
Power Frequency Magnetic Fields Affect the p38 MAPK-Mediated Regulation of NB69 Cell Proliferation Implication of Free Radicals

Abstract
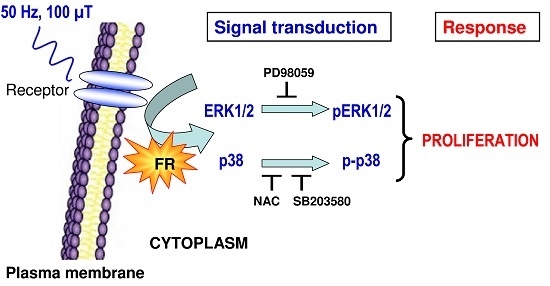
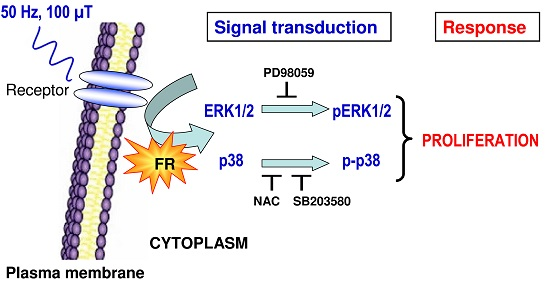
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
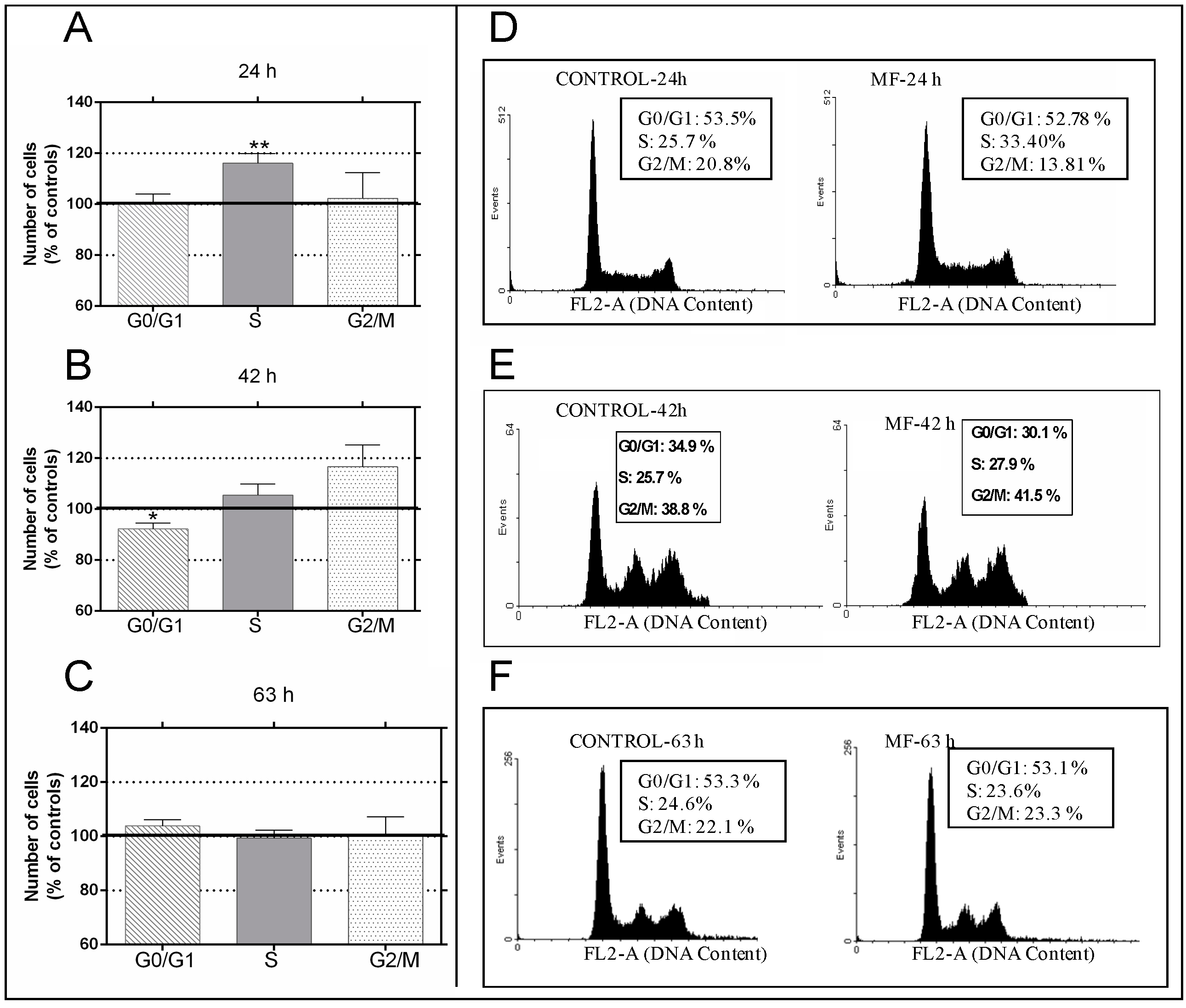
2.1. Effects of the 100 µT Magnetic Fields (MF) on the Cell Cycle
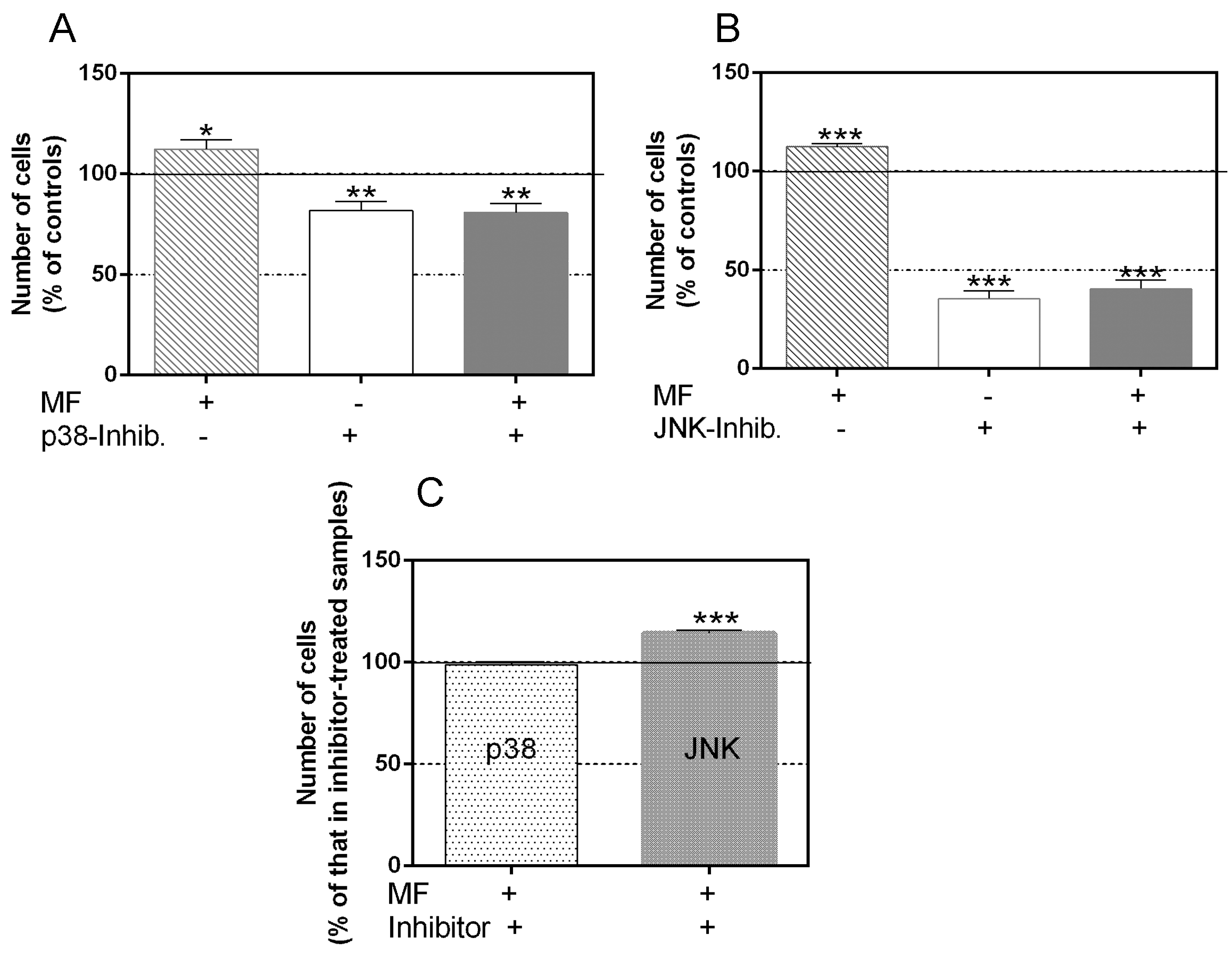
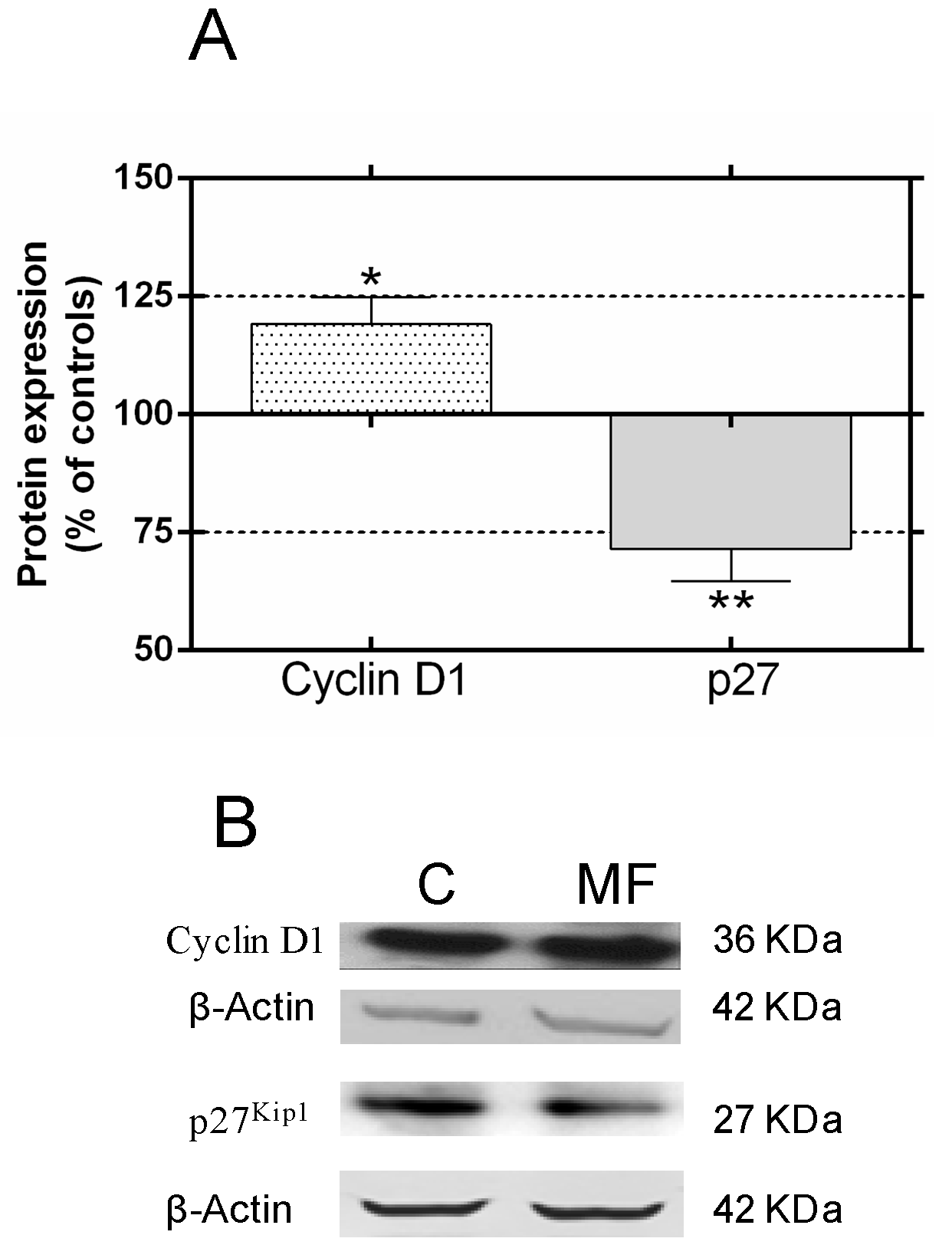
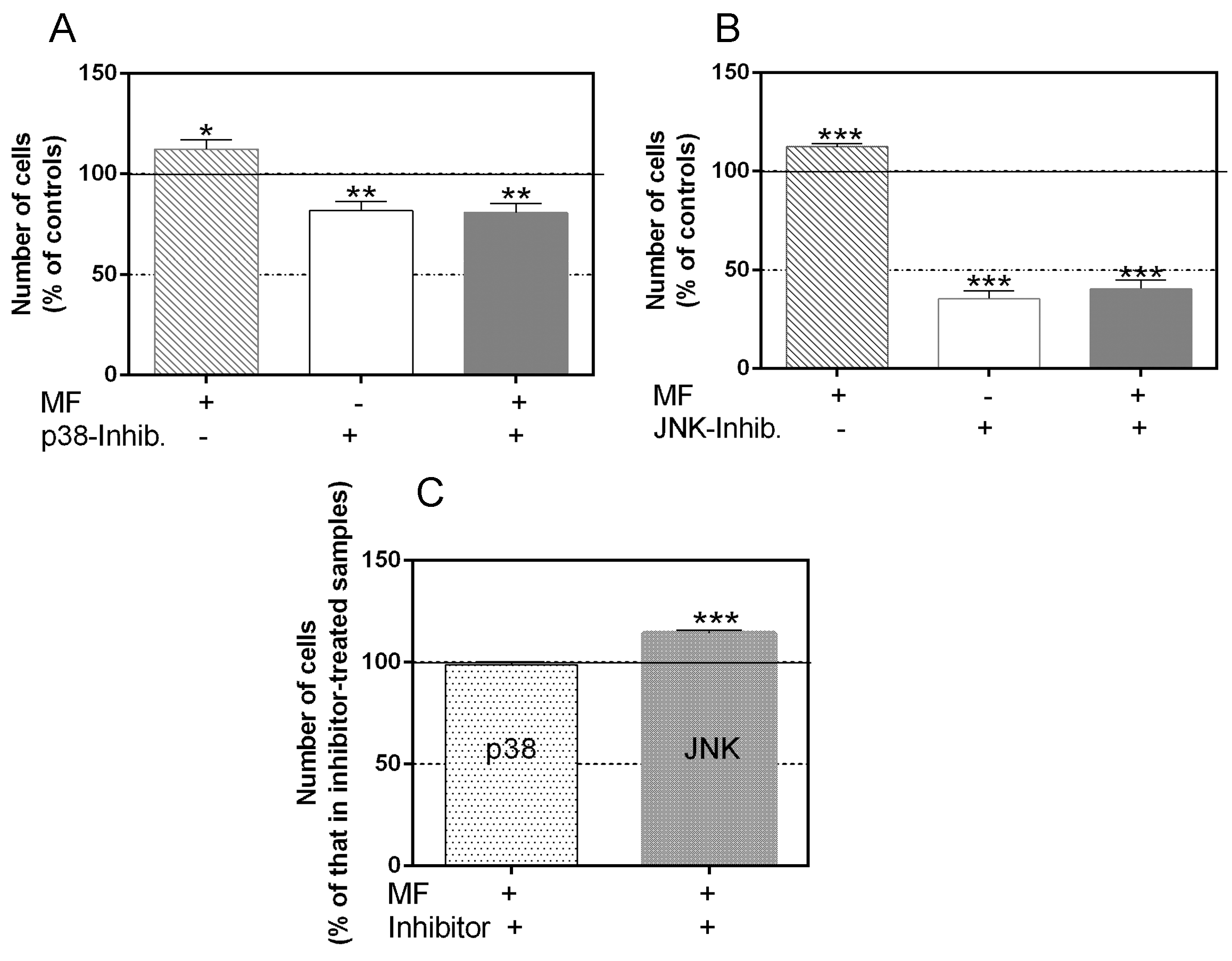
2.2. Effects of p38 and JNK Inhibition on the MF-Induced Proliferative Response
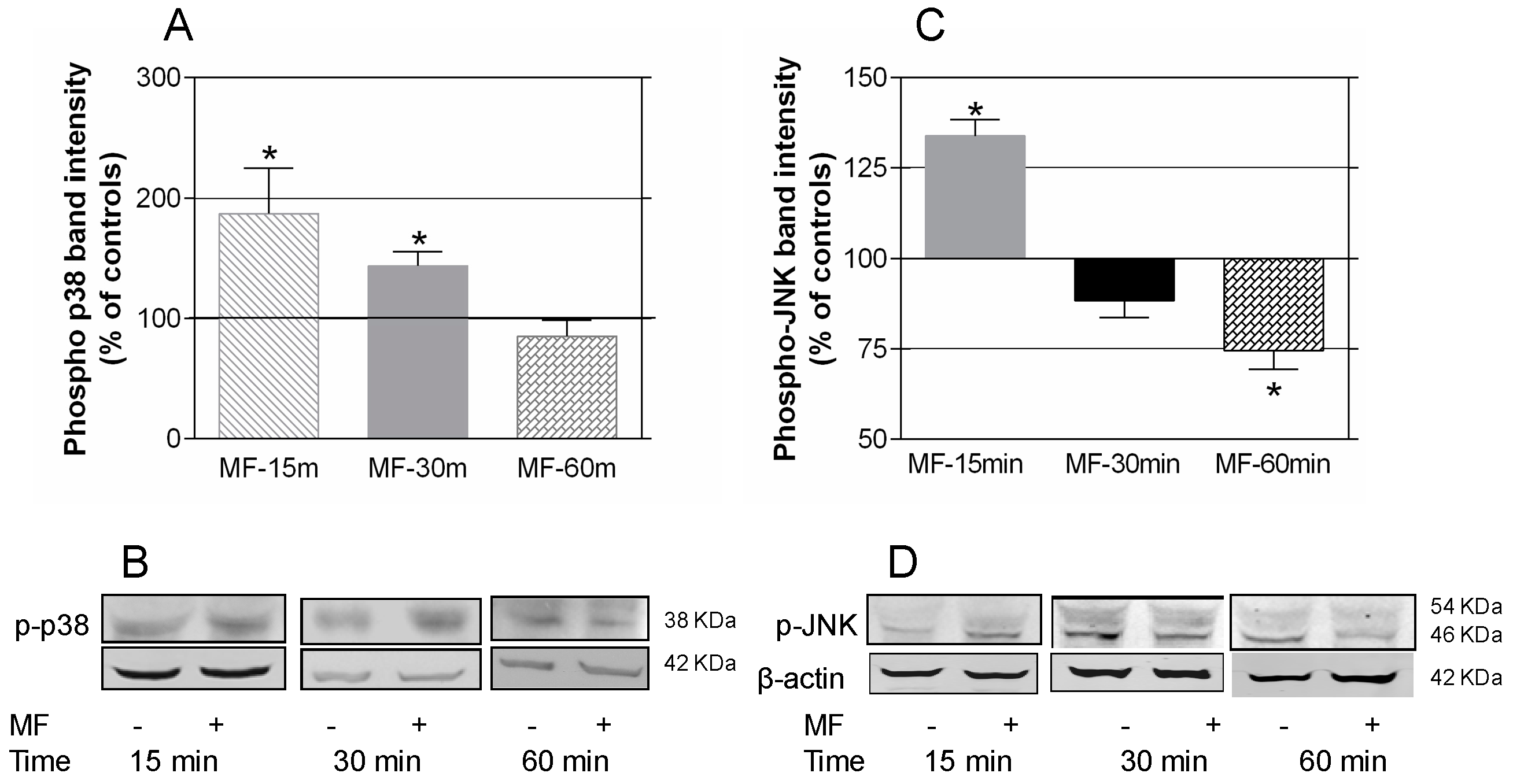
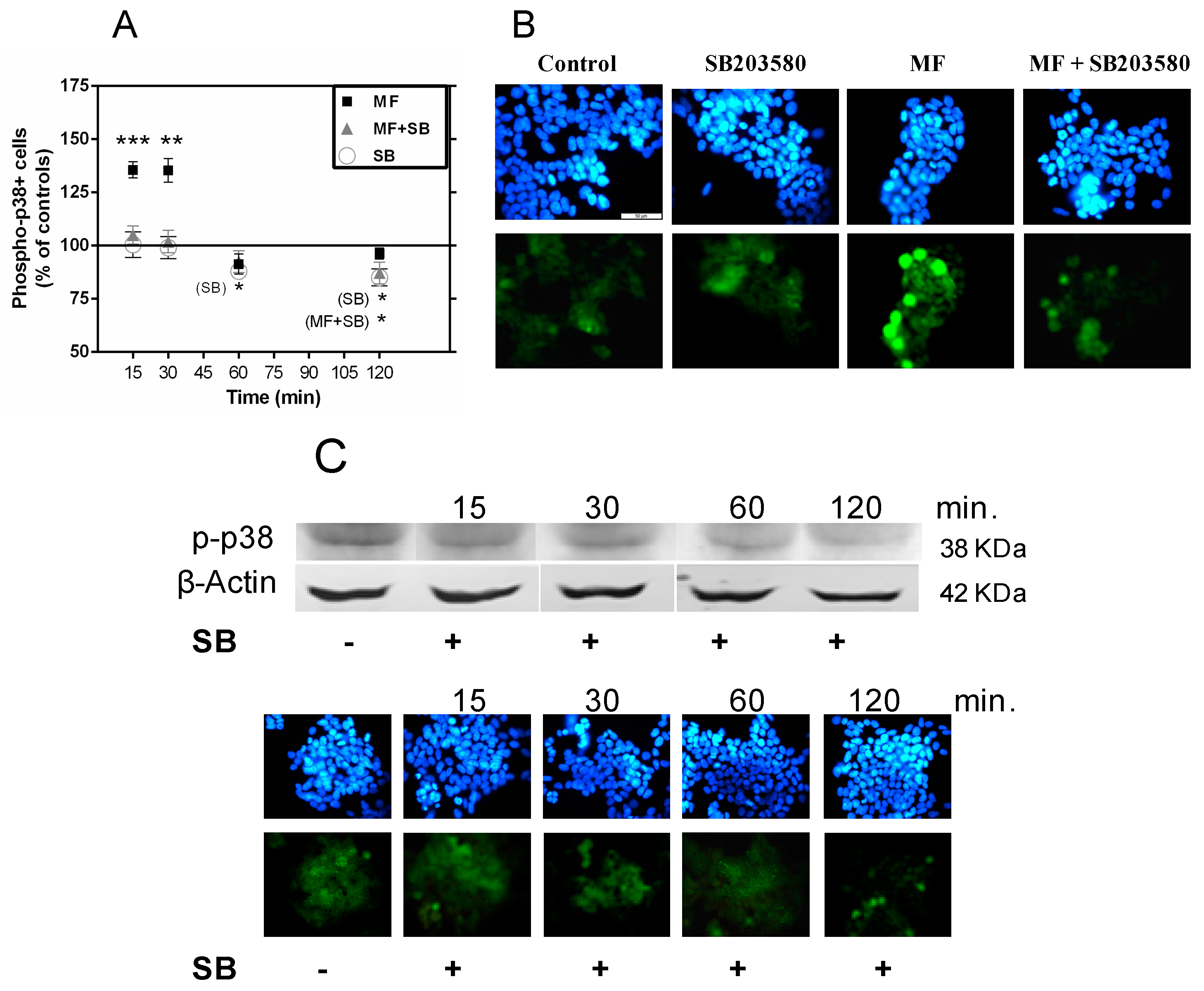
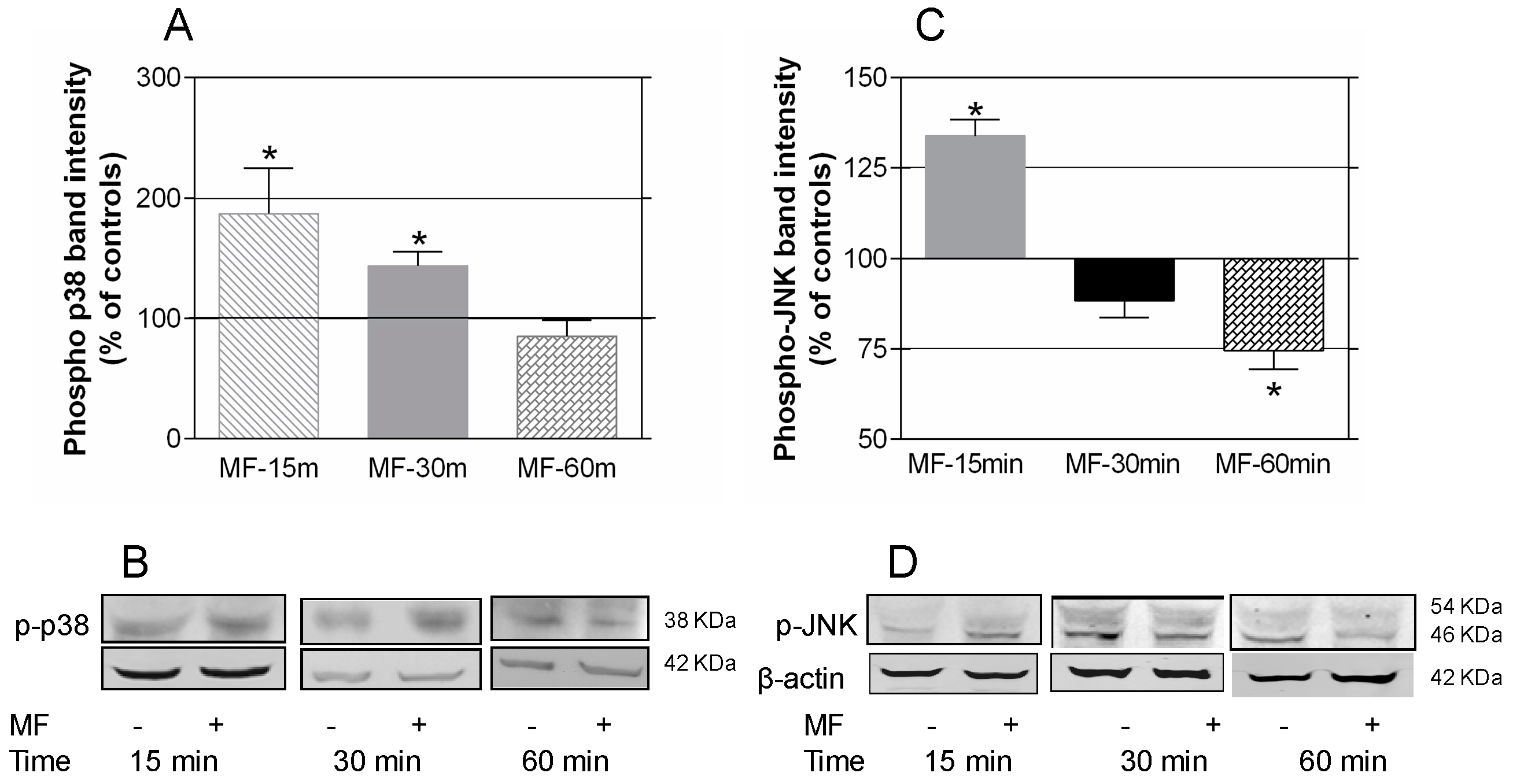
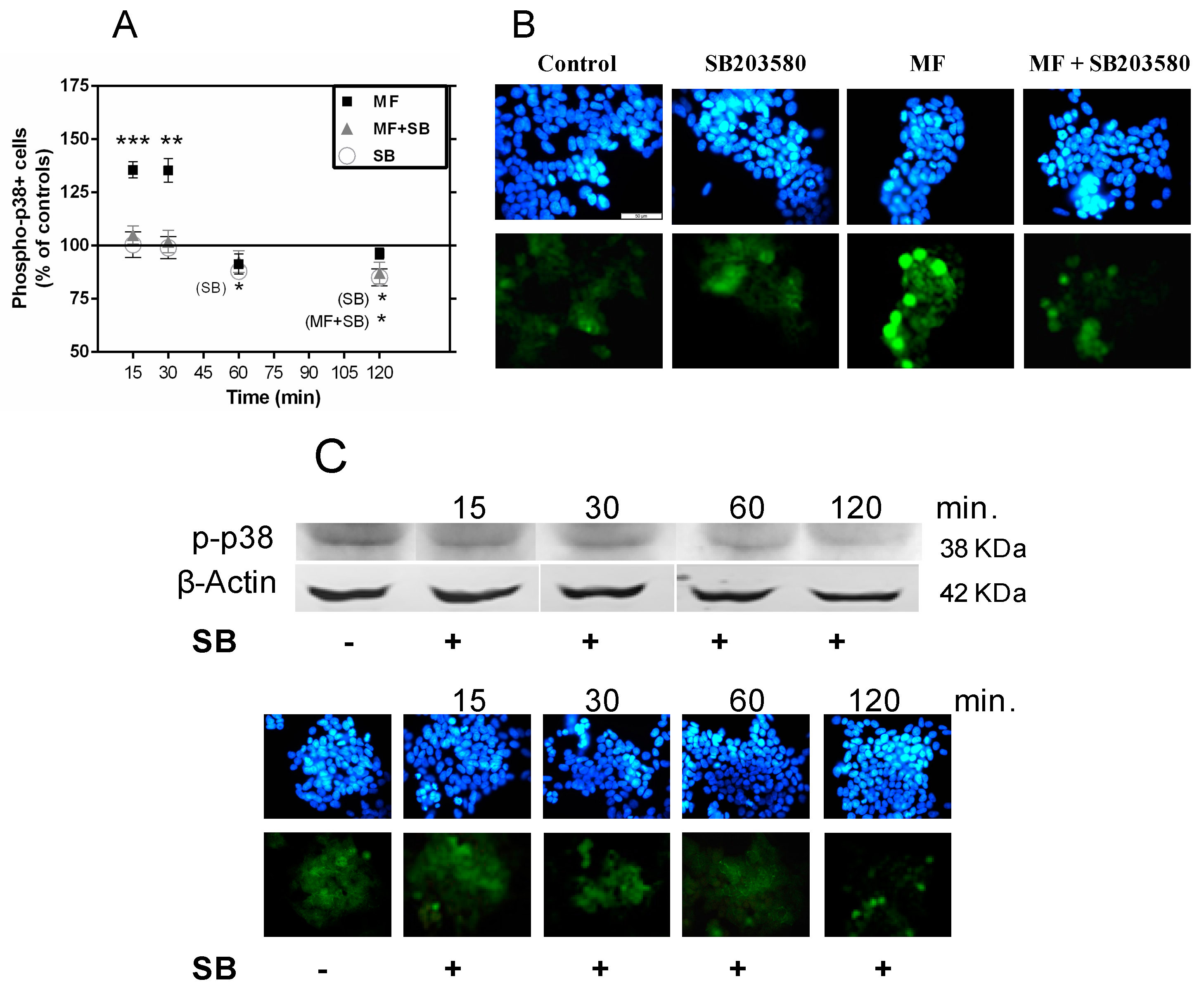
2.3. MF Exposure Induces Early Activation of p38 and JNK
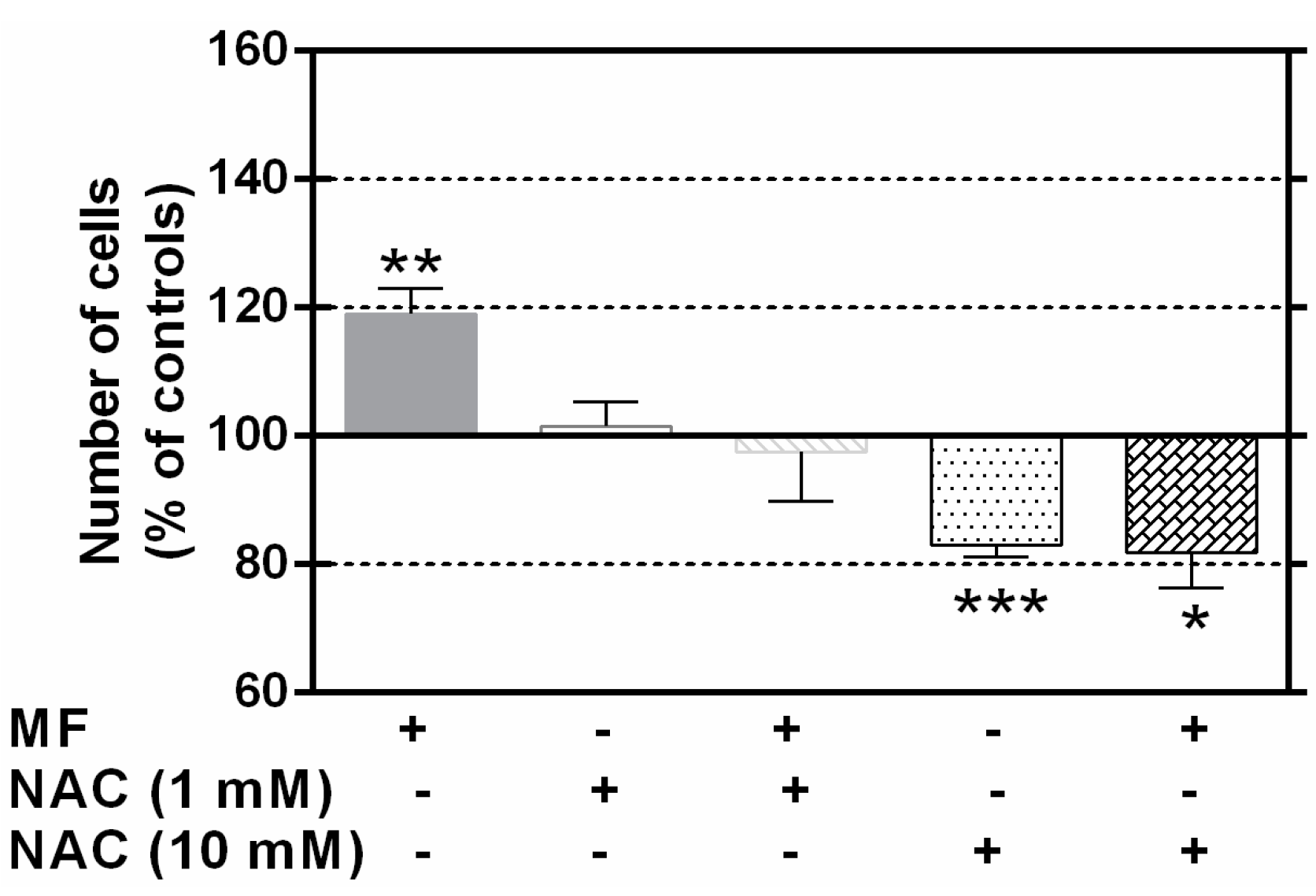
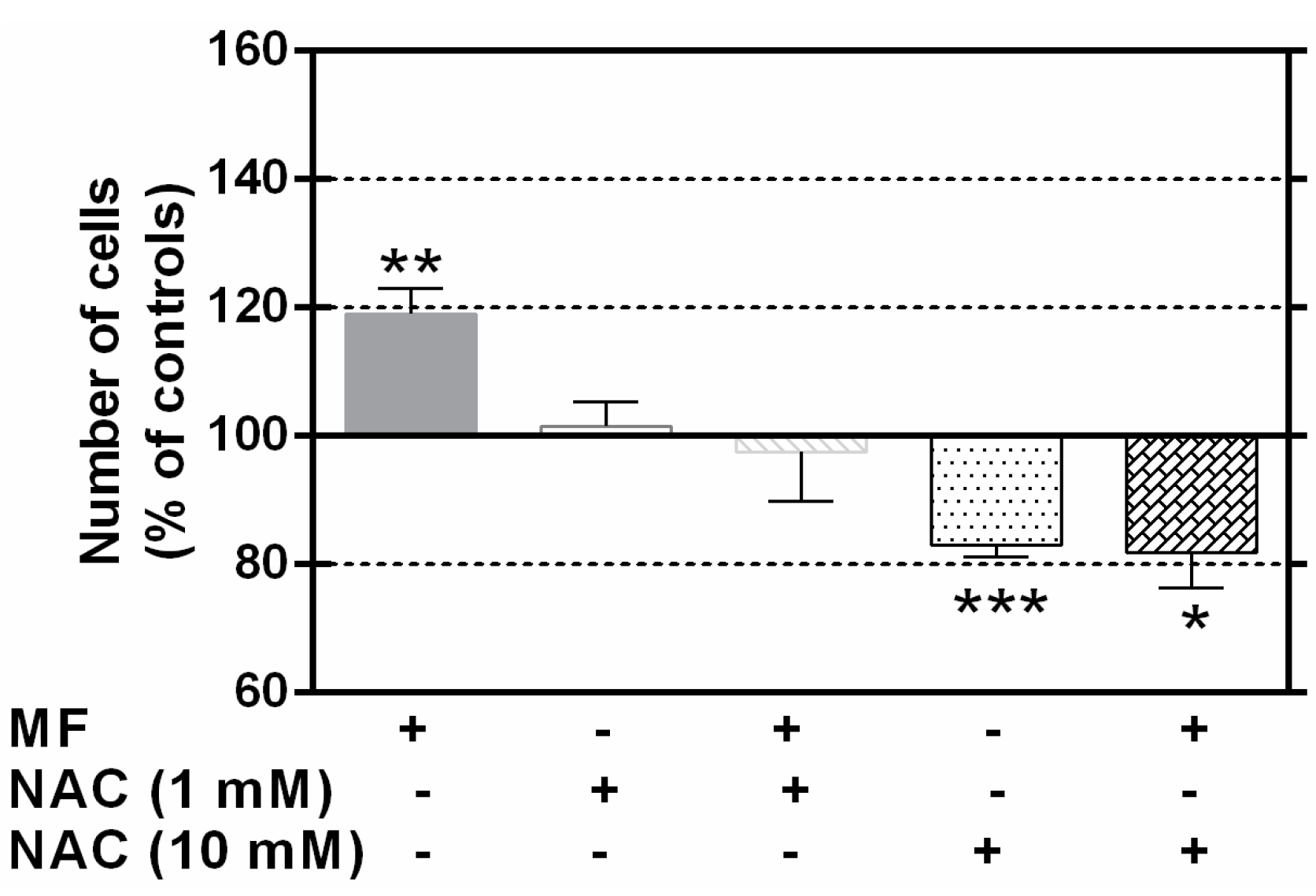
2.4. N-Acetylcysteine Blocks the MF Proliferative Effects
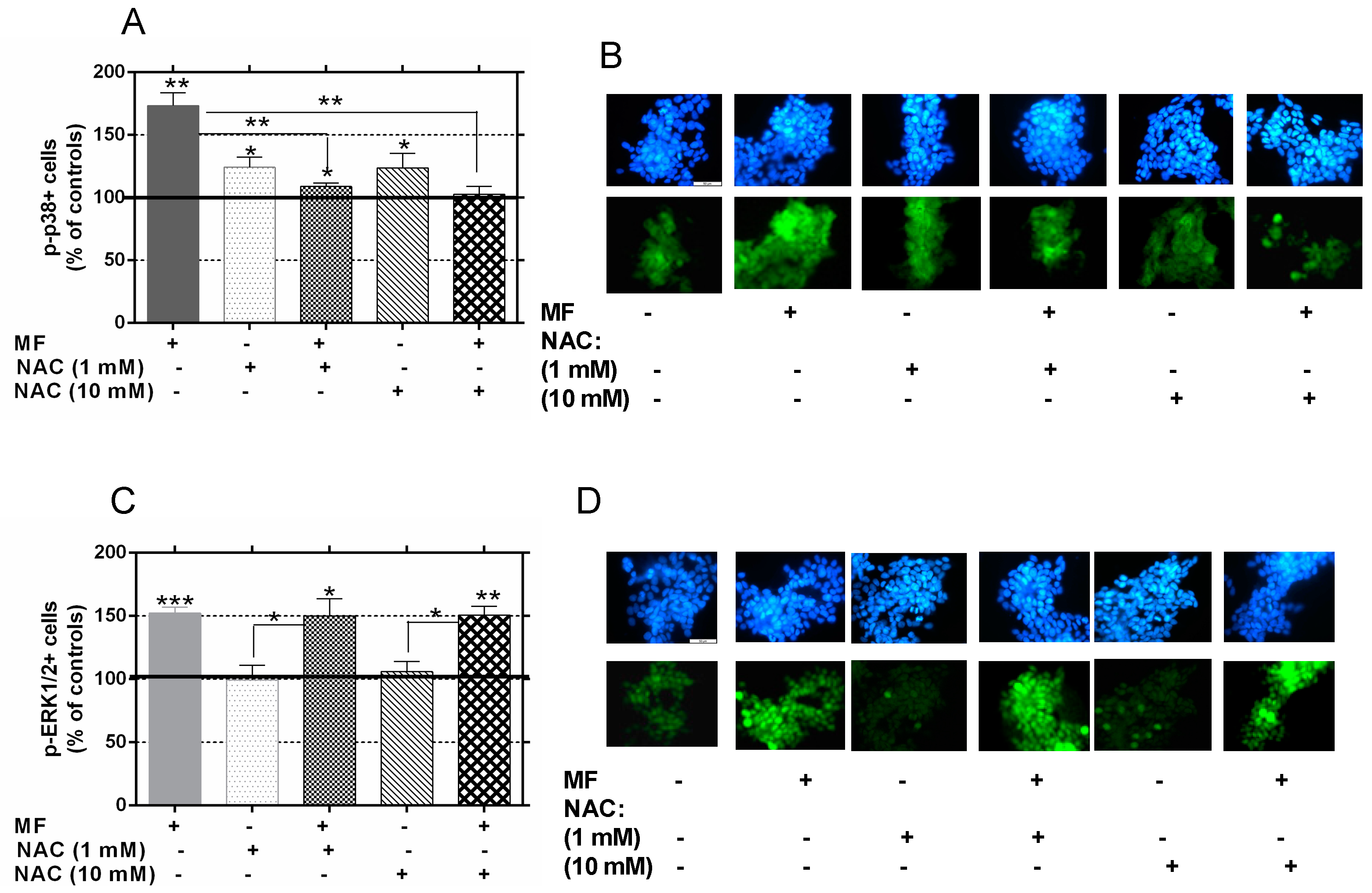
2.5. N-Acetylcysteine Blocks the MF-Induced Activation of p38 But Not ERK1/2 Activation
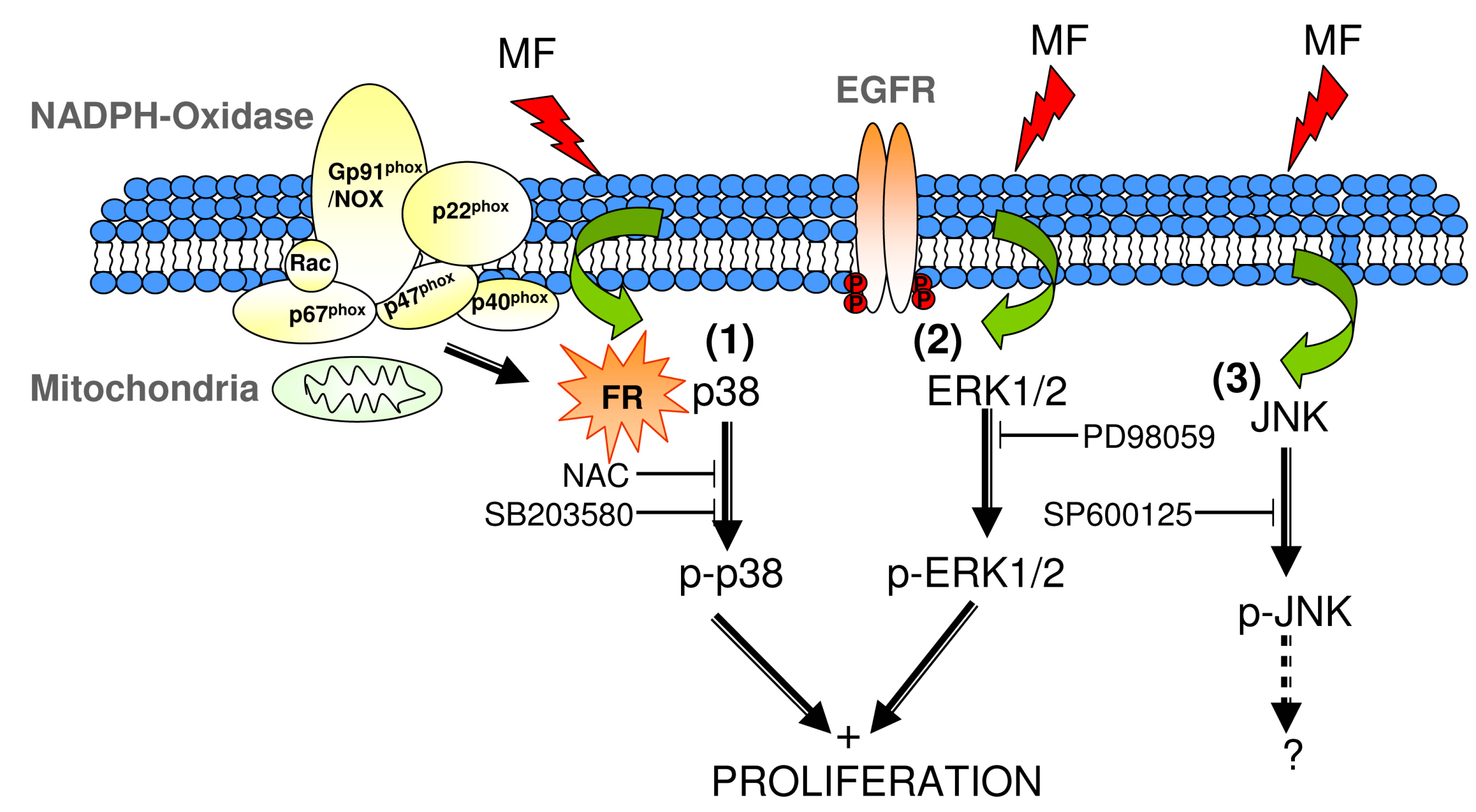
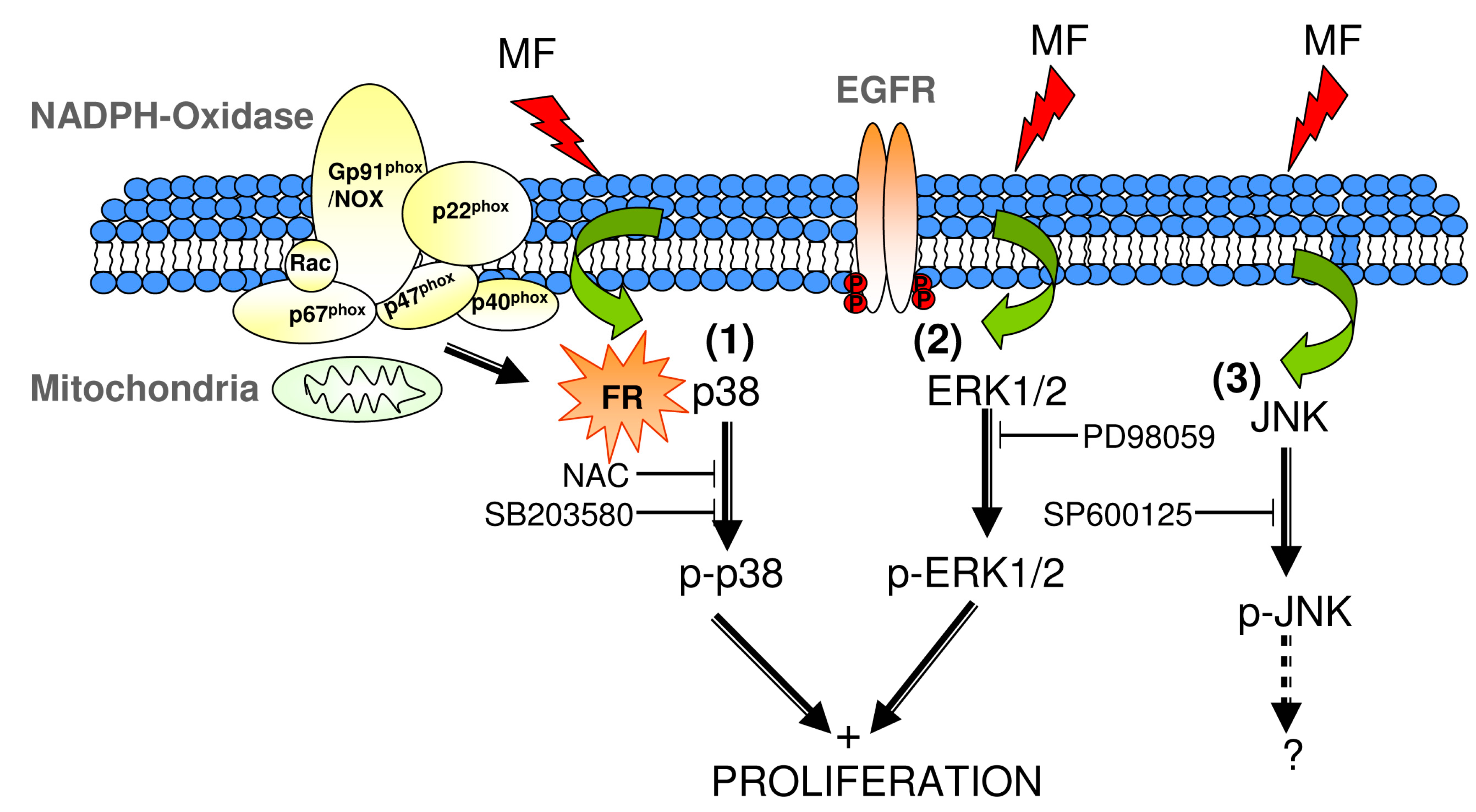
3. Discussion
4. Material and Methods
4.1. Cell Culture
4.2. Treatment with MAPK-p38 and -JNK Inhibitors
4.3. Treatment with N-Acetyl-l-cysteine
4.4. Magnetic Field Exposure
4.5. Analysis of Cell Cycle Progression
4.6. Western Blotting
4.7. Long-Term Exposure for Proliferation Assay. Growth Response at the End of a 63-h Lapse of Intermittent Exposure to MF in the Presence or Absence of p38 or JNK Inhibitors
4.8. Long-Term Exposure for Proliferation Assay. Growth Response at the End of a 63-h Lapse of Intermittent Exposure to MF in the Presence or Absence of NAC
4.9. Immunocytochemistry Assay for Phospho-p38 Expression after Short-Term Exposure to MF
4.10. Changes in the Expression of p-p38 and p-ERK1/2 in Response to Short-Term Exposure to MF in the Presence or Absence of NAC
4.11. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kheifets, L.; Ahlbom, A.; Crespi, C.M.; Tittarelli, A.; Draper, G.; Hagihara, J.; Wunsch Filho, V.; Lowenthal, R.M.; Mezei, G.; Oksuzyan, S.; et al. Pooled analysis of recent studies of magnetic fields and childhood leukemia. Br. J. Cancer 2010, 103, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Nitta, H.; Kubo, O.; Yamamoto, S.; Yamaguchi, N.; Akiba, S.; Honda, Y.; Hagihara, J.; Isaka, K.; Ojima, T.; et al. Power-frequency magnetic fields and childhood brain tumors: A case-control study in Japan. J. Epidemiol. 2010, 20, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Teepen, J.C.; van Dijck, J.A. Impact of high electromagnetic field levels on childhood leukaemia incidence. Int. J. Cancer 2012, 131, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Chen, G.; Chen, C.; Yu, Y.; Xu, Z. Association between extremely low-frequency electromagnetic fields occupations and amyotrophic lateral sclerosis: A meta-analysis. PLoS ONE 2012, 7, e48354. [Google Scholar] [CrossRef] [PubMed]
- Bunch, K.J.; Keegan, T.J.; Swanson, J.; Vincent, T.J.; Murphy, M.F. Residential distance at birth from overhead high-voltage powerlines childhood cancer risk in Britain 1962–2008. Br. J. Cancer 2014, 110, 14082–14088. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.C.; Benke, G.; Bowman, J.D.; Figuerola, J.; Fleming, S.; Hours, M.; Kincl, L.; Krewski, D.; McLean, D.; Parent, M.E.; et al. Occupational exposure to extremely low-frequency magnetic fields and brain tumor risks in the INTEROCC study. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research of Cancer (IARC). IARC monograph on the evaluation of carcinogenic risks to humans, Vol. 80. In Non-Ionizing Radiation, Part 1: Static and Extremely Low-Frequency (ELF) Electric and Magnetic Fields; IARC Press: Lyon, France, 2002. [Google Scholar]
- Santini, M.T.; Rainaldi, G.; Indovina, P.L. Cellular effects of extremely low frequency (ELF) electromagnetic fields. Int. J. Radiat. Biol. 2009, 85, 294–313. [Google Scholar] [CrossRef] [PubMed]
- Adair, R.K. Extremely low frequency electromagnetic fields do not interact directly with DNA. Bioelectromagnetics 1998, 19, 136–138. [Google Scholar] [CrossRef]
- Focke, F.; Schuermann, D.; Kuster, N.; Schär, P. DNA fragmentation in human fibroblasts under extremely low frequency electromagnetic field exposure. Mutat. Res. 2010, 683, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Rageh, M.M.; El-Gebaly, R.H.; El-Bialy, N.S. Assessment of genotoxic and cytotoxic hazards in brain and bone marrow cells of newborn rats exposed to extremely low-frequency magnetic field. J. Biomed. Biotechnol. 2012, 2012, 716023. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, J.; Liimatainen, A.; Juutilainen, J.; Naarala, J. Induction of genomic instability, oxidative processes, and mitochondrial activity by 50Hz magnetic fields in human SH-SY5Y neuroblastoma cells. Mutat. Res. 2014, 760, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Fiebig, T.; Schiemann, O.; Barton, J.K.; Zewail, A.H. Femtosecond direct observation of charge transfer between bases in DNA. Proc. Natl. Acad. Sci. USA 2000, 97, 14052–14055. [Google Scholar] [CrossRef] [PubMed]
- Porath, D.; Bezryadin, A.; de Vries, S.; Dekker, C. Direct measurement of electrical transport through DNA molecules. Nature 2000, 403, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Giese, B. Electron transfer through DNA and peptides. Bioorg. Med. Chem. 2006, 14, 6139–6143. [Google Scholar] [CrossRef] [PubMed]
- Falone, S.; Santini, S.; di Loreto, S.; Cordone, V.; Grannonico, M.; Cesare, P.; Cacchio, M.; Amicarelli, F. Improved mitochondrial and methylglyoxal-related metabolisms support hyperproliferation induced by 50 Hz magnetic field in neuroblastoma cells. J. Cell. Physiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.I.; Torsello, A.; Tedesco, B.; Fasanella, S.; Boninsegna, A.; D’Ascenzo, M.; Grassi, C.; Azzena, G.B.; Cittadini, A. 50-Hz extremely low frequency electromagnetic fields enhance cell proliferation and DNA damage: Possible involvement of a redox mechanism. Biochim. Biophys. Acta 2005, 1743, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Trillo, M.A.; Martínez, M.A.; Cid, M.A.; Leal, J.; Ubeda, A. Influence of a 50 Hz magnetic field and of all-trans-retinol on the proliferation of human cancer cell lines. Int. J. Oncol. 2012, 40, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, X.; Bai, L.; Uchida, K.; Bai, W.; Wu, B.; Xu, W.; Zhu, H.; Huang, H. Effects of low frequency electromagnetic field on proliferation of human epidermal stem cells: An in vitro study. Bioelectromagnetics 2013, 34, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Blank, M.; Goodman, R. ERK1/2 phosphorylation, induced by electromagnetic fields, diminishes during neoplastic transformation. J. Cell. Biochem. 2000, 78, 371–379. [Google Scholar] [CrossRef]
- Sun, W.; Yu, Y.; Chiang, H.; Fu, Y.; Lu, D. Exposure to power-frequency magnetic fields can induce activation of p38 mitogen-activated protein kinase. Chin. J. Ind. Occup. Dis. 2002, 20, 252–255. [Google Scholar]
- Sun, W.; Yu, Y.; Fu, Y.; Chiang, H.; Xie, H.; Lu, D. Effects of power-frequency magnetic fields exposure on phosphorylation and enzymatic activity of stress-activated protein kinase and its upstream kinase. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 2002, 20, 256–259. [Google Scholar] [PubMed]
- Nie, K.; Henderson, A. MAP kinase activation in cells exposed to 60 Hz electromagnetic field. J. Cell. Biochem. 2003, 90, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK signal transduction pathways activated by stress and inflammation:A 10-year update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [PubMed]
- Sulpizio, M.; Falone, S.; Amicarelli, F.; Marchisio, M.; di Giuseppe, F.; Eleuterio, E.; di Ilio, C.; Angelucci, S. Molecular basis underlying the biological effects elicited by extremely low-frequency magnetic field (ELF-MF) on neuroblastoma cells. J. Cell. Biochem. 2011, 112, 3797–3806. [Google Scholar] [CrossRef] [PubMed]
- Tofani, S.; Barone, D.; Berardelli, M.; Berno, E.; Cintorino, M.; Foglia, L.; Ossola, P.; Ronchetto, F.; Toso, E.; Eandi, M. Static and ELF magnetic fields enhance the in vivo anti-tumor efficacy of cis-platin against lewis lung carcinoma, but not of cyclophosphamide against B16 melanotic melanoma. Pharmacol. Res. 2003, 18, 83–90. [Google Scholar] [CrossRef]
- Jung, I.S.; Kim, H.J.; Noh, R.; Kim, S.C.; Kim, C.W. Effects of extremely low frequency magnetic fields on NGF induced neuronal differentiation of PC12 cells. Bioelectromagnetics 2014, 35, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Destefanis, M.; Viano, M.; Leo, C.; Gervino, G.; Ponzetto, A.; Silvagno, F. Extremely low frequency electromagnetic fields affect proliferation and mitochondrial activity of human cancer cell lines. Int. J. Radiat. Biol. 2015, 91, 964–972. [Google Scholar] [CrossRef] [PubMed]
- REFLEX-Project 2004: Risk Evaluation of Potential Environmental Hazards from Low Energy Electromagnetic Field Exposure Using Sensitive in Vitro Methods. Final Report. Available online: http://www.powerwatch.org.uk/pdfs/20041222_reflex.pdf (accessed on 16 January 2016).
- Cid, M.A.; Ubeda, A.; Hernández-Bule, M.L.; Martínez, M.A.; Trillo, M.Á. Antagonistic effects of a 50 Hz magnetic field and melatonin in the proliferation and differentiation of hepatocarcinoma cells. Cell. Physiol. Biochem. 2012, 30, 1502–1516. [Google Scholar] [CrossRef] [PubMed]
- Martínez, M.A.; Úbeda, A.; Cid, M.A.; Trillo, M.A. The proliferative response of NB69 human neuroblastoma cells to a 50 Hz magnetic field is mediated by ERK1/2 signaling. Cell. Physiol. Biochem. 2012, 29, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Trillo, M.Á.; Martínez, M.A.; Cid, M.A.; Úbeda, A. Retinoic acid inhibits the cytoproliferative response to weak 50‑Hz magnetic fields in neuroblastoma cells. Oncol. Rep. 2013, 29, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Tarn, W.Y.; Lai, M.C. Translational control of cyclins. Cell. Div. 2011, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Chiao, C.Y.; Enzer, K.G.; Stankiewicz, A.J.; Faller, D.V.; Dai, Y. SIRT1 Inactivation evokes antitumor activities in NSCLC through the tumor suppressor p27. Mol. Cancer Res. 2015, 13, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Bubici, C.; Papa, S. JNK signalling in cancer: In need of new, smarter therapeutic targets. Br. J. Pharmacol. 2014, 171, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Yumoto, H.; Hirao, K.; Tominaga, T.; Bando, N.; Takahashi, K.; Matsuo, T. Electromagnetic wave irradiation promotes osteoblastic cell proliferation and up-regulates growth factors via activation of the ERK1/2 and p38 MAPK pathways. Cell. Physiol. Biochem. 2015, 35, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, M.O.; Simkó, M. Grouping of experimental conditions as an approach to evaluate effects of extremely low-frequency magnetic fields on oxidative response in vitro studies. Front. Public Health 2014, 2, 132. [Google Scholar] [CrossRef] [PubMed]
- Patruno, A.; Amerio, P.; Pesce, M.; Vianale, G.; di Luzio, S.; Tulli, A.; Franceschelli, S.; Grilli, A.; Muraro, R.; Reale, M. Extremely low frequency electromagnetic fields modulate expression of inducible nitric oxide synthase, endothelial nitric oxide synthase and cyclooxygenase-2 in the human keratinocyte cell line HaCat: Potential therapeutic effects in wound healing. Br. J. Dermatol. 2010, 162, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Martín, E.; Canals, S.; Casarejos, M.J.; de Bernardo, S.; Handler, A.; Mena, M.A. L-DOPA and glia-conditioned medium have additive effects on tyrosine hydroxylase expression in human catecholamine-rich neuroblastoma NB69 cells. J. Neurochem. 2001, 78, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Directive 2013/35/EU of the European Parliament and of the Council of 26 June 2013 on the Minimum Health and Safety Requirements Regarding the Exposure of Workers to the Risks Arising From Physical Agents (Electromagnetic Fields). Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32013L0035 (accessed on 4 April 2016).
- Grassi, C.; D’Ascenzo, M.; Torsello, A.; Martinotti, G.; Wolf, F.; Cittadini, A.; Azzena, G.B. Effects of 50 Hz electromagnetic fields on voltage-gated Ca2+ channels and their role in modulation of neuroendocrine cell proliferation and death. Cell Calcium 2004, 35, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Héroux, P. Extra-low-frequency magnetic fields alter cancer cells through metabolic restriction. Electromagn. Biol. Med. 2014, 33, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Pirozzoli, M.C.; Marino, C.; Lovisolo, G.A.; Laconi, C.; Mosiello, L.; Negroni, A. Effects of 50 Hz electromagnetic field exposure on apoptosis and differentiation in a neuroblastoma cell line. Bioelectromagnetics 2003, 24, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Cuenda, A.; Rousseau, S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Faust, D.; Schmitt, C.; Oesch, F.; Oesch-Bartlomowicz, B.; Schreck, I.; Weiss, C.; Dietrich, C. Differential p38-dependent signalling in response to cellular stress and mitogenic stimulation in fibroblasts. Cell Commun. Signal. 2012, 10. [Google Scholar] [CrossRef] [PubMed]
- Koul, H.K.; Pal, M.; Koul, S. Role of p38 MAP kinase signal transduction in solid tumors. Genes Cancer 2013, 4, 342–359. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, S.E.; Olson, E.R.; Zhang, J.; Cooper, S.J.; Melton, T.; Criswell, P.J.; Casanova, A.; Dong, Z.; Hu, C.; Saboda, K.; et al. p38 MAP kinase plays a functional role in UVB-induced mouse skin carcinogenesis. Mol. Carcinog. 2011, 50, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.S.; Kim, J.M.; Nam, S.Y.; Yang, K.H.; Jeong, M.; Kim, H.S.; Lim, Y.K.; Kim, C.S.; Jin, Y.W.; Kim, J. Low-dose of ionizing radiation enhances cell proliferation via transient ERK1/2 and p38 activation in normal human lung fibroblasts. J. Radiat. Res. 2007, 48, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Soda, A.; Ikehara, T.; Kinouchi, Y.; Yoshizaki, K. Effect of exposure to an extremely low frequency-electromagnetic field on the cellular collagen with respect to signaling pathways in osteoblast-like cells. J. Med. Investig. 2008, 55, 267–278. [Google Scholar] [CrossRef]
- Kim, J.; Ha, C.S.; Lee, H.J.; Song, K. Repetitive exposure to a 60-Hz time-varying magnetic field induces DNA double-strand breaks and apoptosis in human cells. Biochem. Biophys. Res. Commun. 2010, 400, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cui, J.; Zhu, H. Suppression of type I collagen in human scleral fibroblasts treated with extremely low-frequency electromagnetic fields. Mol. Vis. 2013, 19, 885–893. [Google Scholar] [PubMed]
- Halawani, D.; Mondeh, R.; Stanton, L.A.; Beier, F. p38 MAP kinase signaling is necessary for rat chondrosarcoma cell proliferation. Oncogene 2004, 23, 3726–3731. [Google Scholar] [CrossRef] [PubMed]
- Recio, J.A.; Merlino, G. Hepatocyte growth factor/scatter factor activates proliferation in melanoma cells through p38 MAPK, ATF-2 and cyclin D1. Oncogene 2002, 21, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
- Chambard, J.C.; Lefloch, R.; Pouysségur, J.; Lenormand, P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta 2007, 1773, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.; Tanner, K.G.; Cheung, W.L.; Sassone-Corsi, P.; Denu, J.M.; Allis, C.D. Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol. Cell 2000, 5, 905–915. [Google Scholar] [CrossRef]
- Clayton, A.L.; Rose, S.; Barratt, M.J.; Mahadevan, L.C. Phosphoacetylation of histone H3 on c-fos- and c-jun-associated nucleosomes upon gene activation. EMBO J. 2000, 19, 3714–3726. [Google Scholar] [CrossRef] [PubMed]
- Simkó, M.; Mattsson, M.O. Extremely low frequency electromagnetic fields as effectors of cellular responses in vitro: Possible immune cell activation. J. Cell. Biochem. 2004, 93, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Vanderstraeten, J.; Gillis, P. Theoretical evaluation of magnetoreception of power-frequency fields. Bioelectromagnetics 2010, 31, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-activated protein kinases and reactive oxygen species: How can ROS activate MAPK pathways? J. Signal Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef] [PubMed]
- De Flora, S.; Cesarone, C.F.; Balansky, R.M.; Albini, A.; D’Agostini, F.; Bennicelli, C.; Bagnasco, M.; Camoirano, A.; Scatolini, L.; Rovida, A.; et al. Chemopreventive properties and mechanisms of N-acetylcysteine. The experimental background. J. Cell. Biochem. 1995, 22, 33–41. [Google Scholar] [CrossRef]
- De Flora, S.; Izzotti, A.; D’Agostini, F.; Balansky, R.M. Mechanisms of N-acetylcysteine in the prevention of DNA damage and cancer, with special reference to smoking-related end-points. Carcinogenesis 2001, 22, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Parasassi, T.; Brunelli, R.; Costa, G.; Spirito, M.D.; Krasnowska, E.K.; Lundeberg, T.; Pittaluga, E.; Ursini, F. Thiol redox transitions in cell signaling: A lesson from N-acetylcysteine. Sci. World J. 2010, 10, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Morini, M.; D’Agostini, F.; Ferrari, F.; Campelli, F.; Arena, G.; Noonan, D.M.; Pesce, C.; Flora, S.D. Inhibition of angiogenesis-driven Kaposi’s sarcoma tumor growth in nude mice by oral N-acetylcysteine. Cancer Res. 2001, 61, 8171–8178. [Google Scholar] [PubMed]
- Swain, W.A.; Faux, S.P. Activation of p38 MAP kinase by crocidolite in mesothelial cells is dependent upon oxidative stress. Ann. Occup. Hyg. 2002, 46, 136–139. [Google Scholar] [CrossRef]
- Löhrke, B.; Xu, J.; Weitzel, J.M.; Krüger, B.; Goldammer, T.; Viergutz, T. N-acetylcysteine impairs survival of luteal cells through mitochondrial dysfunction. Cytom. A 2010, 77, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Sagara, J.; Bannai, S.; Shikano, N.; Makino, N. Conflicting effects of N-acetylcysteine on purified neurons derived from rat cortical culture. NeuroReport 2010, 21, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.G.; Sarsour, E.H.; Kalen, A.L.; Venkataraman, S.; Hitchler, M.J.; Domann, F.E.; Oberley, L.W.; Goswami, P.C. Superoxide signaling mediates N-acetyl-l-cysteine-induced G1 arrest: Regulatory role of cyclin D1 and manganese superoxide dismutase. Cancer Res. 2007, 67, 6392–6399. [Google Scholar] [CrossRef] [PubMed]
- Morabito, C.; Rovetta, F.; Bizzarri, M.; Mazzoleni, G.; Fanò, G.; Mariggiò, M.A. Modulation of redox status and calcium handling by extremely low frequency electromagnetic fields in C2C12 muscle cells: A real-time, single-cell approach. Free Radic. Biol. Med. 2010, 48, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Frahm, J.; Mattsson, M.O.; Simkó, M. Exposure to ELF magnetic fields modulate redox related protein expression in mouse macrophages. Toxicol. Lett. 2010, 192, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Murillo-Carretero, M.; Ruano, M.J.; Matarredona, E.R.; Villalobo, A.; Estrada, C. Antiproliferative effect of nitric oxide on epidermal growth factor-responsive human neuroblastoma cells. J. Neurochem. 2002, 83, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Zhou, Z.; Liu, R.; Chen, S.; Xia, R. EGF receptor clustering is induced by a 0.4 mT power frequency magnetic field and blocked by the EGF receptor tyrosine kinase inhibitor PD153035. Bioelectromagnetics 2007, 28, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Gan, Y.; Fu, Y.; Lu, D.; Chiang, H. An incoherent magnetic field inhibited EGF receptor clustering and phosphorylation induced by a 50-Hz magnetic field in cultured FL cells. Cell. Physiol. Biochem. 2008, 22, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.Q.; Sun, W.J.; Lu, D.Q.; Fu, Y.T.; Chiang, H. 50-Hz magnetic field induces EGF receptor clustering and activates RAS. Int. J. Radiat. Biol. 2008, 84, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Tonini, R.; Baroni, M.D.; Masala, E.; Micheletti, M.; Ferroni, A.; Mazzanti, M. Calcium protects differentiating neuroblastoma cells during 50 Hz electromagnetic radiation. Biophys. J. 2001, 81, 2580–2589. [Google Scholar] [CrossRef]
- Lacy-Hulbert, A.; Metcalfe, J.C.; Hesketh, R. Biological responses to electromagnetic fields. FASEB J. 1998, 12, 395–420. [Google Scholar] [PubMed]
- Russo, V.C.; Andaloro, E.; Fornaro, S.A.; Najdovska, S.; Newgreen, D.F.; Bach, L.A.; Werther, G.A. Fibroblast growth factor-2 over-rides insulin-like growth factor-I induced proliferation and cell survival in human neuroblastoma cells. J. Cell. Physiol. 2004, 199, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.J.; Meyerowitz, J.; James, J.L.; Liddell, J.R.; Nonaka, T.; Hasegawa, M.; Kanninen, K.M.; Lim, S.; Paterson, B.M.; Donnelly, P.S.; et al. Inhibition of TDP-43 accumulation by bis(thiosemicarbazonato)-copper complexes. PLoS ONE 2012, 7, e42277. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Hamprecht, B. N-acetylcysteine, but not methionine or 2-oxothiazolidine-4-carboxylate, serves as cysteine donor for the synthesis of glutathione in cultured neurons derived from embryonal rat brain. Neurosci. Lett. 1999, 259, 79–82. [Google Scholar] [CrossRef]
- Olivieri, G.; Baysang, G.; Meier, F.; Muller-Spahn, F.; Stahelin, H.B.; Brockhaus, M.; Brack, C. N-acetyl-l-cysteine protects SHSY5Y neuroblastoma cells from oxidative stress and cell cytotoxicity: Effects on β-amyloid secretion and tau phosphorylation. J. Neurochem. 2001, 76, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Martínez, M.A.; Trillo, M.Á.; Cid, M.A.; Paíno, C.L.; Úbeda, A. Power-frequency magnetic field inhibits adipogenic differentiation in human ADSC. Cell. Physiol. Biochem. 2015, 37, 2297–2310. [Google Scholar] [CrossRef] [PubMed]
- Aktas, H.; Cai, H.; Cooper, G.M. Ras links growth factor signaling to the cell cycle machinery via regulation of cyclin D1 and the CDK inhibitor p27KIP1. Mol. Cell. Biol. 1997, 17, 3850–3857. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of proteins utilizing the principle of protein dye-binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez, M.A.; Úbeda, A.; Moreno, J.; Trillo, M.Á. Power Frequency Magnetic Fields Affect the p38 MAPK-Mediated Regulation of NB69 Cell Proliferation Implication of Free Radicals. Int. J. Mol. Sci. 2016, 17, 510. https://doi.org/10.3390/ijms17040510
Martínez MA, Úbeda A, Moreno J, Trillo MÁ. Power Frequency Magnetic Fields Affect the p38 MAPK-Mediated Regulation of NB69 Cell Proliferation Implication of Free Radicals. International Journal of Molecular Sciences. 2016; 17(4):510. https://doi.org/10.3390/ijms17040510
Chicago/Turabian StyleMartínez, María Antonia, Alejandro Úbeda, Jorge Moreno, and María Ángeles Trillo. 2016. "Power Frequency Magnetic Fields Affect the p38 MAPK-Mediated Regulation of NB69 Cell Proliferation Implication of Free Radicals" International Journal of Molecular Sciences 17, no. 4: 510. https://doi.org/10.3390/ijms17040510
APA StyleMartínez, M. A., Úbeda, A., Moreno, J., & Trillo, M. Á. (2016). Power Frequency Magnetic Fields Affect the p38 MAPK-Mediated Regulation of NB69 Cell Proliferation Implication of Free Radicals. International Journal of Molecular Sciences, 17(4), 510. https://doi.org/10.3390/ijms17040510