Focus on Extracellular Vesicles: Development of Extracellular Vesicle-Based Therapeutic Systems


Abstract
:1. Introduction
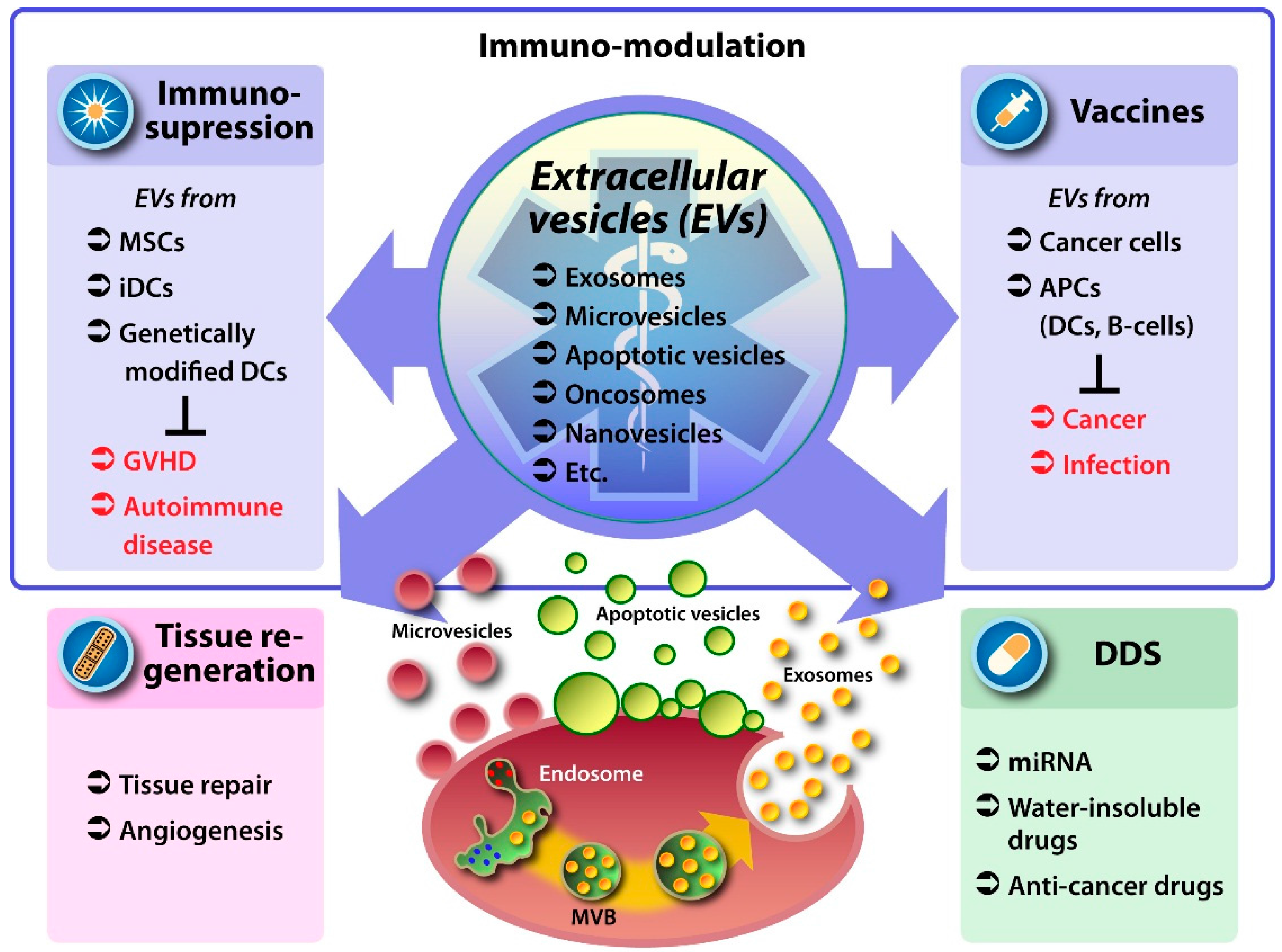
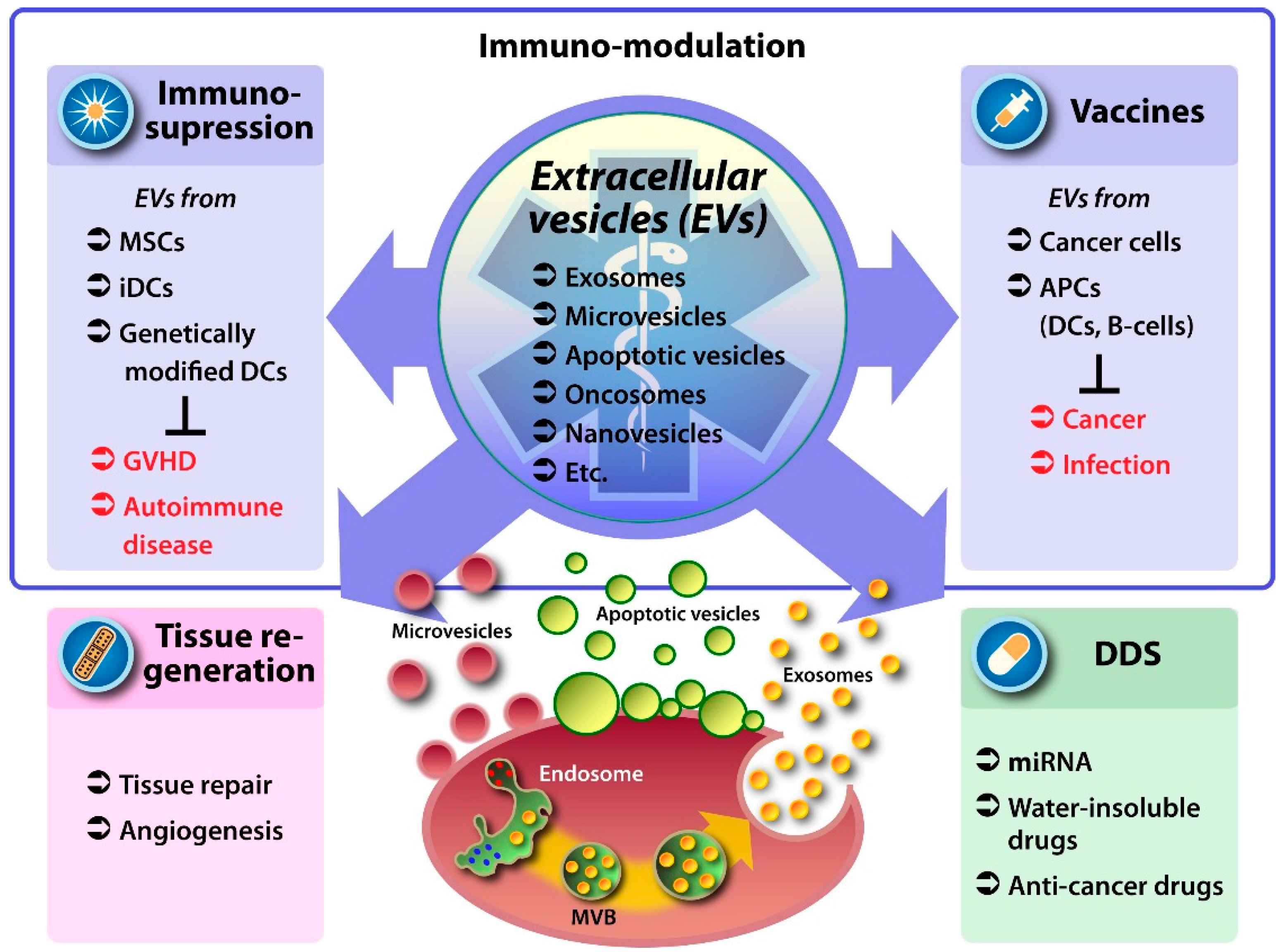
2. EVs as Therapeutic Vehicles
2.1. Liposomes vs. EVs
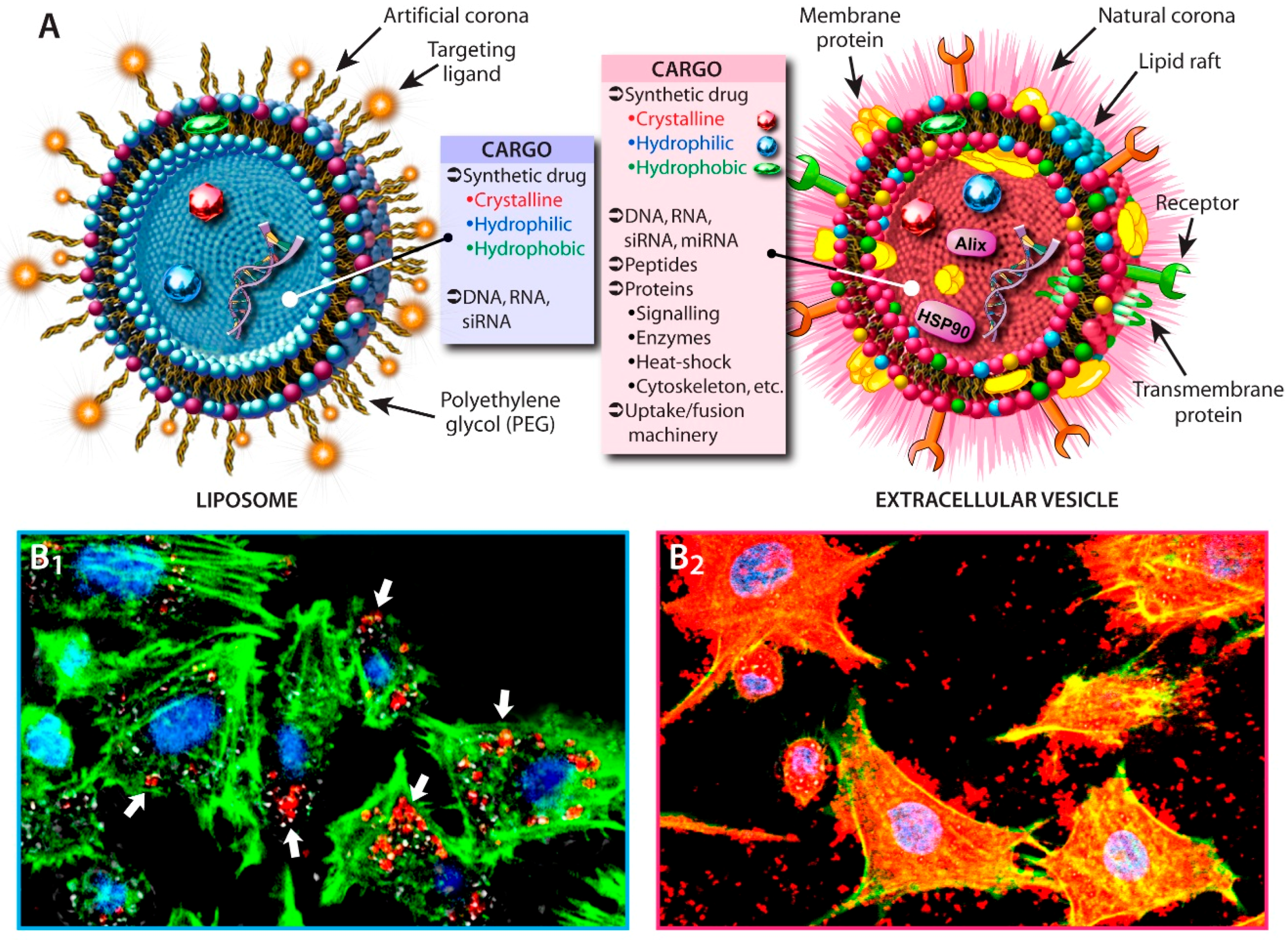
2.2. Cellular Uptake and Drug-Loading
3. Medicinal Use of Native EVs
3.1. EVs from Mesenchymal Stem Cells
3.2. EVs from Antigen Presenting and Other Immune Cells
4. EVs as Drug-Delivery System Carriers
4.1. EVs as Carriers for Small Molecular Drugs
4.2. EV-Encapsulated Curcumin
4.3. Nucleic Acid-Based Drugs and EVs
5. EVs in Clinical Trials

{kind=link}
{kind=link}
| Vesicle Type | Disease | Drug | EV Source | Admin.1 | Patients | Therapeutic Results 2 | Side-Effects 2 | Status 3 | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Phase I clinical trials | |||||||||
| Autologous dexosomes | Metastatic melanoma | Melanoma peptide antigens | DCs | i.d./s.c. | 15 | MART1–HLA-A2 T-cell response and tumor shrinkage (1); minor response (1); mixed response (1); stabilization (2) | No major toxicity; minor inflammation; grade 1 fever (5) | C | [82] |
| Autologous dexosomes | Non-small cell lung cancer | MAGE peptides | DCs | i.d./s.c. | 9 | MAGE-specific T cell responses (3); NK cell lysis (2) | No major toxicity; moderate pain (1), swelling at injection site (8); mild fever (1) | C | [83] |
| Autologous exosomes | Colorectal cancer | EVs, EVs + GM-CSF | Ascites fluid | s.c. | 37 4 | EVs+GM-CSF: cytotoxic T cell resp. to CAP-1 (76.9%); stabilization (1); minor response (1) | No major toxicity; moderate pain, swelling, pruritus at injection site (37); fever (1), fatigue (3) and nausea (1) | C | [84] |
| Autologous exosomes | Malignant Glioma | EVs + AS-ODN | Glioma | Implanted biodiffusion chamber | 12 | N.D. | N.D. | C | [85] |
| Allogeneic exosomes/MVs | Type I diabetes mellitus | EVs | Umbilical cord-blood derived MSC | i.v. | 20 | N.D | N.D | E | [87] |
| Allogeneic exosomes | GVHD | EVs | MSC | i.v. | 1 | GVHD symptoms improved; stabilization for several months. Patient died of pneumonia 7 months post exosome application | No major side-effects | C | [35] |
| OMVs | Meningitis | Vaccine | B:4:P1.7-2,4; B:15:P1.7,16 strains | i.m. | 91 | MenBVac/MeNZB vaccines: high efficacy | No major toxicity; moderate pain, swelling, induration at the injection site; mild fever, malaise/headache | C | [88] |
| OMVs | Meningitis | Vaccine | FetA modified strain 44/76 | i.m. | 52 | MenPF-1 vaccine:high additive efficacy | No major toxicity; moderate pain, swelling at injection site; mild fever, malaise, nausea/headache | C | [89] |
| Plexosomes 5 | Colon cancer | Curcumin | Fruit | oral | 35 | N.D. | N.D. | R | [90] |
| Plexosomes 5 | Mucositis | Curcumin | Fruit | oral | 60 | N.D. | N.D | R | [91] |
| Phase II clinical trials | |||||||||
| Autologous dexosomes | Non-small cell lung cancer | IFN-γ, MAGE peptides | DCs | i.d./s.c. | 41 | N.D | N.D | E | [86] |
| OMVs | Meningitis | rMenB vaccine, NadA/fHBP/NHBA | B:4:P1.7-2,4 strains | i.m. | 147 | rMenB+OMV: high additive efficacy | Moderate pain, swelling, induration at the injection site; mild fever; serious adverse events (18) | C | [92] |
6. EVs Caveats and Challenges
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [PubMed]
- Kalra, H.; Drummen, G.P.C.; Mathivanan, S. Focus on extracellular vesicles: Exosomes, the next small big thing. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006. [Google Scholar] [CrossRef]
- Camussi, G.; Deregibus, M.C.; Bruno, S.; Grange, C.; Fonsato, V.; Tetta, C. Exosome/microvesicle-mediated epigenetic reprogramming of cells. Am. J. Cancer Res. 2011, 1, 98–110. [Google Scholar] [PubMed]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Boussac, M.; Véron, P.; Ricciardi-Castagnoli, P.; Raposo, G.; Garin, J.; Amigorena, S. Proteomic analysis of dendritic cell-derived exosomes: A secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 2001, 166, 7309–7318. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration (US), Drugs@FDA, Silver Spring (MD). Available online: www.fda.gov (accessed on 17 January 2016).
- European Medicines Agency (EU), Find Medicines, London (UK). Available online: www.ema.europa.eu (accessed on 17 January 2016).
- National Library of Medicine (US), ClinicalTrials.gov, Bethesda (MD). Available online: www.clinicaltrials.gov (accessed on 17 January 2016).
- Szebeni, J. Complement activation-related pseudoallergy caused by liposomes, micellar carriers of intravenous drugs, and radiocontrast agents. Crit. Rev. Ther. Drug Carrier Syst. 2001, 18, 567–606. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J. Complement activation-related pseudoallergy: A new class of drug-induced acute immune toxicity. Toxicology 2005, 216, 106–121. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson's disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Nagasaki, Y. Construction of a densely poly(ethylene glycol)-chain-tethered surface and its performance. Polym. J. 2011, 43, 949–958. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kima, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Leger, R.M.; Arndt, P.; Garratty, G.; Armstrong, J.; Meiselman, H.; Fisher, T. Normal donor sera can contain antibodies to polyethylene glycol (PEG). Transfusion 2001, 41, 29S. [Google Scholar]
- Armstrong, J.; Leger, R.; Wenby, R.; Meiselman, H.; Garratty, G.; Fisher, T. Occurrence of an antibody to poly(ethylene glycol) in normal donors. Blood 2003, 102, 566A. [Google Scholar]
- Armstrong, J.K.; Hempel, G.; Koling, S.; Chan, L.S.; Fisher, T.; Meiselman, H.J.; Garratty, G. Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer 2007, 110, 103–111. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration, Select Committee on GRAS Substances opinion: Propylene glycol and propylene glycol monostearate. In SCOGS Database; 1973. Available online: www.fda.gov (accessed on 5 January 2016).
- Fuhrmann, G.; Serio, A.; Mazo, M.; Nair, R.; Stevens, M.M. Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins. J. Control. Release 2015, 205, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Kooijmans, S.; Stremersch, S.; Braeckmans, K.; de Smedt, S.; Hendrix, A.; Wood, M.; Schiffelers, R.M.; Raemdonck, K.; Vader, P. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J. Control. Release 2013, 172, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Zeelenberg, I.S.; Ostrowski, M.; Krumeich, S.; Bobrie, A.; Jancic, C.; Boissonnas, A.; Delcayre, A.; Le Pecq, J.B.; Combadiere, B.; Amigorena, S.; et al. Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res. 2008, 68, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.J.; Zhao, Y.; Harrison, E.B.; Mahajan, V.; Ahmed, S.; He, Z.; Suresh, P.; Hingtgen, S.D.; Klyachko, N.L.; Mosley, R.L.; et al. Specific transfection of inflamed brain by macrophages: A new therapeutic strategy for neurodegenerative diseases. PLoS ONE 2013, 8, e61852. [Google Scholar] [CrossRef] [PubMed]
- Maguire, C.A.; Balaj, L.; Sivaraman, S.; Crommentuijn, M.H.; Ericsson, M.; Mincheva-Nilsson, L.; Baranov, V.; Gianni, D.; Tannous, B.A.; Sena-Esteves, M.; et al. Microvesicle-associated AAV vector as a novel gene delivery system. Mol. Ther. 2012, 20, 960–971. [Google Scholar] [CrossRef] [PubMed]
- DiMarino, A.M.; Caplan, A.; Bonfield, T.L. Mesenchymal stem cells in tissue repair. Front. Immunol. 2013, 4, 201. [Google Scholar] [CrossRef] [PubMed]
- Frenette, P.S.; Pinho, S.; Lucas, D.; Scheiermann, C. Mesenchymal stem cell: Keystone of the hematopoietic stem cell niche and a stepping-stone for regenerative medicine. Annu. Rev. Immunol. 2013, 31, 285–316. [Google Scholar] [CrossRef] [PubMed]
- Curley, G.F.; Ansari, B.; Hayes, M.; Devaney, J.; Masterson, C.; Ryan, A.; Barry, F.; O’Brien, T.; Toole, D.O.; Laffey, J.G. Effects of intratracheal mesenchymal stromal cell therapy during recovery and resolution after ventilator-induced lung injury. Anesthesiology 2013, 118, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yuan, L.; Ren, X.; Nian, H.; Zhang, L.; Han, Z.C.; Li, X.; Zhang, X. The effect of mesenchymal stem cells on dynamic changes of T cell subsets in experimental autoimmune uveoretinitis. Clin. Exp. Immunol. 2013, 173, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.V.; Bull, N.D.; Hunt, D.P.; Marina, N.; Tomarev, S.I.; Martin, K.R. Neuroprotective effects of intravitreal mesenchymal stem cell transplantation in experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Bouchez, G.; Sensebé, L.; Vourc’h, P.; Garreau, L.; Bodard, S.; Rico, A.; Guilloteau, D.; Charbord, P.; Besnard, J.C.; Chalon, S. Partial recovery of dopaminergic pathway after graft of adult mesenchymal stem cells in a rat model of Parkinson's disease. Neurochem. Int. 2008, 52, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Mazzini, L.; Mareschi, K.; Ferrero, I.; Miglioretti, M.; Stecco, A.; Servo, S.; Carriero, A.; Monaco, F.; Fagioli, F. Mesenchymal stromal cell transplantation in amyotrophic lateral sclerosis: A long-term safety study. Cytotherapy 2012, 14, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Mazzini, L.; Vercelli, A.; Mareschi, K.; Ferrero, I.; Testa, L.; Fagioli, F. Mesenchymal stem cells for ALS patients. Amyotroph. Lateral Scler. 2009, 10, 123–124. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zhang, H.; Hua, B.; Wang, H.; Wang, J.; Han, Z.; Sun, L. Allogeneic mesenchymal stem cells transplantation in treatment of multiple sclerosis. Mult. Scl. 2009, 15, 644–646. [Google Scholar] [CrossRef] [PubMed]
- Kordelas, L.; Rebmann, V.; Ludwig, A.K.; Radtke, S.; Ruesing, J.; Doeppner, T.R.; Epple, M.; Horn, P.A.; Beelen, D.W.; Giebel, B. MSC-derived exosomes: A novel tool to treat therapy-refractory graft-versus-host disease. Leukemia 2014, 28, 970–973. [Google Scholar] [CrossRef] [PubMed]
- Schafer, A.; Pahnke, A.; Schaffert, D.; van Weerden, W.M.; de Ridder, C.M.; Rodl, W.; Vetter, A.; Spitzweg, C.; Kraaij, R.; Wagner, E.; et al. Disconnecting the yin and yang relation of epidermal growth factor receptor (EGFR)-mediated delivery: A fully synthetic, EGFR-targeted gene transfer system avoiding receptor activation. Hum. Gene Ther. 2011, 22, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; An, R.; Liu, Z.J.; Wang, J.J.; Chen, S.Z.; Hong, M.M.; Liu, J.H.; Xiao, M.Y.; Chen, Y.F. Therapeutic effects of mesenchymal stem cell-derived microvesicles on pulmonary arterial hypertension in rats. Acta Pharmacol. Sin. 2014, 35, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.G.; Feng, X.M.; Abbott, J.; Fang, X.H.; Hao, Q.; Monsel, A.; Qu, J.M.; Matthay, M.A.; Lee, J.W. Human mesenchymal stem cell microvesicles for treatment of Escherichia coli endotoxin-induced acute lung injury in mice. Stem Cells 2014, 32, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Blin, G.; Nury, D.; Stefanovic, S.; Neri, T.; Guillevic, O.; Brinon, B.; Bellamy, V.; Rücker-Martin, C.; Barbry, P.; Bel, A.; et al. A purified population of multipotent cardiovascular progenitors derived from primate pluripotent stem cells engrafts in postmyocardial infarcted nonhuman primates. J. Clin. Investig. 2010, 120, 1125–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspi, O.; Huber, I.; Kehat, I.; Habib, M.; Arbel, G.; Gepstein, A.; Yankelson, L.; Aronson, D.; Beyar, R.; Gepstein, L. Transplantation of human embryonic stem cell-derived cardiomyocytes improves myocardial performance in infarcted rat hearts. J. Am. Coll. Cardiol. 2007, 50, 1884–1893. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Nickoloff, E.; Abramova, T.; Johnson, J.; Verma, S.K.; Krishnamurthy, P.; Mackie, A.R.; Vaughan, E.; Garikipati, V.N.; Benedict, C.; et al. Embryonic stem cell-derived exosomes promote endogenous repair mechanisms and enhance cardiac function following myocardial infarction. Circ. Res. 2015, 117, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Grange, C.; Deregibus, M.C.; Calogero, R.A.; Saviozzi, S.; Collino, F.; Morando, L.; Busca, A.; Falda, M.; Bussolati, B.; et al. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J. Am. Soc. Nephrol. 2009, 20, 1053–1067. [Google Scholar] [CrossRef] [PubMed]
- Gatti, S.; Bruno, S.; Deregibus, M.C.; Sordi, A.; Cantaluppi, V.; Tetta, C.; Camussi, G. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol. Dial. Transplant. 2011, 26, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Zhang, X.; Li, X. Exosomes derived from mesenchymal stem cells. Int. J. Mol. Sci. 2014, 15, 4142–4157. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Nolte’t Hoen, E.N.; Buschow, S.I.; Anderton, S.M.; Stoorvogel, W.; Wauben, M.H. Activated T cells recruit exosomes secreted by dendritic cells via LFA-1. Blood 2009, 113, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Muntasell, A.; Berger, A.C.; Roche, P.A. T cell-induced secretion of MHC class II-peptide complexes on B cell exosomes. EMBO J. 2007, 26, 4263–4272. [Google Scholar] [CrossRef] [PubMed]
- Martin-Jaular, L.; Nakayasu, E.S.; Ferrer, M.; Almeida, I.C.; Del Portillo, H.A. Exosomes from Plasmodium yoelii-infected reticulocytes protect mice from lethal infections. PLoS ONE 2011, 6, e26588. [Google Scholar] [CrossRef] [PubMed]
- Näslund, T.I.; Gehrmann, U.; Qazi, K.R.; Karlsson, M.C.; Gabrielsson, S. Dendritic cell-derived exosomes need to activate both T and B cells to induce antitumor immunity. J. Immunol. 2013, 190, 2712–2719. [Google Scholar] [CrossRef] [PubMed]
- Qazi, K.R.; Gehrmann, U.; Domange Jordö, E.; Karlsson, M.C.; Gabrielsson, S. Antigen-loaded exosomes alone induce Th1-type memory through a B-cell-dependent mechanism. Blood 2009, 113, 2673–2683. [Google Scholar] [CrossRef] [PubMed]
- Damo, M.; Wilson, D.S.; Simeoni, E.; Hubbell, J.A. TLR-3 stimulation improves anti-tumor immunity elicited by dendritic cell exosome-based vaccines in a murine model of melanoma. Sci. Rep. 2015, 5, 17622. [Google Scholar] [CrossRef] [PubMed]
- Bianco, N.R.; Kim, S.H.; Ruffner, M.A.; Robbins, P.D. Therapeutic effect of exosomes from indoleamine 2,3-dioxygenase-positive dendritic cells in collagen-induced arthritis and delayed-type hypersensitivity disease models. Arthritis Rheum. 2009, 60, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Prado, N.; Marazuela, E.G.; Segura, E.; Fernández-García, H.; Villalba, M.; Théry, C.; Rodríguez, R.; Batanero, E. Exosomes from bronchoalveolar fluid of tolerized mice prevent allergic reaction. J. Immunol. 2008, 181, 1519–1525. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Bianco, N.R.; Shufesky, W.J.; Morelli, A.E.; Robbins, P.D. Effective treatment of inflammatory disease models with exosomes derived from dendritic cells genetically modified to express IL-4. J. Immunol. 2007, 179, 2242–2249. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Bianco, N.; Menon, R.; Lechman, E.R.; Shufesky, W.J.; Morelli, A.E.; Robbins, P.D. Exosomes derived from genetically modified DC expressing FasL are anti-inflammatory and immunosuppressive. Mol. Ther. 2006, 13, 289–300. [Google Scholar] [PubMed]
- Admyre, C.; Johansson, S.M.; Qazi, K.R.; Filén, J.J.; Lahesmaa, R.; Norman, M.; Neve, E.P.; Scheynius, A.; Gabrielsson, S. Exosomes with immune modulatory features are present in human breast milk. J. Immunol. 2007, 179, 1969–1978. [Google Scholar] [CrossRef] [PubMed]
- Aucher, A.; Rudnicka, D.; Davis, D.M. MicroRNAs transfer from human macrophages to hepato-carcinoma cells and inhibit proliferation. J. Immunol. 2013, 191, 6250–6260. [Google Scholar] [CrossRef] [PubMed]
- Squadrito, M.L.; Baer, C.; Burdet, F.; Maderna, C.; Gilfillan, G.D.; Lyle, R.; Ibberson, M.; De Palma, M. Endogenous RNAs modulate microRNA sorting to exosomes and transfer to acceptor cells. Cell. Rep. 2014, 8, 1432–1446. [Google Scholar] [CrossRef] [PubMed]
- Buck, A.H.; Coakley, G.; Simbari, F.; McSorley, H.J.; Quintana, J.F.; Le Bihan, T.; Kumar, S.; Abreu-Goodger, C.; Lear, M.; Harcus, Y.; et al. Exosomes secreted by nematode parasites transfer small RNAs to mammalian cells and modulate innate immunity. Nat. Commun. 2014, 5, 5488. [Google Scholar] [CrossRef] [PubMed]
- Nono, J.K.; Pletinckx, K.; Lutz, M.B.; Brehm, K. Excretory/secretory-products of Echinococcus multilocularis larvae induce apoptosis and tolerogenic properties in dendritic cells in vitro. PLoS Negl. Trop. Dis. 2012, 6, e1516. [Google Scholar] [CrossRef] [PubMed]
- Marcilla, A.; Trelis, M.; Cortes, A.; Sotillo, J.; Cantalapiedra, F.; Minguez, M.T.; Valero, M.L.; Sanchez del Pino, M.M.; Munoz-Antoli, C.; Toledo, R.; et al. Extracellular vesicles from parasitic helminths contain specific excretory/secretory proteins and are internalized in intestinal host cells. PLoS ONE 2012, 7, e45974. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.C.; Kim, O.Y.; Yoon, C.M.; Choi, D.S.; Roh, T.Y.; Park, J.; Nilsson, J.; Lötvall, J.; Kim, Y.K.; Gho, Y.S. Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS Nano 2013, 7, 7698–7710. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Chang, C.K.; Fan, H.H.; Nie, X.X.; Ren, Y.N.; Liu, Y.Y.; Zhao, L.H. Anti-tumour effects of exosomes in combination with cyclophosphamide and polyinosinic-polycytidylic acid. J. Int. Med. Res. 2008, 36, 1342–1353. [Google Scholar] [CrossRef] [PubMed]
- Pascucci, L.; Coccè, V.; Bonomi, A.; Ami, D.; Ceccarelli, P.; Ciusani, E.; Viganò, L.; Locatelli, A.; Sisto, F.; Doglia, S.M.; et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: A new approach for drug delivery. J. Control. Release 2014, 192, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Federici, C.; Petrucci, F.; Caimi, S.; Cesolini, A.; Logozzi, M.; Borghi, M.; D’llio, S.; Lugini, L.; Violante, N.; Azzarito, T.; et al. Exosome release and low pH belong to a framework of resistance of human melanoma cells to cisplatin. PLoS ONE 2014, 9, e88193. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Uechi, S.; Takara, K.; Asikin, Y.; Wada, K. Evaluation of an oral carrier system in rats: Bioavailability and antioxidant properties of liposome-encapsulated curcumin. J. Agric. Food Chem. 2009, 57, 9141–9146. [Google Scholar] [CrossRef] [PubMed]
- Ray, B.; Bisht, S.; Maitra, A.; Maitra, A.; Lahiri, D.K. Neuroprotective and neurorescue effects of a novel polymeric nanoparticle formulation of curcumin (NanoCurc) in the neuronal cell culture and animal model: Implications for Alzheimer's disease. J. Alzheimers Dis. 2011, 23, 61–77. [Google Scholar] [PubMed]
- Tomren, M.A.; Masson, M.; Loftsson, T.; Tonnesen, H.H. Studies on curcumin and curcuminoids XXXI. Symmetric and asymmetric curcuminoids: Stability, activity and complexation with cyclodextrin. Int. J. Pharm. 2007, 338, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, F.; Rizvi, M.M.; Kar, S.K. Oral delivery of curcumin bound to chitosan nanoparticles cured Plasmodium yoelii infected mice. Biotechnol. Adv. 2012, 30, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, J.; Ankola, D.D.; Beniwal, V.; Singh, D.; Kumar, M.N. Nanoparticle encapsulation improves oral bioavailability of curcumin by at least 9-fold when compared to curcumin administered with piperine as absorption enhancer. Eur. J. Pharm. Sci. 2009, 37, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.G. A novel nanoparticle drug delivery system: The anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Patchva, S.; Aggarwal, B.B. Therapeutic roles of curcumin: Lessons learned from clinical trials. AAPS J. 2013, 15, 195–218. [Google Scholar] [CrossRef] [PubMed]
- Osterman, C.J.; Lynch, J.C.; Leaf, P.; Gonda, A.; Ferguson Bennit, H.R.; Griffiths, D.; Wall, N.R. Curcumin modulates pancreatic adenocarcinoma cell-derived exosomal function. PLoS ONE 2015, 10, e0132845. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Li, Y.; Buller, B.; Katakowski, M.; Zhang, Y.; Wang, X.; Shang, X.; Zhang, Z.G.; Chopp, M. Exosome-mediated transfer of miR-133b from multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells 2012, 30, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Yeo, R.W.; Lai, R.C.; Zhang, B.; Tan, S.S.; Yin, Y.; Teh, B.J.; Lim, S.K. Mesenchymal stem cell: An efficient mass producer of exosomes for drug delivery. Adv. Drug Deliv. Rev. 2013, 65, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Bryniarski, K.; Ptak, W.; Jayakumar, A.; Püllmann, K.; Caplan, M.J.; Chairoungdua, A.; Lu, J.; Adams, B.D.; Sikora, E.; Nazimek, K.; et al. Antigen-specific, antibody-coated, exosome-like nanovesicles deliver suppressor T-cell microRNA-150 to effector T cells to inhibit contact sensitivity. J. Allergy Clin. Immunol. 2013, 132, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Dorval, T.; Chaput, N.; André, F.; Caby, M.P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of thefirst phase I clinical trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J. Transl. Med. 2005, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Wei, D.; Wu, Z.; Zhou, X.; Wei, X.; Huang, H.; Li, G. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol. Ther. 2008, 16, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Thomas Jefferson University Hospital, Pilot Immunotherapy Trial for Recurrent Malignant Gliomas. In ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US); 2000. Available online: https://clinicaltrials.gov/show/NCT01550523 (accesssed on 12 January 2016).
- Viaud, S.; Ploix, S.; Lapierre, V.; Thery, C.; Commere, P.H.; Tramalloni, D.; Gorrichon, K.; Virault-Rocroy, P.; Tursz, T.; Lantz, O.; et al. Updated technology to produce highly immunogenic dendritic cell-derived exosomes of clinical grade: A critical role of interferon-gamma. J. Immunother. 2011, 34, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Sahel Teaching Hospital; Faculty of Medicine Cairo University, Effect of Microvesicles and Exosomes Therapy on β-cell Mass in Type I Diabetes Mellitus (T1DM). In ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US); 2000. Available online: https://clinicaltrials.gov/show/NCT02138331 (accesssed on 12 January 2016).
- Sandbu, S.; Feiring, B.; Oster, P.; Helland, O.S.; Bakke, H.S.; Naess, L.M.; Aase, A.; Aaberge, I.S.; Kristoffersen, A.C.; Rydland, K.M.; et al. Immunogenicity and safety of a combination of two serogroup B meningococcal outer membrane vesicle vaccines. Clin. Vaccine Immunol. 2007, 14, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Marsay, L.; Dold, C.; Green, C.A.; Rollier, C.S.; Norheim, G.; Sadarangani, M.; Shanyinde, M.; Brehony, C.; Thompson, A.J.; Sanders, H.; et al. A novel meningococcal outer membrane vesicle vaccine with constitutive expression of FetA: A phase I clinical trial. J. Infect. 2015, 71, 326–337. [Google Scholar] [CrossRef] [PubMed]
- James Graham Brown Cancer Center; University of Louisville, Study Investigating the Ability of Plant Exosomes to Deliver Curcumin to Normal and Colon Cancer Tissue. In ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US); 2000. Available online: https://clinicaltrials.gov/show/NCT01294072 (accesssed on 12 January 2016).
- James Graham Brown Cancer Center; University of Louisville, Edible Plant Exosome Ability to Prevent Oral Mucositis Associated with Chemoradiation Treatment of Head and Neck Cancer. In ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US); 2000. Available online: https://clinicaltrials.gov/show/NCT01668849 (accesssed on 12 January 2016).
- Findlow, J.; Borrow, R.; Snape, M.D.; Dawson, T.; Holland, A.; John, T.M.; Evans, A.; Telford, K.L.; Ypma, E.; Toneatto, D.; et al. Multicenter, open-label, randomized phase II controlled trial of an investigational recombinant Meningococcal serogroup B vaccine with and without outer membrane vesicles, administered in infancy. Clin. Infect. Dis. 2010, 51, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Harshyne, L.A.; Hooper, K.M.; Andrews, E.G.; Nasca, B.J.; Kenyon, L.C.; Andrews, D.W.; Hooper, D.C. Glioblastoma exosomes and IGF-1R/AS-ODN are immunogenic stimuli in a translational research immunotherapy paradigm. Cancer Immunol. Immunother. 2015, 64, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Morin-Brureau, M.; Hooper, K.M.; Prosniak, M.; Sauma, S.; Harshyne, L.A.; Andrews, D.W.; Hooper, D.C. Enhancement of glioma-specific immunity in mice by “NOBEL”, an insulin-like growth factor 1 receptor antisense oligodeoxynucleotide. Cancer Immunol. Immunother. 2015, 64, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Andrews, D.W. Thomas Jefferson University Hospital, Pilot immunotherapy trial for recurrent malignant gliomas. Personal communication, 20 January 2016. [Google Scholar]
- Navabi, H.; Croston, D.; Hobot, J.; Clayton, A.; Zitvogel, L.; Jasani, B.; Bailey-Wood, R.; Wilson, K.; Tabi, Z.; Mason, M.D.; et al. Preparation of human ovarian cancer ascites-derived exosomes for a clinical trial. Blood Cells Mol. Dis. 2005, 35, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, L.; Stork, M.; van der Ley, P. Outer membrane vesicles as platform vaccine technology. Biotechnol. J. 2015, 10, 1689–1706. [Google Scholar] [CrossRef]
- Gujrati, V.; Kim, S.; Kim, S.H.; Min, J.J.; Choy, H.E.; Kim, S.C.; Jon, S. Bioengineered bacterial outer membrane vesicles as cell-specific drug-delivery vehicles for cancer therapy. ACS Nano 2014, 8, 1525–1537. [Google Scholar] [CrossRef] [PubMed]
- Knight, S.D.; Parrish, C.A. Recent progress in the identification and clinical evaluation of inhibitors of the mitotic kinesin KSP. Curr. Top. Med. Chem. 2008, 8, 888–904. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.K.; South, V.J.; Zhang, Y.; Davide, J.P.; Farrell, L.; Kohl, N.E.; Sepp-Lorenzino, L.; Lobell, R.B. Induction of apoptosis by an inhibitor of the mitotic kinesin KSP requires both activation of the spindle assembly checkpoint and mitotic slippage. Cancer Cell 2005, 8, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kim, K.S.; Lee, S.R.; Kim, E.; Kim, M.S.; Lee, E.Y.; Gho, Y.S.; Kim, J.W.; Bishop, R.E.; Chang, K.T. Structural modifications of outer membrane vesicles to refine them as vaccine delivery vehicles. Biochim. Biophys. Acta 2009, 1788, 2150–2059. [Google Scholar] [CrossRef] [PubMed]
- Somerville, J.E., Jr.; Cassiano, L.; Darveau, R.P. Escherichia coli msbB gene as a virulence factor and a therapeutic target. Infect. Immun. 1999, 67, 6583–6590. [Google Scholar] [PubMed]
- Mathivanan, S.; Lim, J.W.; Tauro, B.J.; Ji, H.; Moritz, R.L.; Simpson, R.J. Proteomics analysis of A33 immunoaffinity-purified exosomes released from the human colon tumor cell line LIM1215 reveals a tissue-specific protein signature. Mol. Cell. Proteom. 2010, 9, 197–208. [Google Scholar] [CrossRef] [PubMed]
- McCready, J.; Sims, J.D.; Chan, D.; Jay, D.G. Secretion of extracellular hsp90α via exosomes increases cancer cell motility: A role for plasminogen activation. BMC Cancer 2010, 10, 294. [Google Scholar] [CrossRef] [PubMed]
- Ochieng, J.; Pratap, S.; Khatua, A.K.; Sakwe, A.M. Anchorage-independent growth of breast carcinoma cells is mediated by serum exosomes. Exp. Cell Res. 2009, 315, 1875–1888. [Google Scholar] [CrossRef] [PubMed]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Lagana, A.; Russo, F.; Veneziano, D.; Bella, S.D.; Giugno, R.; Pulvirenti, A.; Croce, C.M.; Ferro, A. Extracellular circulating viral microRNAs: Current knowledge and perspectives. Front. Genet. 2013, 4, 120. [Google Scholar] [CrossRef] [PubMed]
- Delabranche, X.; Berger, A.; Boisrame-Helms, J.; Meziani, F. Microparticles and infectious diseases. Med. Mal. Infect. 2012, 42, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Schwab, A.; Meyering, S.S.; Lepene, B.; Iordanskiy, S.; van Hoek, M.L.; Hakami, R.M.; Kashanchi, F. Extracellular vesicles from infected cells: Potential for direct pathogenesis. Front. Microbiol. 2015, 6, 1132. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.W.; Li, Q.; Niu, X.; Hu, B.; Liu, J.; Zhou, S.M.; Guo, S.C.; Lang, H.L.; Zhang, C.Q.; Wang, Y.; Deng, Z.F. Exosomes secreted by human-induced pluripotent stem cell-derived mesenchymal stem cells attenuate limb ischemia by promoting angiogenesis in mice. Stem Cell Res. Ther. 2015, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H.; Odorizzi, G. Get on the exosome bus with ALIX. Nat. Cell Biol. 2012, 14, 654–655. [Google Scholar] [CrossRef] [PubMed]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Vilette, D.; Laulagnier, K.; Huor, A.; Alais, S.; Simoes, S.; Maryse, R.; Provansal, M.; Lehmann, S.; Andreoletti, O.; Schaeffer, L.; et al. Efficient inhibition of infectious prions multiplication and release by targeting the exosomal pathway. Cell. Mol. Life Sci. 2015, 72, 4409–4427. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnaiah, V.; Thumann, C.; Fofana, I.; Habersetzer, F.; Pan, Q.; de Ruiter, P.E.; Willemsen, R.; Demmers, J.A.; Stalin Raj, V.; Jenster, G.; et al. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. USA 2013, 110, 13109–13113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Tan, K.H.; Lim, S.K. Focus on extracellular vesicles: Therapeutic efficacy of stem cell-derived extracellular vesicles. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Vella, L.J.; Hill, A.F.; Cheng, L. Focus on extracellular vesicles: Exosomes and their role in protein trafficking in Alzheimer’s and Parkinson’s disease. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Iraci, N.; Leonardi, T.; Gessler, F.; Vega, B.; Pluchino, S. Focus on extracellular vesicles: Physiological role and signaling properties of extracellular membrane vesicles. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, C.; Cavallini, L.; Spinelli, C.; Yang, J.; Reis-Sobreiro, M.; de Candia, P.; Rene’ Minciacchi, V.; di Vizio, D. Focus on extracellular vesicles: New frontiers of cell-to-cell communication in cancer. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohno, S.-i.; Drummen, G.P.C.; Kuroda, M. Focus on Extracellular Vesicles: Development of Extracellular Vesicle-Based Therapeutic Systems. Int. J. Mol. Sci. 2016, 17, 172. https://doi.org/10.3390/ijms17020172
Ohno S-i, Drummen GPC, Kuroda M. Focus on Extracellular Vesicles: Development of Extracellular Vesicle-Based Therapeutic Systems. International Journal of Molecular Sciences. 2016; 17(2):172. https://doi.org/10.3390/ijms17020172
Chicago/Turabian StyleOhno, Shin-ichiro, Gregor P. C. Drummen, and Masahiko Kuroda. 2016. "Focus on Extracellular Vesicles: Development of Extracellular Vesicle-Based Therapeutic Systems" International Journal of Molecular Sciences 17, no. 2: 172. https://doi.org/10.3390/ijms17020172