
Selenomethionine Ameliorates Neuropathology in the Olfactory Bulb of a Triple Transgenic Mouse Model of Alzheimer’s Disease



Abstract
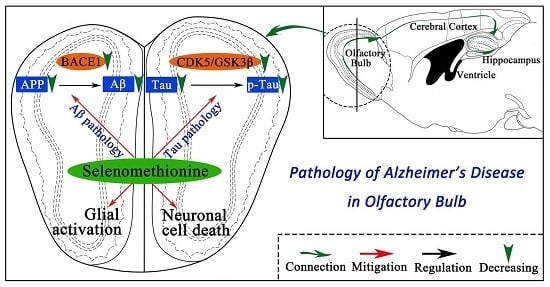
:
1. Introduction
2. Results
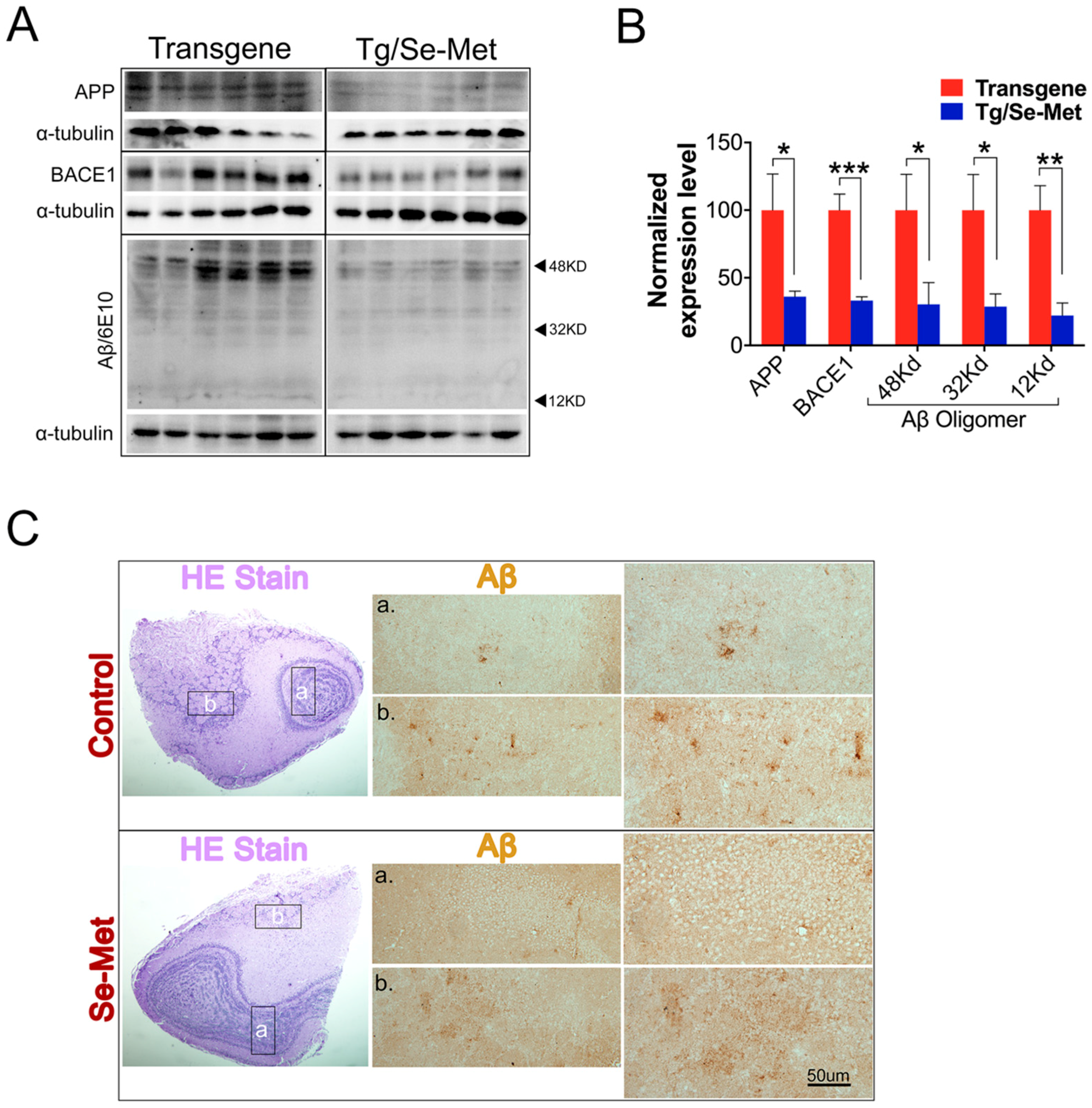
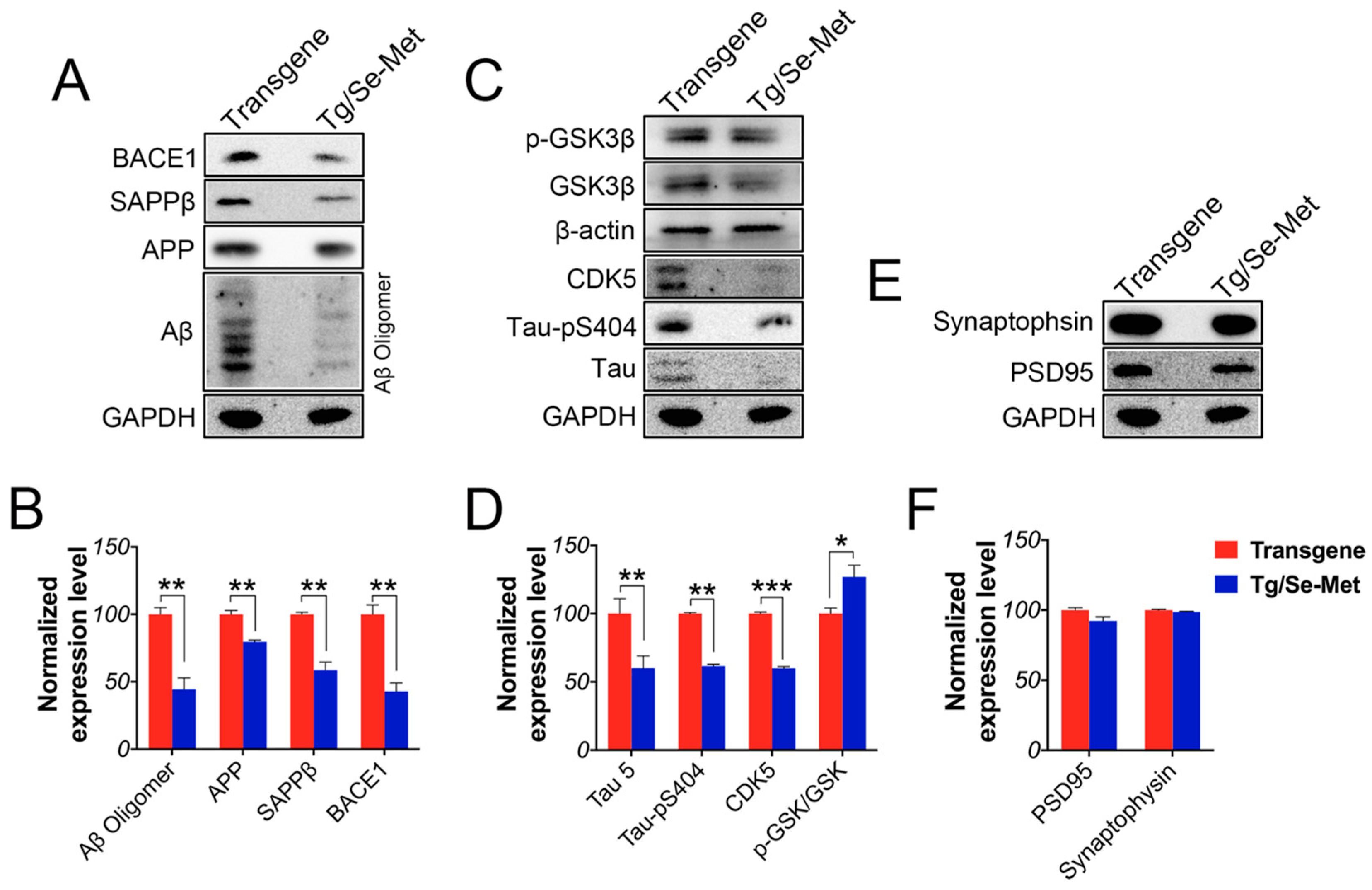
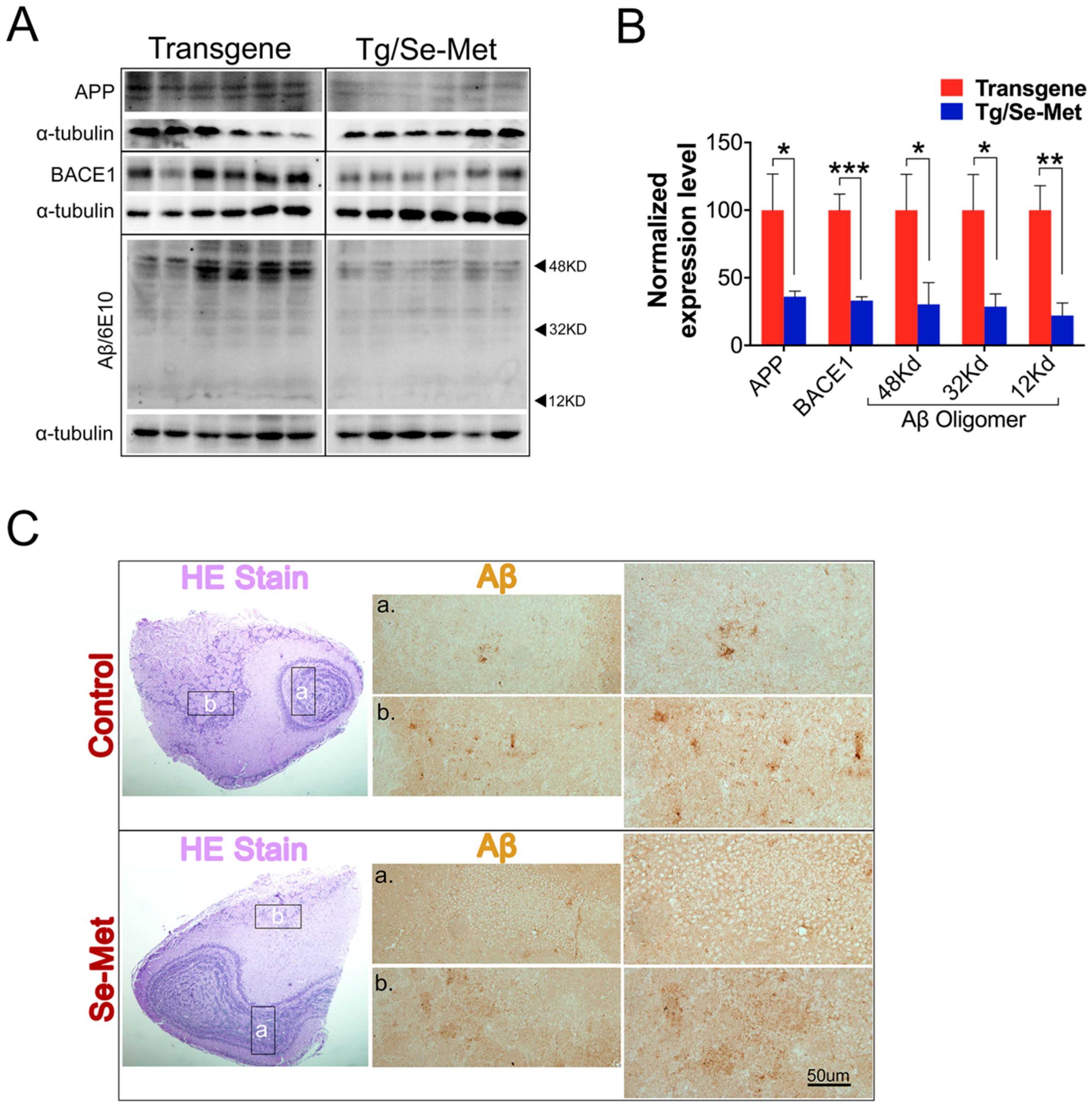
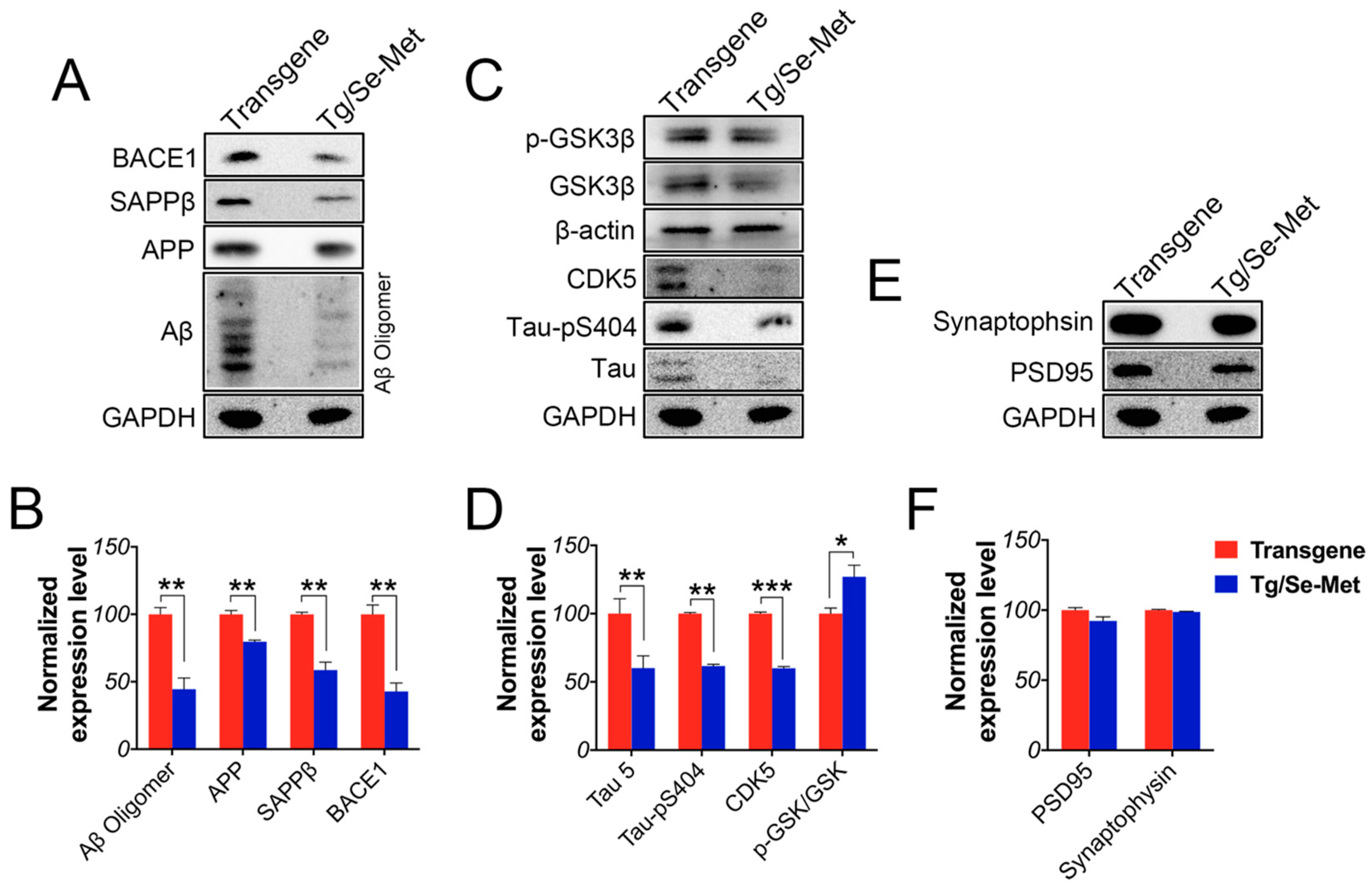
2.1. Se-Met Decreased the Production and Deposition of Aβ by Inhibiting Level of APP and BACE1-Mediated APP Processing
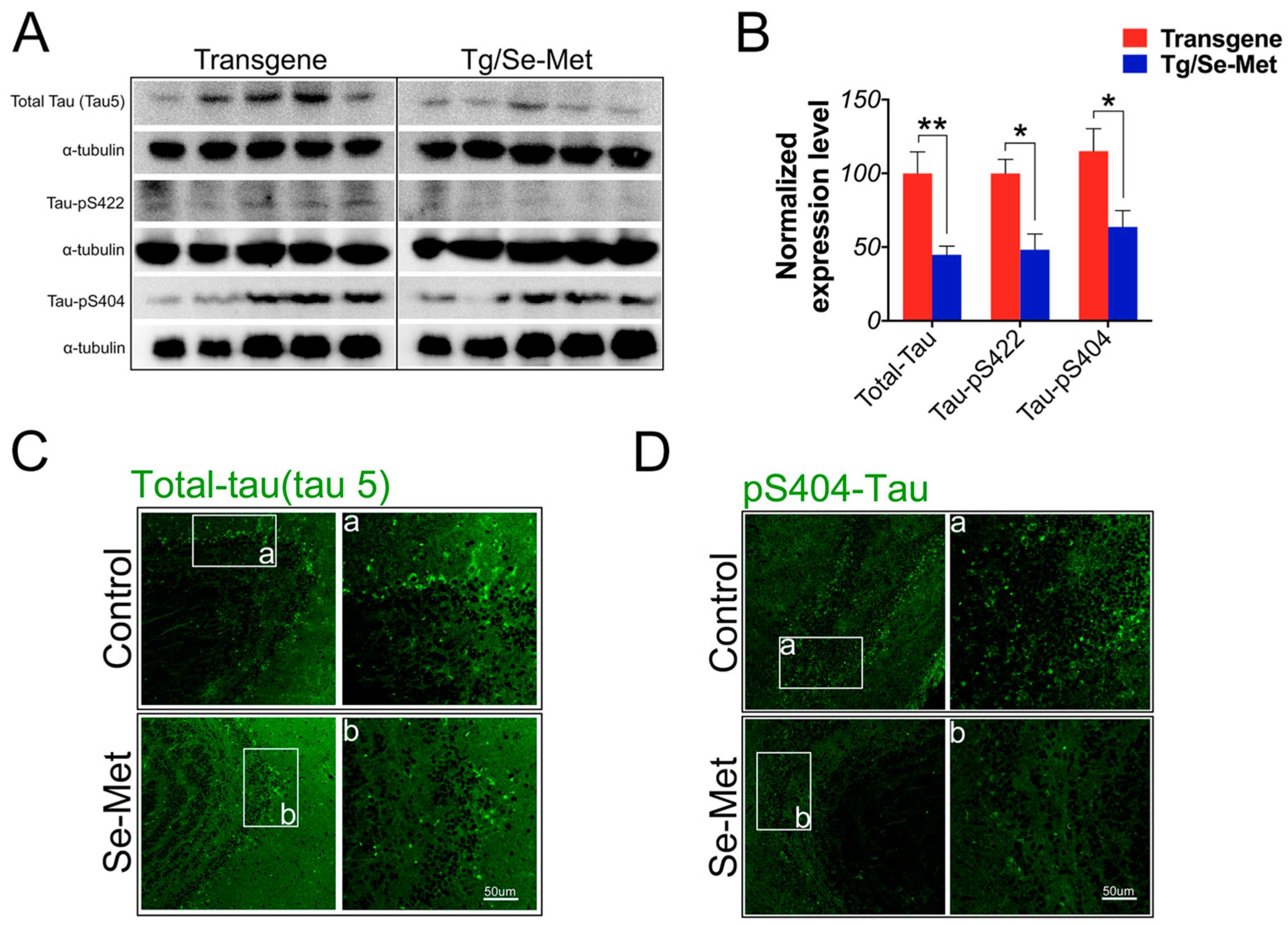
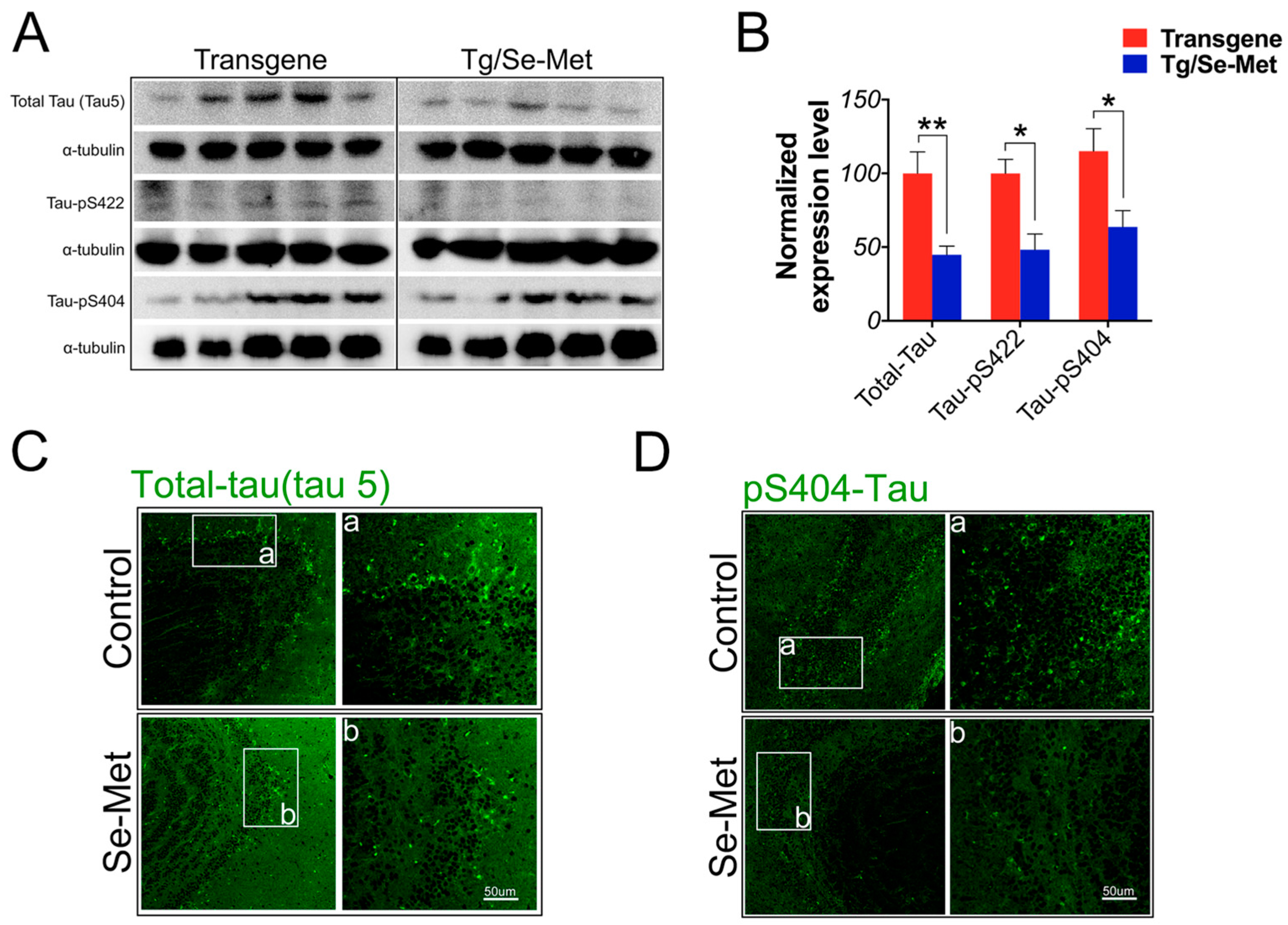
2.2. Se-Met Reduced the Level of Total Tau and Phosphorylation of Tau at Ser404, Ser422 in the OB
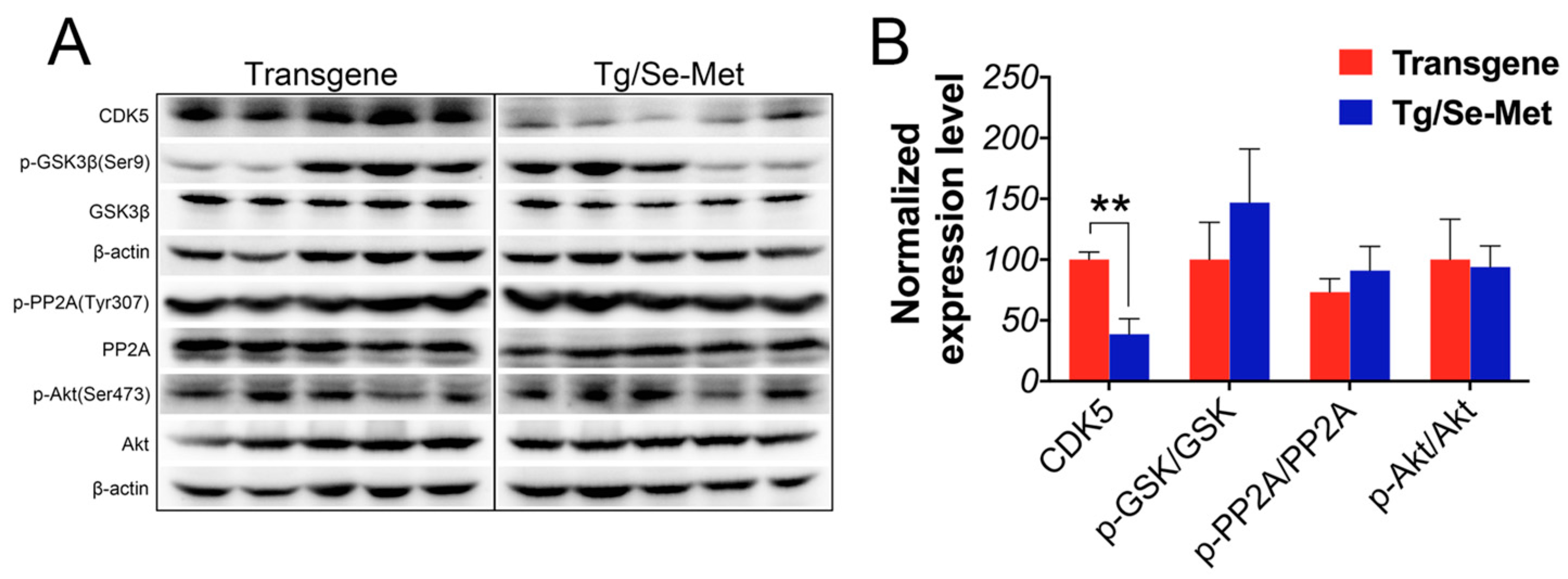
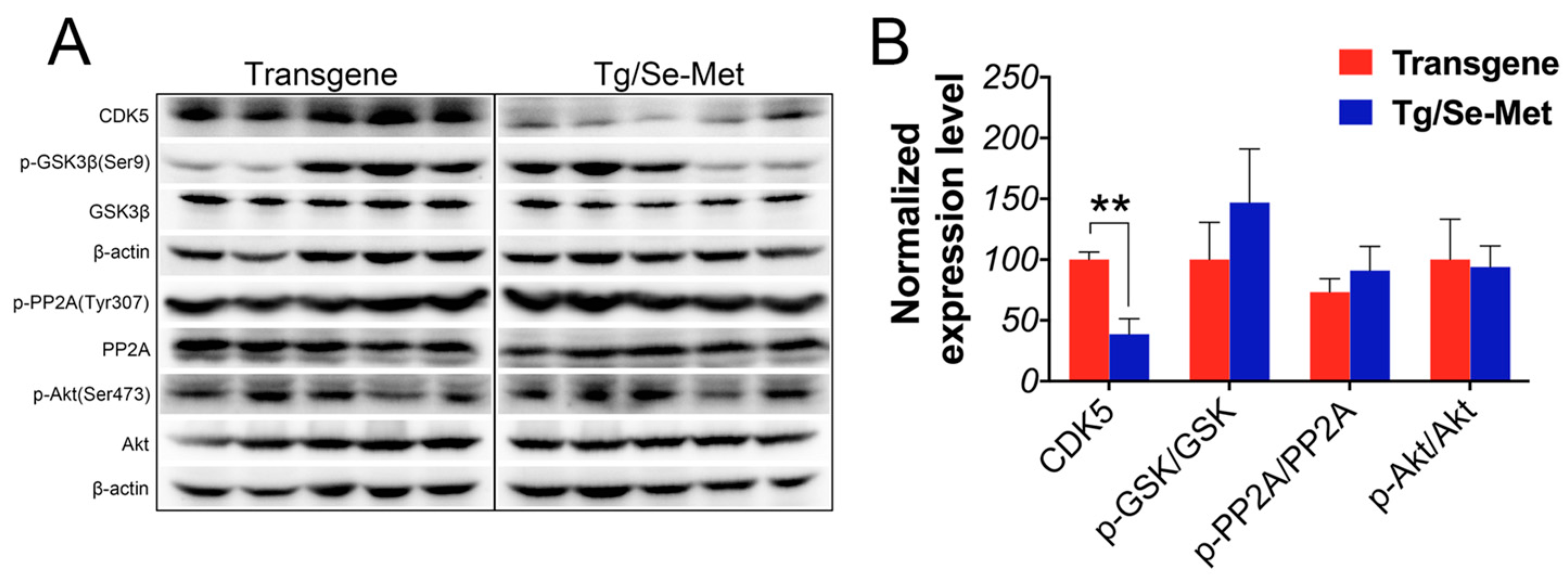
2.3. Effect of Se-Met on the Activity of CDK5, GSK-3β, PP2A, and Akt in the OB
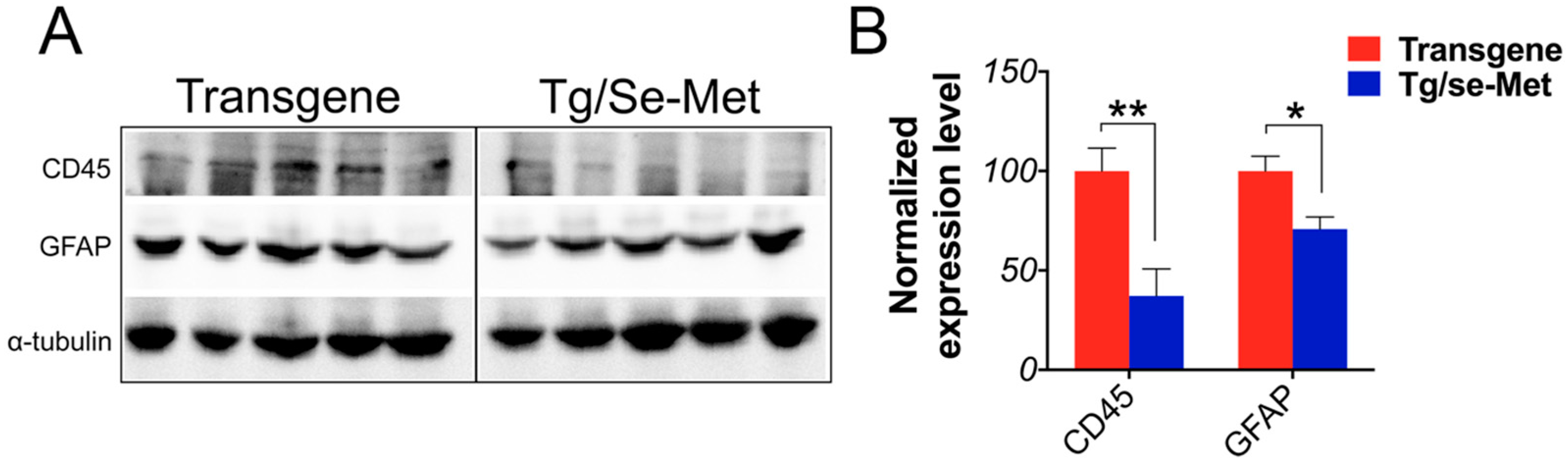
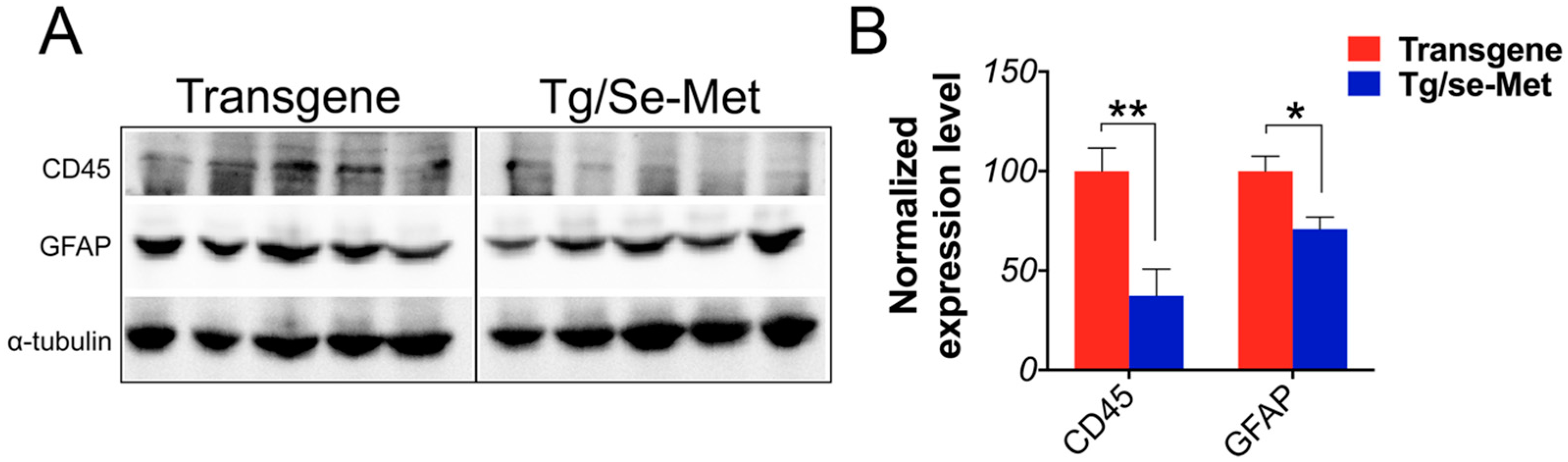
2.4. Se-Met Treatment Inhibited the Activation of Glials in OB
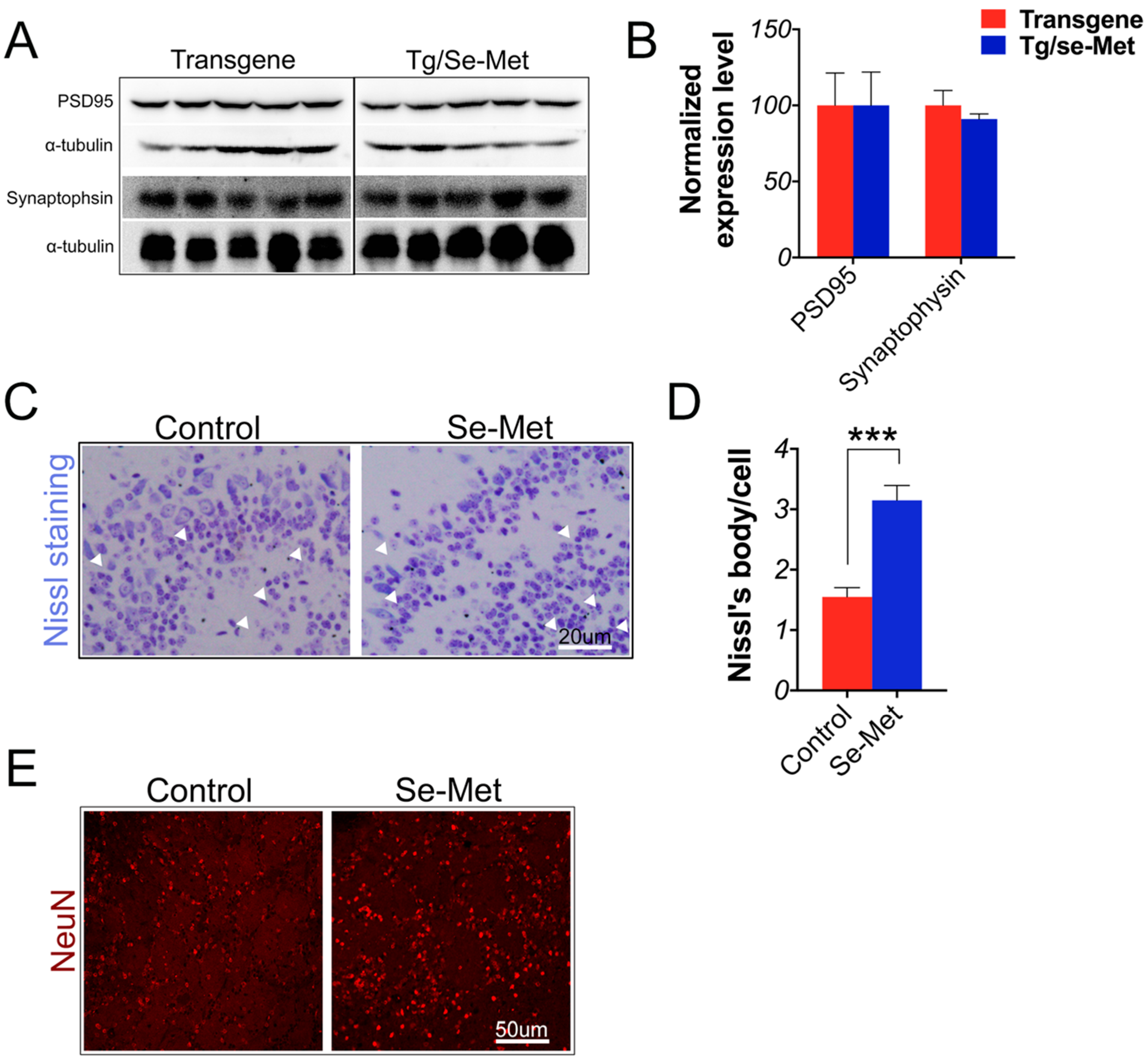
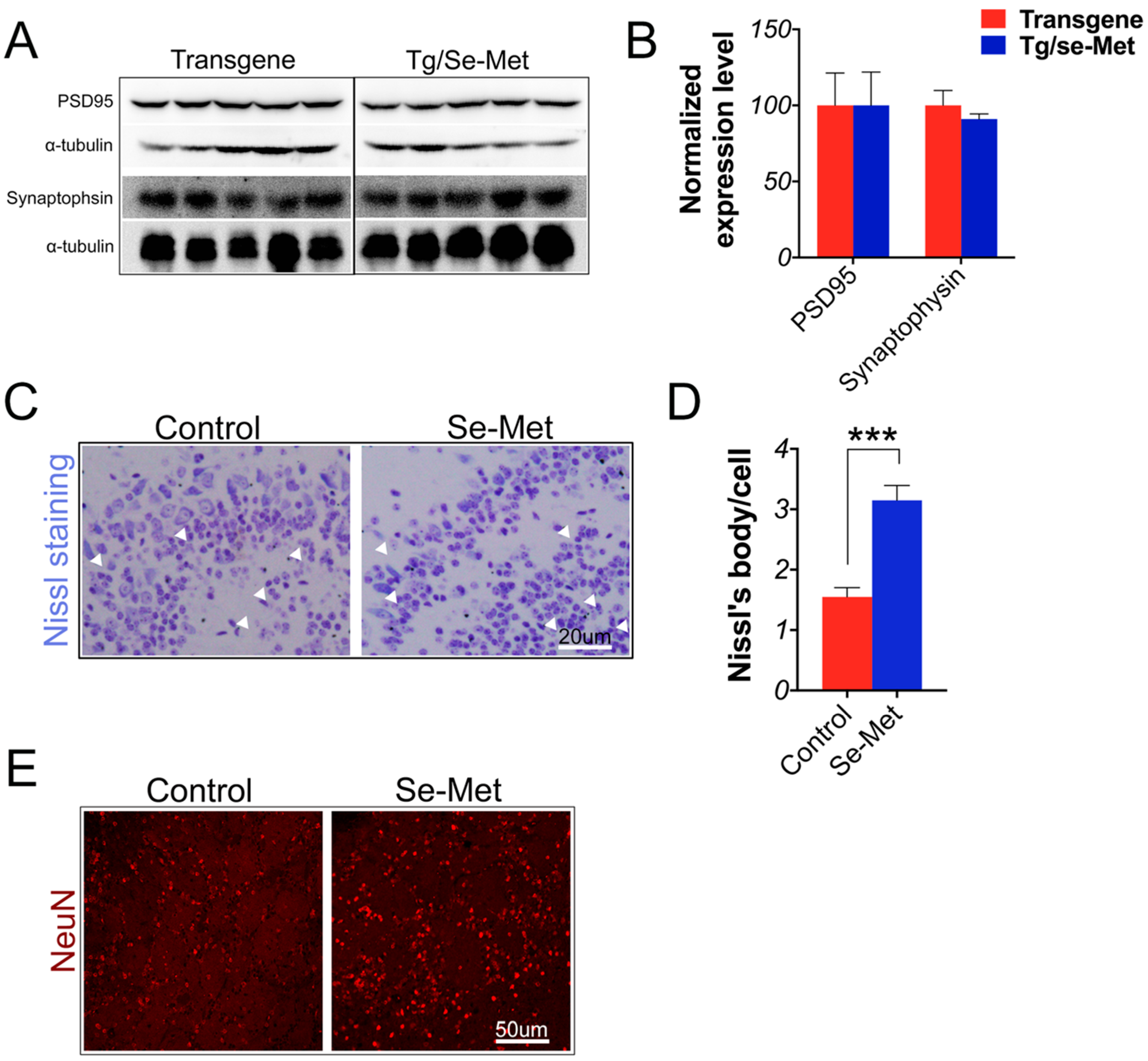
2.5. Se-Met Attenuated Neuronal Cell Death in the OB of 3× Tg-AD Mice
2.6. Se-Met Improved AD Related Pathology in the Primary Neuron of OB
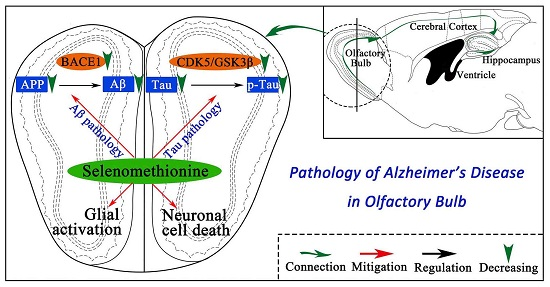
3. Discussion
4. Materials and Methods
4.1. Animals and Treatment
4.2. Immunofluorescent Staining and Histological Analysis
4.3. Immunoblot Analysis
4.4. Primary Neuron Cultures
4.5. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| APP | amyloid precursor protein |
| CDK5 | cyclin-dependent kinase 5 |
| EP | The external plexiform layer |
| Gr | The granule cell layer |
| GSK-3β | glycogen synthase kinase-3β |
| IP | The inner plexiform layer |
| MT | The mitral and tufted cell layer |
| OB | Olfactory bulb |
| PHF | paired helical filaments |
| Se-Met | Selenomethionine |
| 3× Tg-AD mice | Triple transgenic mouse model of AD |
References
- Wesson, D.W.; Levy, E.; Nixon, R.A.; Wilson, D.A. Olfactory dysfunction correlates with amyloid-β burden in an Alzheimer′s disease mouse model. J. Neurosci. 2010, 30, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mucke, L. Alzheimer mechanisms and therapeutic strategies. Cell 2012, 148, 1204–1222. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C. Olfaction in neurodegenerative disorder. Mov. Disord. 2003, 18, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Lee, E.B.; Moberg, P.J.; Stutzbach, L.; Kazi, H.; Han, L.Y.; Lee, V.M.; Trojanowski, J.Q. Olfactory epithelium amyloid-β and paired helical filament-tau pathology in Alzheimer disease. Ann. Neurol. 2010, 67, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Young, J.W.; Sharkey, J.; Finlayson, K. Progressive impairment in olfactory working memory in a mouse model of mild cognitive impairment. Neurobiol. Aging 2009, 30, 1430–1443. [Google Scholar] [CrossRef] [PubMed]
- Van Dijck, A.; Vloeberghs, E.; van Dam, D.; Staufenbiel, M.; de Deyn, P.P. Evaluation of the APP23-model for Alzheimer′s disease in the odour paired-associate test for hippocampus-dependent memory. Behav. Brain Res. 2008, 190, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Guerin, D.; Sacquet, J.; Mandairon, N.; Jourdan, F.; Didier, A. Early locus coeruleus degeneration and olfactory dysfunctions in Tg2576 mice. Neurobiol. Aging 2009, 30, 272–283. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, L.G.; Mannava, S.; Camalier, C.R.; Folley, B.S.; Albritton, A.; Konrad, P.E.; Charles, D.; Park, S.; Neimat, J.S. Emotion recognition in early Parkinson′s disease patients undergoing deep brain stimulation or dopaminergic therapy: A comparison to healthy participants. Front. Aging Neurosci. 2014. [Google Scholar] [CrossRef]
- Kovacs, T.; Cairns, N.J.; Lantos, P.L. β-amyloid deposition and neurofibrillary tangle formation in the olfactory bulb in ageing and Alzheimer′s disease. Neuropathol. Appl. Neurobiol. 1999, 25, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.W.; Wilson, D.A.; Nixon, R.A. Should olfactory dysfunction be used as a biomarker of Alzheimer′s disease? Expert Rev. Neurother. 2010, 10, 633–635. [Google Scholar] [CrossRef] [PubMed]
- Rey, N.L.; Jardanhazi-Kurutz, D.; Terwel, D.; Kummer, M.P.; Jourdan, F.; Didier, A.; Heneka, M.T. Locus coeruleus degeneration exacerbates olfactory deficits in APP/PS1 transgenic mice. Neurobiol. Aging 2012, 33, 426 e1–426 e11. [Google Scholar] [CrossRef] [PubMed]
- Brennan, P.A.; Keverne, E.B. Neural mechanisms of mammalian olfactory learning. Prog. Neurobiol. 1997, 51, 457–481. [Google Scholar] [CrossRef]
- Attems, J.; Jellinger, K.A. Olfactory tau pathology in Alzheimer disease and mild cognitive impairment. Clin. Neuropathol. 2006, 25, 265–271. [Google Scholar] [PubMed]
- Price, J.L.; Davis, P.B.; Morris, J.C.; White, D.L. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer′s disease. Neurobiol. Aging 1991, 12, 295–312. [Google Scholar] [CrossRef]
- Attems, J.; Walker, L.; Jellinger, K.A. Olfactory bulb involvement in neurodegenerative diseases. Acta Neuropathol. 2014, 127, 459–475. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, S.; Berry, M.J. Selenium: Role of the essential metalloid in health. Met. Ions Life Sci. 2013, 13, 499–534. [Google Scholar] [PubMed]
- Song, G.; Zhang, Z.; Wen, L.; Chen, C.; Shi, Q.; Zhang, Y.; Ni, J.; Liu, Q. Selenomethionine ameliorates cognitive decline, reduces tau hyperphosphorylation, and reverses synaptic deficit in the triple transgenic mouse model of Alzheimer′s disease. J. Alzheimers Dis. 2014, 41, 85–99. [Google Scholar] [PubMed]
- Zhang, Z.; Song, M.; Liu, X.; Kang, S.S.; Kwon, I.S.; Duong, D.M.; Seyfried, N.T.; Hu, W.T.; Liu, Z.; Wang, J.Z.; et al. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer′s disease. Nat. Med. 2014, 20, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer′s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Di Meco, A.; Lauretti, E.; Vagnozzi, A.N.; Pratico, D. Zileuton restores memory impairments and reverses amyloid and tau pathology in aged Alzheimer′s disease mice. Neurobiol. Aging 2014, 35, 2458–2464. [Google Scholar] [CrossRef] [PubMed]
- Cassano, T.; Romano, A.; Macheda, T.; Colangeli, R.; Cimmino, C.S.; Petrella, A.; LaFerla, F.M.; Cuomo, V.; Gaetani, S. Olfactory memory is impaired in a triple transgenic model of Alzheimer disease. Behav. Brain Res. 2011, 224, 408–412. [Google Scholar] [CrossRef] [PubMed]
- Doucette, W.; Milder, J.; Restrepo, D. Adrenergic modulation of olfactory bulb circuitry affects odor discrimination. Learn. Mem. 2007, 14, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Saiz-Sanchez, D.; de la Rosa-Prieto, C.; Ubeda-Banon, I.; Martinez-Marcos, A. Interneurons and β-amyloid in the olfactory bulb, anterior olfactory nucleus and olfactory tubercle in APPxPS1 transgenic mice model of Alzheimer′s disease. Anat. Rec. 2013, 296, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Lopez-Guzman, M.; Schoen, C.; Fitzgerald, S.; Lauer, S.L.; Nixon, R.A.; Levy, E.; Wilson, D.A. Spared piriform cortical single-unit odor processing and odor discrimination in the Tg2576 mouse model of Alzheimer′s disease. PLoS ONE 2014, 9, e106431. [Google Scholar]
- Saiz-Sanchez, D.; Ubeda-Banon, I.; de la Rosa-Prieto, C.; Martinez-Marcos, A. Differential expression of interneuron populations and correlation with amyloid-β deposition in the olfactory cortex of an AβPP/PS1 transgenic mouse model of Alzheimer′s disease. J. Alzheimers Dis. 2012, 31, 113–129. [Google Scholar] [PubMed]
- Hu, Y.; Ding, W.; Zhu, X.; Chen, R.; Wang, X. Olfactory dysfunctions and decreased nitric oxide production in the brain of human P301L tau transgenic mice. Neurochem. Res. 2016, 41, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Rao, X.; Gao, Y.; Wang, J.; Xu, F. Amyloid-β deposition and olfactory dysfunction in an Alzheimer′s disease model. J. Alzheimers Dis. 2013, 37, 699–712. [Google Scholar] [PubMed]
- Lei, M.; Xu, H.; Li, Z.; Wang, Z.; O′Malley, T.T.; Zhang, D.; Walsh, D.M.; Xu, P.; Selkoe, D.J.; Li, S. Soluble Aβ oligomers impair hippocampal LTP by disrupting glutamatergic/GABAergic balance. Neurobiol. Dis. 2016, 85, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Martinez, R.; Salgado-Puga, K.; Pena-Ortega, F. Amyloid β inhibits olfactory bulb activity and the ability to smell. PLoS ONE 2013, 8, e75745. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer′s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Maurya, S.K.; Mishra, J.; Abbas, S.; Bandyopadhyay, S. Cypermethrin stimulates GSK3β-dependent Aβ and p-tau proteins and cognitive loss in young rats: Reduced HB-EGF signaling and downstream neuroinflammation as critical regulators. Mol. Neurobiol. 2016, 53, 968–982. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.T.; Wu, Y.; Zou, H.; Wang, R.; Zhou, W.; Kinoshita, A.; Zhang, M.; Yang, Y.; Cai, F.; Woodgett, J.; et al. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Investig. 2013, 123, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Lalioti, V.; Pulido, D.; Sandoval, I.V. Cdk5, the multifunctional surveyor. Cell Cycle 2010, 9, 284–311. [Google Scholar] [CrossRef] [PubMed]
- Angelo, M.; Plattner, F.; Giese, K.P. Cyclin-dependent kinase 5 in synaptic plasticity, learning and memory. J. Neurochem. 2006, 99, 353–370. [Google Scholar] [CrossRef] [PubMed]
- Kusakawa, G.; Saito, T.; Onuki, R.; Ishiguro, K.; Kishimoto, T.; Hisanaga, S. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J. Biol. Chem. 2000, 275, 17166–17172. [Google Scholar] [CrossRef] [PubMed]
- Su, S.C.; Seo, J.; Pan, J.Q.; Samuels, B.A.; Rudenko, A.; Ericsson, M.; Neve, R.L.; Yue, D.T.; Tsai, L.H. Regulation of N-type voltage-gated calcium channels and presynaptic function by cyclin-dependent kinase 5. Neuron 2012, 75, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Hamdane, M.; Sambo, A.V.; Delobel, P.; Begard, S.; Violleau, A.; Delacourte, A.; Bertrand, P.; Benavides, J.; Buee, L. Mitotic-like tau phosphorylation by p25-Cdk5 kinase complex. J. Biol. Chem. 2003, 278, 34026–34034. [Google Scholar] [CrossRef] [PubMed]
- Peterson, D.W.; Ando, D.M.; Taketa, D.A.; Zhou, H.; Dahlquist, F.W.; Lew, J. No difference in kinetics of tau or histone phosphorylation by CDK5/p25 versus CDK5/p35 in vitro. Proc. Natl. Acad. Sci. USA 2010, 107, 2884–2889. [Google Scholar] [CrossRef] [PubMed]
- Castro-Alvarez, J.F.; Uribe-Arias, S.A.; Mejia-Raigosa, D.; Cardona-Gomez, G.P. Cyclin-dependent kinase 5, a node protein in diminished tauopathy: A systems biology approach. Front. Aging Neurosci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Castro-Alvarez, J.F.; Uribe-Arias, A.; Cardona-Gomez, G.P. Cyclin-dependent kinase 5 targeting prevents β-Amyloid aggregation involving glycogen synthase kinase 3β and phosphatases. J. Neurosci. Res. 2015, 93, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer′s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Gylys, K.H.; Fein, J.A.; Yang, F.; Wiley, D.J.; Miller, C.A.; Cole, G.M. Synaptic changes in Alzheimer′s disease: Increased amyloid-β and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am. J. Pathol. 2004, 165, 1809–1817. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, B.; Liu, D.; Li, J.J.; Xue, Y.; Sakata, K.; Zhu, L.Q.; Heldt, S.A.; Xu, H.; Liao, F.F. Hsp90 chaperone inhibitor 17-AAG attenuates Aβ-induced synaptic toxicity and memory impairment. J. Neurosci. 2014, 34, 2464–2470. [Google Scholar] [CrossRef] [PubMed]
- Vedagiri, A.; Thangarajan, S. Mitigating effect of chrysin loaded solid lipid nanoparticles against Amyloid β23–25 induced oxidative stress in rat hippocampal region: An efficient formulation approach for Alzheimer′s disease. Neuropeptides 2016, 58, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.U.; Shah, S.A.; Ali, T.; Chung, J.I.; Kim, M.O. Anthocyanins reversed d-galactose-induced oxidative stress and neuroinflammation mediated cognitive impairment in adult rats. Mol. Neurobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, R.; Stolmeier, D.; Yang, X.; Anantharam, P.; Zaheer, A. Expression of glia maturation factor in neuropathological lesions of Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Furuta, T.; Ohshima, C.; Matsumura, M.; Takebayashi, N.; Hirota, E.; Mawaribuchi, T.; Nishida, K.; Nagasawa, K. Oxidative stress upregulates zinc uptake activity via Zrt/Irt-like protein 1 (ZIP1) in cultured mouse astrocytes. Life Sci. 2016, 151, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Kitazawa, M.; Tseng, B.P.; LaFerla, F.M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Aβ causes the onset of early Alzheimer′s disease-related cognitive deficits in transgenic mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Host | Application | Source |
|---|---|---|---|
| APP | Mouse | IMB | Millipore (Billerica, MA, USA) |
| 6E10 | Mouse | IMB/IMF | Covance (Princeton, NJ, USA) |
| sAPPβ | Mouse | IMB/IMF | Covance |
| BACE1 | Rabbit | IMB | Abcam (Cambridge, UK) |
| Tau5 | Mouse | IMB/IMF | Abcam |
| Tau-pS404 | Rabbit | IMB/IMF | Abcam |
| Tau-pS422 | Rabbit | IMB | Abcam |
| GFAP | Rabbit | IMB/IMF | Abcam |
| CD45 | Rabbit | IMB/IMF | Abcam |
| PSD95 | Rabbit | IMB | Cell signaling (Danvers, MA, USA) |
| Synaptophysin | Rabbit | IMB | Cell signaling |
| Akt | Rabbit | IMB | Cell signaling |
| p-Akt | Rabbit | IMB | Cell signaling |
| CDK5 | Rabbit | IMB | Cell signaling |
| GSK-3β | Mouse | IMB | Cell signaling |
| p-GSK-3β | Rabbit | IMB | Cell signaling |
| PP2A-C | Rabbit | IMB | Cell signaling |
| NeuN | Rabbit | IMF | Abcam |
| p-PP2A | Rabbit | IMB | Abcam |
| β-actin | Rabbit | IMB | Sigma (Santa clara, CA, USA) |
| α-tubulin | Mouse | IMB | Sigma |
| GAPDH | Rabbit | IMB | Proteintech (Rosemont, CA, USA) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.-H.; Chen, C.; Wu, Q.-Y.; Zheng, R.; Chen, Y.; Liu, Q.; Ni, J.-Z.; Song, G.-L. Selenomethionine Ameliorates Neuropathology in the Olfactory Bulb of a Triple Transgenic Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2016, 17, 1595. https://doi.org/10.3390/ijms17101595
Zhang Z-H, Chen C, Wu Q-Y, Zheng R, Chen Y, Liu Q, Ni J-Z, Song G-L. Selenomethionine Ameliorates Neuropathology in the Olfactory Bulb of a Triple Transgenic Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences. 2016; 17(10):1595. https://doi.org/10.3390/ijms17101595
Chicago/Turabian StyleZhang, Zhong-Hao, Chen Chen, Qiu-Yan Wu, Rui Zheng, Yao Chen, Qiong Liu, Jia-Zuan Ni, and Guo-Li Song. 2016. "Selenomethionine Ameliorates Neuropathology in the Olfactory Bulb of a Triple Transgenic Mouse Model of Alzheimer’s Disease" International Journal of Molecular Sciences 17, no. 10: 1595. https://doi.org/10.3390/ijms17101595
APA StyleZhang, Z.-H., Chen, C., Wu, Q.-Y., Zheng, R., Chen, Y., Liu, Q., Ni, J.-Z., & Song, G.-L. (2016). Selenomethionine Ameliorates Neuropathology in the Olfactory Bulb of a Triple Transgenic Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences, 17(10), 1595. https://doi.org/10.3390/ijms17101595