
The Role of ERK1/2 in the Development of Diabetic Cardiomyopathy

Abstract
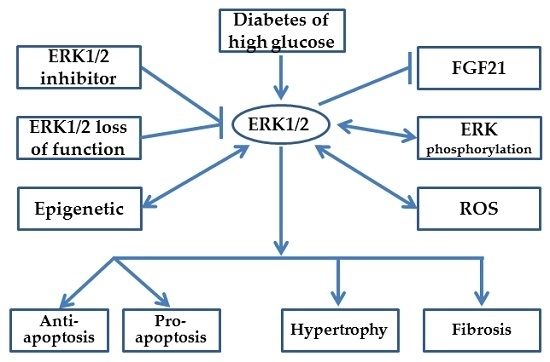
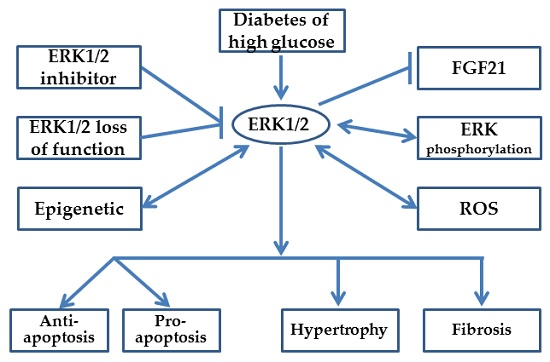
:
1. Introduction
2. Mitogen Activated Protein Kinase (MAPK) Pathway
3. ERK1/2 Signaling Pathway
4. ERK1/2, a Two-Edged Sword in DCM Development
4.1. Oxidative Stress
4.2. Anti-Apoptosis vs. Pro-Apoptosis Influence of ERK1/2
4.3. Hypertrophy
4.4. Fibrosis
4.5. FGF21 Functions on ERK1/2 Signaling
5. Inhibition of MEK/ERK to Study the Function of ERK
5.1. Exploiting Pharmacologic Agents to Study ERK1/2 Function
5.2. Exploiting Loss of Function Mutation of ERK1/2
6. ERK Phosphorylation Site
7. HDAC Inhibitors Regulate ERK1/2 Activity in the Heart
8. MicroRNAs Regulates ERK1/2 Activity in Diabetic Heart
9. Conclusions
Acknowledgments
Author Contributions
Conflict of Interest
Abbreviations
| AGEs | advanced glycation end products |
| Ang II | angiotensin II |
| DCM | diabetic cardiomyopathy |
| DUSPs | dual-specificity phosphatases |
| ERK1/2 | extracellular signal-regulated kinase 1/2 |
| ET-1 | endothelin 1 |
| FGF21 | fibroblast growth factor 21 |
| FN | fibronectin |
| FS | fractional shortening |
| GPCR | G-protein coupled receptor |
| HDAC | histone deacetylase |
| HG | high glucose |
| HUVECs | human umbilical vein endothelial cells |
| I/R | ischemia/reperfusion |
| ISO | isoproterenol |
| JNK | c-Jun N-terminal protein kinase |
| MAPK | mitogen-activated protein kinase |
| MEK | MAPK kinase |
| MI | myocardial infarction |
| miRNA | microRNA |
| MKPs | MAPK phosphatases |
| MMP | mitochondrial membrane potential |
| NO | nitric oxide |
| NRVCs | neonatal rat ventricular cardiomyocytes |
| Pak-1 | p21-activated kinase-1 |
| PE | phenylephrine |
| PP2A | protein phosphatase 2A |
| ROS | reactive oxygen species |
| SFAs | saturated fatty acids |
| SOD | superoxide dismutase |
| STZ | streptozocin |
| T1DM | type 1 diabetes mellitus |
| T2DM | type 2 diabetes mellitus |
| TAC | transverse aortic constriction |
| TGF-β | transforming growth factor beta |
| TSA | trichostatin A |
| UFAs | unsaturated fatty acids |
| VEGF | vascular endothelial growth factor |
References
- International Diabetes Federation. IDF Diabetes ATLAS, 7th ed.; Karakas Print: Brussels, Belgium, 2015; p. 13. [Google Scholar]
- Shi, Y.; Hu, F.B. The global implications of diabetes and cancer. Lancet 2014, 383, 1947–1948. [Google Scholar] [CrossRef]
- The Top 10 Causes of Death. World Health Organization Media center. Available online: http://www.who.int/mediacentre/factsheets/fs310/en/ (accessed on 13 October 2013).
- Boudina, S.; Abel, E.D. Diabetic cardiomyopathy revisited. Circulation 2007, 115, 3213–3223. [Google Scholar] [CrossRef] [PubMed]
- Monkemann, H.; de Vriese, A.S.; Blom, H.J.; Kluijtmans, L.A.; Heil, S.G.; Schild, H.H.; Golubnitschaja, O. OCEarly molecular events in the development of the diabetic cardiomyopathy. Amino Acids 2002, 23, 331–336. [Google Scholar] [PubMed]
- Miki, T.; Yuda, S.; Kouzu, H.; Miura, T. Diabetic cardiomyopathy: Pathophysiology and clinical features. Heart Fail. Rev. 2013, 18, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Abel, E.D. Diabetic cardiomyopathy, causes and effects. Rev. Endocr. Metab. Disord. 2010, 11, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Severson, D.L. Diabetic cardiomyopathy: Recent evidence from mouse models of type 1 and type 2 diabetes. Can. J. Physiol. Pharmacol. 2004, 82, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Gallo, S.; Sala, V.; Gatti, S.; Crepaldi, T. Cellular and molecular mechanisms of HGF/MET in the cardiovascular system. Clin. Sci. 2015, 129, 1173–1193. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Jang, S.; Agostini, B. Crosstalk between mitogen-activated protein kinases and mitochondria in cardiac diseases: Therapeutic perspectives. Pharmacol. Ther. 2014, 144, 202–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Luo, M.; Zhang, Z.; Gu, J.; Chen, J.; Payne, K.M.; Tan, Y.; Wang, Y.; Yin, X.; Zhang, X.; et al. Zinc deficiency exacerbates while zinc supplement attenuates cardiac hypertrophy in high-fat diet-induced obese mice through modulating p38 MAPK-dependent signaling. Toxicol. Lett. 2016, 258, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.Y.; Ma, Z.G.; Xu, S.C.; Zhang, N.; Tang, Q.Z. Pioglitazone protected against cardiac hypertrophy via inhibiting AKT/GSK3B and MAPK signaling pathways. PPAR Res. 2016, 2016, 9174190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yang, Z.; Yuan, Y.; Li, F.; Liu, Y.; Ma, Z.; Liao, H.; Bian, Z.; Zhang, Y.; Zhou, H.; et al. Naringenin attenuates pressure overload-induced cardiac hypertrophy. Exp. Ther. Med. 2015, 10, 2206–2212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ding, W.Y.; Wang, Z.H.; Tang, M.X.; Wang, F.; Li, Y.; Zhong, M.; Zhang, Y.; Zhang, W. Early administration of trimetazidine attenuates diabetic cardiomyopathy in rats by alleviating fibrosis, reducing apoptosis and enhancing autophagy. J. Transl. Med. 2016, 14, 109. [Google Scholar] [CrossRef] [PubMed]
- Fei, A.H.; Wang, F.C.; Wu, Z.B.; Pan, S.M. Phosphocreatine attenuates angiotensin ii-induced cardiac fibrosis in rat cardiomyocytes through modulation of MAPK and NF-κB pathway. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2726–2733. [Google Scholar] [PubMed]
- Jiang, S.; Jiang, D.; Zhao, P.; He, X.; Tian, S.; Wu, X.; Tao, Y. Activation of amp-activated protein kinase reduces collagen production via p38 MAPK in cardiac fibroblasts induced by coxsackievirus b3. Mol. Med. Rep. 2016, 14, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Sager, H.B.; Hulsmans, M.; Lavine, K.J.; Beltrami Moreira, M.B.; Heidt, T.; Courties, G.; Sun, Y.; Iwamoto, Y.; Tricot, B.; Khan, O.F.; et al. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ. Res. 2016, 119, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.F.; Bonomo, C.; Guizoni, D.M.; Junior, S.A.; Damatto, R.L.; Cezar, M.D.; Lima, A.R.; Pagan, L.U.; Seiva, F.R.; Bueno, R.T.; et al. Modulation of MAPK and NF-κB signaling pathways by antioxidant therapy in skeletal muscle of heart failure rats. Cell. Physiol. Biochem. 2016, 39, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Thandavarayan, R.A.; Giridharan, V.V.; Arumugam, S.; Suzuki, K.; Ko, K.M.; Krishnamurthy, P.; Watanabe, K.; Konishi, T. Schisandrin b prevents doxorubicin induced cardiac dysfunction by modulation of DNA damage, oxidative stress and inflammation through inhibition of MAPK/p53 signaling. PLoS ONE 2015, 10, e0119214. [Google Scholar] [CrossRef] [PubMed]
- Kehat, I.; Molkentin, J.D. Extracellular signal-regulated kinase 1/2 (ERK1/2) signaling in cardiac hypertrophy. Ann. N. Y. Acad. Sci. 2010, 1188, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Mutlak, M.; Kehat, I. Extracellular signal-regulated kinases 1/2 as regulators of cardiac hypertrophy. Front. Pharmacol. 2015, 6, 149. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, A.P.; Harima, M.; Sukumaran, V.; Soetikno, V.; Thandavarayan, R.A.; Suzuki, K.; Kodama, M.; Nagata, M.; Takagi, R.; Watanabe, K. Modulation of AT-1R/AMPK-MAPK cascade plays crucial role for the pathogenesis of diabetic cardiomyopathy in transgenic type 2 diabetic (spontaneous diabetic torii) rats. Biochem. Pharmacol. 2012, 83, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Huang, Z.; Gu, J.; Yan, X.; Lu, X.; Zhou, S.; Wang, S.; Shao, M.; Zhang, F.; Cheng, P.; et al. Fibroblast growth factor 21 protects the heart from apoptosis in a diabetic mouse model via extracellular signal-regulated kinase 1/2-dependent signaling pathway. Diabetologia 2015, 58, 1937–1948. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Ichikawa, T.; Li, J.; Si, Q.; Yang, H.; Chen, X.; Goldblatt, C.S.; Meyer, C.J.; Li, X.; Cai, L.; et al. Diabetic downregulation of NRF2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes 2011, 60, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Cobb, M.H. Map kinase pathways. Prog. Biophys. Mol. Biol. 1999, 71, 479–500. [Google Scholar] [CrossRef]
- McKay, M.M.; Morrison, D.K. Integrating signals from RTKs to ERK/MAPK. Oncogene 2007, 26, 3113–3121. [Google Scholar] [PubMed]
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef] [PubMed]
- Miao, R.; Lu, Y.; Xing, X.; Li, Y.; Huang, Z.; Zhong, H.; Huang, Y.; Chen, A.F.; Tang, X.; Li, H.; et al. Regulator of G-protein signaling 10 negatively regulates cardiac remodeling by blocking mitogen-activated protein kinase-extracellular signal-regulated protein kinase 1/2 signaling. Hypertension 2016, 67, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Han, J. The p38 signal transduction pathway: Activation and function. Cell Signal. 2000, 12, 1–13. [Google Scholar] [CrossRef]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; Li, S.P.; Westermarck, J. Phosphatase-mediated crosstalk between mapk signaling pathways in the regulation of cell survival. FASEB J. 2008, 22, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Ravingerova, T.; Barancik, M.; Strniskova, M. Mitogen-activated protein kinases: A new therapeutic target in cardiac pathology. Mol. Cell. Biochem. 2003, 247, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Gerits, N.; Kostenko, S.; Moens, U. In vivo functions of mitogen-activated protein kinases: Conclusions from knock-in and knock-out mice. Transgenic Res. 2007, 16, 281–314. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J. Signal transduction by the jnk group of map kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Ramos, J.W. The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int. J. Biochem. Cell Biol. 2008, 40, 2707–2719. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tournier, C. Regulation of cellular functions by the ERK5 signaling pathway. Cell Signal. 2006, 18, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.J.; Tanaka, N.; Rockman, H.A.; Ross, J., Jr.; Chien, K.R. Ventricular expression of a MLC-2V-RAS fusion gene induces cardiac hypertrophy and selective diastolic dysfunction in transgenic mice. J. Biol. Chem. 1995, 270, 23173–23178. [Google Scholar] [CrossRef] [PubMed]
- Ferrell, J.E., Jr. Tripping the switch fantastic: How a protein kinase cascade can convert graded inputs into switch-like outputs. Trends Biochem. Sci. 1996, 21, 460–466. [Google Scholar] [CrossRef]
- Lloyd, A.C. Distinct functions for ERKS? J. Biol. 2006, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Erk1/2 map kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 2004, 5, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.R.; Hsu, A.K.; Gross, G.J. Diabetes abolishes morphine-induced cardioprotection via multiple pathways upstream of glycogen synthase kinase-3β. Diabetes 2007, 56, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Cho, J.E.; Hwang, K.C.; Shim, Y.H.; Lee, J.H.; Kwak, Y.L. Diabetes mellitus mitigates cardioprotective effects of remifentanil preconditioning in ischemia-reperfused rat heart in association with anti-apoptotic pathways of survival. Eur. J. Pharmacol. 2010, 628, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.P.; Nicholson, C.K.; Amin, H.; Amin, S.; Calvert, J.W. Hydrogen sulfide provides cardioprotection against myocardial/ischemia reperfusion injury in the diabetic state through the activation of the risk pathway. Med. Gas Res. 2014, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Ebisuya, M.; Kondoh, K.; Nishida, E. The duration, magnitude and compartmentalization of ERK map kinase activity: Mechanisms for providing signaling specificity. J. Cell Sci. 2005, 118, 2997–3002. [Google Scholar] [CrossRef] [PubMed]
- Fjeld, C.C.; Rice, A.E.; Kim, Y.; Gee, K.R.; Denu, J.M. Mechanistic basis for catalytic activation of mitogen-activated protein kinase phosphatase 3 by extracellular signal-regulated kinase. J. Biol. Chem. 2000, 275, 6749–6757. [Google Scholar] [CrossRef] [PubMed]
- Purcell, N.H.; Wilkins, B.J.; York, A.; Saba-El-Leil, M.K.; Meloche, S.; Robbins, J.; Molkentin, J.D. Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 14074–14079. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, K.; Schmitt, J.P.; Schmitteckert, E.M.; Lohse, M.J. A new type of ERK1/2 auto-phosphorylation causes cardiac hypertrophy. Nat. Med. 2009, 15, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Honda, M.; Takabatake, T. Redox regulation of mapk pathways and cardiac hypertrophy in adult rat cardiac myocyte. J. Am. Coll. Cardiol. 2001, 37, 676–685. [Google Scholar] [CrossRef]
- Kim, D.E.; Kim, B.; Shin, H.S.; Kwon, H.J.; Park, E.S. The protective effect of hispidin against hydrogen peroxide-induced apoptosis in H9c2 cardiomyoblast cells through AKT/GSK-3β and ERK1/2 signaling pathway. Exp. Cell Res. 2014, 327, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.C.; Lacefield, J.C.; Lu, Y.; Le Tissier, S.; Peng, T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 90, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Li, R.; Ma, Y.; Wang, X.; Li, C.; Zhang, X.; Ma, R.; Ding, Z.; Liu, L. α-Lipoic acid increases tolerance of cardiomyoblasts to glucose/glucose oxidase-induced injury via ROS-dependent ERK1/2 activation. Biochim. Biophys. Acta 2012, 1823, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.K.; Lu, Q.H.; Zhang, J.N.; Wang, B.; Liu, X.J.; An, F.S.; Qin, W.D.; Chen, X.Y.; Dong, W.Q.; Zhang, C.; et al. HMGB1 mediates hyperglycaemia-induced cardiomyocyte apoptosis via ERK/ETS-1 signalling pathway. J. Cell. Mol. Med. 2014, 18, 2311–2320. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; Shi, E.; Yan, L.; Jiang, X.; Ma, H.; Ai, C. Diabetes abolishes the cardioprotection induced by sevoflurane postconditioning in the rat heart in vivo: Roles of glycogen synthase kinase-3beta and its upstream pathways. J. Surg. Res. 2012, 178, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.T.; Yang, C.X.; Li, H.; Zhang, C.J.; Wen, X.J.; Zhou, J.; Fan, Y.L.; Huang, T.; Zeng, Y.M. Cardioprotection of sevoflurane postconditioning by activating extracellular signal-regulated kinase 1/2 in isolated rat hearts. Acta Pharmacol. Sin. 2008, 29, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Y.; Tao, L.; Liu, H.; Christopher, T.A.; Lopez, B.L.; Ma, X.L. Role of ERK1/2 in the anti-apoptotic and cardioprotective effects of nitric oxide after myocardial ischemia and reperfusion. Apoptosis 2006, 11, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Bourke, L.; McCormick, J.; Taylor, V.; Pericleous, C.; Blanchet, B.; Costedoat-Chalumeau, N.; Stuckey, D.; Lythgoe, M.F.; Stephanou, A.; Ioannou, Y. Hydroxychloroquine protects against cardiac ischaemia/reperfusion injury in vivo via enhancement of ERK1/2 phosphorylation. PLoS ONE 2015, 10, e0143771. [Google Scholar] [CrossRef] [PubMed]
- Peake, B.F.; Nicholson, C.K.; Lambert, J.P.; Hood, R.L.; Amin, H.; Amin, S.; Calvert, J.W. Hydrogen sulfide preconditions the db/db diabetic mouse heart against ischemia-reperfusion injury by activating NRF2 signaling in an ERK-dependent manner. Am. J. Physiol. Heart C 2013, 304, H1215–H1224. [Google Scholar] [CrossRef] [PubMed]
- Mebratu, Y.; Tesfaigzi, Y. How erk1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Xu, S. ERK1/2 map kinases in cell survival and apoptosis. IUBMB Life 2006, 58, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Yu, L.; Wang, M.; Xu, S.; Xia, Q.; Fu, G. O-glcnacylation involvement in high glucose-induced cardiac hypertrophy via ERK1/2 and cyclin D2. Amino Acids 2013, 45, 339–349. [Google Scholar] [PubMed]
- Ko, S.Y.; Lin, I.H.; Shieh, T.M.; Ko, H.A.; Chen, H.I.; Chi, T.C.; Chang, S.S.; Hsu, Y.C. Cell hypertrophy and MEK/ERK phosphorylation are regulated by glyceraldehyde-derived ages in cardiomyocyte H9c2 cells. Cell Biochem. Biophys. 2013, 66, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.C.; Chang, C.K.; Ou, H.Y.; Cheng, K.C.; Cheng, J.T. Decrease of peroxisome proliferator-activated receptor delta expression in cardiomyopathy of streptozotocin-induced diabetic rats. Cardiovasc. Res. 2008, 80, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Li, G.N.; Xie, J.; Li, R.; Chen, Q.H.; Chen, J.Z.; Wei, Z.H.; Kang, L.N.; Xu, B. Resveratrol ameliorates myocardial fibrosis by inhibiting ROS/ERK/TGF-β/periostin pathway in STZ-induced diabetic mice. BMC Cardiovasc. Disord. 2016, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Zhang, W.; Lin, H.; Jiang, H.; Dai, H.; Zhang, Y. High glucose promotes the production of collagen types I and III by cardiac fibroblasts through a pathway dependent on extracellular-signal-regulated kinase 1/2. Mol. Cell. Biochem. 2007, 301, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Song, S.E.; Kim, Y.W.; Kim, J.Y.; Lee, D.H.; Kim, J.R.; Park, S.Y. IGFBP5 mediates high glucose-induced cardiac fibroblast activation. J. Mol. Endocrinol. 2013, 50, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Q.; Roberts, D.; Kharitonenkov, A.; Zhang, B.; Hanson, S.M.; Li, Y.C.; Zhang, L.Q.; Wu, Y.H. Endocrine protection of ischemic myocardium by FGF21 from the liver and adipose tissue. Sci. Rep. 2013, 3, 2767. [Google Scholar] [CrossRef] [PubMed]
- Cong, W.T.; Ling, J.; Tian, H.S.; Ling, R.; Wang, Y.; Huang, B.B.; Zhao, T.; Duan, Y.M.; Jin, L.T.; Li, X.K. Proteomic study on the protective mechanism of fibroblast growth factor 21 to ischemia-reperfusion injury. Can. J. Physiol. Pharmacol. 2013, 91, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Planavila, A.; Redondo, I.; Hondares, E.; Vinciguerra, M.; Munts, C.; Iglesias, R.; Gabrielli, L.A.; Sitges, M.; Giralt, M.; van Bilsen, M.; et al. Fibroblast growth factor 21 protects against cardiac hypertrophy in mice. Nat. Commun. 2013, 4, 2019. [Google Scholar] [CrossRef] [PubMed]
- Rose, B.A.; Force, T.; Wang, Y. Mitogen-activated protein kinase signaling in the heart: Angels vs. demons in a heart-breaking tale. Physiol. Rev. 2010, 90, 1507–1546. [Google Scholar] [CrossRef] [PubMed]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, S.; Nishida, E. MAPK signaling: ERK5 vs. ERK1/2. EMBO Rep. 2006, 7, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Ohori, M.; Kinoshita, T.; Okubo, M.; Sato, K.; Yamazaki, A.; Arakawa, H.; Nishimura, S.; Inamura, N.; Nakajima, H.; Neya, M.; et al. Identification of a selective ERK inhibitor and structural determination of the inhibitor-ERK2 complex. Biochem. Biophys. Res. Commun. 2005, 336, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Taglieri, D.M.; Monasky, M.M.; Knezevic, I.; Sheehan, K.A.; Lei, M.; Wang, X.; Chernoff, J.; Wolska, B.M.; Ke, Y.; Solaro, R.J. Ablation of p21-activated kinase-1 in mice promotes isoproterenol-induced cardiac hypertrophy in association with activation of ERK1/2 and inhibition of protein phosphatase 2A. J. Mol. Cell. Cardiol. 2011, 51, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Chaikuad, A.; Tacconi, E.M.; Zimmer, J.; Liang, Y.; Gray, N.S. A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat. Chem. Biol. 2014, 10, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Sun, H.; Xu, J.; Xiao, F.; Wang, H.; Yang, Y.; Ren, H.; Wu, C.T.; Gao, C.; Wang, L. Kinesin spindle protein inhibitor sb743921 induces mitotic arrest and apoptosis and overcomes imatinib resistance of chronic myeloid leukemia cells. Leuk. Lymphoma 2015, 56, 1813–1820. [Google Scholar]
- Ji, S.; Qin, Y.; Shi, S.; Liu, X.; Hu, H.; Zhou, H.; Gao, J.; Zhang, B.; Xu, W.; Liu, J.; et al. ERK kinase phosphorylates and destabilizes the tumor suppressor FBW7 in pancreatic cancer. Cell Res. 2015, 25, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Krepler, C.; Xiao, M.; Sproesser, K.; Brafford, P.A.; Shannan, B.; Beqiri, M.; Liu, Q.; Xu, W.; Garman, B.; Nathanson, K.L.; et al. Personalized preclinical trials in BRAF inhibitor-resistant patient-derived XENO graft models identify second-line combination therapies. Clin. Cancer Res. 2016, 22, 1592–1602. [Google Scholar] [CrossRef] [PubMed]
- Mateu, A.; de Dios, I.; Manso, M.A.; Ramudo, L. Unsaturated but not saturated fatty acids induce transcriptional regulation of CCL2 in pancreatic ACINI. A potential role in acute pancreatitis. Biochim. Biophys. Acta 2015, 1852, 2671–2677. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Kang, L.; Li, C.; Wang, X.; Sun, C.; Li, Q.; Liu, R.; Wang, J. Resveratrol ameliorates diabetes-induced cardiac dysfunction through AT1R-ERK/p38 MAPK signaling pathway. Cardiovasc. Toxicol. 2016, 16, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Saba-El-Leil, M.K.; Vella, F.D.; Vernay, B.; Voisin, L.; Chen, L.; Labrecque, N.; Ang, S.L.; Meloche, S. An essential function of the mitogen-activated protein kinase ERK2 in mouse trophoblast development. EMBO Rep. 2003, 4, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Ulm, S.; Liu, W.; Zi, M.; Tsui, H.; Chowdhury, S.K.; Endo, S.; Satoh, Y.; Prehar, S.; Wang, R.; Cartwright, E.J.; et al. Targeted deletion of ERK2 in cardiomyocytes attenuates hypertrophic response but provokes pathological stress induced cardiac dysfunction. J. Mol. Cell. Cardiol. 2014, 72, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Lips, D.J.; Bueno, O.F.; Wilkins, B.J.; Purcell, N.H.; Kaiser, R.A.; Lorenz, J.N.; Voisin, L.; Saba-El-Leil, M.K.; Meloche, S.; Pouyssegur, J.; et al. MEK1-ERK2 signaling pathway protects myocardium from ischemic injury in vivo. Circulation 2004, 109, 1938–1941. [Google Scholar] [CrossRef] [PubMed]
- Kehat, I.; Davis, J.; Tiburcy, M.; Accornero, F.; Saba-El-Leil, M.K.; Maillet, M.; York, A.J.; Lorenz, J.N.; Zimmermann, W.H.; Meloche, S.; et al. Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ. Res. 2011, 108, 176–183. [Google Scholar] [PubMed]
- Lorenz, K.; Schmitt, J.P.; Vidal, M.; Lohse, M.J. Cardiac hypertrophy: Targeting RAF/MEK/ERK1/2-signaling. Int. J. Biochem. Cell Biol. 2009, 41, 2351–2355. [Google Scholar] [CrossRef] [PubMed]
- Heineke, J.; Molkentin, J.D. Regulation of cardiac hypertrophy by intracellular signaling pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, C.; Deiss, K.; Herrmann, S.; Vidal, M.; Oezkur, M.; Gorski, A.; Weidemann, F.; Lohse, M.J.; Lorenz, K. Interference with ERK(THR188) phosphorylation impairs pathological but not physiological cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 7440–7445. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M.; Wieland, T.; Lohse, M.J.; Lorenz, K. β-Adrenergic receptor stimulation causes cardiac hypertrophy via a Gβγ/ERK-dependent pathway. Cardiovasc. Res. 2012, 96, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Bassett, S.A.; Barnett, M.P. The role of dietary histone deacetylases (HDACs) inhibitors in health and disease. Nutrients 2014, 6, 4273–4301. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.K.; Kern, C.B.; Kimbrough, D.; Addy, B.; Kasiganesan, H.; Rivers, W.T.; Patel, R.K.; Chou, J.C.; Spinale, F.G.; Mukherjee, R.; et al. Inhibition of class I histone deacetylase activity represses matrix metalloproteinase-2 and -9 expression and preserves lv function postmyocardial infarction. Am. J. Physiol. Heart C 2015, 308, H1391–H1401. [Google Scholar] [CrossRef] [PubMed]
- Aune, S.E.; Herr, D.J.; Mani, S.K.; Menick, D.R. Selective inhibition of class I but not class IIb histone deacetylases exerts cardiac protection from ischemia reperfusion. J. Mol. Cell. Cardiol. 2014, 72, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, J.P.; Sriramula, S.; Pariaut, R.; Guggilam, A.; Mariappan, N.; Elks, C.M.; Francis, J. Hdac inhibition attenuates inflammatory, hypertrophic, and hypertensive responses in spontaneously hypertensive rats. Hypertension 2010, 56, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Du, J.; Zhao, Y.T.; Zhang, L.; Lv, G.; Zhuang, S.; Qin, G.; Zhao, T.C. Histone deacetylase (HDAC) inhibition improves myocardial function and prevents cardiac remodeling in diabetic mice. Cardiovasc. Diabetol. 2015, 14, 99. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Kao, Y.H. HDAC inhibition modulates cardiac PPARS and fatty acid metabolism in diabetic cardiomyopathy. PPAR Res. 2016, 2016, 5938740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cheng, Y.; Gu, J.; Wang, S.; Zhou, S.; Wang, Y.; Tan, Y.; Feng, W.; Fu, Y.; Mellen, N.; et al. Fenofibrate increases cardiac autophagy via FGF21/SIRT1 and prevents fibrosis and inflammation in the hearts of type 1 diabetic mice. Clin. Sci. 2016, 130, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, G.; Adris, P.; Bhargava, N.; Chen, H.; Raghow, R. Pan-histone deacetylase inhibitors regulate signaling pathways involved in proliferative and pro-inflammatory mechanisms in H9c2 cells. BMC Genom. 2012, 13, 709. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.S.; Harrison, B.C.; Jeong, M.Y.; Reid, B.G.; Wempe, M.F.; Wagner, F.F.; Holson, E.B.; McKinsey, T.A. Signal-dependent repression of DUSP5 by class I HDACs controls nuclear ERK activity and cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 9806–9811. [Google Scholar] [CrossRef] [PubMed]
- Barter, M.J.; Pybus, L.; Litherland, G.J.; Rowan, A.D.; Clark, I.M.; Edwards, D.R.; Cawston, T.E.; Young, D.A. HDAC-mediated control of ERK- and PI3K-dependent TGF-β-induced extracellular matrix-regulating genes. Matrix Biol. 2010, 29, 602–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, S.M.; Golden-Mason, L.; Ferguson, B.S.; Schuetze, K.B.; Cavasin, M.A.; Demos-Davies, K.; Yeager, M.E.; Stenmark, K.R.; McKinsey, T.A. Class I HDACs regulate angiotensin II-dependent cardiac fibrosis via fibroblasts and circulating fibrocytes. J. Mol. Cell. Cardiol. 2014, 67, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Park, J.T.; Kato, M.; Yuan, H.; Castro, N.; Lanting, L.; Wang, M.; Natarajan, R. Fog2 protein down-regulation by transforming growth factor-β1-induced microRNA-200b/c leads to AKT kinase activation and glomerular mesangial hypertrophy related to diabetic nephropathy. J. Biol. Chem. 2013, 288, 22469–22480. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Chakrabarti, S. MiR-320 regulates glucose-induced gene expression in diabetes. ISRN Endocrinol. 2012, 2012, 549875. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, M. The role of microRNA-133 in cardiac hypertrophy uncovered. Circ. Res. 2010, 106, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.X.; Lin, Q.X.; Deng, C.Y.; Zhu, J.N.; Mai, L.P.; Liu, J.L.; Fu, Y.H.; Liu, X.Y.; Li, Y.X.; Zhang, Y.Y.; et al. MiR-1/miR-206 regulate hsp60 expression contributing to glucose-mediated apoptosis in cardiomyocytes. FEBS Lett. 2010, 584, 3592–3600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, J.; Qu, D.; Wang, L.; Luo, J.Y.; Lau, C.W.; Liu, P.; Gao, Z.; Tipoe, G.L.; Lee, H.K.; et al. Inhibition of miR-200c restores endothelial function in diabetic mice through suppression of COX-2. Diabetes 2016, 65, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Puthanveetil, P.; Feng, B.; Matkovich, S.J.; Dorn, G.W., 2nd; Chakrabarti, S. Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J. Cell. Mol. Med. 2014, 18, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Y.; Song, Y.H.; Geng, Y.J.; Lin, Q.X.; Shan, Z.X.; Lin, S.G.; Li, Y. Glucose induces apoptosis of cardiomyocytes via microRNA-1 and IGF-1. Biochem. Biophys. Res. Commun. 2008, 376, 548–552. [Google Scholar] [CrossRef] [PubMed]
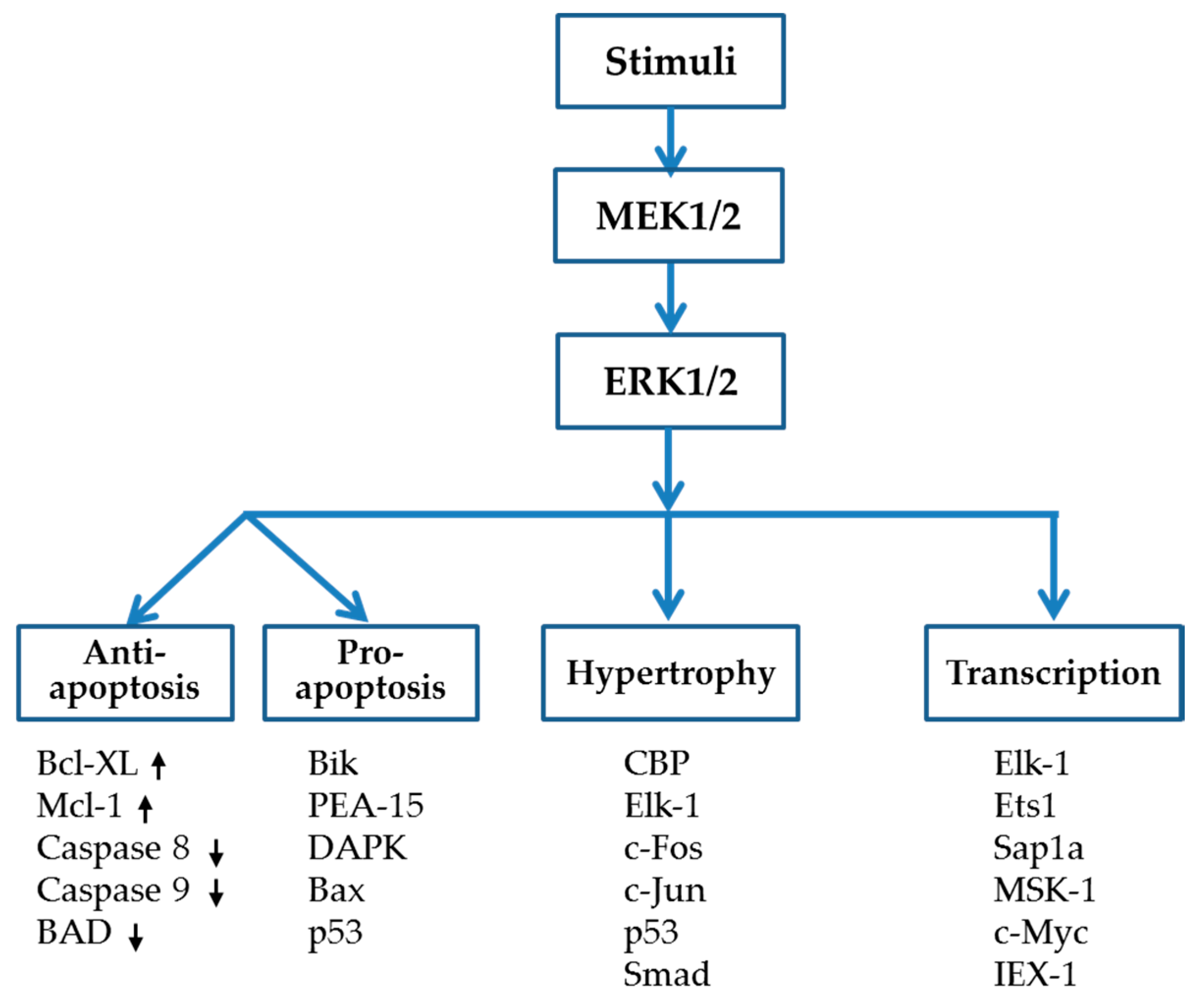

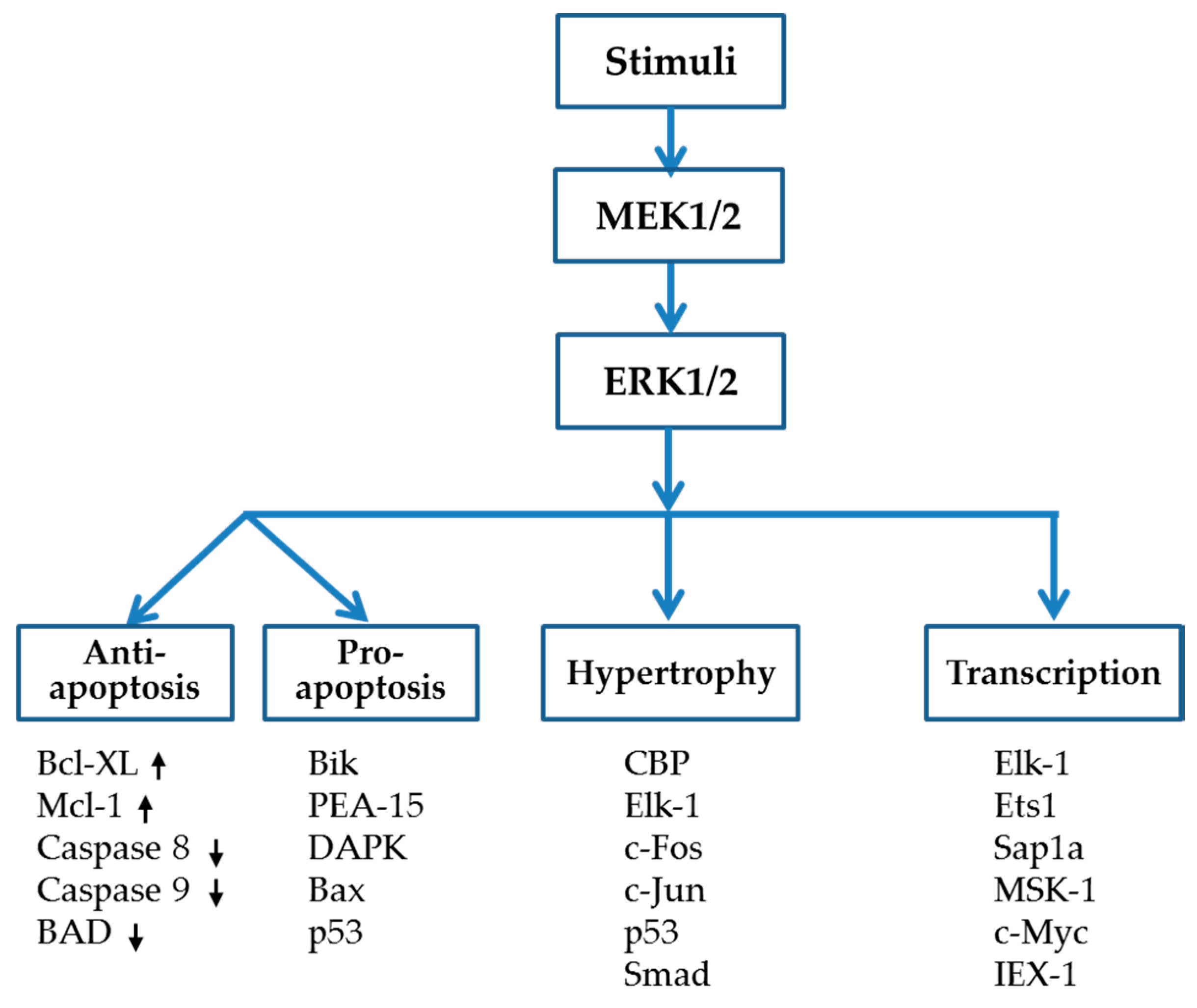
{kind=link}
{kind=link}
| Inhibitors | Isoforms | Model | Response | References |
|---|---|---|---|---|
| U0126 | MEK/ERK | Cardiomyocyte treated with HG and animal model of myocardial I/R injury | Alleviated the HG and I/R-induced cardiomyocyte injury | [58,81] |
| PD98059 | MEK/ERK | H9c2 cell treated with AGEs and diabetic mice induced by STZ | Prevented cardiomyocyte hypertrophy and cardiac remodeling and apoptosis of diabetic mice | [23,63] |
| FR 180204 | ERK1/2 | WT and Pak-1-KO mice treated with ISO | Inhibited the cardiac hypertrophy | [75] |
| SCH772984 | ERK1/2 | pancreatic cancer clinical samples | Anti-cancer | [78] |
| VX-11e | ERK1/2 | Patient derived xenograft models | Anti-cancer | [79] |
| GDC-0994 | ERK1/2 | Rats treated with SFAs or UFAs | Reduced inflammatory response | [80] |
| MicroRNA | Location | Model | Response | References |
|---|---|---|---|---|
| miR-200c | Upstream | db/db T2DM | Increase aorta endothelial dysfunction | [101] |
| miR-320 | Upstream | High glucose treated HUVECs | Reduced expression of ET-1, VEGF, and FN | [102] |
| miR-133a | Upstream | STZ-induced diabetes | Prevent cardiac fibrosis | [103] |
| miR-1 and 6 | Up stream | STZ-induced diabetes and high glucose treated NRVCs | Increase cardiomyocyte apoptosis | [104] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Z.; Sun, J.; Tong, Q.; Lin, Q.; Qian, L.; Park, Y.; Zheng, Y. The Role of ERK1/2 in the Development of Diabetic Cardiomyopathy. Int. J. Mol. Sci. 2016, 17, 2001. https://doi.org/10.3390/ijms17122001
Xu Z, Sun J, Tong Q, Lin Q, Qian L, Park Y, Zheng Y. The Role of ERK1/2 in the Development of Diabetic Cardiomyopathy. International Journal of Molecular Sciences. 2016; 17(12):2001. https://doi.org/10.3390/ijms17122001
Chicago/Turabian StyleXu, Zheng, Jian Sun, Qian Tong, Qian Lin, Lingbo Qian, Yongsoo Park, and Yang Zheng. 2016. "The Role of ERK1/2 in the Development of Diabetic Cardiomyopathy" International Journal of Molecular Sciences 17, no. 12: 2001. https://doi.org/10.3390/ijms17122001