
Synthesis of Non-Toxic Silica Particles Stabilized by Molecular Complex Oleic-Acid/Sodium Oleate



 ,
,  ,
,  and
and
Abstract
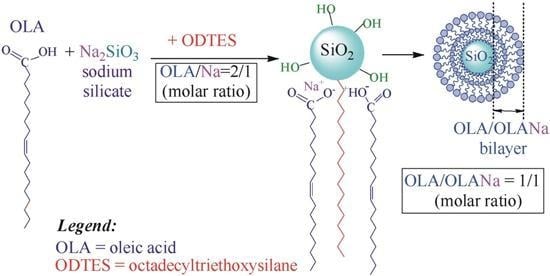
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Aqueous Dispersion
3.3. Characterization Methods
3.3.1. Dynamic Light Scattering (DLS) and Laser Doppler Velocimetry (LDV)
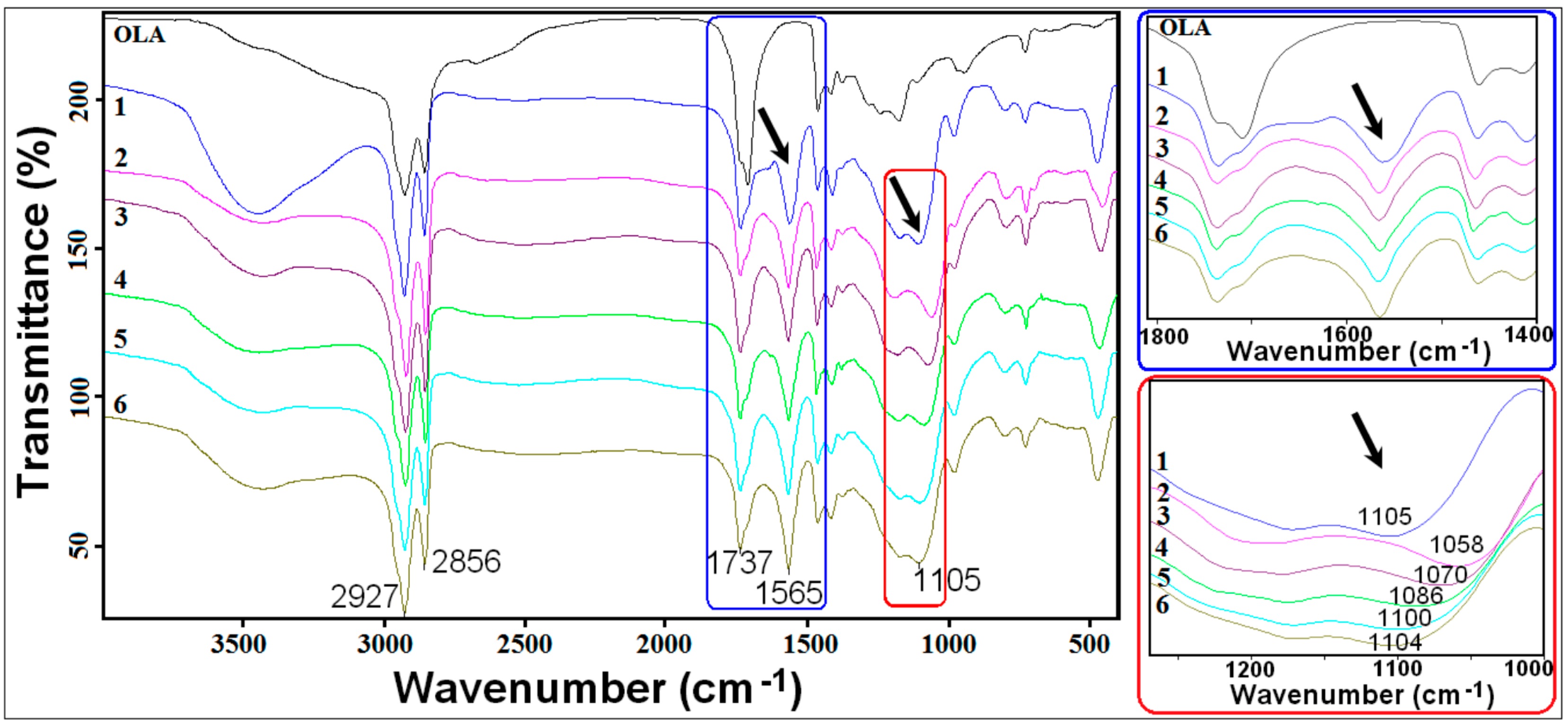
3.3.2. Fourier Transformed Infrared (FTIR)
3.3.3. Simultaneous Thermal Analyses (STA)
3.3.4. Transmission Electron Microscopy (TEM)
3.3.5. N2 Adsorption-Desorption
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fameau, C.L.; Arnold, A.; Saint-Jalmes, A. Responsive self-assemblies on fatty acids. Curr. Opin. Colloid Interface Sci. 2014, 19, 471–479. [Google Scholar] [CrossRef] [Green Version]
- Walde, P.; Namani, T.; Morigaki, K.; Hauser, H. Formation and Properties of Fatty Acid Vesicules (Liposomes), Liposome Technology. In Liposome Preparation and Related Technologies, 3rd ed.; Gregoriadis, G., Ed.; Informa Healthcare: London, UK, 2006; Volume I, pp. 1–19. [Google Scholar]
- Delampe, M.; Jerome, F.; Barrault, J.; Douliez, J.P. Self-assembly and emulsions of oleic acid-oleate mixture in glycerol. Green Chem. 2011, 13, 64–68. [Google Scholar] [CrossRef]
- Suga, K.; Yokoi, T.; Kondo, D.; Hayashi, K.; Morita, S.; Okamoto, Y.; Shimauchi, T.; Umakoshi, H. Systematic characterization of phase behaviors and membrane properties of fatty acid/didecyldimethylammonium bromide vesicles. Langmuir 2014, 30, 12721–12728. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.M.; Jani, R.H.; Patel, C.N. Ufasomes: A vesicular drug delivery. Syst. Rev. Pharm. 2011, 2, 72–78. [Google Scholar] [CrossRef]
- Kanicky, J.R.; Shah, D.O. Effect of degree type and position of unsaturation on the pKa of long-chain fatty acids. J. Colloid Interface Sci. 2002, 256, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Cistola, D.P.; Hamilton, J.A.; Jackson, D.; Small, D.M. Ionization and phase behavior of fatty acids in water: Application of the Gibbs phase rule. Biochemistry 1988, 27, 1881–1888. [Google Scholar] [CrossRef] [PubMed]
- Cistola, D.P.; Atkinson, D.; Hamilton, J.A.; Small, D.M. Phase behavior and bilayer properties of fatty acids: Hydrated 1:1 acid-soap. Biochemistry 1986, 25, 2804–2812. [Google Scholar] [CrossRef] [PubMed]
- Apel, C.L.; Deamer, D.W.; Mautner, M.N. Self-assembled vesicles of monocarboxylic acids and alcohols: Condition for stability and for encapsulation of biopolymers. Biochim. Biophys. Acta 2002, 1559, 1–9. [Google Scholar] [CrossRef]
- Rendon, A.; Carton, D.G.; Sot, J.; Garcia-Pacios, M.; Montes, L.R.; Valle, M. Model systems of precursor cellular membranes: Long-chain alcohols stabilize spontaneously formed oleic acid vesicles. Biophys. J. 2012, 102, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Hentrich, C.J.; Szostak, W. Controlled growth of filamentous fatty acid vesicles under flow. Langmuir 2014, 30, 14016–14923. [Google Scholar] [CrossRef] [PubMed]
- Janke, J.J.; Bennet, W.F.D.; Tieleman, D.P. Oleic acid behavior from molecular dynamic simulations. Langmuir 2014, 30, 10661–10667. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Fang, Y.; Ma, L. The self-crosslinkedufasome of conjugated linoleic acid: Investigation of morphology, bilayer membrane and stability. Colloid Surf. B Biointerfaces 2014, 123, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Vivero-Escoto, J.L.; Huang, Y.T. Inorganic-organic hybrid nanomaterials for therapeutic and diagnostic imaging applications. Int. J. Mol. Sci. 2011, 12, 3888–3927. [Google Scholar] [CrossRef] [PubMed]
- Slowing, I.I.; Vivero-Escoto, J.L.; Wu, C.W.; Lin, V.S.Y. Mesoporous silica nanoparticles as controlled release drug delivery and gene transfection carriers. Adv. Drug Deliv. Rev. 2008, 60, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Yague, C.; Moros, M.; Grazu, V.; Arruebo, M.; Santamaria, J. Synthesis and stealthing study of bare and PEGylated silica micro-and nanoparticles as potential drug-delivery vectors. Chem. Eng. J. 2008, 137, 45–53. [Google Scholar] [CrossRef]
- Gerardin, C.; Reboult, J.; Bonne, M.; Lebeau, B. Ecodesign of ordered mesoporous silica materials. Chem. Soc. Rev. 2013, 42, 4217–4255. [Google Scholar] [CrossRef] [PubMed]
- Nicole, L.; Boissiere, C.; Grosso, D.; Quach, A.; Sanchez, C. Mesostructured hybrid organic-inorganic films. J. Mater. Chem. 2005, 15, 3598–3627. [Google Scholar] [CrossRef]
- Shimojima, A.; Kuroda, K. Designed synthesis of nanostructured siloxane-organic hybrids from amphiphilic silicon-based precursors. Chem. Rec. 2006, 6, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Brambila, R.; Pires, G.P.; dos Santos, J.H.Z.; Miranda, M.S.L. Octadecylsilane hybrid silicas prepared by the sol–gel method: Morphological and textural aspects. J. Colloid Interface Sci. 2007, 312, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Brambila, R.; dos Santos, J.H.Z.; Miranda, M.S.L.; Frost, R.L. Thermal stability of octadecylsilane hybrid silicas prepared by grafting and sol–gel methods. Thermochim. Acta 2008, 469, 91–97. [Google Scholar] [CrossRef]
- Brambila, R.; Pires, G.P.; da Silveira, N.P.; dos Santos, J.H.Z.; Miranda, M.S.L.; Frost, R.L. Spherical and lamellar octadecylsilanesilicas. J. NonCryst. Solids 2008, 354, 5033–5040. [Google Scholar] [CrossRef] [Green Version]
- Nistor, C.L.; Ianchis, R.; Ghiurea, M.; Nicolae, C.A.; Spataru, C.I.; Culita, D.C.; Pandele-Cusu, J.; Fruth, V.; Oancea, F.; Donescu, D. Aqueous dispersions of silica stabilized with oleic acid obtained by green chemistry. Nanomaterials 2016, 6, 9. [Google Scholar] [CrossRef]
- Bansal, V.; Ahmad, A.; Sastry, M. Fungus-mediated biotransformation of amorphous silica in rice husk to nanocrystalline silica. J. Am. Chem. Soc. 2006, 128, 14059–14066. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Epasinghe, D.J.; Zhang, W.; Zhou, B.; Niu, L.; Ryou, H.; Eid, A.A.; Frassetto, A.; Yiu, C.K.Y.; Arola, D.D.; et al. Synthesis of antimicrobial silsesquioxane-silica hybrids by hydrolytic co-condensation of alkoxysilanes. Polym. Chem. 2014, 5, 454–462. [Google Scholar] [CrossRef]
- Wang, W.; Efrima, S.; Regev, O. Directing oleate stabilized nanosized silver colloids into organic phases. Langmuir 1998, 14, 602–610. [Google Scholar] [CrossRef]
- Wang, W.; Chen, X.; Efrima, S. Silver nanoparticles capped by long-chain unsaturated carboxylates. J. Phys. Chem. B 1999, 103, 7238–7246. [Google Scholar] [CrossRef]
- Presswala, L.; Mattheus, M.E.; Atkinson, I.; Najjar, O.; Gerhardstein, N.; Moran, J.; Wei, R.; Riga, A.T. Discovery of bound and unbound waters in crystalline amino acids revealed by thermal analysis. J. Therm. Anal. Calorim. 2008, 93, 295–300. [Google Scholar] [CrossRef]
- Laporta, M.; Pegoraro, M.; Zanderighi, L. Perfluorosulfonated membrane (Nafion): FT-IR study of the state of water with increasing humidity. Phys. Chem. Chem. Phys. 1999, 1, 4619–4528. [Google Scholar] [CrossRef]
- Staszczuk, P.; Cabriezo-Vilchez, M.A.; Hidalgo-Alvarez, R. Differential thermal analysis of negatively charged polystyrene lattices. Colloid Polym. Sci. 1990, 271, 759–765. [Google Scholar] [CrossRef]
- Pshezhetskii, V.S.; Rakhnyanskaya, A.A.; Gaponenko, I.M.; Nablandyan, Y.E. A differential scanning calorimetry study of polyvinyl alcohol. Polym. Sci. USSR 1990, 32, 722–726. [Google Scholar] [CrossRef]
- Zelenka, T. Adsorption and desorption of nitrogen at 77 K on micro- and mesoporous materials: Study of transport kinetics. Microporous Mesoporous Mater. 2016, 227, 202–209. [Google Scholar] [CrossRef]

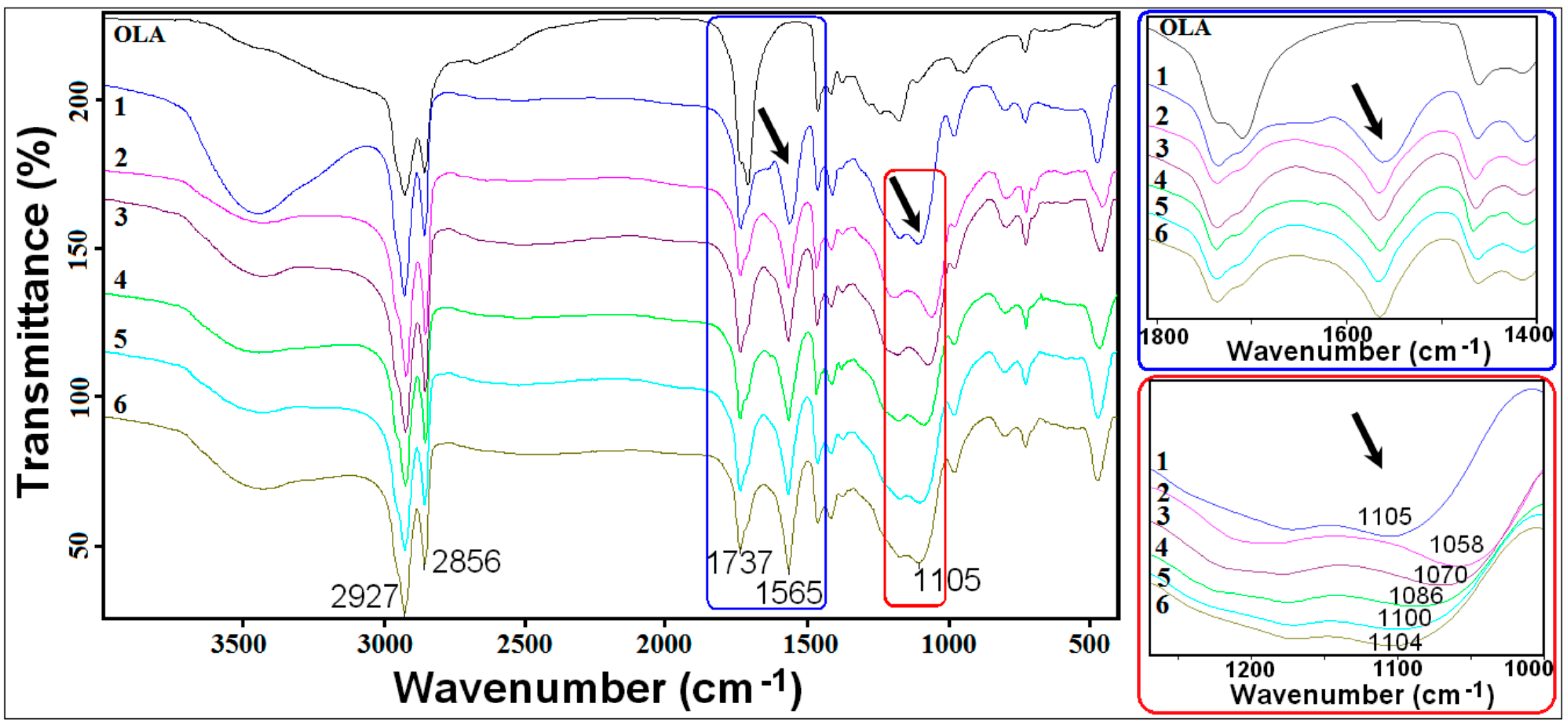

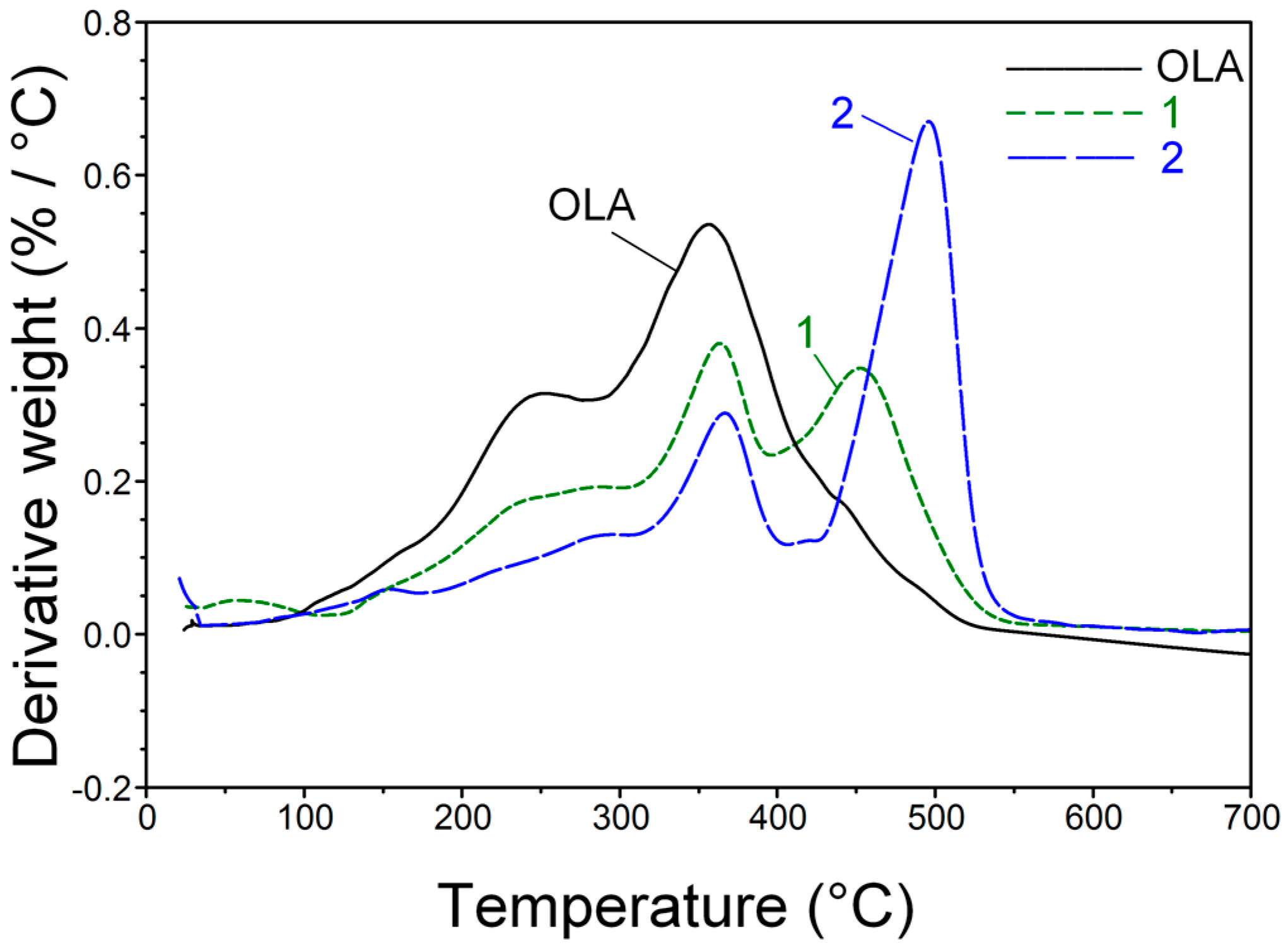
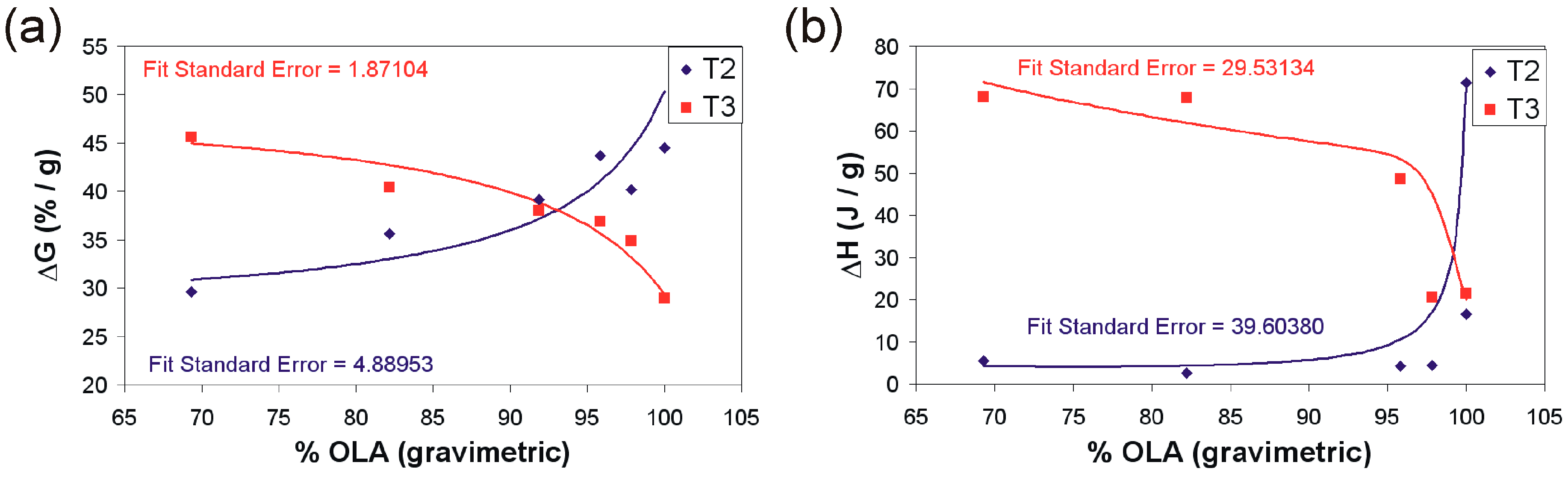


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
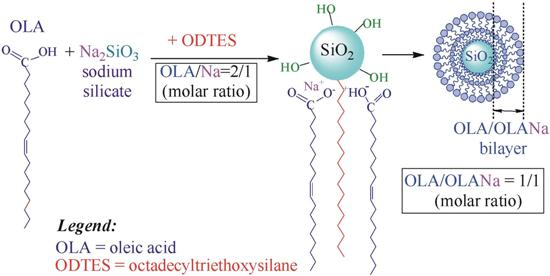
| Sample | ODTES (g) | ODTES/Sodium Silicate (mol/mol) | OLA/OLA + ODTES (mol/mol) | D a (nm) | Z b (mV) | Appearance |
|---|---|---|---|---|---|---|
| 1 | 0 | Onlysodium silicate | 1 | 140 ± 1.124 | −63.3 ± 1.35 | opaque fluid |
| 2 | 3.07 | 1:1 | 0.67 | 163 ± 0.845 | −67.8 ± 1.53 | opaque gel |
| 3 | 1.5 | 1:2 | 0.805 | 217 ± 1.567 | −60.5 ± 0.948 | opaque gel |
| 4 | 0.61 | 1:5 | 0.91 | 311 ± 1.499 | −87.2 ± 2.21 | opaque gel |
| 5 | 0.3 | 1:10 | 0.954 | 307 ± 3.303 | −63.0 ± 2.56 | opaque fluid |
| 6 | 0.15 | 1:20 | 0.968 | 147 ± 1.130 | −64.8 ± 1.73 | opaque fluid |
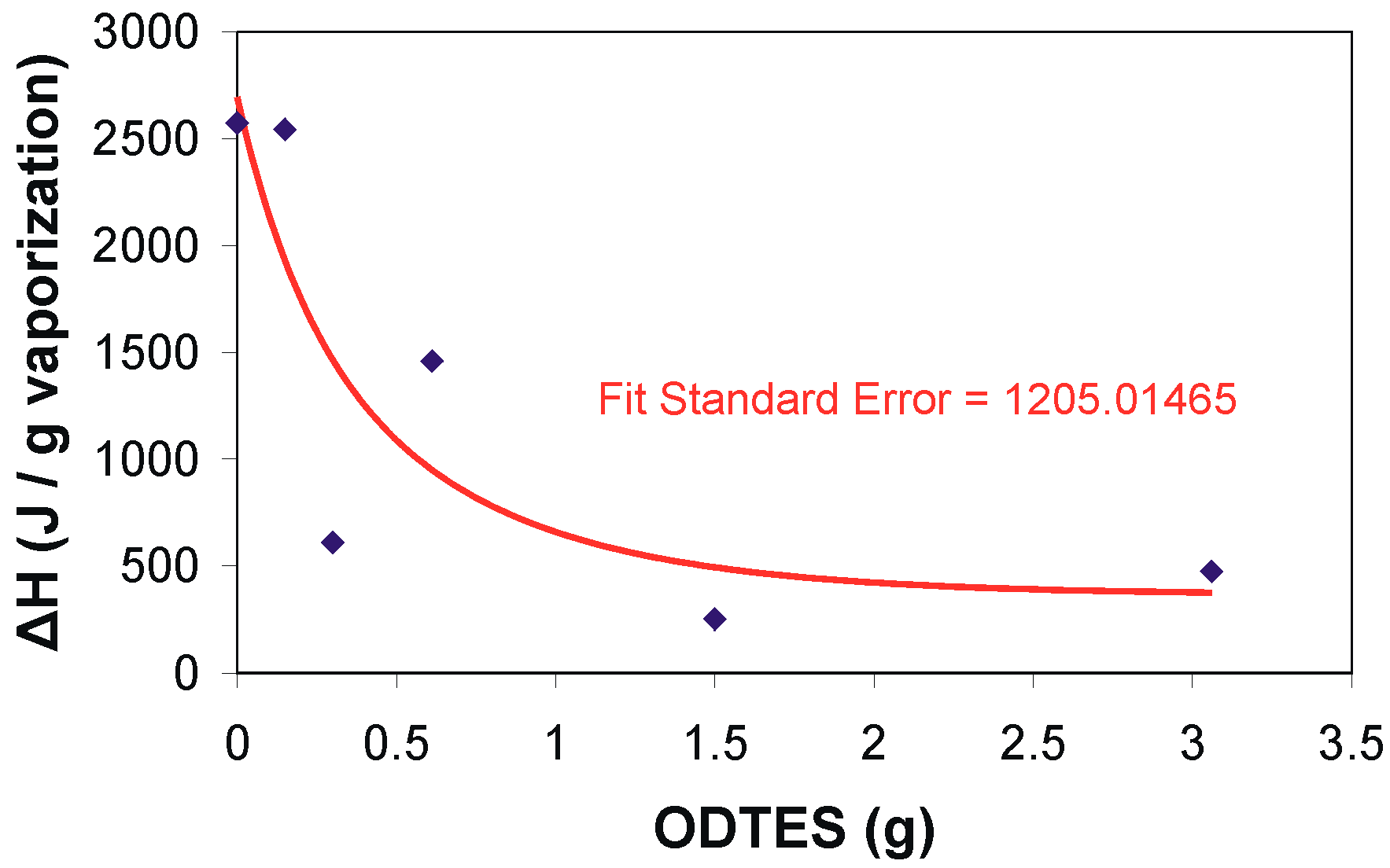
| Sample No. | Water Dispersions | Dried Hybrids | |||||
|---|---|---|---|---|---|---|---|
| DSC | TGA/DTG (ΔG%/Tmax °C) | DSC (ΔH (J/g)/Ti (°C)) | |||||
| 10–200 °C | 0–200 °C | 200–400 °C | 400–700 °C | Residue at 700 °C % | 200–400 °C | 400–700 °C | |
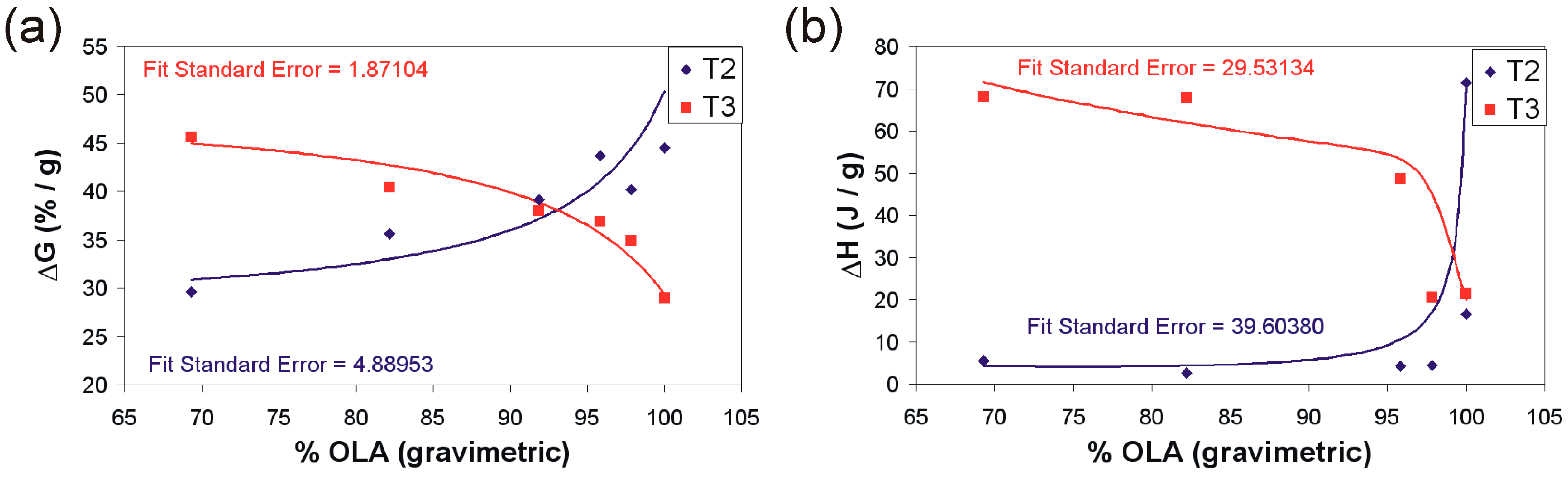
| ΔH (J/g) | ΔG | ΔG/Tmax 2 | ΔG/Tmax 3 | ΔH/Ti2 | ΔH/Ti3 | ||
| 1 | 2573 | 8.69 | 44.50:364.0 | 28.94:457.0 | 17.88 | 16.49:337 | 21.45:426 |
| 2 | 472 | 6.59 | 29.63:366.7 | 45.64:495.0 | 18.25 | 5.56:338 | 68.1:466 |
| 3 | 249 | 5.46 | 35.60:369.1 | 40.43:488.6 | 18.51 | 2.63:385 | 67.92:448.3 |
| 4 | 1458 | 6.94 | 39.14:367.1 | 38.99:471.7 | 14.96 | 5.29:322.9 | 12.28:480.3 |
| 5 | 608 | 7.14 | 43.70:367.8 | 36.92:470.7 | 12.25 | 4.22:382.2 | 48.7:454.7 |
| 6 | 2543 | 6.48 | 40.15:363.8 | 34.90:488.6 | 18.51 | 4.43:375.3 | 20.82:452.5 |
| OLA | – | 10 | 73.6:356 | 16.3:– | 0 | 71.3:370 | – |
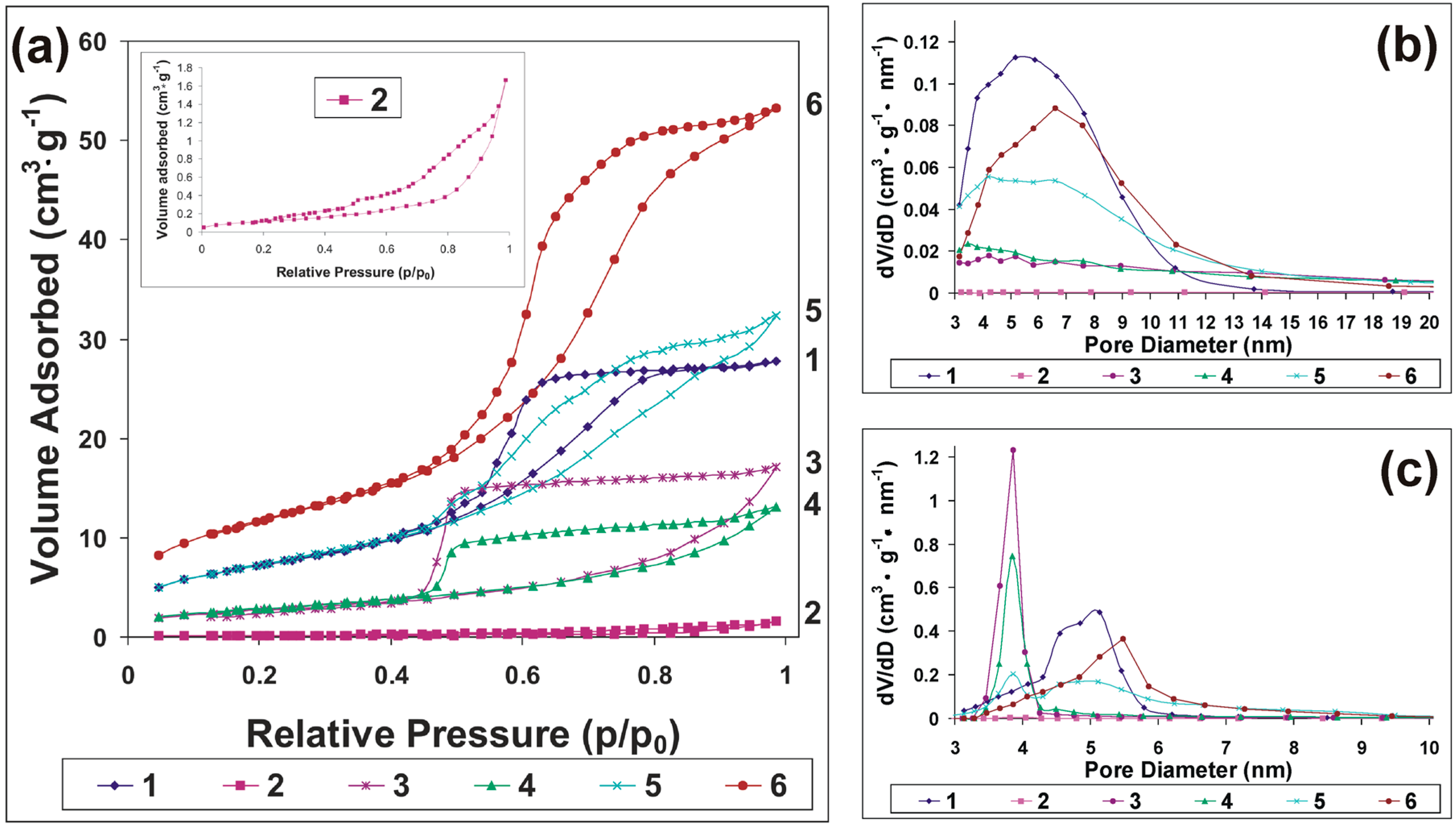
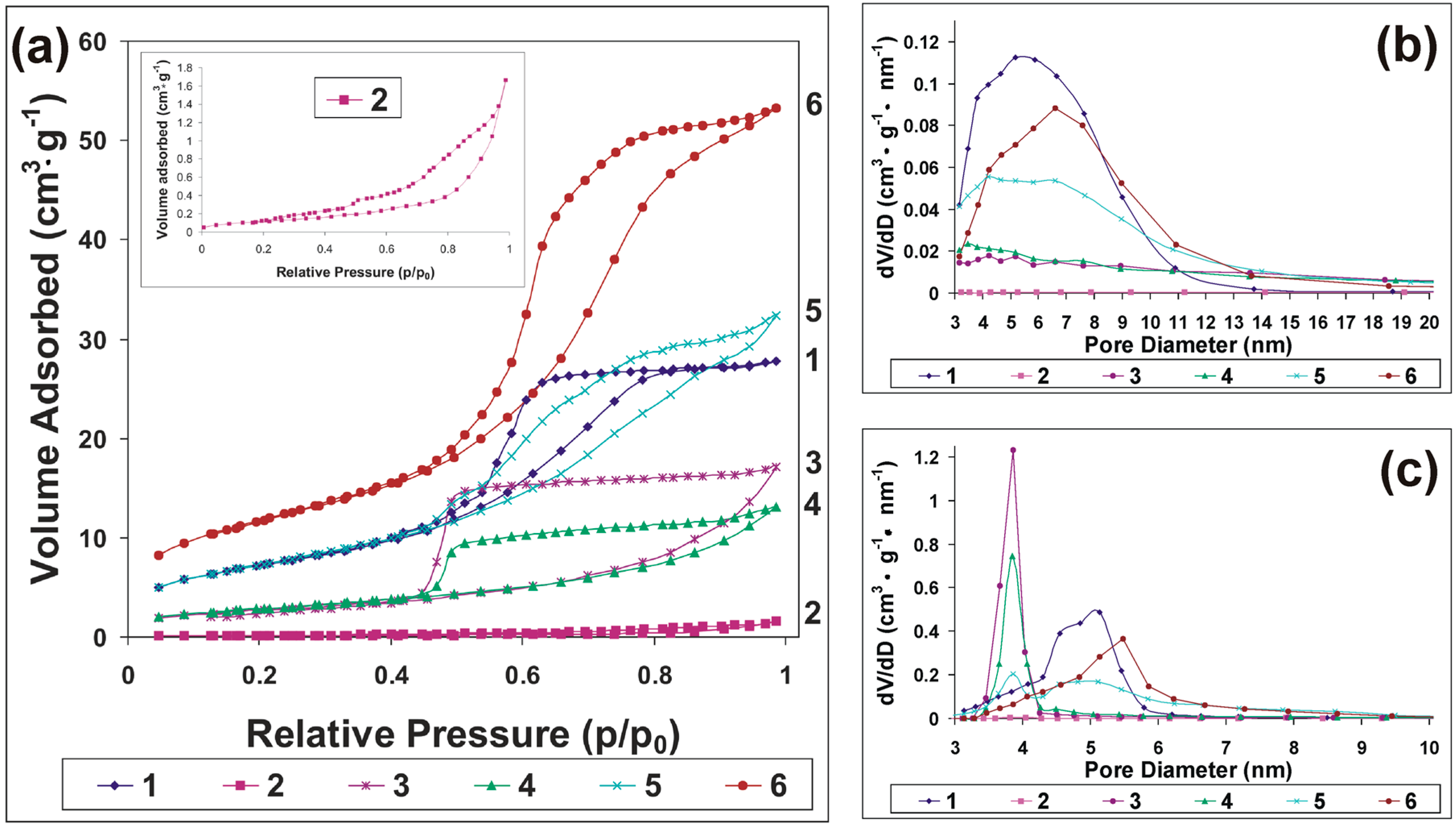
| Sample | SBET (m2·g−1) | SBJH ads. (m2·g−1) | Da a (nm) | Dd b (nm) | Pore Volume c (m3·g−1) | Vt d (m3·g−1) |
|---|---|---|---|---|---|---|
| 1 | 380.202 | 416.509 | 5.19 | 5.14 | 0.621 | 0.624 |
| 2 | 2.868 | 3.560 | 3.54 | 4.05 | 0.017 | 0.0167 |
| 3 | 114.364 | 104.409 | 4.22 | 3.86 | 0.308 | 0.318 |
| 4 | 165.261 | 115.717 | 3.47 | 3.86 | 0.293 | 0.325 |
| 5 | 291.300 | 268.893 | 4.20 | 3.86 | 0.524 | 0.551 |
| 6 | 294.393 | 319.468 | 6.60 | 5.49 | 0.572 | 0.571 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spataru, C.I.; Ianchis, R.; Petcu, C.; Nistor, C.L.; Purcar, V.; Trica, B.; Nitu, S.G.; Somoghi, R.; Alexandrescu, E.; Oancea, F.; et al. Synthesis of Non-Toxic Silica Particles Stabilized by Molecular Complex Oleic-Acid/Sodium Oleate. Int. J. Mol. Sci. 2016, 17, 1936. https://doi.org/10.3390/ijms17111936
Spataru CI, Ianchis R, Petcu C, Nistor CL, Purcar V, Trica B, Nitu SG, Somoghi R, Alexandrescu E, Oancea F, et al. Synthesis of Non-Toxic Silica Particles Stabilized by Molecular Complex Oleic-Acid/Sodium Oleate. International Journal of Molecular Sciences. 2016; 17(11):1936. https://doi.org/10.3390/ijms17111936
Chicago/Turabian StyleSpataru, Catalin Ilie, Raluca Ianchis, Cristian Petcu, Cristina Lavinia Nistor, Violeta Purcar, Bogdan Trica, Sabina Georgiana Nitu, Raluca Somoghi, Elvira Alexandrescu, Florin Oancea, and et al. 2016. "Synthesis of Non-Toxic Silica Particles Stabilized by Molecular Complex Oleic-Acid/Sodium Oleate" International Journal of Molecular Sciences 17, no. 11: 1936. https://doi.org/10.3390/ijms17111936
APA StyleSpataru, C. I., Ianchis, R., Petcu, C., Nistor, C. L., Purcar, V., Trica, B., Nitu, S. G., Somoghi, R., Alexandrescu, E., Oancea, F., & Donescu, D. (2016). Synthesis of Non-Toxic Silica Particles Stabilized by Molecular Complex Oleic-Acid/Sodium Oleate. International Journal of Molecular Sciences, 17(11), 1936. https://doi.org/10.3390/ijms17111936