
Study on the Application of the Combination of TMD Simulation and Umbrella Sampling in PMF Calculation for Molecular Conformational Transitions

Abstract
:
1. Introduction
2. Results and Discussion
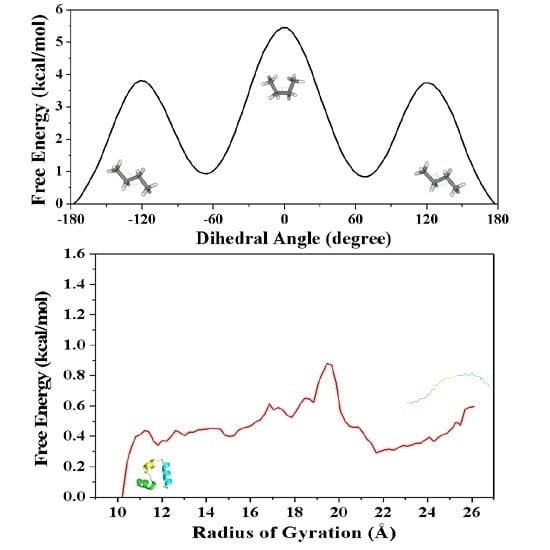
2.1. Potential of Mean Force (PMF) Calculations Based on the Combination of Targeted Molecular Dynamics (TMD) Simulation and Umbrella Samplings along Different Coordinates for the Dihedral Rotation of Butane
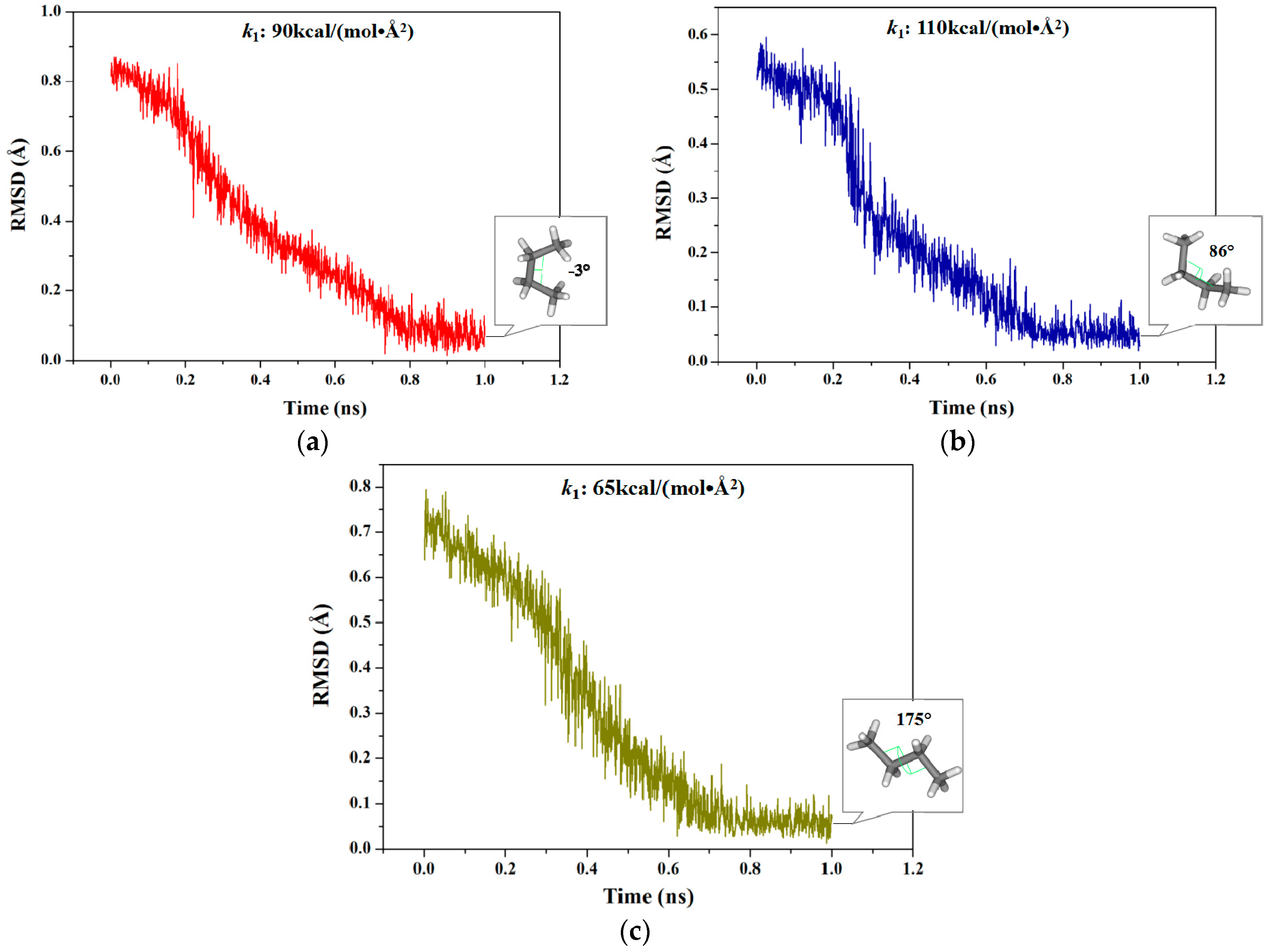
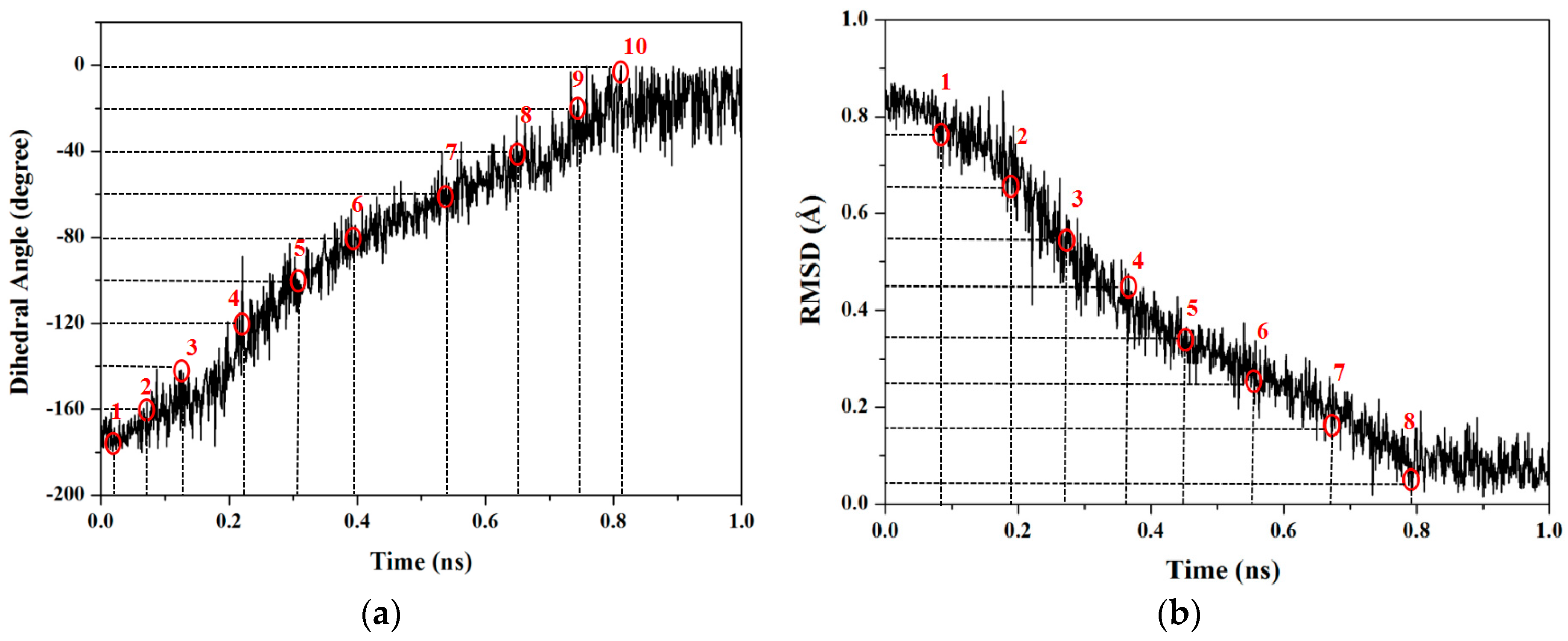
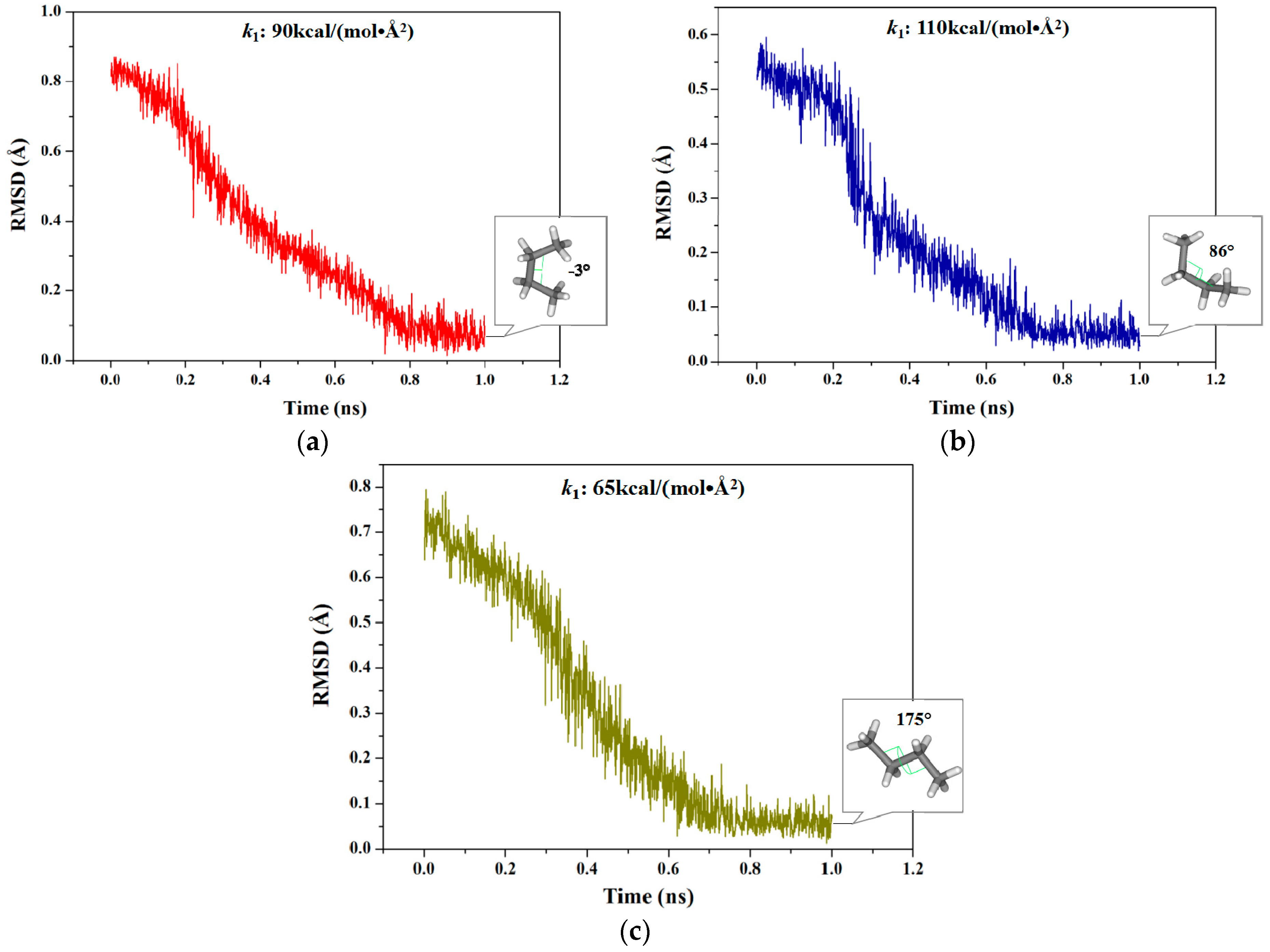
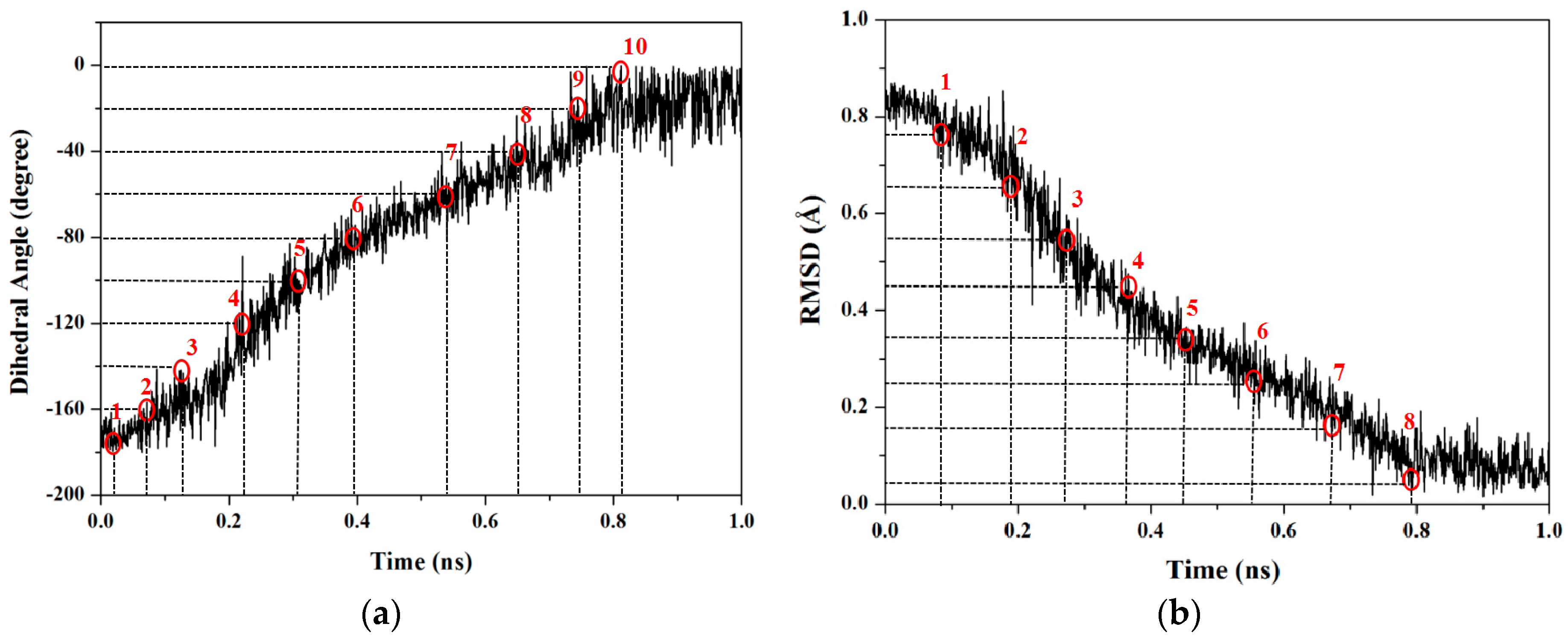
2.1.1. Targeted Molecular Dynamics Simulations for Rotations of Butane
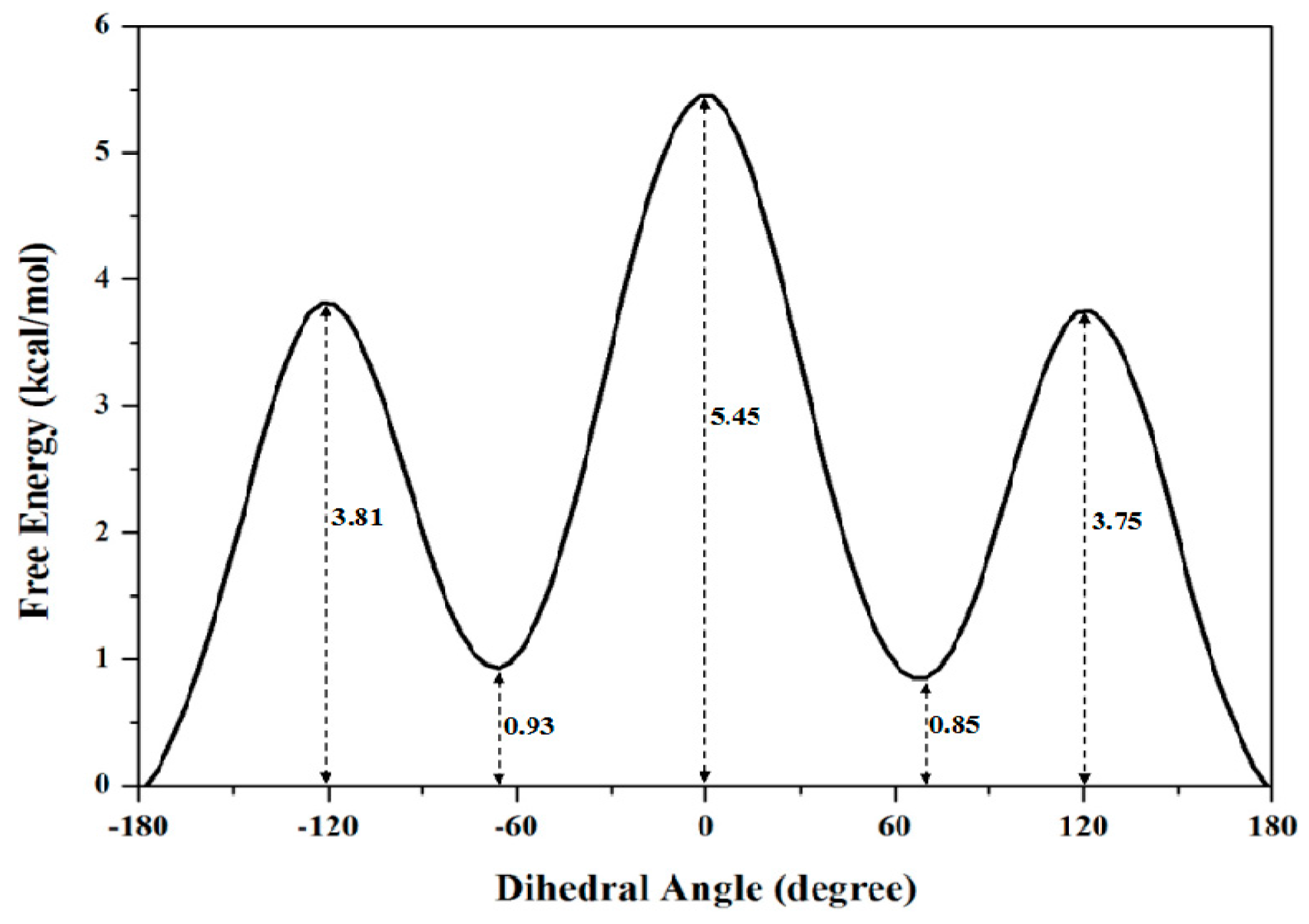
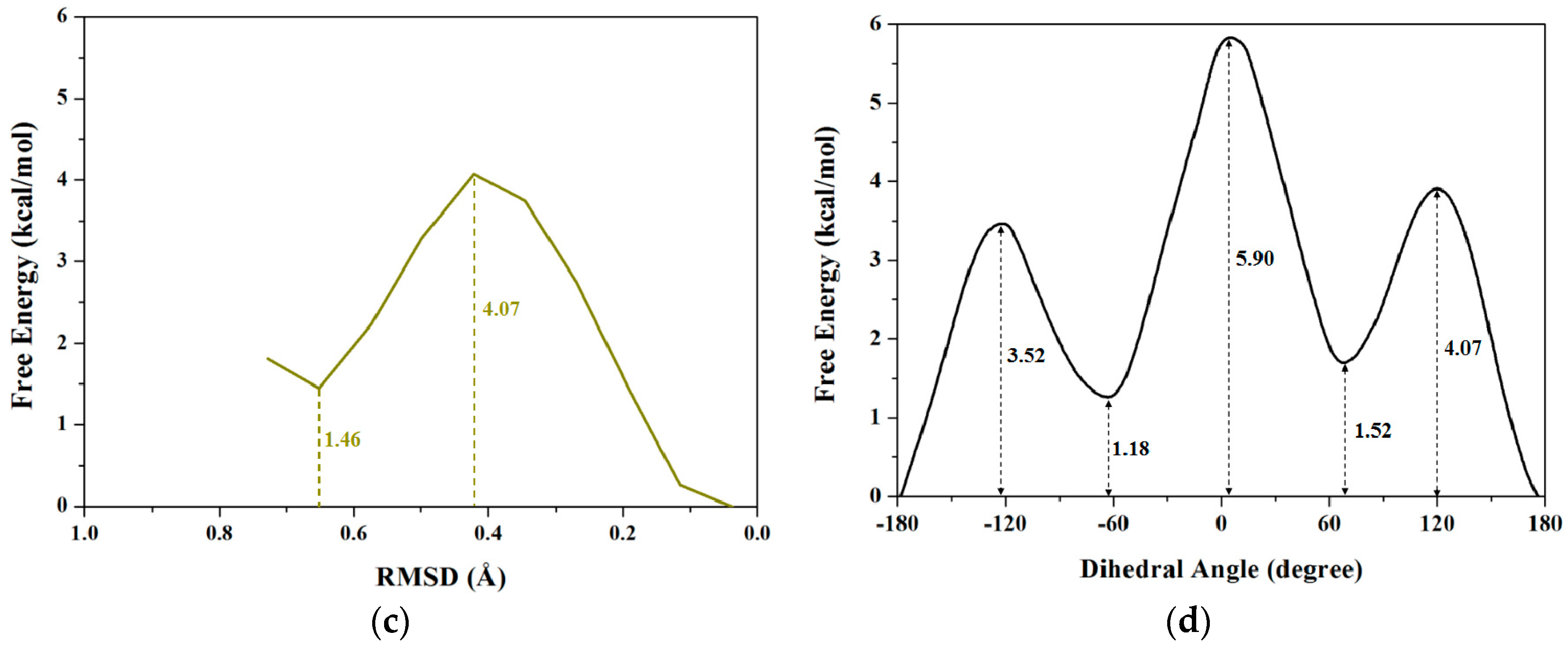
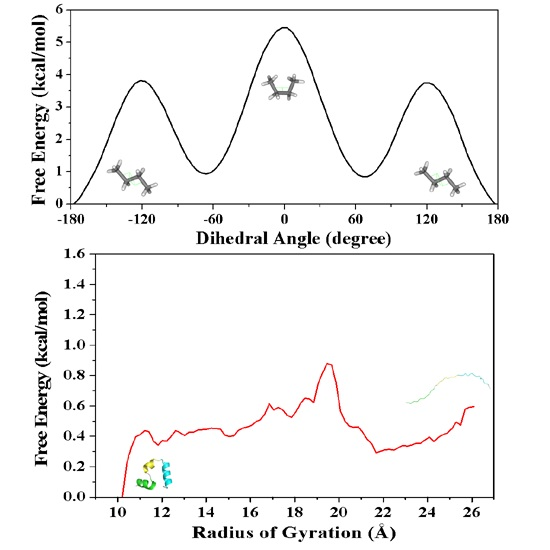
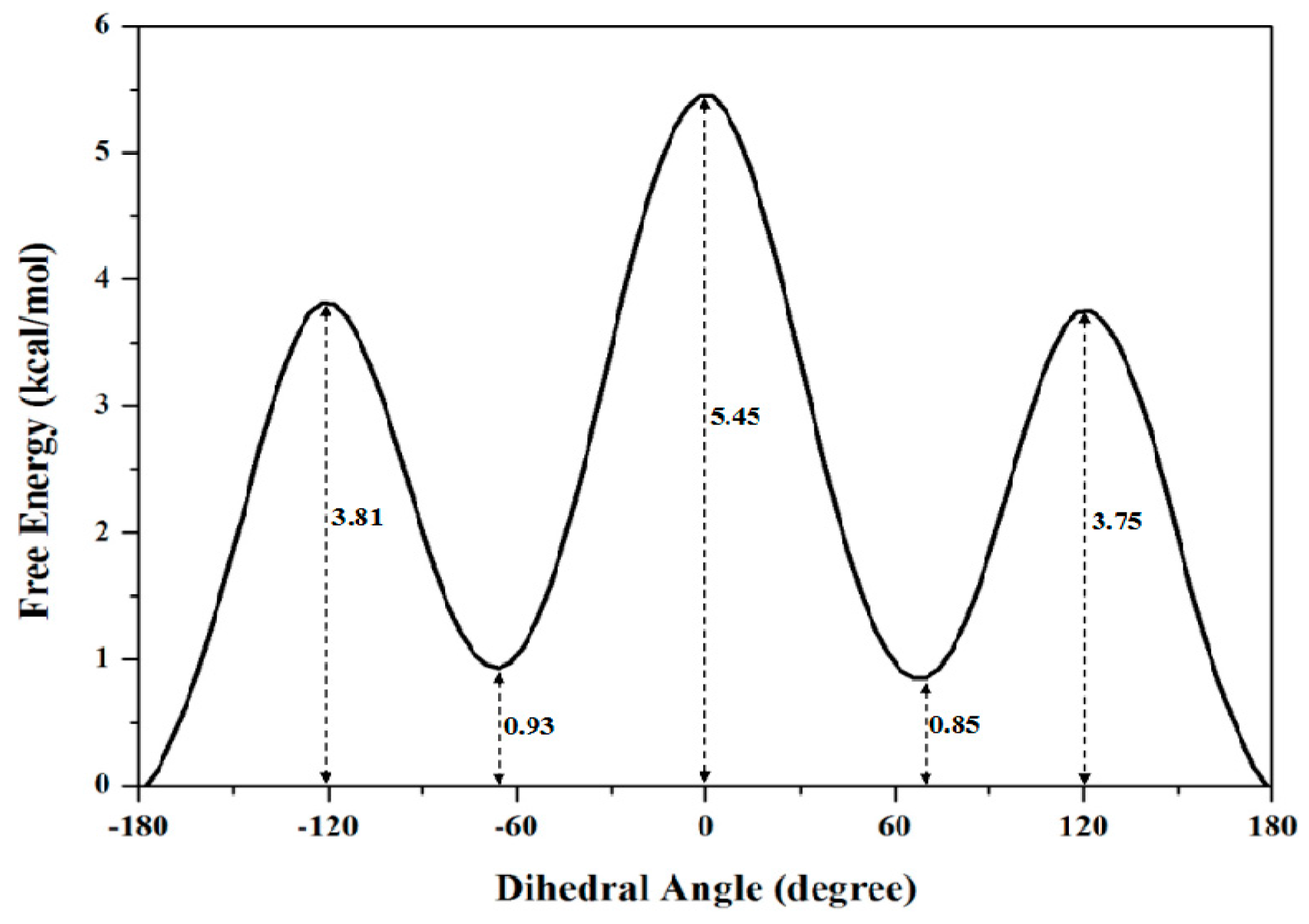
2.1.2. PMF Calculation along with the Coordinate of Dihedral Angle
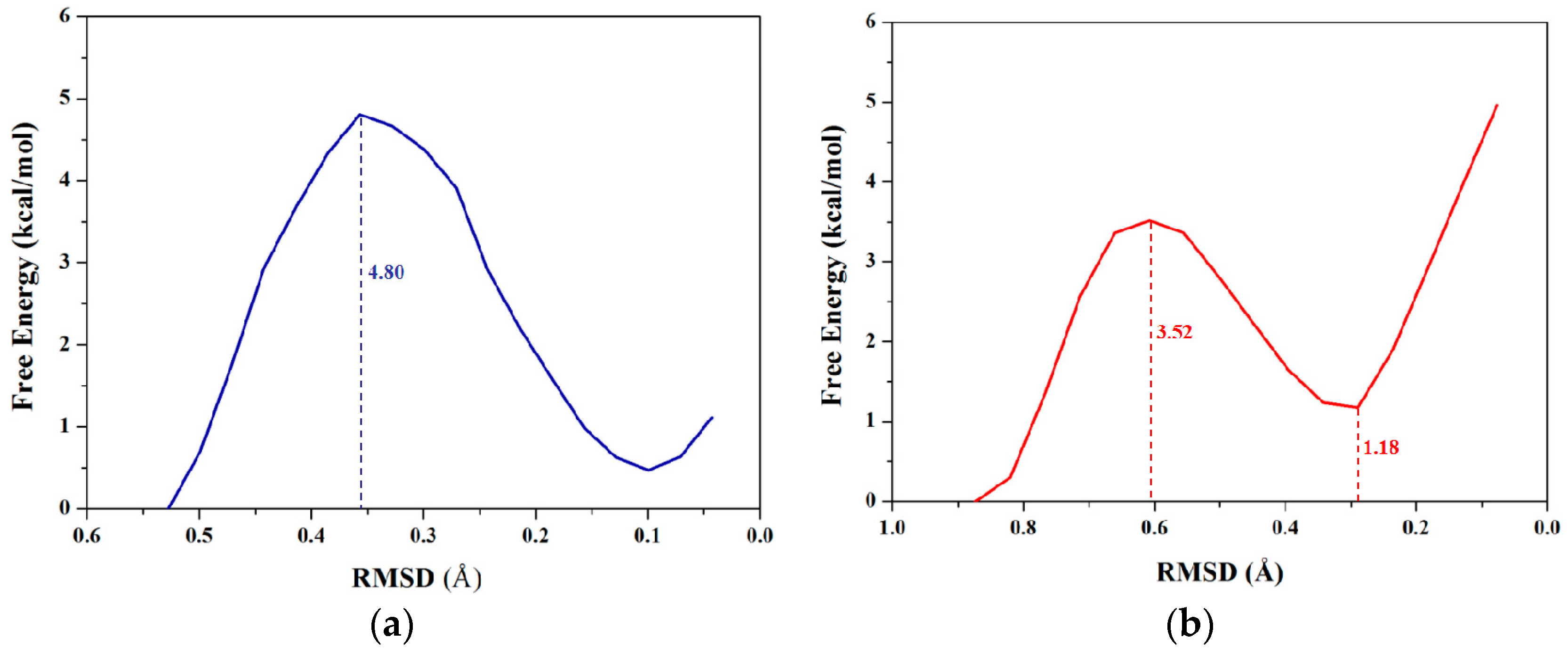
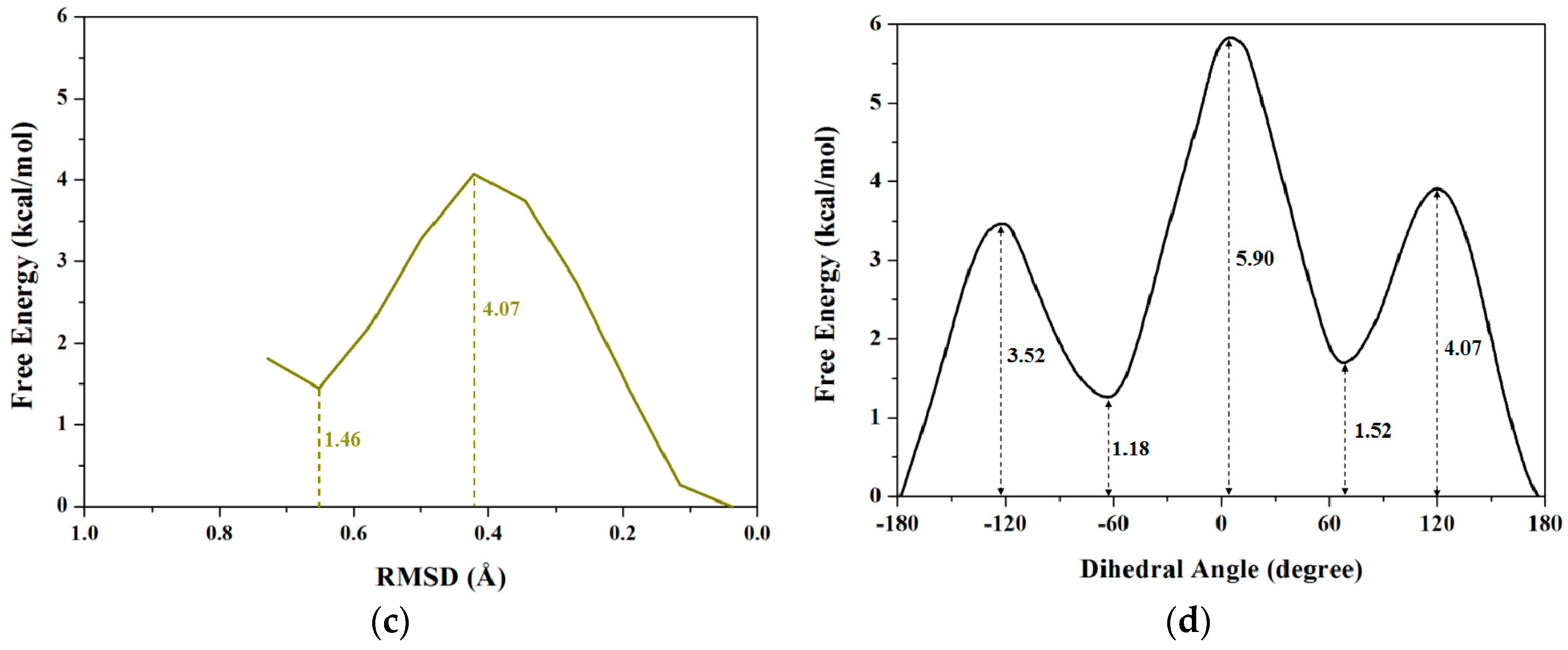
2.1.3. PMF Calculation along with the Coordinate of Root Mean Square Deviation (RMSD)
2.2. PMF Calculation Based on the Combination of TMD Simulation and Umbrella Samplings for the Unfolding Process of 35-Residue Villin Headpiece Subdomain (HP35) Protein
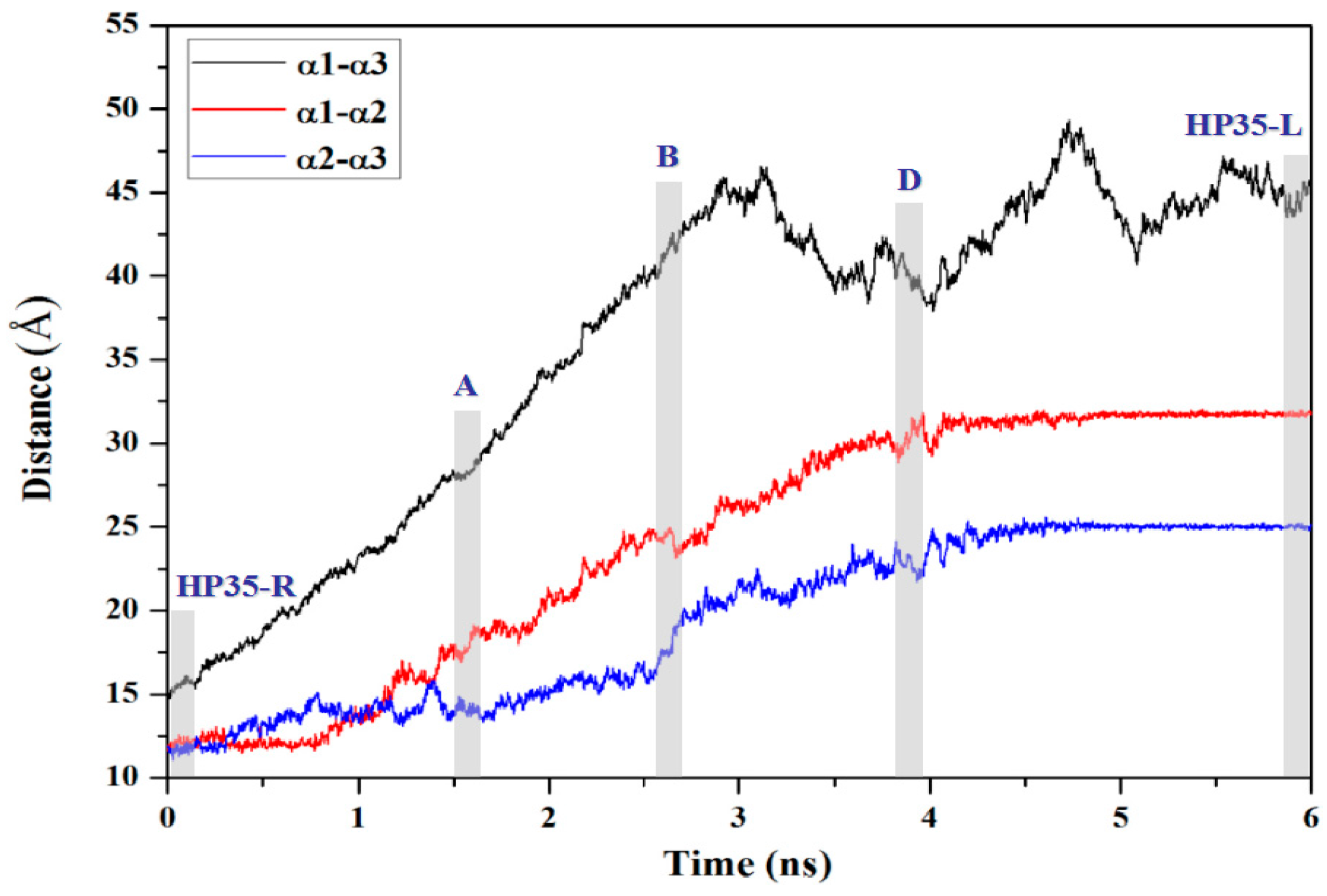
2.2.1. Targeted Molecular Dynamics Simulation for the Unfolding Process
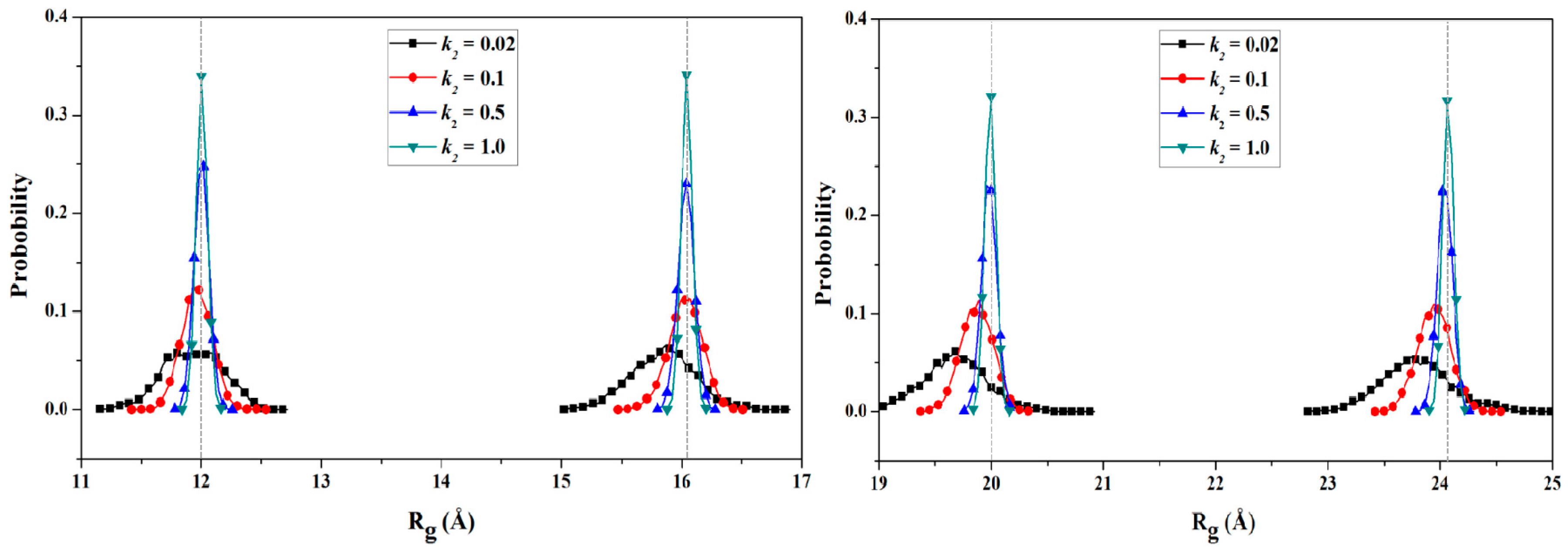
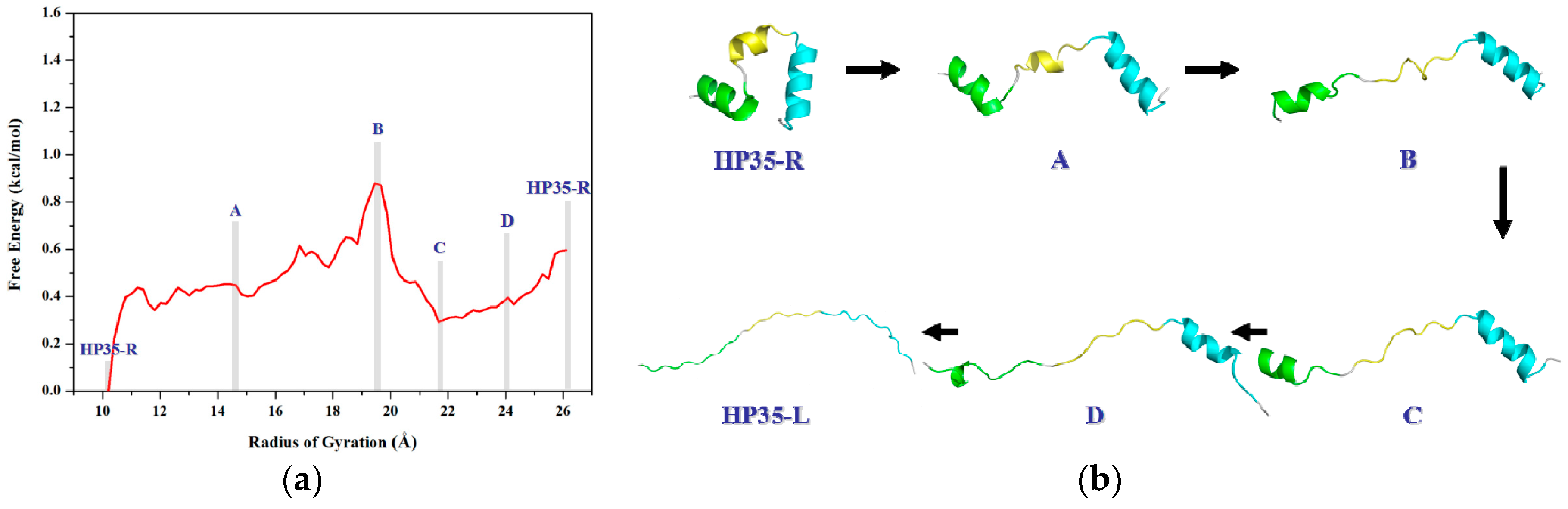
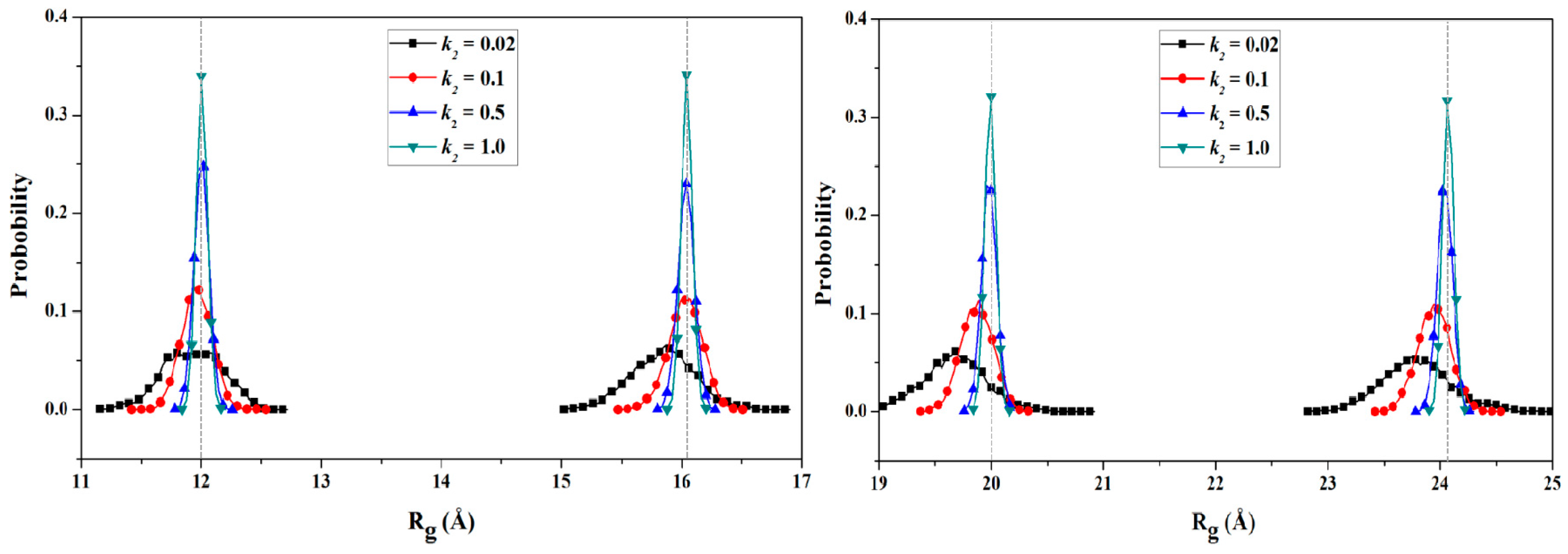
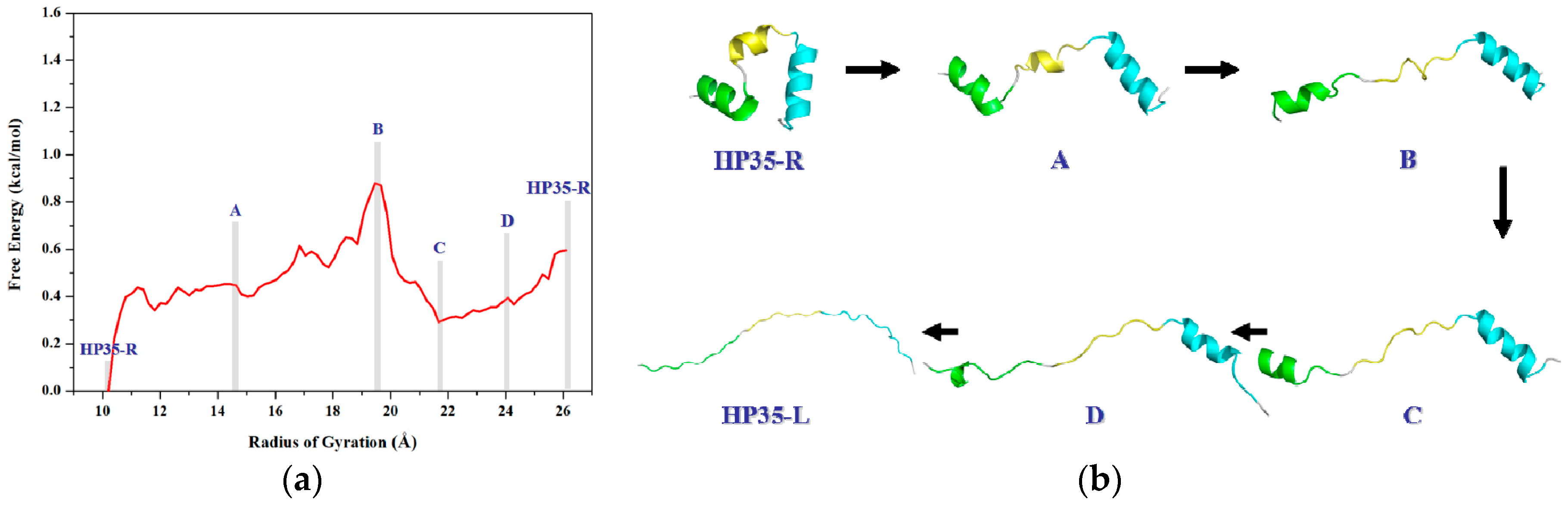
2.2.2. PMF Calculation along with the Coordinate of the Radius of Gyration
3. Models and Methods
3.1. Conventional Molecular Dynamics Simulation
3.2. Targeted Molecular Dynamics Simulation
3.3. Umbrella Sampling and the Potential of Mean Force
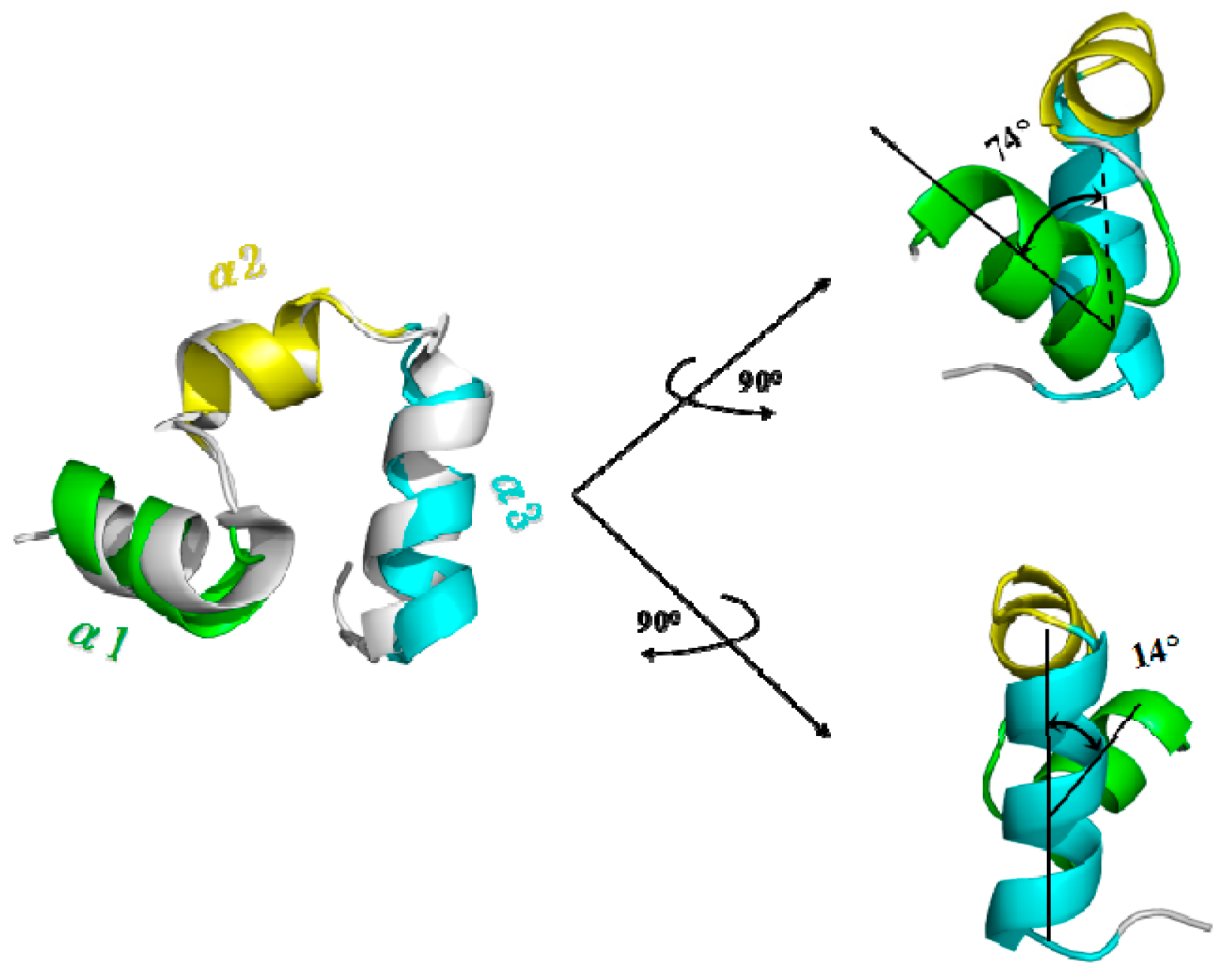
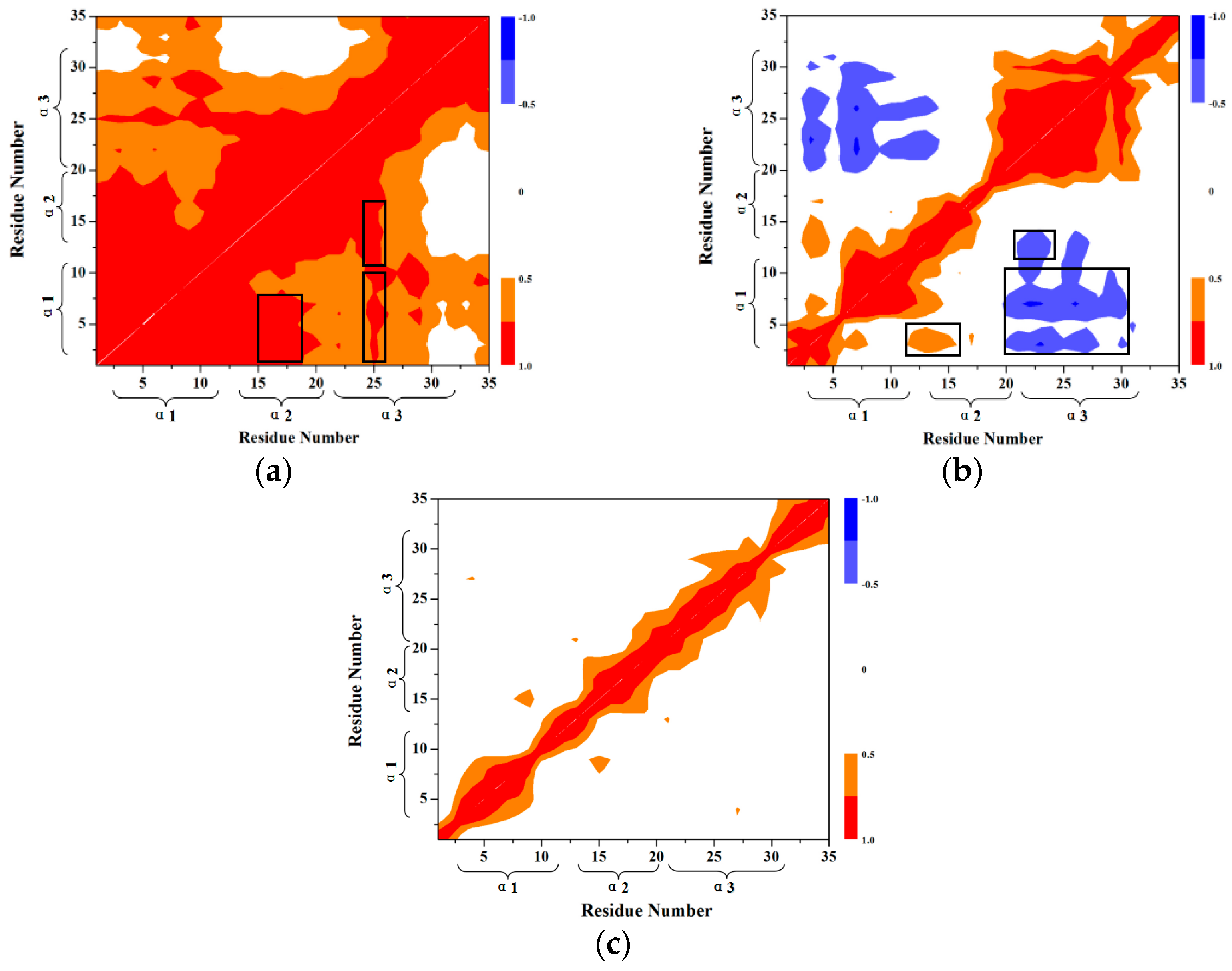
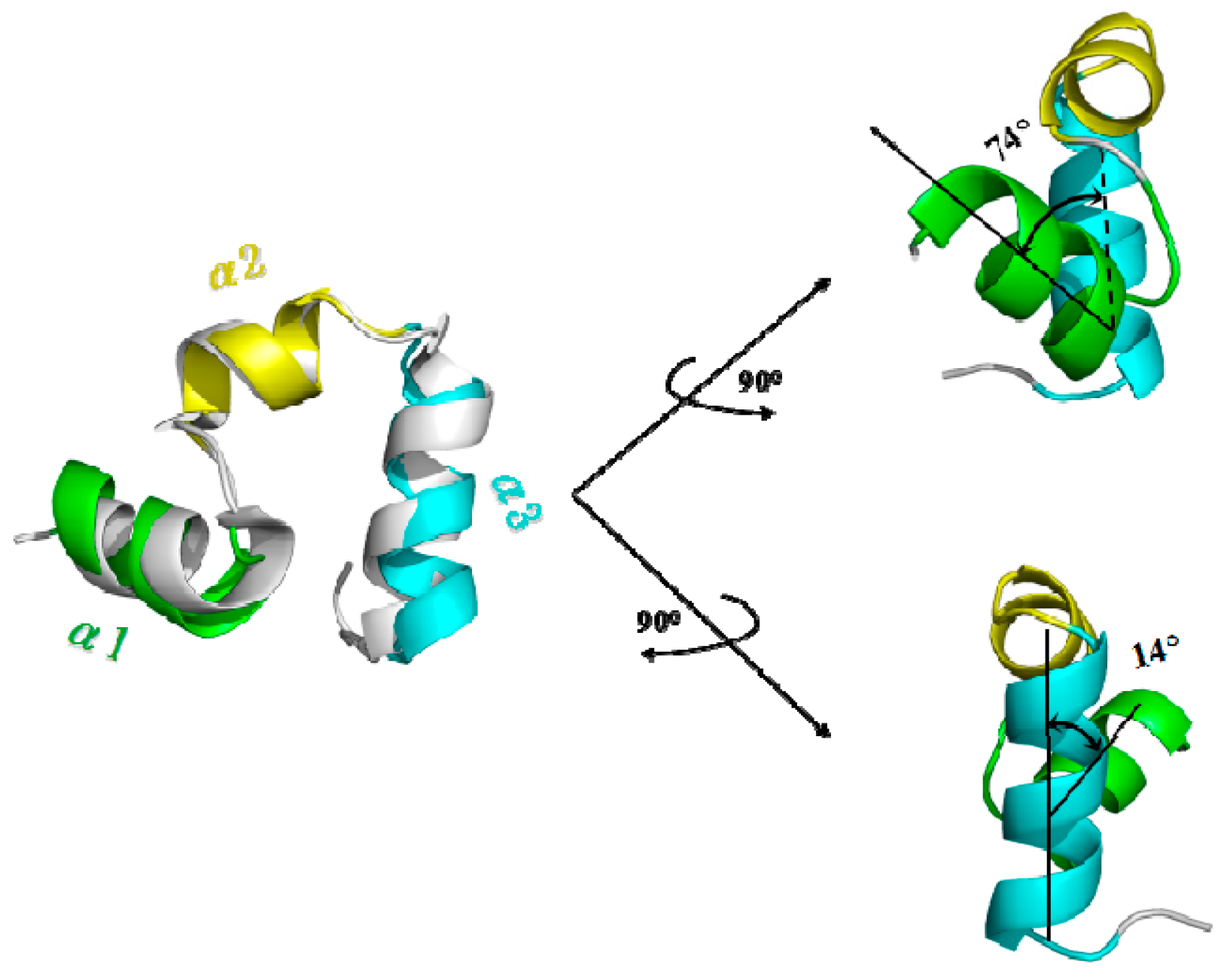
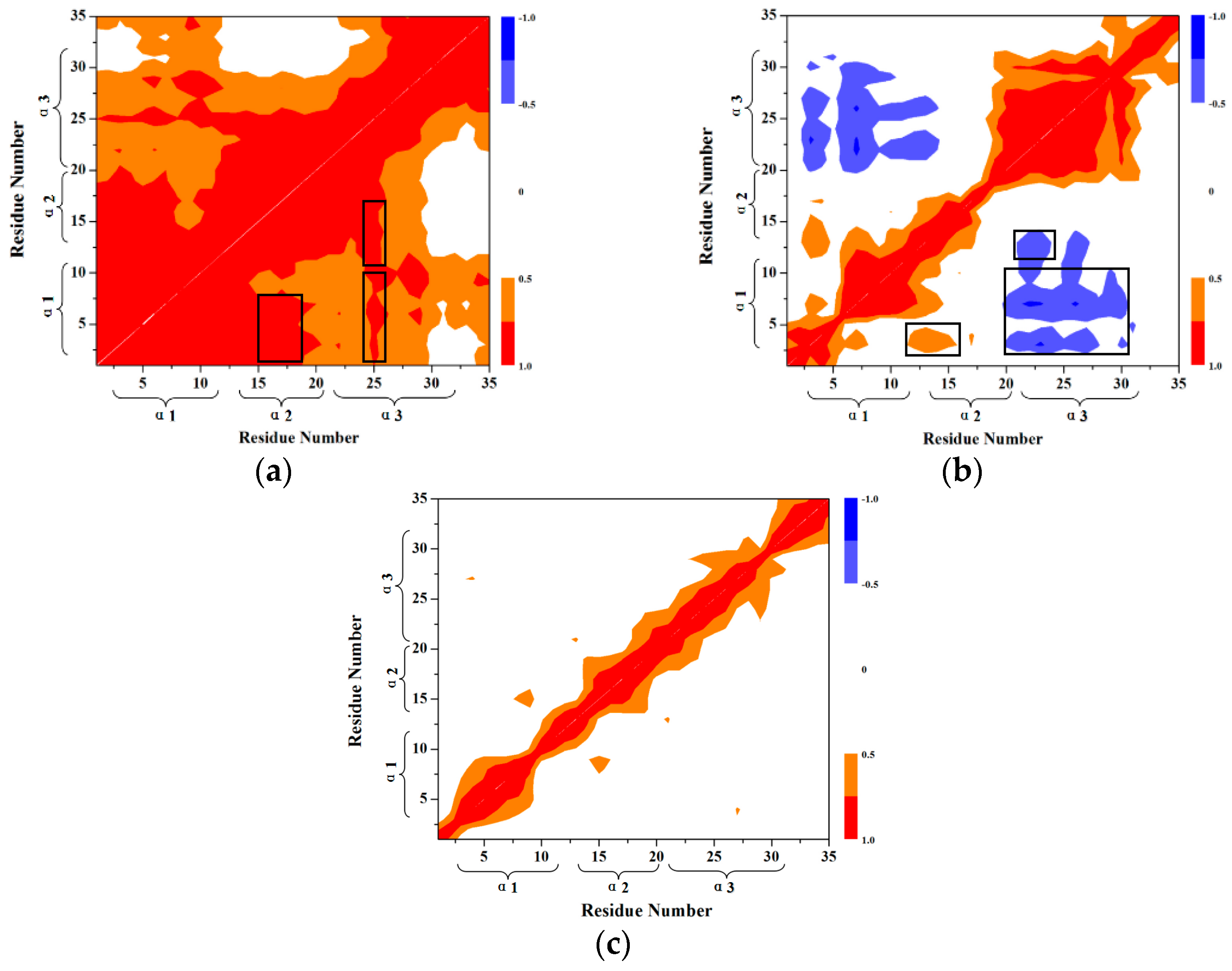
3.4. Calculations of Interhelical Angle and Correlation of Atomic Motions
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brandsdal, B.O.; Österberg, F.; Almlöf, M.; Feierberg, I.; Luzhkov, V.B.; Åqvist, J. Free energy calculations and ligand binding. Adv. Protein Chem. 2003, 66, 123–158. [Google Scholar] [PubMed]
- Fenton, A.W. Allostery: An illustrated definition for the ‘second secret of life’. Trends Biochem. Sci. 2008, 33, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Hilbert, M.; Kebbel, F.; Gubaev, A.; Klostermeier, D. eIF4G stimulates the activity of the DEAD box protein eIF4A by a conformational guidance mechanism. Nucleic Acids Res. 2011, 39, 2260–2270. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T.; Miller, L.J. Seven transmembrane receptors as shapeshifting proteins: The impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol. Rev. 2010, 62, 265–304. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Tang, Z.; Xia, G.; Wassmann, K.; Matsumoto, T.; Rizo, J.; Yu, H. The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat. Struct. Mol. Biol. 2004, 11, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Khalili-Araghi, F.; Tajkhorshid, E.; Schulten, K. Free energy calculation from steered molecular dynamics simulations using Jarzynski’s equality. J. Chem. Phys. 2003, 119, 3559–3566. [Google Scholar] [CrossRef]
- Hénin, J.; Chipot, C. Overcoming free energy barriers using unconstrained molecular dynamics simulations. J. Chem. Phys. 2004, 121, 2904–2914. [Google Scholar] [CrossRef] [PubMed]
- Kofke, D.A. Free energy methods in molecular simulation. Fluid Phase Equilib. 2005, 228, 41–48. [Google Scholar] [CrossRef]
- Pearlman, D.A. A comparison of alternative approaches to free energy calculations. J. Phys. Chem. 1994, 98, 1487–1493. [Google Scholar] [CrossRef]
- Smith, D.E.; Haymet, A. Free energy, entropy, and internal energy of hydrophobic interactions: Computer simulations. J. Chem. Phys. 1993, 98, 6445–6454. [Google Scholar] [CrossRef]
- Tobias, D.J.; Brooks, C.L. Calculation of free energy surfaces using the methods of thermodynamic perturbation theory. Chem. Phys. Lett. 1987, 142, 472–476. [Google Scholar] [CrossRef]
- Kirkwood, J.G. Statistical mechanics of fluid mixtures. J. Chem. Phys. 1935, 3, 300–313. [Google Scholar] [CrossRef]
- Roux, B. The calculation of the potential of mean force using computer simulations. Comput. Phys. Commun. 1995, 91, 275–282. [Google Scholar] [CrossRef]
- Allen, T.W.; Andersen, O.S.; Roux, B. Molecular dynamics—Potential of mean force calculations as a tool for understanding ion permeation and selectivity in narrow channels. Biophys. Chem. 2006, 124, 251–267. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.W.; Baştuğ, T.; Kuyucak, S.; Chung, S.-H. Gramicidin A channel as a test ground for molecular dynamics force fields. Biophys. J. 2003, 84, 2159–2168. [Google Scholar] [CrossRef]
- Baştuğ, T.; Chen, P.-C.; Patra, S.M.; Kuyucak, S. Potential of mean force calculations of ligand binding to ion channels from Jarzynski’s equality and umbrella sampling. J. Chem. Phys. 2008, 128, 155104. [Google Scholar] [CrossRef] [PubMed]
- Baştuğ, T.; Kuyucak, S. Application of Jarzynski’s equality in simple versus complex systems. Chem. Phys. Lett. 2007, 436, 383–387. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Ma, S.; Stankovich, M.T.; Truhlar, D.G.; Gao, J. Potential of mean force calculation for the proton and hydride transfer reactions catalyzed by medium-chain acyl-CoA dehydrogenase: Effect of mutations on enzyme catalysis. Biochemistry 2005, 44, 16549–16562. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-C.; Kuyucak, S. Accurate determination of the binding free energy for KcsA-charybdotoxin complex from the potential of mean force calculations with restraints. Biophys. J. 2011, 100, 2466–2474. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Tsao, D.; Nie, H.; Dokholyan, N.V. Ab initio folding of proteins with all-atom discrete molecular dynamics. Structure 2008, 16, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Doudou, S.; Burton, N.A.; Henchman, R.H. Standard free energy of binding from a one-dimensional potential of mean force. J. Chem. Theory Comput. 2009, 5, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Forney, M.W.; Janosi, L.; Kosztin, I. Calculating free-energy profiles in biomolecular systems from fast nonequilibrium processes. Phys. Rev. E 2008, 78, 051913. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhao, X.; Zheng, F.; Zhan, C.-G. Cocaine esterase–cocaine binding process and the free energy profiles by molecular dynamics and potential of mean force simulations. J. Phys. Chem. B 2012, 116, 3361–3368. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Luo, Y.; Maragliano, L.; Roux, B. Calculation of free energy landscape in multi-dimensions with Hamiltonian-exchange umbrella sampling on petascale supercomputer. J. Chem. Theory Comput. 2012, 8, 4672–4680. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Im, W. Revisiting hydrophobic mismatch with free energy simulation studies of transmembrane helix tilt and rotation. Biophys. J. 2010, 99, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Kosztin, I.; Barz, B.; Janosi, L. Calculating potentials of mean force and diffusion coefficients from nonequilibrium processes without Jarzynski’s equality. J. Chem. Phys. 2006, 124, 064106. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Im, W. Restraint potential and free energy decomposition formalism for helical tilting. Chem. Phys. Lett. 2007, 441, 132–135. [Google Scholar] [CrossRef]
- Lee, M.S.; Olson, M.A. Calculation of absolute protein-ligand binding affinity using path and endpoint approaches. Biophys. J. 2006, 90, 864–877. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Feixas, F.; Eun, C.; McCammon, J.A. Accelerated molecular dynamics simulations of protein folding. J. Comput. Chem. 2015, 36, 1536–1549. [Google Scholar] [CrossRef] [PubMed]
- Okada, O.; Odai, K.; Sugimoto, T.; Ito, E. Molecular dynamics simulations for glutamate-binding and cleft-closing processes of the ligand-binding domain of GluR2. Biophys. Chem. 2012, 162, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Pu, J.; Gao, J.; Truhlar, D.G.; Allemann, R.K. Hydride transfer reaction catalyzed by hyperthermophilic dihydrofolate reductase is dominated by quantum mechanical tunneling and is promoted by both inter-and intramonomeric correlated motions. J. Am. Chem. Soc. 2006, 128, 8015–8023. [Google Scholar] [CrossRef] [PubMed]
- Raghav, N.; Chakraborty, S.; Maiti, P.K. Molecular mechanism of water permeation in a helium impermeable graphene and graphene oxide membrane. Phys. Chem. Chem. Phys. 2015, 17, 20557–20562. [Google Scholar] [CrossRef] [PubMed]
- Roca, M.; de Maria, L.; Wodak, S.J.; Moliner, V.; Tunon, I.; Giraldo, J. Coupling of the guanosine glycosidic bond conformation and the ribonucleotide cleavage reaction: Implications for barnase catalysis. Proteins Struct. Funct. Bioinform. 2008, 70, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Chan, H.S. Anti-cooperativity and cooperativity in hydrophobic interactions: Three-body free energy landscapes and comparison with implicit-solvent potential functions for proteins. Proteins Struct. Funct. Bioinform. 2002, 48, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Vashisth, H.; Abrams, C.F. Ligand escape pathways and (un)binding free energy calculations for the hexameric insulin-phenol complex. Biophys. J. 2008, 95, 4193–4204. [Google Scholar] [CrossRef] [PubMed]
- Zeller, F.; Zacharias, M. Adaptive biasing combined with Hamiltonian replica exchange to improve umbrella sampling free energy simulations. J. Chem. Theory Comput. 2014, 10, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Zeller, F.; Zacharias, M. Efficient calculation of relative binding free energies by umbrella sampling perturbation. J. Comput. Chem. 2014, 35, 2256–2262. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tong, M.; Liu, C.; Song, C.; Wei, D.; Zhao, Q.; Tang, M. Molecular dynamics simulations on inclusion complexes for chiral enantiomers with heterocyclic cyclodecapeptide. Comput. Theor. Chem. 2014, 1027, 46–52. [Google Scholar] [CrossRef]
- Bagai, S.; Sun, C.; Tang, T. Potential of mean force of polyethylenimine-mediated DNA attraction. J. Phys. Chem. B 2012, 117, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Banavali, N.K.; MacKerell, A.D. Free energy and structural pathways of base flipping in a DNA GCGC containing sequence. J. Mol. Biol. 2002, 319, 141–160. [Google Scholar] [CrossRef]
- Baştuğ, T.; Kuyucak, S. Energetics of ion permeation, rejection, binding, and block in gramicidin A from free energy simulations. Biophys. J. 2006, 90, 3941–3950. [Google Scholar] [CrossRef] [PubMed]
- Berneche, S.; Roux, B. Energetics of ion conduction through the K+ channel. Nature 2001, 414, 73–77. [Google Scholar] [PubMed]
- Gumbart, J.C.; Roux, B.; Chipot, C. Standard binding free energies from computer simulations: What is the best strategy? J. Chem. Theory Comput. 2012, 9, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Gumbart, J.C.; Roux, B.; Chipot, C. Efficient determination of protein–protein standard binding free energies from first principles. J. Chem. Theory Comput. 2013, 9, 3789–3798. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, Y.; Tian, S.; Wang, J.; Hou, T. P-loop conformation governed crizotinib resistance in G2032R-mutated ROS1 tyrosine kinase: Clues from free energy landscape. PLoS Comput. Biol. 2014, 10, e1003729. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Deng, Y.; Roux, B. Absolute binding free energy calculations using molecular dynamics simulations with restraining potentials. Biophys. J. 2006, 91, 2798–2814. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.-J.; Roux, B. Calculation of absolute protein–ligand binding free energy from computer simulations. Proc. Natl. Acad. Sci. USA 2005, 102, 6825–6830. [Google Scholar] [CrossRef] [PubMed]
- Zeller, F.; Zacharias, M. Evaluation of generalized born model accuracy for absolute binding free energy calculations. J. Phys. Chem. B 2014, 118, 7467–7474. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Wang, H.; Grant, B.; Sine, S.M.; McCammon, J.A. Targeted molecular dynamics study of C-loop closure and channel gating in nicotinic receptors. PLoS Comput. Biol. 2006, 2, e134. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Predeus, A.V.; Burton, Z.F.; Feig, M. Energetic and structural details of the trigger-loop closing transition in RNA polymerase II. Biophys. J. 2013, 105, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Fujitani, H. On accurate calculation of the potential of mean force between antigen and antibody: A case of the HyHEL-10-hen egg white lysozyme system. Chem. Phys. Lett. 2014, 609, 50–53. [Google Scholar] [CrossRef]
- Banavali, N.K.; Roux, B. Free energy landscape of A-DNA to B-DNA conversion in aqueous solution. J. Am. Chem. Soc. 2005, 127, 6866–6876. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, Y.-G.; Kim, K.K.; Seok, C. Transition between B-DNA and Z-DNA: Free energy landscape for the B–Z junction propagation. J. Phys. Chem. B 2010, 114, 9872–9881. [Google Scholar] [CrossRef] [PubMed]
- Noy, A.; Perez, A.; Laughton, C.A.; Orozco, M. Theoretical study of large conformational transitions in DNA: The B↔A conformational change in water and ethanol/water. Nucleic Acids Res. 2007, 35, 3330–3338. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Pan, Y.; Zheng, F.; Zhan, C.-G. Reaction pathway and free energy profile for prechemical reaction step of human butyrylcholinesterase-catalyzed hydrolysis of (−)-cocaineby combined targeted molecular dynamics and potential of mean force simulations. J. Phys. Chem. B 2010, 114, 13545–13554. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Schatz, G.C. Free energy profile and mechanism of self-assembly of peptide amphiphiles based on a collective assembly coordinate. J. Phys. Chem. B 2013, 117, 9004–9013. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Wang, Q.; Wang, Y.; Chen, G. Structures and energies of the transition between two conformations of the alternate frame folding calbindin-D9k protein: A theoretical study. RSC Adv. 2015, 5, 65798–65810. [Google Scholar] [CrossRef]
- Chiu, T.K.; Kubelka, J.; Herbst-Irmer, R.; Eaton, W.A.; Hofrichter, J.; Davies, D.R. High-resolution X-ray crystal structures of the villin headpiece subdomain, an ultrafast folding protein. Proc. Natl. Acad. Sci. USA 2005, 102, 7517–7522. [Google Scholar] [CrossRef] [PubMed]
- Kubelka, J.; Henry, E.R.; Cellmer, T.; Hofrichter, J.; Eaton, W.A. Chemical, physical, and theoretical kinetics of an ultrafast folding protein. Proc. Natl. Acad. Sci. USA 2008, 105, 18655–18662. [Google Scholar] [CrossRef] [PubMed]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular-orbital methods. IX. An extended gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Sousa, J.; Silva, P.; Machado, A.; Reis, M.; Romanielo, L.; Hori, C. Application of computational chemistry methods to obtain thermodynamic data for hydrogen production from liquefied petroleum gas. Braz. J. Chem. Eng. 2013, 30, 83–93. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09, Revision A. 02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Habenschuss, A.; Narten, A. X-ray diffraction study of liquid n-butane at 140 and 267 K. J. Chem. Phys. 1989, 91, 4299–4306. [Google Scholar] [CrossRef]
- Herrebout, W.; van der Veken, B.; Wang, A.; Durig, J. Enthalpy difference between conformers of n-butane and the potential function governing conformational interchange. J. Phys. Chem. 1995, 99, 578–585. [Google Scholar] [CrossRef]
- Ota, N.; Brünger, A.T. Overcoming barriers in macromolecular simulations: Non-Boltzmann thermodynamic integration. Theor. Chem. Acc. 1998, 98, 171–181. [Google Scholar] [CrossRef]
- Case, D.; Darden, T.; Cheatham, T., III; Simmerling, C.; Wang, J.; Duke, R.; Luo, R.; Merz, K.; Pearlman, D.; Crowley, M.; et al. AMBER 9; University of California: San Francisco, CA, USA, 2006. [Google Scholar]
- Potential of Mean Force (PMF) Calculations with AMBER and WHAM. Available online: https://sites.google.com/site/wangtingpage/home/tutorials/pmf (accessed on 4 July 2013).
- Murcko, M.A.; Castejon, H.; Wiberg, K.B. Carbon–carbon rotational barriers in butane, 1-butene, and 1,3-butadiene. J. Phys. Chem. 1996, 100, 16162–16168. [Google Scholar] [CrossRef]
- Kubelka, J.; Eaton, W.A.; Hofrichter, J. Experimental tests of villin subdomain folding simulations. J. Mol. Biol. 2003, 329, 625–630. [Google Scholar] [CrossRef]
- Lei, H.; Wu, C.; Liu, H.; Duan, Y. Folding free-energy landscape of villin headpiece subdomain from molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2007, 104, 4925–4930. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Shaw, D.E. How fast-folding proteins fold. Science 2011, 334, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Cellmer, T.; Henry, E.R.; Hofrichter, J.; Eaton, W.A. Measuring internal friction of an ultrafast-folding protein. Proc. Natl. Acad. Sci. USA 2008, 105, 18320–18325. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Ruiz, R.; Henry, E.R.; Kubelka, J.; Hofrichter, J.; Muñoz, V.; Sanchez-Ruiz, J.M.; Eaton, W.A. Estimating free-energy barrier heights for an ultrafast folding protein from calorimetric and kinetic data. J. Phys. Chem. B 2008, 112, 5938–5949. [Google Scholar] [CrossRef] [PubMed]
- Kubelka, J.; Chiu, T.K.; Davies, D.R.; Eaton, W.A.; Hofrichter, J. Sub-microsecond protein folding. J. Mol. Biol. 2006, 359, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.C.; Duan, Y. Distinguish protein decoys by using a scoring function based on a new AMBER force field, short molecular dynamics simulations, and the generalized born solvent model. Proteins Struct. Funct. Bioinform. 2004, 55, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Schlitter, J.; Engels, M.; Krüger, P. Targeted molecular dynamics: A new approach for searching pathways of conformational transitions. J. Mol. Graph. 1994, 12, 84–89. [Google Scholar] [CrossRef]
- Schlitter, J.; Engels, M.; Krüger, P.; Jacoby, E.; Wollmer, A. Targeted molecular dynamics simulation of conformational change-application to the T↔R transition in insulin. Mol. Simul. 1993, 10, 291–308. [Google Scholar] [CrossRef]
- Chipot, C.; Jaffe, R.; Maigret, B.; Pearlman, D.A.; Kollman, P.A. Benzene dimer: A good model for π–π interactions in proteins? A comparison between the benzene and the toluene dimers in the gas phase and in an aqueous solution. J. Am. Chem. Soc. 1996, 118, 11217–11224. [Google Scholar] [CrossRef]
- Kumar, S.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A.; Rosenberg, J.M. The weighted histogram analysis method for free-energy calculations on biomolecules. I: The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Patey, G.; Valleau, J. A Monte Carlo method for obtaining the interionic potential of mean force in ionic solution. J. Chem. Phys. 1975, 63, 2334–2339. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Weighted Histogram Analysis Method for Analyzing Umbrella Sampling Simulation Data. Available online: http://membrane.urmc.rochester.edu/content/wham (accessed on 7 November 2003).
- Interhelical Angle Program (with Sign Designation). Available online: http://nmr.uhnres.utoronto.ca/ikura/resources/data+sw/interhlx (accessed on 20 October 1998).
- Sadiq, S.K.; de Fabritiis, G. Explicit solvent dynamics and energetics of HIV-1 protease flap opening and closing. Proteins Struct. Funct. Bioinform. 2010, 78, 2873–2885. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Kollman, P.A. SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Yap, K.L.; Ames, J.B.; Swindells, M.B.; Ikura, M. Diversity of conformational states and changes within the EF-hand protein superfamily. Proteins Struct. Funct. Bioinform. 1999, 37, 499–507. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coordinates | Free Energy (kcal/mol) | |||
|---|---|---|---|---|
| anti | 120° | gauche | syn | |
| Dihedral Angle | 0 | 3.75 | 0.85 | 5.45 |
| RMSD | 0 | 4.07 | 1.52 | 5.9 |
| References | 0 a | 4.22 a | 1.44 a | 5.86 a |
| 0 b | 3.83 b | 0.66 b | 6.48 b | |
| Helices | Hydrogen Bond | HP35-R | B | D | HP35-L |
|---|---|---|---|---|---|
| α1 | (Ala8)N–H···O(Glu4) | 95.32 | – | – | – |
| (Val9)N–H···O(Asp5) | 99.96 | – | – | – | |
| (Asp5)N–H···O(Leu1) | 99.56 | 99.72 | – | – | |
| (Phe6)N–H···O(Ser2) | 95.56 | 97.20 | – | – | |
| (Lys7)N–H···O(Glu4) | 97.92 | 69.64 | 65.92 | – | |
| (Phe10)N–H···O(Phe6) | 99.72 | – | – | – | |
| (Glu4)N–H···O(Leu1) | 67.40 | – | – | – | |
| (Gly11)N–H···O(Lys7) | 52.60 | – | – | – | |
| α2 | (Ala18)N–H···O(Agr14) | 99.76 | 99.68 | – | – |
| (Leu20)N–H···O(Phe17) | 97.48 | – | – | – | |
| (Phe17)N–H···O(Thr13) | 99.32 | – | – | – | |
| (Asn19)N–H···O(Ala16) | 71.00 | – | – | – | |
| (Asn19)N–H···O(Ser15) | 71.80 | 68.56 | – | – | |
| (Ala16)N–H···O(Thr13) | 63.76 | – | – | – | |
| α3 | (Gln26)N–H···O(Leu22) | 100 | 99.92 | 99.28 | – |
| (Leu34)N–H···O(Lys29) | 98.88 | 99.80 | – | – | |
| (Glu31)N–H···O(Hie27) | 99.48 | 98.88 | 98.40 | – | |
| (Lys32)N–H···O(Leu28) | 99.72 | 99.88 | – | – | |
| (Lys30)N–H···O(Gln26) | 99.80 | 87.48 | 90.08 | – | |
| (Gly33)N–H···O(Lys30) | 91.88 | 88.64 | – | – | |
| (Lys29)N–H···O(Gln25) | 99.92 | 98.88 | – | – | |
| (Leu28)N–H···O(Lys24) | 98.68 | 96.56 | 99.84 | – | |
| (Hie27)N–H···O(Trp23) | 96.44 | 84.12 | 80.80 | – | |
| (Gly25)N–H···O(Leu22) | 96.24 | 54.12 | 84.60 | – |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Xue, T.; Song, C.; Wang, Y.; Chen, G. Study on the Application of the Combination of TMD Simulation and Umbrella Sampling in PMF Calculation for Molecular Conformational Transitions. Int. J. Mol. Sci. 2016, 17, 692. https://doi.org/10.3390/ijms17050692
Wang Q, Xue T, Song C, Wang Y, Chen G. Study on the Application of the Combination of TMD Simulation and Umbrella Sampling in PMF Calculation for Molecular Conformational Transitions. International Journal of Molecular Sciences. 2016; 17(5):692. https://doi.org/10.3390/ijms17050692
Chicago/Turabian StyleWang, Qing, Tuo Xue, Chunnian Song, Yan Wang, and Guangju Chen. 2016. "Study on the Application of the Combination of TMD Simulation and Umbrella Sampling in PMF Calculation for Molecular Conformational Transitions" International Journal of Molecular Sciences 17, no. 5: 692. https://doi.org/10.3390/ijms17050692