
Influence of Secondary-Structure Folding on the Mutually Exclusive Folding Process of GL5/I27 Protein: Evidence from Molecular Dynamics Simulations

Abstract
:
1. Introduction
2. Results
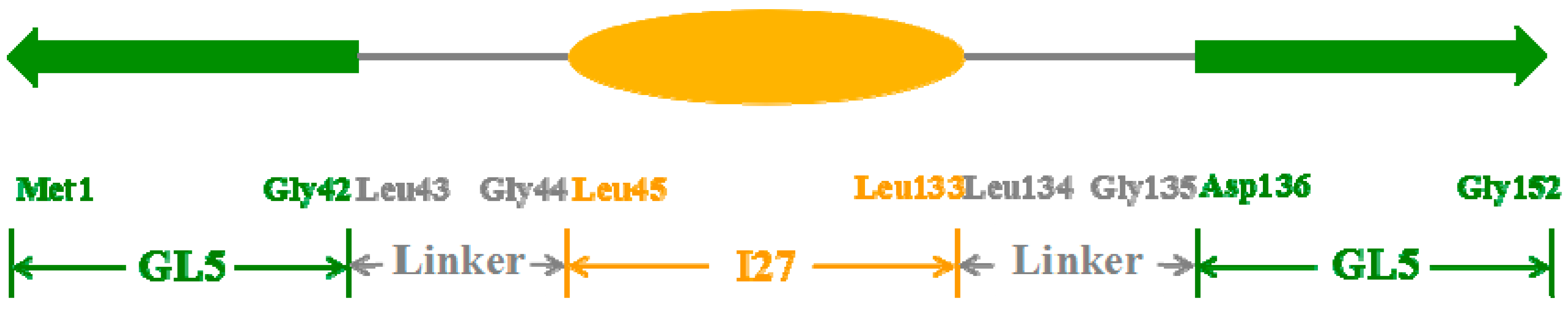
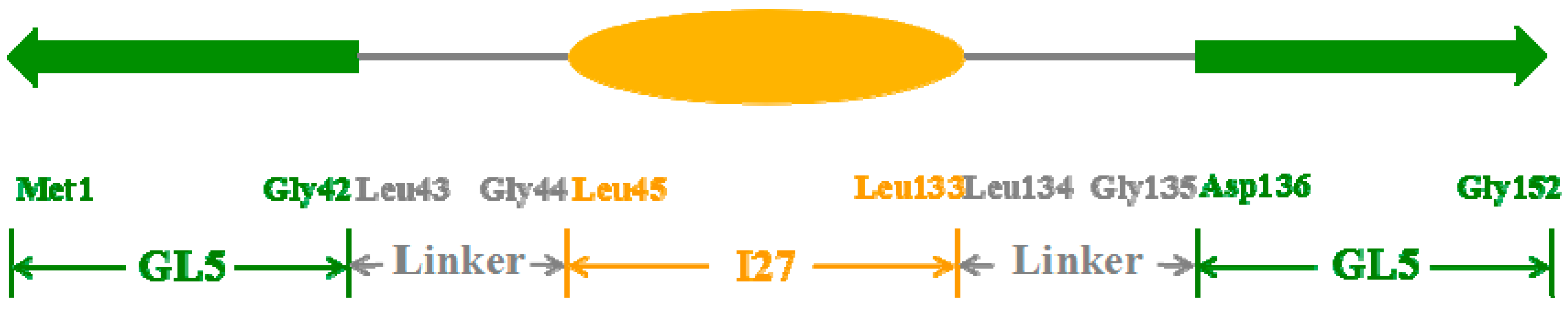
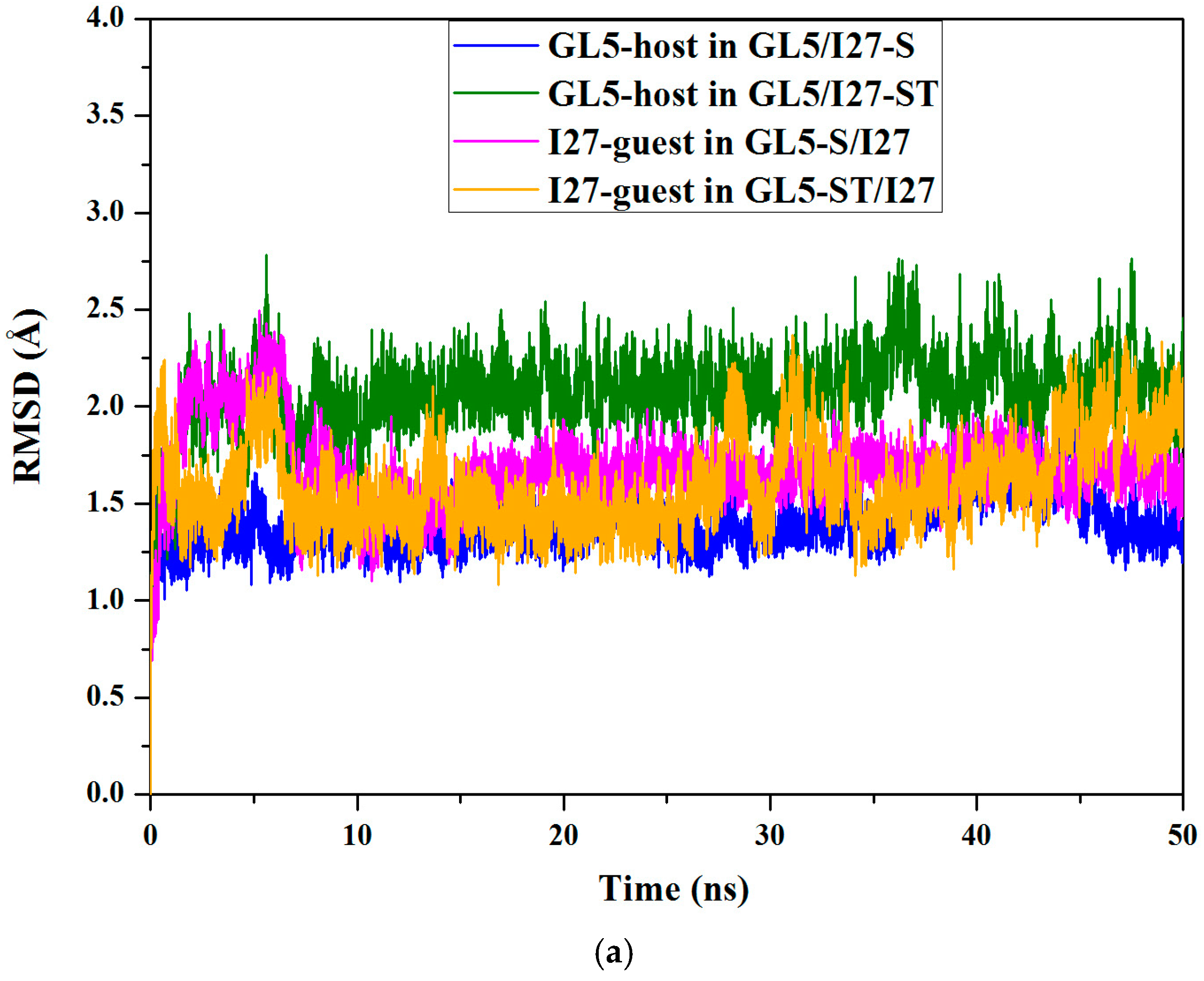
2.1. Initial Structure Construction
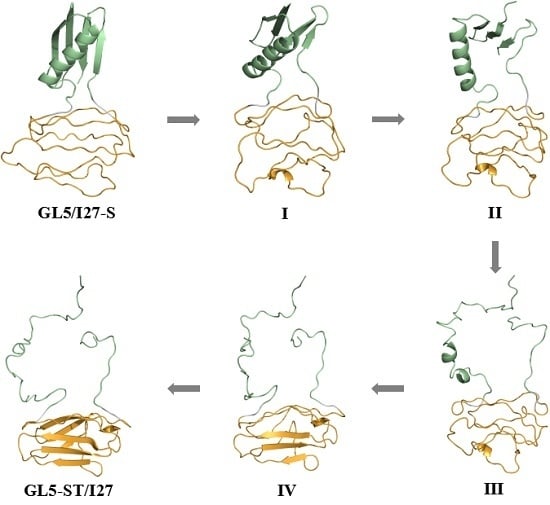
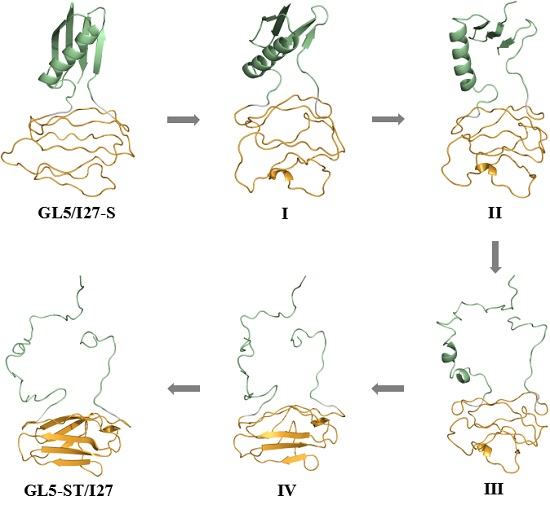
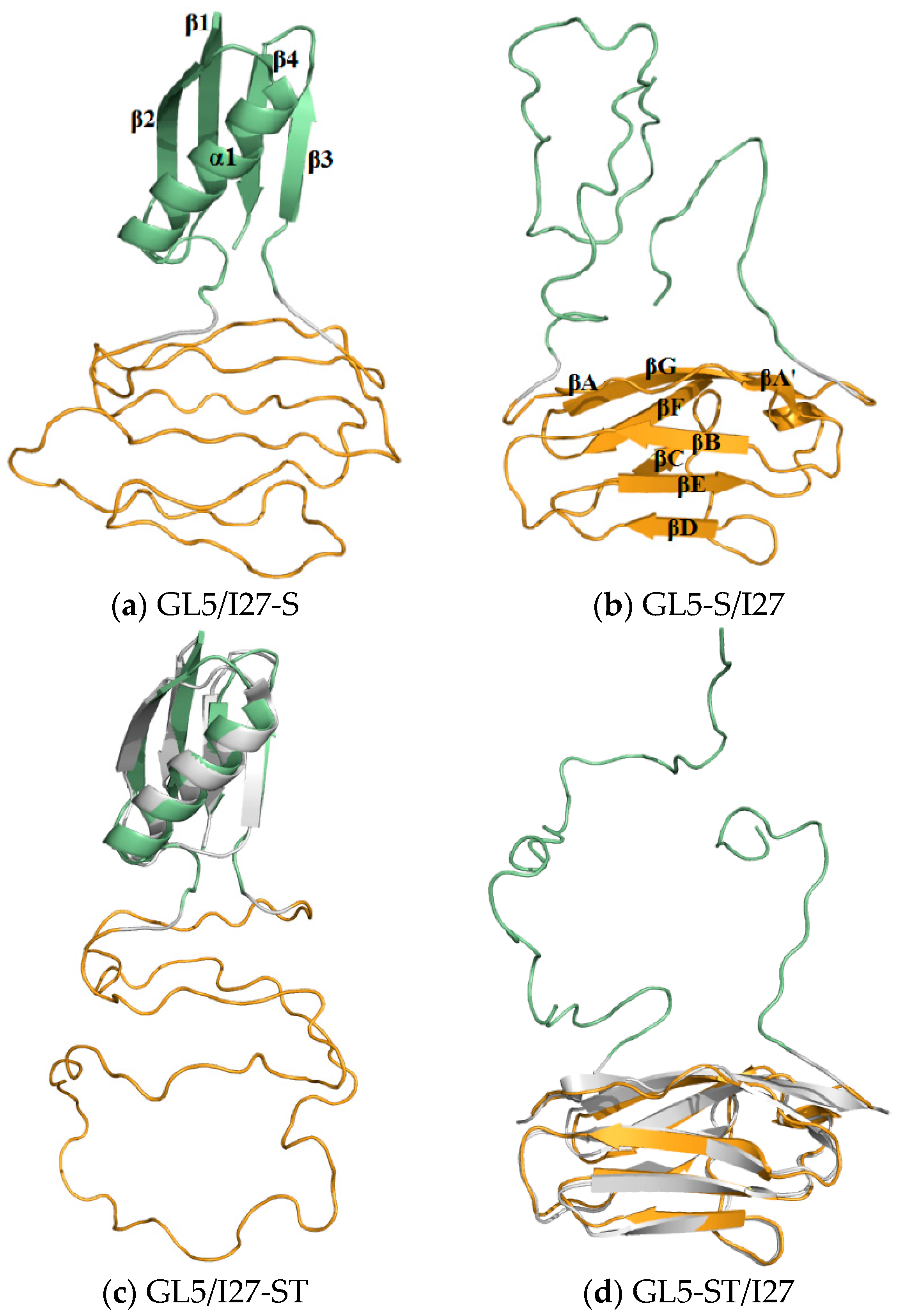
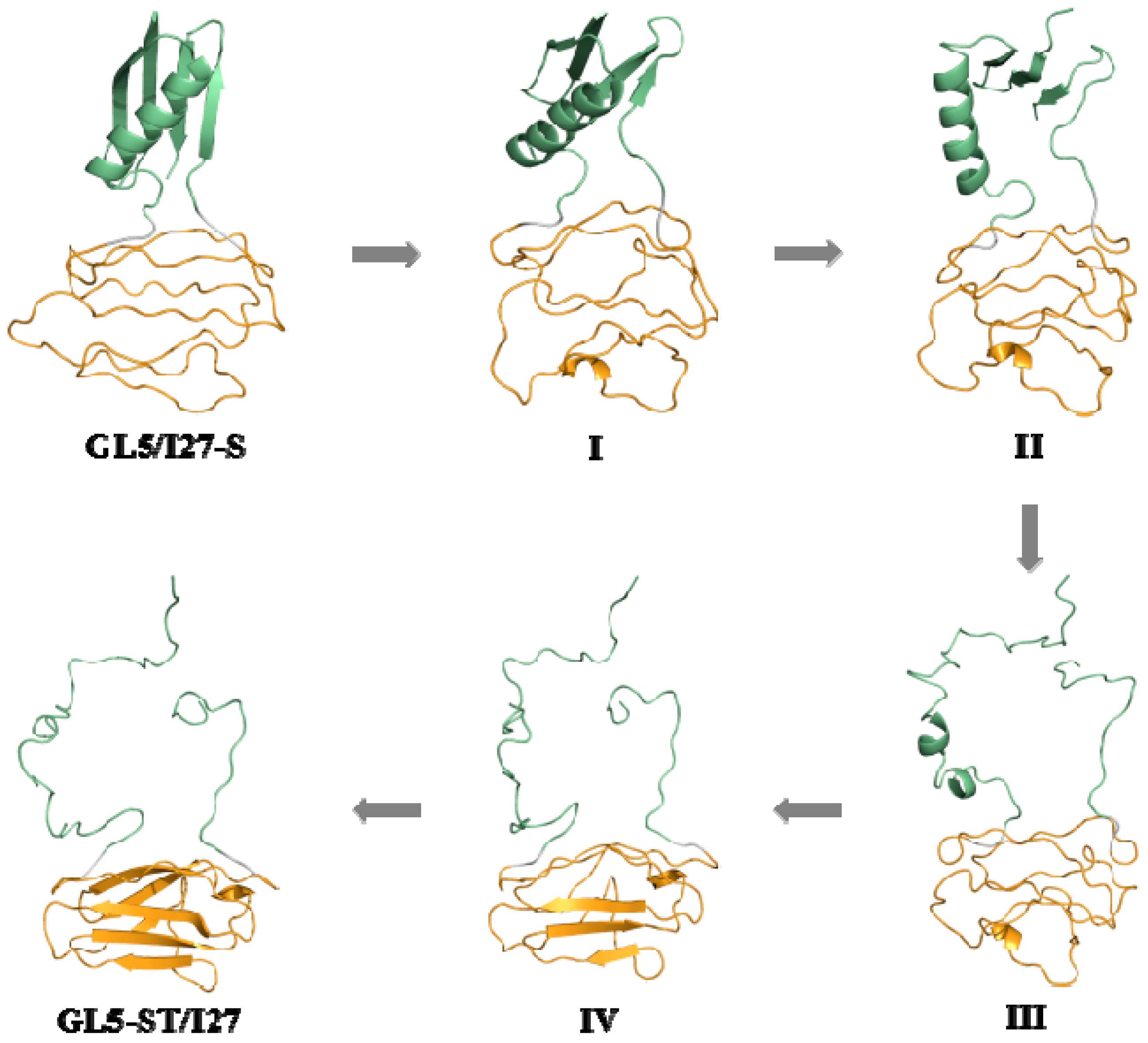
2.2. Structures of the Mutually Exclusive Folding Models of GL5i/I27i
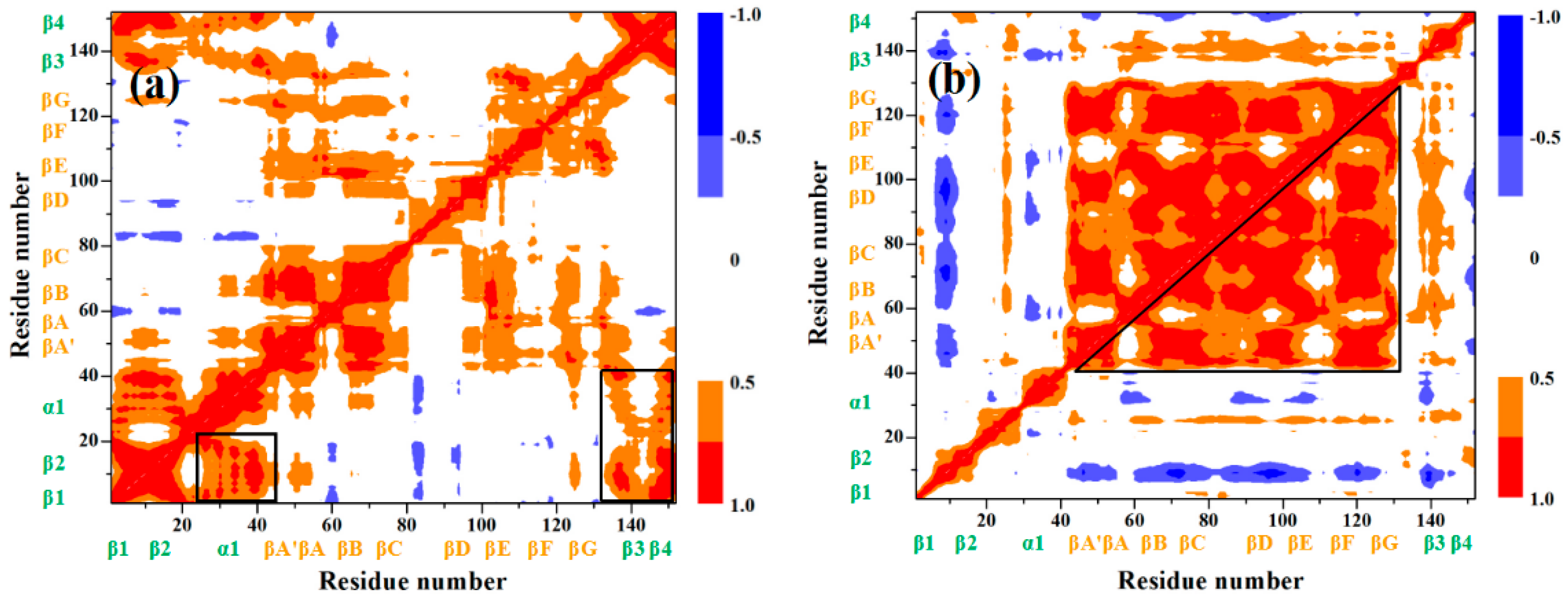
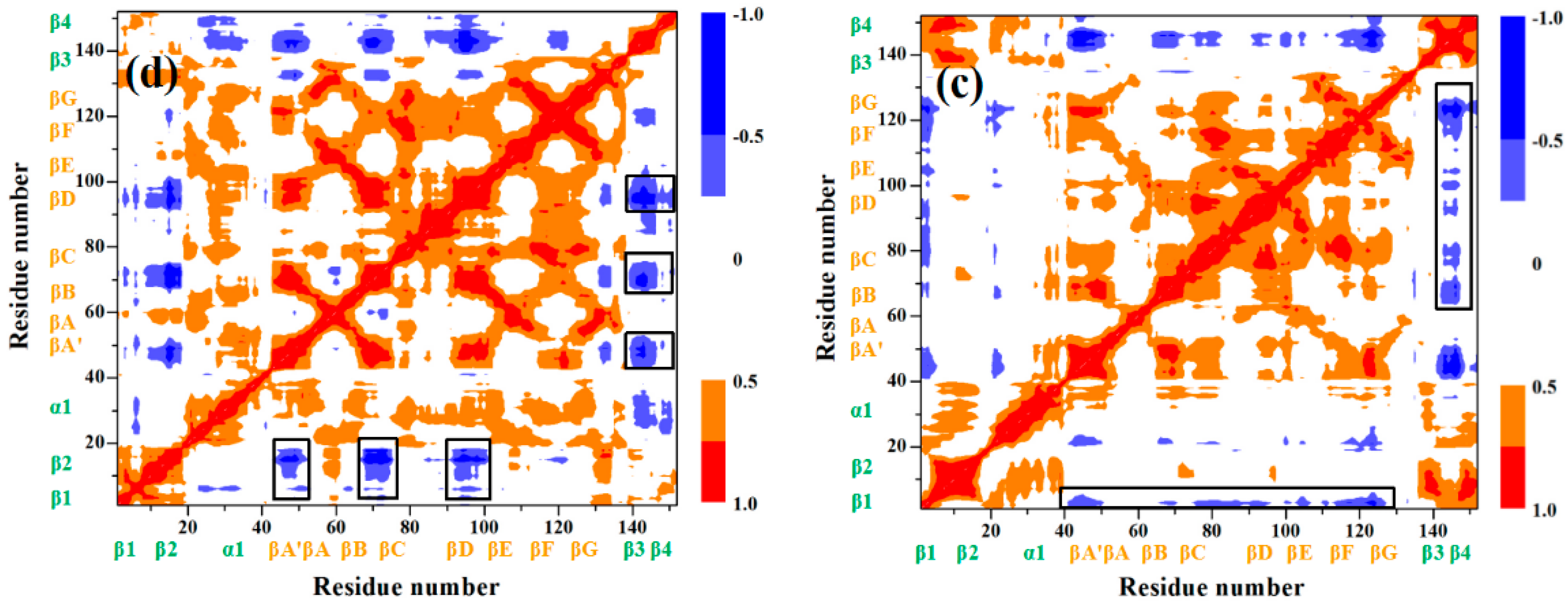
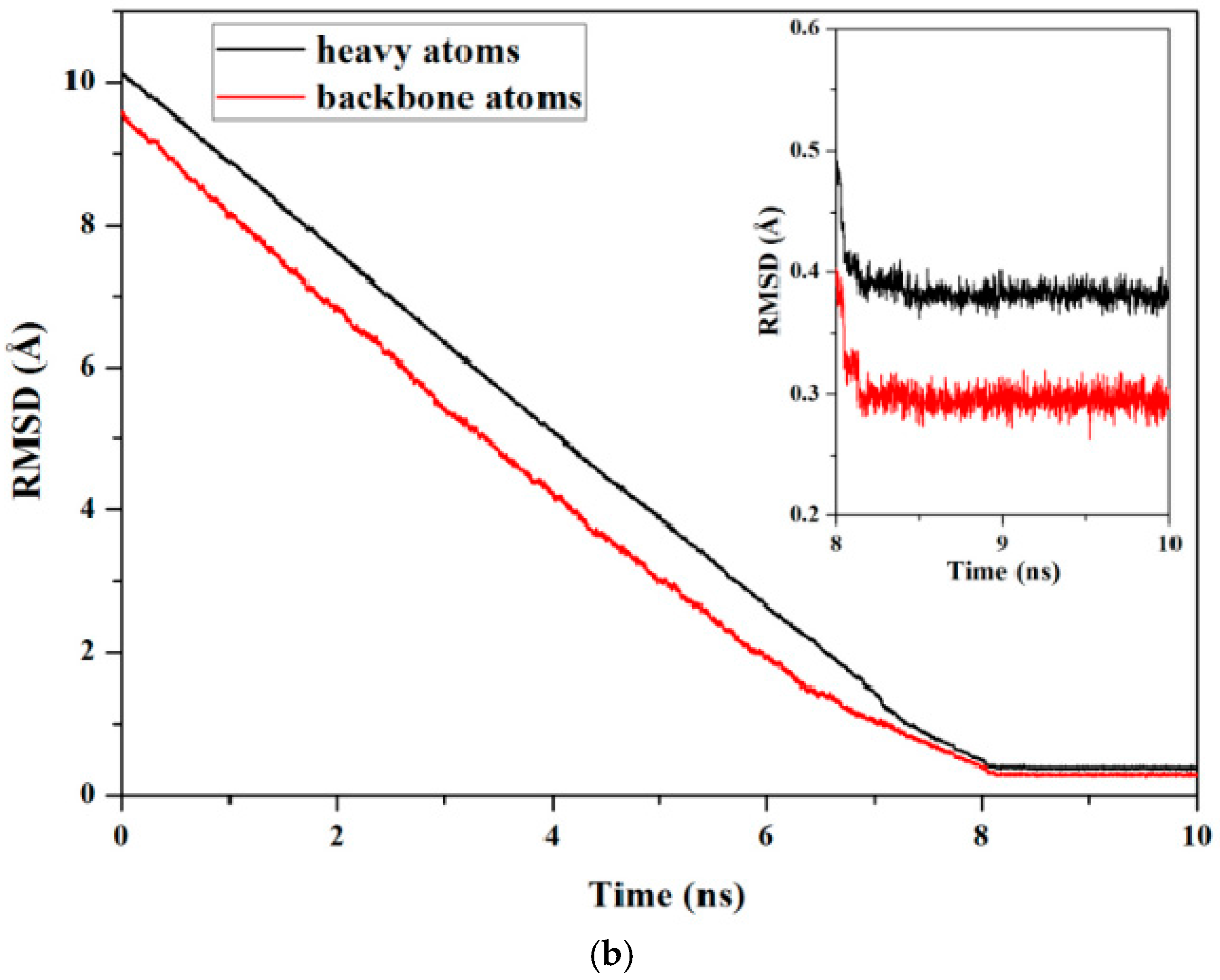
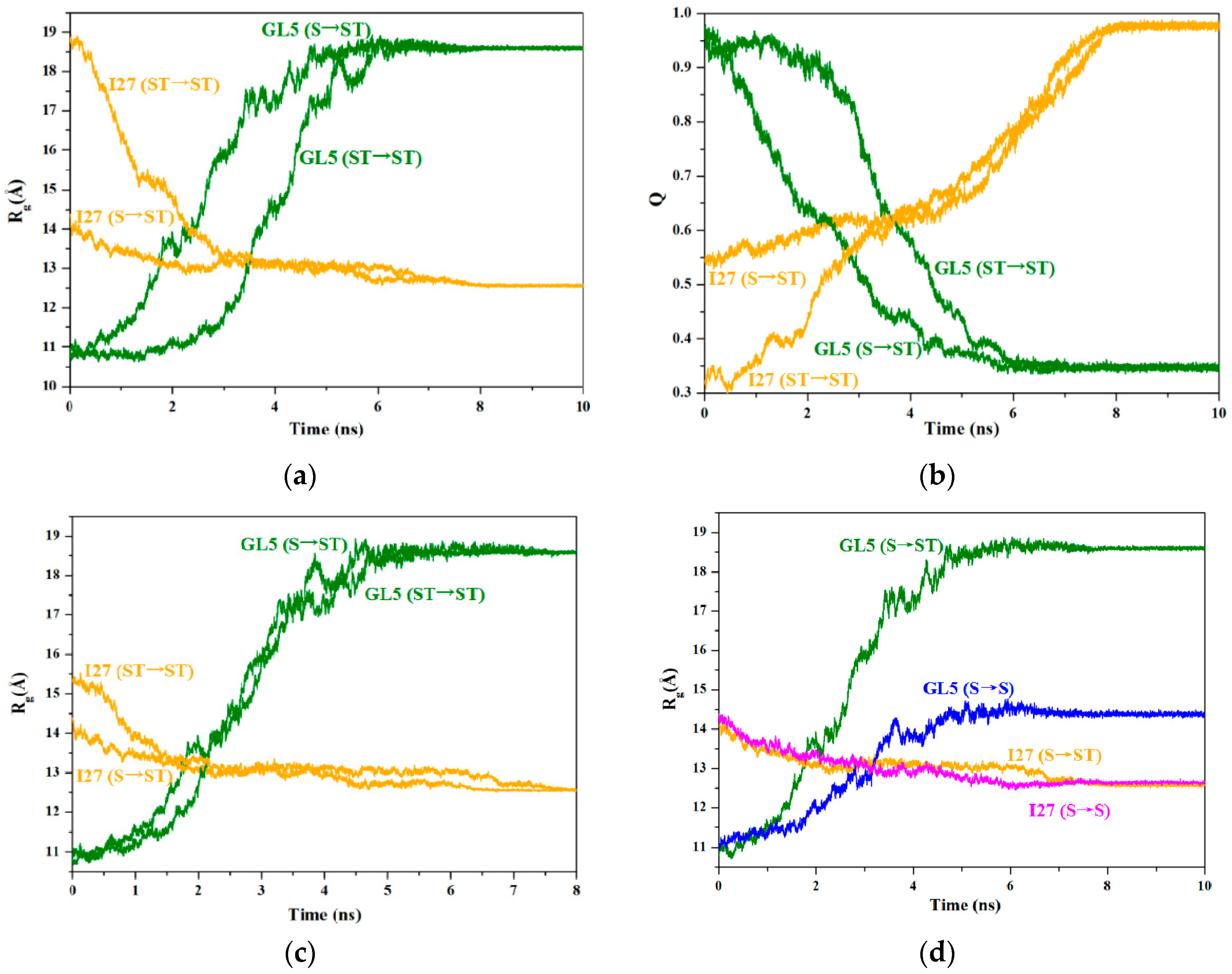
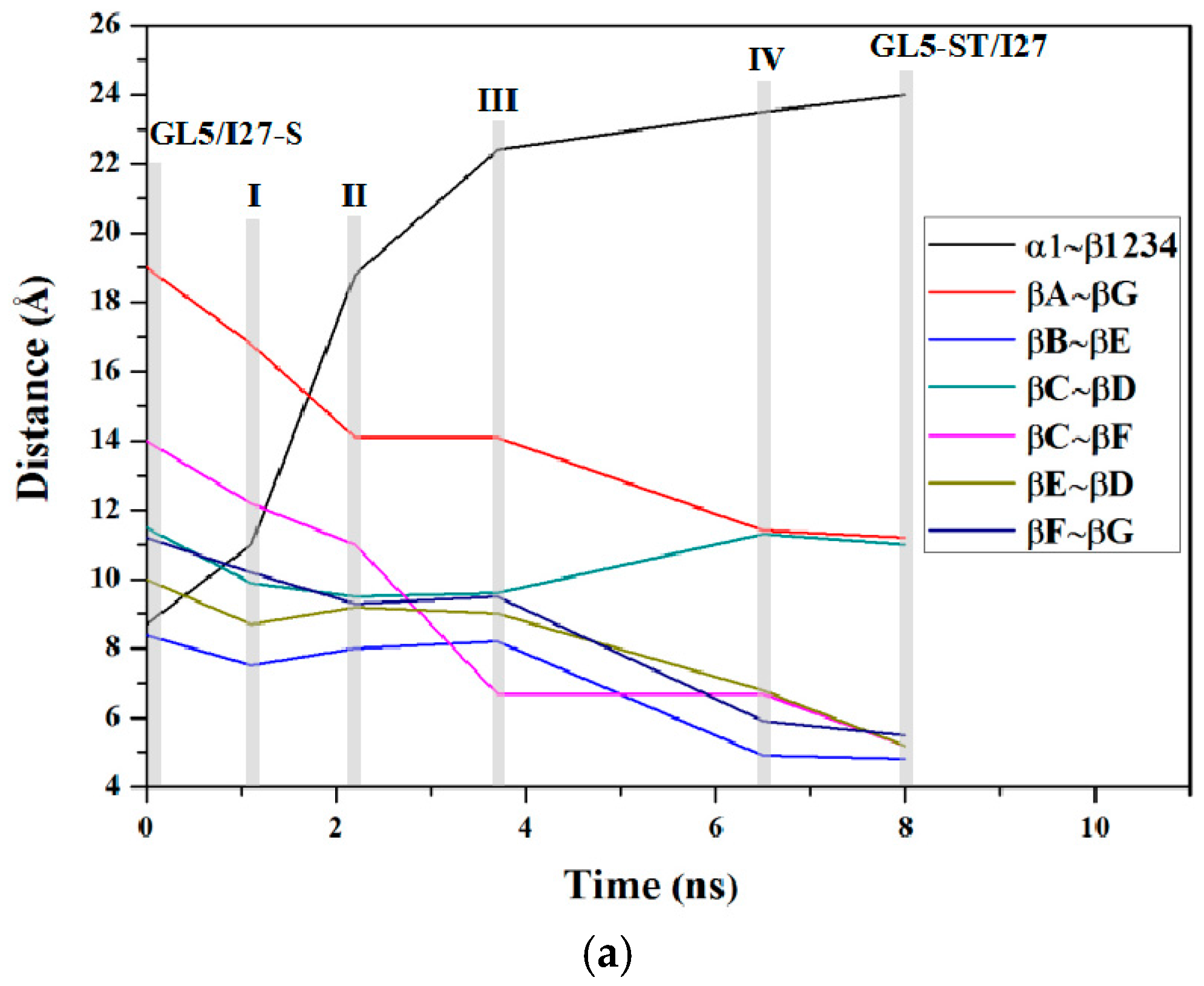
2.3. Characteristics of the Induced Full Extending Processes of the Mutually Exclusive Folding Models GL5i/I27i
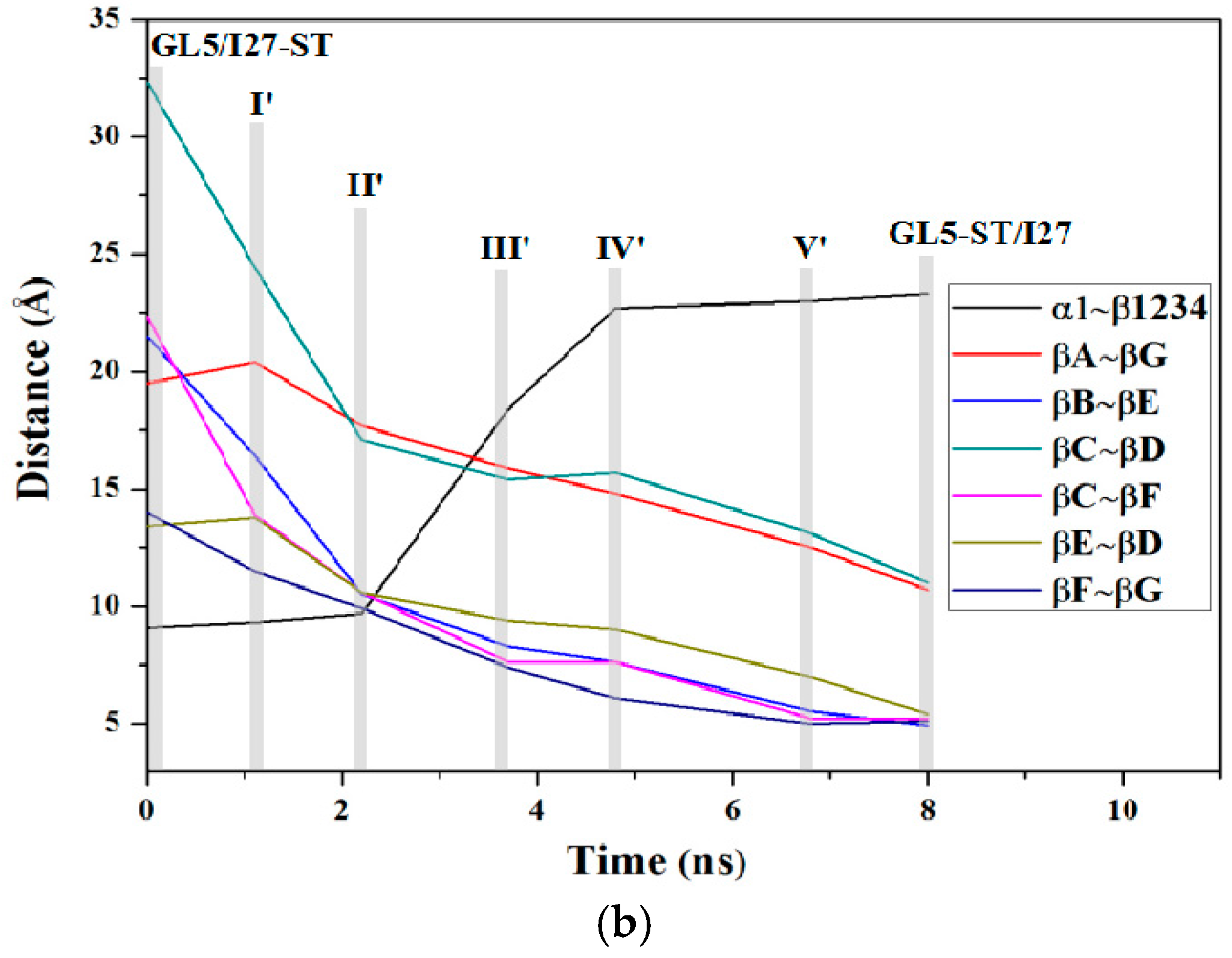
2.4. Characteristics of the Induced Secondary-Structure Unfolding Processes of the Mutually Exclusive Folding Models GL5i/I27i
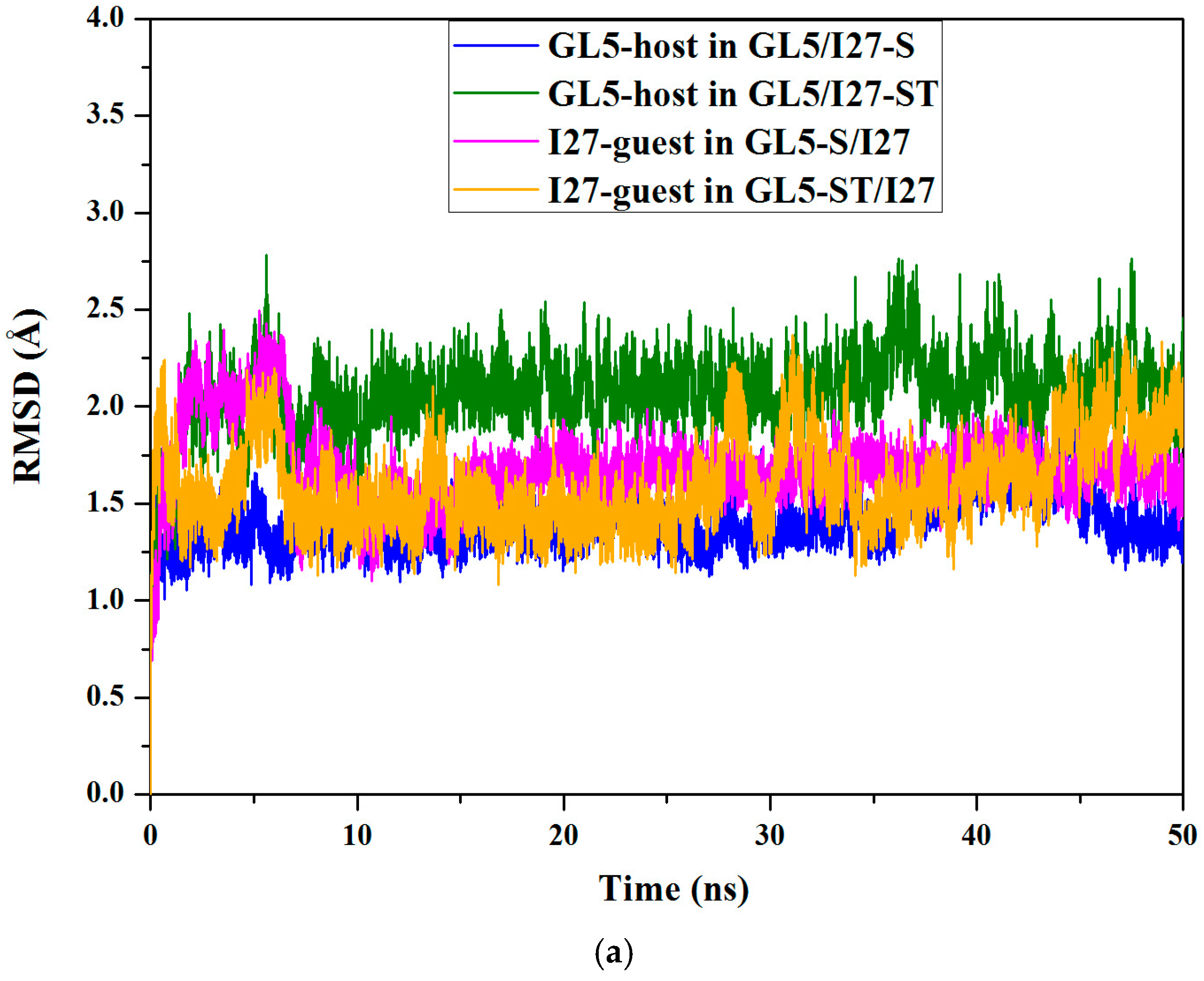
2.5. Comparison between the Induced Full Extending and Secondary-Structure Unfolding Processes
3. Discussion
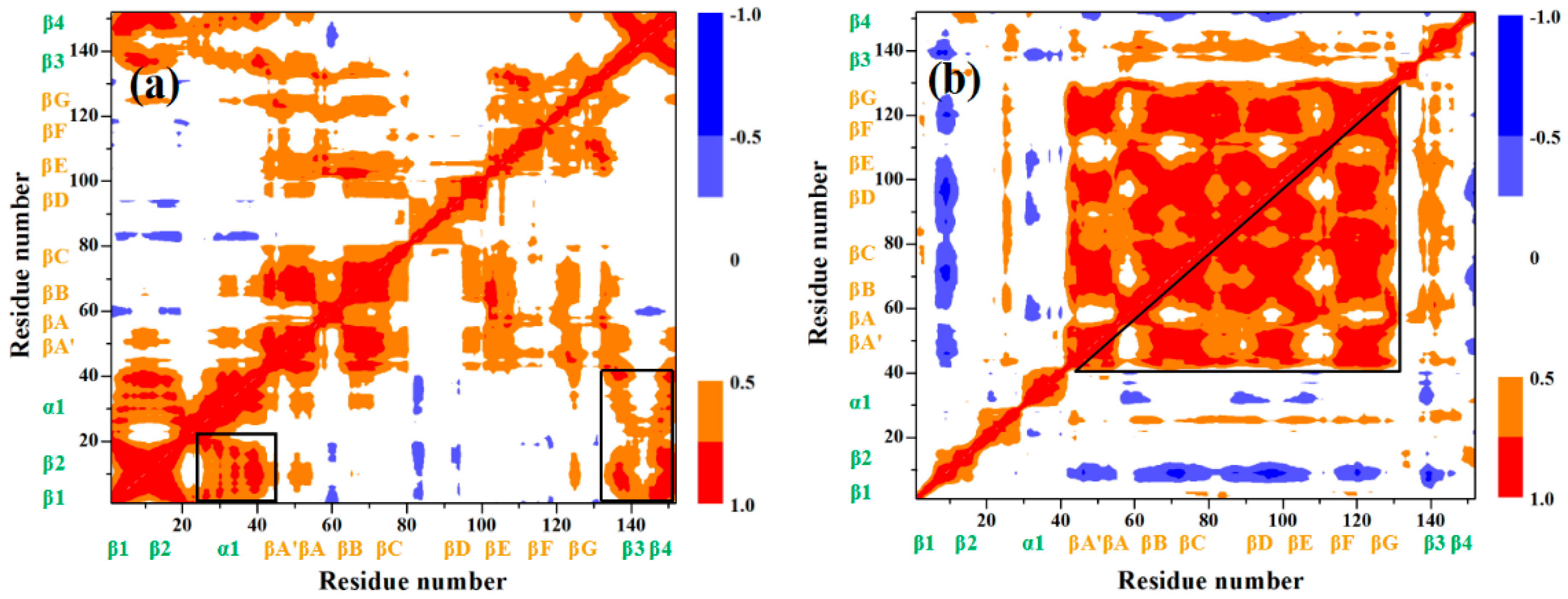
3.1. Analysis of the Structural Changes for the Induced Full Extending Processes
3.2. Mutually Exclusive Correlation Network in the Induced Full Extending Process
4. Methodology
4.1. Conventional Molecular Dynamics Simulation
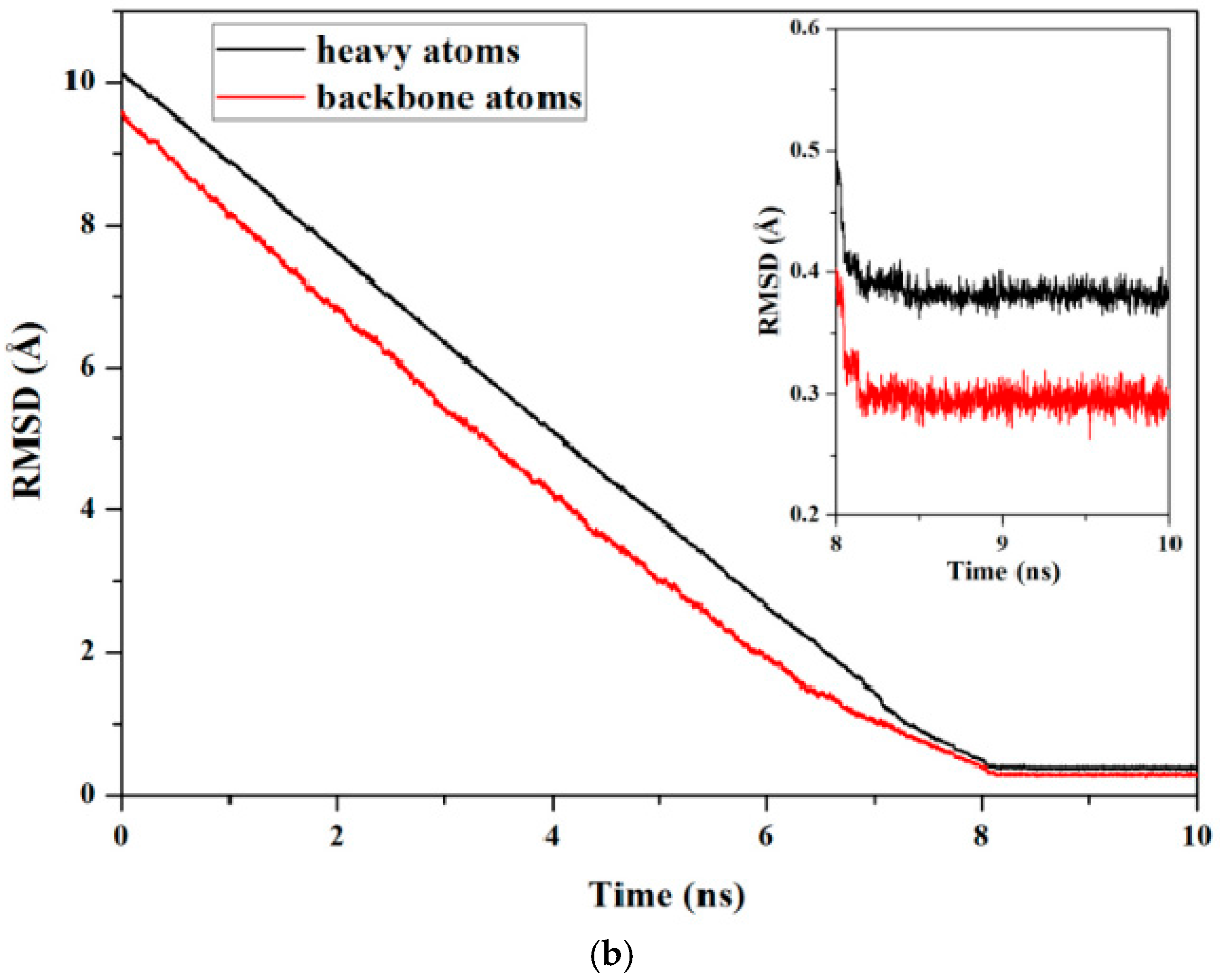
4.2. Targeted Molecular Dynamics Simulation
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fenton, A.W. Allostery: An illustrated definition for the “second secret of life”. Trends Biochem. Sci. 2008, 33, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Hilbert, M.; Kebbel, F.; Gubaev, A.; Klostermeier, D. eIF4G stimulates the activity of the DEAD box protein eIF4A by a conformational guidance mechanism. Nucleic Acids Res. 2011, 39, 2260–2270. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Tang, Z.; Xia, G.; Wassmann, K.; Matsumoto, T.; Rizo, J.; Yu, H. The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat. Struct. Mol. Biol. 2004, 11, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Björkman, A.J.; Mowbray, S.L. Multiple open forms of ribose-binding protein trace the path of its conformational change. J. Mol. Biol. 1998, 279, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Ravindranathan, K.P.; Gallicchio, E.; Levy, R.M. Conformational equilibria and free energy profiles for the allosteric transition of the ribose-binding protein. J. Mol. Biol. 2005, 353, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.M.; Loh, S.N. Converting a protein into a switch for biosensing and functional regulation. Protein Sci. 2011, 20, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Aroul-Selvam, R.; Hubbard, T.; Sasidharan, R. Domain insertions in protein structures. J. Mol. Biol. 2004, 338, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Selvam, R.A.; Sasidharan, R. Domins: A web resource for domain insertions in known protein structures. Nucleic Acids Res. 2004, 32, 193–195. [Google Scholar] [CrossRef] [PubMed]
- Guntas, G.; Mansell, T.J.; Kim, J.R.; Ostermeier, M. Directed evolution of protein switches and their application to the creation of ligand-binding proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 11224–11229. [Google Scholar] [CrossRef] [PubMed]
- Guntas, G.; Mitchell, S.F.; Ostermeier, M. A molecular switch created by in vitro recombination of nonhomologous genes. Chem. Biol. 2004, 11, 1483–1487. [Google Scholar] [CrossRef] [PubMed]
- Guntas, G.; Ostermeier, M. Creation of an allosteric enzyme by domain insertion. J. Mol. Biol. 2004, 336, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Cutler, T.A.; Loh, S.N. Thermodynamic analysis of an antagonistic folding–unfolding equilibrium between two protein domains. J. Mol. Biol. 2007, 371, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Cutler, T.A.; Mills, B.M.; Lubin, D.J.; Chong, L.T.; Loh, S.N. Effect of interdomain linker length on an antagonistic folding–unfolding equilibrium between two protein domains. J. Mol. Biol. 2009, 386, 854–868. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.-H.; Karchin, J.M.; Walker-Kopp, N.; Huang, L.S.; Berry, E.A.; Loh, S.N. Engineering domain-swapped binding interfaces by mutually exclusive folding. J. Mol. Biol. 2012, 416, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Radley, T.L.; Markowska, A.I.; Bettinger, B.T.; Ha, J.H.; Loh, S.N. Allosteric switching by mutually exclusive folding of protein domains. J. Mol. Biol. 2003, 332, 529–536. [Google Scholar] [CrossRef]
- Adikaram, P.R.; Beckett, D. Protein: Protein interactions in control of a transcriptional switch. J. Mol. Biol. 2013, 425, 4584–4594. [Google Scholar] [CrossRef] [PubMed]
- Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. Circular permutation and receptor insertion within green fluorescent proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 11241–11246. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T.; Miller, L.J. Seven transmembrane receptors as shapeshifting proteins: The impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol. Rev. 2010, 62, 265–304. [Google Scholar] [CrossRef] [PubMed]
- Kong, N.; Peng, Q.; Li, H. Rationally designed dynamic protein hydrogels with reversibly tunable mechanical properties. Adv. Funct. Mater. 2014, 24, 7310–7317. [Google Scholar] [CrossRef]
- Ostermeier, M. Designing switchable enzymes. Curr. Opin. Struct. Biol. 2009, 19, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Kong, N.; Wang, H.C.E.; Li, H. Designing redox potential-controlled protein switches based on mutually exclusive proteins. Protein Sci. 2012, 21, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Sallee, N.A.; Yeh, B.J.; Lim, W.A. Engineering modular protein interaction switches by sequence overlap. J. Am. Chem. Soc. 2007, 129, 4606–4611. [Google Scholar] [CrossRef] [PubMed]
- Streaker, E.D.; Beckett, D. The biotin regulatory system: Kinetic control of a transcriptional switch. Biochemistry 2006, 45, 6417–6425. [Google Scholar] [CrossRef] [PubMed]
- Strickland, D.; Moffat, K.; Sosnick, T.R. Light-activated DNA binding in a designed allosteric protein. Proc. Natl. Acad. Sci. USA 2008, 105, 10709–10714. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.-H.; Butler, J.S.; Mitrea, D.M.; Loh, S.N. Modular enzyme design: Regulation by mutually exclusive protein folding. J. Mol. Biol. 2006, 357, 1058–1062. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, H.-C.; Cao, Y.; Sharma, D.; Wang, M. Configurational entropy modulates the mechanical stability of protein GB1. J. Mol. Biol. 2008, 379, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Gronenborn, A.M.; Filpula, D.R.; Essig, N.Z.; Achari, A.; Whitlow, M.; Wingfield, P.T.; Clore, G.M. A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein g. Science 1991, 253, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Improta, S.; Politou, A.S.; Pastore, A. Immunoglobulin-like modules from titin I-band: Extensible components of muscle elasticity. Structure 1996, 4, 323–337. [Google Scholar] [CrossRef]
- Carrion-Vazquez, M.; Oberhauser, A.F.; Fowler, S.B.; Marszalek, P.E.; Broedel, S.E.; Clarke, J.; Fernandez, J.M. Mechanical and chemical unfolding of a single protein: A comparison. Proc. Natl. Acad. Sci. USA 1999, 96, 3694–3699. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Li, H. Direct observation of tug-of-war during the folding of a mutually exclusive protein. J. Am. Chem. Soc. 2009, 131, 13347–13354. [Google Scholar] [CrossRef] [PubMed]
- Kong, N.; Li, H. Protein fragment reconstitution as a driving force for self-assembling reversible protein hydrogels. Adv. Funct. Mater. 2015, 25, 5593–5601. [Google Scholar] [CrossRef]
- Kopeček, J.; Yang, J. Smart self-assembled hybrid hydrogel biomaterials. Angew. Chem. Int. Ed. 2012, 51, 7396–7417. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.-H.; Shinsky, S.A.; Loh, S.N. Stepwise conversion of a binding protein to a fluorescent switch: Application to thermoanaerobacter tengcongensis ribose binding protein. Biochemistry 2013, 52, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Mitrea, D.M.; Parsons, L.S.; Loh, S.N. Engineering an artificial zymogen by alternate frame protein folding. Proc. Natl. Acad. Sci. USA 2010, 107, 2824–2829. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.M.; Loh, S.N. On the mechanism of protein fold-switching by a molecular sensor. Proteins Struct. Funct. Bioinform. 2010, 78, 3260–3269. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.M.; McClendon, S.; Eliezer, D.; Loh, S.N. Structural characterization of two alternate conformations in a calbindin D9k-based molecular switch. Biochemistry 2011, 50, 5583–5589. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.M.; Mitrea, D.M.; Loh, S.N. A Ca2+-sensing molecular switch based on alternate frame protein folding. ACS Chem. Biol. 2008, 3, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Bi, J.; Krendel, M.; Loh, S.N. Converting a binding protein into a biosensing conformational switch using protein fragment exchange. Biochemistry 2014, 53, 5505–5514. [Google Scholar] [CrossRef] [PubMed]
- Perilla, J.R.; Goh, B.C.; Cassidy, C.K.; Liu, B.; Bernardi, R.C.; Rudack, T.; Yu, H.; Wu, Z.; Schulten, K. Molecular dynamics simulations of large macromolecular complexes. Curr. Opin. Struct. Biol. 2015, 31, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, M.; Mortier, J.; Rakers, C.; Sydow, D.; Wolber, G. More than a look into a crystal ball: Protein structure elucidation guided by molecular dynamics simulations. Drug Discov. Today 2016, 21, 1799–1805. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Dirks, R.M.; Grossman, J.; Xu, H.; Shaw, D.E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Mills, B.M.; Chong, L.T. Molecular simulations of mutually exclusive folding in a two-domain protein switch. Biophys. J. 2011, 100, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Wang, Q.; Wang, Y.; Chen, G. Structures and energies of the transition between two conformations of the alternate frame folding calbindin-D9k protein: A theoretical study. RSC Adv. 2015, 5, 65798–65810. [Google Scholar] [CrossRef]
- Duan, Y.; Kollman, P.A. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science 1998, 282, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, P.; Caflisch, A. Native topology or specific interactions: What is more important for protein folding? J. Mol. Biol. 2001, 306, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Knott, M.; Chan, H.S. Criteria for downhill protein folding: Calorimetry, chevron plot, kinetic relaxation, and single-molecule radius of gyration in chain models with subdued degrees of cooperativity. Proteins Struct. Funct. Bioinform. 2006, 65, 373–391. [Google Scholar] [CrossRef] [PubMed]
- Kuwajima, K.; Hiraoka, Y.; Ikeguchi, M.; Sugai, S. Comparison of the transient folding intermediates in lysozyme and α-lactalbumin. Biochemistry 1985, 24, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Mark, A.E.; Van Gunsteren, W.F. Simulation of the thermal denaturation of hen egg white lysozyme: Trapping the molten globule state. Biochemistry 1992, 31, 7745–7748. [Google Scholar] [CrossRef] [PubMed]
- Case, D.; Darden, T.; Cheatham, T., III; Simmerling, C.; Wang, J.; Duke, R.; Luo, R.; Merz, K.; Pearlman, D.; Crowley, M.; et al. Amber 9; University of California: San Francisco, CA, USA, 2006. [Google Scholar]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.C.; Duan, Y. Distinguish protein decoys by using a scoring function based on a new amber force field, short molecular dynamics simulations, and the generalized born solvent model. Proteins Struct. Funct. Bioinform. 2004, 55, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Apostolakis, J.; Ferrara, P.; Caflisch, A. Calculation of conformational transitions and barriers in solvated systems: Application to the alanine dipeptide in water. J. Chem. Phys. 1999, 110, 2099–2108. [Google Scholar] [CrossRef]
- Ovchinnikov, V.; Karplus, M. Analysis and elimination of a bias in targeted molecular dynamics simulations of conformational transitions: Application to calmodulin. J. Phys. Chem. B 2012, 116, 8584–8603. [Google Scholar] [CrossRef] [PubMed]
- Schlitter, J.; Engels, M.; Krüger, P. Targeted molecular dynamics: A new approach for searching pathways of conformational transitions. J. Mol. Graph. Model. 1994, 12, 84–89. [Google Scholar] [CrossRef]
- Schlitter, J.; Engels, M.; Krüger, P.; Jacoby, E.; Wollmer, A. Targeted molecular dynamics simulation of conformational change-application to the T ↔ R transition in insulin. Mol. Simul. 1993, 10, 291–308. [Google Scholar] [CrossRef]
- Fairchild, S.Z.; Peterson, M.W.; Hamza, A.; Zhan, C.-G.; Cerasoli, D.M.; Chang, W.E. Computational characterization of how the VX nerve agent binds human serum paraoxonase 1. J. Mol. Model. 2011, 17, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Hao, G.F.; Yang, S.G.; Yang, G.F.; Zhan, C.G. Computational gibberellin-binding channel discovery unraveling the unexpected perception mechanism of hormone signal by gibberellin receptor. J. Comput. Chem. 2013, 34, 2055–2064. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
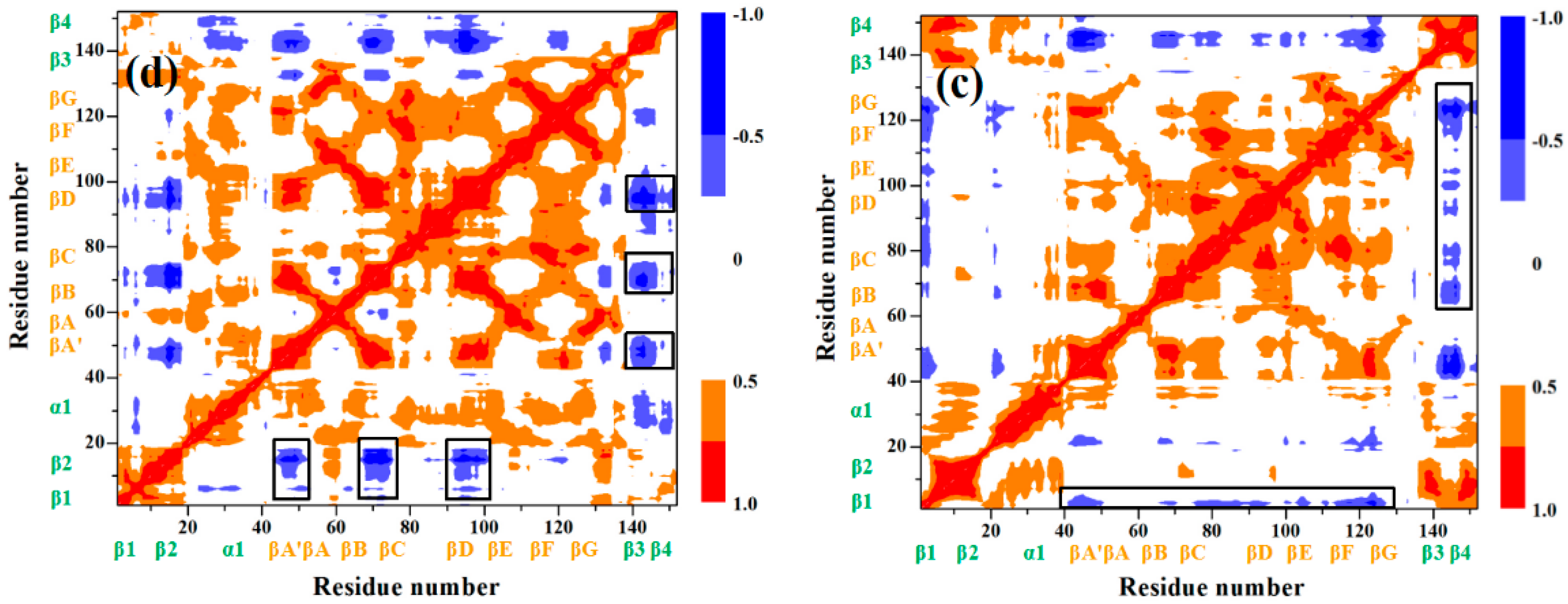{kind=link}
| Hydrogen Bond | GL5/I27-S | I | II | III | IV | GL5-ST/I27 | |
|---|---|---|---|---|---|---|---|
| β1 | (39)N–H···O(34,37) | 99.84 | 96.6 | 86.12 | – | – | – |
| (33)N–H···O(29) | 94.32 | 93.56 | 89.96 | – | – | – | |
| (31)N–H···O(27) | 99.8 | 95.64 | 93.68 | – | – | – | |
| (29)N–H···O(25) | 96.92 | 63.48 | 99.96 | 84 | – | – | |
| (34)N–H···O(30) | 99.72 | 97.76 | 96.48 | 57.12 | – | – | |
| (26)N–H···O(22) | 92.88 | 94.28 | 97.52 | 65.56 | – | – | |
| (30)N–H···O(26) | 99.48 | 78.8 | 88.68 | 46.36 | – | – | |
| (35)N–H···O(31) | 87.08 | 72.64 | 58.24 | 89.16 | – | – | |
| (27)N–H···O(23) | 99.28 | 96.68 | 91.88 | 75.36 | – | – | |
| (32)N–H···O(28) | 88 | 89.76 | 97.88 | – | – | – | |
| (38)N–H···O(35) | 79.92 | 28.92 | 25.6 | – | – | – | |
| (28)N–H···O(24) | 89.72 | 54.36 | 86.36 | 59.32 | – | – | |
| (25)N–H···O(22) | 45.12 | 53.76 | 28.72 | – | – | – | |
| β1–β2 | (14)N–H···O(7) | 50.24 | 52.68 | 24.56 | – | – | – |
| (20)N–H···O(1) | 98 | 90.16 | – | – | – | – | |
| (1)N–H···O(20) | 45.64 | 39 | – | – | – | – | |
| (9)N–H···O(12) | 98.64 | 87.92 | 40.68 | – | – | – | |
| (3)N–H···O(18) | 98.64 | 98.52 | 68.44 | – | – | – | |
| (18)N–H···O(3) | 97.44 | 97.16 | 85.2 | – | – | – | |
| β1–β4 | (10)N–H···OE1(152) | 54.08 | 57.72 | 24.88 | – | – | – |
| (152)N–H···O(8) | 90.48 | – | – | – | – | – | |
| (148)N–H···O(4) | 99.16 | 37.32 | 30.48 | – | – | – | |
| (6)N–H···O(148) | 98.32 | 99.76 | 99.48 | – | – | – | |
| (150)N–H···O(6) | 99.88 | 100 | 100 | 99.96 | – | – | |
| (8)N–H···O(150) | 59.68 | 79.04 | 48.72 | 55.44 | – | – | |
| β3–β4 | (138)N–H···O(151) | 98.52 | – | – | – | – | – |
| (151)N–H···O(138) | 97 | – | – | – | – | – | |
| (140)N–H···O(149) | 97.92 | 94.24 | 40.72 | – | – | – | |
| (142)N–H···O(147) | 96.12 | 88.28 | 92.52 | – | – | – |
| Hydrogen Bond | GL5/I27-S | I | II | III | IV | GL5-ST/I27 | |
|---|---|---|---|---|---|---|---|
| βA-βG | (55)N–H···O(127) | – | – | – | – | 75.96 | 99.64 |
| (57)N–H···O(129) | – | – | – | – | 47.44 | 99.4 | |
| (129)N–H···O(55) | – | – | – | – | 64.96 | 91.6 | |
| (123)N–H···O(43,44) | – | – | – | 83.96 | 94.84 | 96.6 | |
| βA-βB | (49,50)N–H···O(68,69) | 88.08 | 76.96 | 97.08 | 81.36 | 98.96 | 97.76 |
| (68)N–H···O(50) | – | – | – | – | – | 97 | |
| (61)N–H···O(58) | – | – | – | – | 81.36 | 70.28 | |
| βB-βE | (63)N–H···O(104) | – | – | – | – | 98.16 | 98.52 |
| (65)N-H···O(102) | – | – | – | – | 90.56 | 99.36 | |
| (100)N–H···O(66,67) | – | – | – | – | 99.56 | 97.12 | |
| (67,69)N–H···O(98,100) | – | – | 68.08 | 95.72 | 95.92 | 98.36 | |
| (107)N–H···O(61) | – | – | – | – | – | 99.64 | |
| βE-βD | (111)N–H···O(108,133) | – | 67.16 | 97.84 | 96.32 | 97.04 | 99.4 |
| (92)N–H···O(103) | – | – | – | – | 90.04 | 99.68 | |
| (103)N–H···O(92) | – | – | – | – | 90.48 | 99.92 | |
| (101)N–H···O(94) | – | – | – | – | 63.48 | 99.84 | |
| (96)N–H···O(93,99) | – | 55.6 | 62.92 | 76.48 | 93.56 | 96.84 | |
| (105)N–H···O(90) | – | – | – | – | 71.04 | 84.24 | |
| βC-βD | (80)N–H···O(83) | – | 44.08 | 61.28 | 78.28 | 85.84 | 81.68 |
| (90,91)N–H···O(87,88) | – | – | – | – | 53.88 | 84.72 | |
| βC-βF | (77)N–H···O(118) | – | – | – | – | – | 91.84 |
| (118)N–H···O(77) | – | – | – | – | – | 98.84 | |
| (117)N–H···O(80) | – | 65.32 | 66.8 | 85.08 | 69.16 | 96.08 | |
| (79)N–H···O(116) | – | – | – | 51.16 | 69.16 | 96.08 | |
| (116)N–H···O(79) | – | – | – | – | – | 99.92 | |
| βF-βG | (128)N–H···O(113) | – | – | – | – | – | 96.4 |
| (113)N–H···O(128) | – | – | – | – | – | 98.56 | |
| (126)N–H···O(114,115) | – | – | – | – | 50.28 | 98.52 | |
| (116,117)N–H···O(124) | – | – | – | – | 69.4 | 99.68 | |
| (124)N–H···O(117) | – | – | – | – | – | 99.08 | |
| (122)N–H···O(119) | – | 33.04 | 96.84 | 82.72 | 94.96 | 96.96 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Wang, Y.; Chen, G. Influence of Secondary-Structure Folding on the Mutually Exclusive Folding Process of GL5/I27 Protein: Evidence from Molecular Dynamics Simulations. Int. J. Mol. Sci. 2016, 17, 1962. https://doi.org/10.3390/ijms17111962
Wang Q, Wang Y, Chen G. Influence of Secondary-Structure Folding on the Mutually Exclusive Folding Process of GL5/I27 Protein: Evidence from Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2016; 17(11):1962. https://doi.org/10.3390/ijms17111962
Chicago/Turabian StyleWang, Qing, Yan Wang, and Guangju Chen. 2016. "Influence of Secondary-Structure Folding on the Mutually Exclusive Folding Process of GL5/I27 Protein: Evidence from Molecular Dynamics Simulations" International Journal of Molecular Sciences 17, no. 11: 1962. https://doi.org/10.3390/ijms17111962