E 2004, 69, 026113. [Google Scholar] [CrossRef] [PubMed]

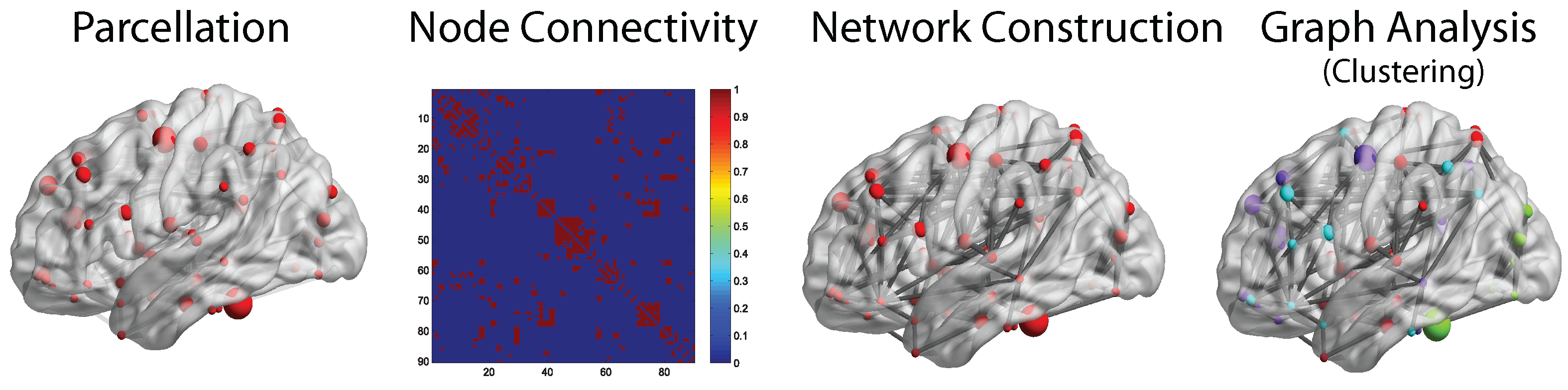

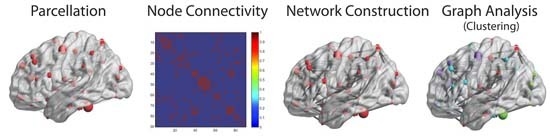
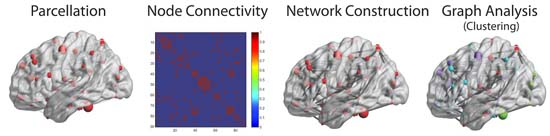{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Measure | Scope | Computation |
|---|---|---|
| Clustering coefficient | Local | , where is the degree and is the number of links between neighbors of the i-th node. |
| Local efficiency | Local | , where , where is the subgraph of G that consists of node i immediate neighbors excluding i and is the shortest path length between nodes i and j. |
| Degree centrality | Local | number of edges emanating from a node |
| Betweenness centrality | Local | where is number of shortest paths between j and k that run through iand is the number of shortest paths between j and k |
| Closeness centrality | Local | |
| Eccentricity | Local | where V is the set of nodes |
| Radiality | Local | where is the value of the diameter |
| Characteristic path length | Global | |
| Global efficiency | Global | |
| Minimum spanning tree | Global | Kruskal’s algorithms [20], etc. |
| Modularity | Global | where is the fraction of edges that connects nodes in module i, is the fraction of edges that connect at least one node in the module i and k is the number of modules |
| Group | Name | Description | Uniform Resource Locator and Reference |
|---|---|---|---|
| PPI | Mint | Collects experimentally-verified PPIs in a binary or complex representation. Merged with InAct since 2013. | http://mint.bio.uniroma2.it/mint/ [27] |
| String | The known and predicted protein interactions. The interactions include direct (physical) and indirect (functional) associations derived from genomic context, high-throughput experiments, coexpression, previous knowledge. | http://string-db.org/ [28,29] | |
| DIP | Manually- and automatically-curated database. Experimentally-determined interactions between proteins. | http://dip.doe-mbi.ucla.edu/dip/Main.cgi [30] | |
| Biological Pathway | HPRD | Human PPI manually extracted from the literature. | http://www.hprd.org/ [31] |
| KEGG | Manually-curated pathway maps representing knowledge of the molecular interaction and reaction networks. | http://www.genome.jp/kegg/ [32] | |
| Reactome | Manually-curated pathway. | http://www.reactome.org/ [33,34] | |
| Alz-Pathway | Manually-curated; comprehensively catalogs signaling pathways for Alzheimer’s disease. | http://alzpathway.org/ [35] | |
| Pathway-Common | Collection of publicly available pathway information from multiple organisms. | http://www.pathwaycommons.org/pc [36] | |
| Gene Disease Network (GDAs) | DisGeNET | Integrated database from various expert-curated databases and text-mining-derived associations, including Mendelian, complex and environmental diseases. | http://www.disgenet.org/web/DisGeNET [37] |
| CTDTM | Integrated chemical-gene, chemical-disease and gene-disease interactions manually-curated from the literature. | http://ctdbase.org/ [38] | |
| Multiple Type | InAct | Standards-compliant repository of molecular interactions, including protein-protein, protein-small molecule and protein-nucleic acid interactions. | https://www.ebi.ac.uk/intact/ [39,40] |
| BioGrid | Curated biological database of protein-protein interactions, genetic interactions, chemical interactions and post-translational modifications. | http://thebiogrid.org/ [41] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, J.; Seo, D.; Sael, L. Review on Graph Clustering and Subgraph Similarity Based Analysis of Neurological Disorders. Int. J. Mol. Sci. 2016, 17, 862. https://doi.org/10.3390/ijms17060862
Thomas J, Seo D, Sael L. Review on Graph Clustering and Subgraph Similarity Based Analysis of Neurological Disorders. International Journal of Molecular Sciences. 2016; 17(6):862. https://doi.org/10.3390/ijms17060862
Chicago/Turabian StyleThomas, Jaya, Dongmin Seo, and Lee Sael. 2016. "Review on Graph Clustering and Subgraph Similarity Based Analysis of Neurological Disorders" International Journal of Molecular Sciences 17, no. 6: 862. https://doi.org/10.3390/ijms17060862