
Innovative Target Therapies Are Able to Block the Inflammation Associated with Dysfunction of the Cholesterol Biosynthesis Pathway

 , , ,
, , ,
Abstract
:1. The Cholesterol Pathway: A Pleiotropic Biochemical System
HMG-CoA Reductase Controls the Cholesterol Pathway
2. Diseases Linked to the Deregulation of the CP

{kind=link}
{kind=link}
| Disease/Syndrome | MIM | Genetics | Gene(s) Involved | Protein Involved | Molecular Features | Main Clinical Features |
|---|---|---|---|---|---|---|
| Mevalonate Kinase Deficiency (MKD) ‡ [15,16] | #260920 #610377 | Autosomal recessive | MVK | Mevalonate kinase | Accumulation of mevalonic acid in urine and plasma | Elevated serum IgD/IgA, periodic fever, vomiting, diarrhea, psychomotor retardation, developmental delay, cerebellar and cerebral atrophy |
| Smith Lemli Opitz Syndrome (SLOS) [17,18,19,20] | #270400 | Autosomal recessive | DHCR7 | 7-dehydrocholesterol reductase | Low cholesterol levels, accumulation of 7-DHC | Failure to thrive, microcephaly, micrognathia, ambiguous genitalia, limb shortening, polydactyly, mental retardation |
| Conradi-Hunermann-Happle [19,21] | #302960 | X-linked dominant | EBP | Sterol-Δ8–Δ7-isomerase | Increased levels of 8-dehydrocholesterol and 8(9)-cholestenol | Growth deficiency, asymmetric limb shortening, mental retardation, ventriculomegaly |
| CHILD syndrome [19,22,23] | #308050 | X-linked dominant | NSDHL | Part of the C-4 sterol demethylase protein complex | Increased levels of 8-dehydrocholesterol and 8(9)-cholestenol | Prenatal growth deficiency, hearing loss, unilateral distribution of abnormalities, skin lesions, erythema, severe skeletal abnormalities |
| Greenberg skeletal dysplasia [19,24] | #215140 | Autosomal recessive | LBR | 3β-hydroxysteroid-Δ14-reductase | Elevated cholesta-8,14-dien-3-β-ol in cultured fibroblasts and cholesta-8,14,24-trien-3β-ol in cartilage | Hydrops-ectopic calcification-moth-eaten (HEM) skeletal dysplasia, fetal death |
| Lathosterolosis [19,25] | #607330 | Autosomal recessive | SC5DL | 3β-hydroxysteroid-Δ5-desaturase | Increased levels of lathosterol in plasma and cultured fibroblast; absent 7-dehydrocholesterol, normal cholesterol | Microcephaly, polysyndactyly, colestatic liver disease, conductive deafness, severe psychomotor retardation |
| Desmosterolosis [19,26,27,28] | #602398 | Autosomal recessive | DHCR24 | 3β-hydroxysterol-Δ24-reductase | Accumulations of desmosterol in plasma, kidney, liver, brain | Failure to thrive, microcephaly, anomalous pulmonary venous drainage, ambiguous genitalia, short limbs, generalized osteosclerosis, delayed psychomotor development, severe spasticity |
3. Convergent Pathogenic Mechanisms on Deregulation of the Cholesterol Pathway
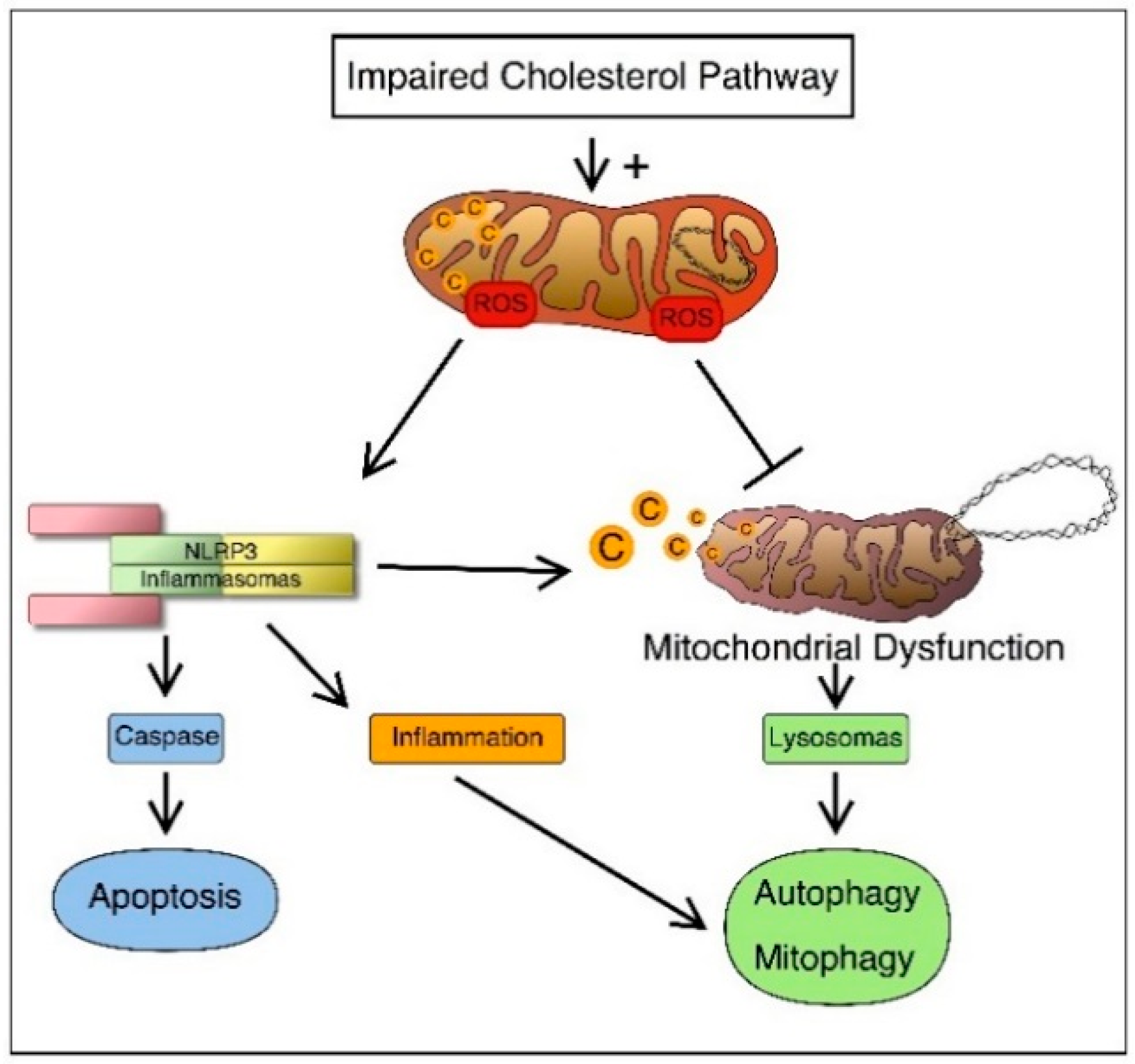
3.1. Inflammatory Mechanisms

3.2. Programmed Cell Death
3.3. Autophagy and Mitophagy
4. Innovative Target Therapies Counteracting Inflammation Caused by Cholesterol Dysfunctions
4.1. Inhibitors of Cholesterol Synthesis
4.2. Mitochondrial-Target Anti-Oxidants
4.3. NLRP3 Inhibitors
5. Outstanding Questions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Thurnher, M.; Gruenbacher, G.; Nussbaumer, O. Regulation of mevalonate metabolism in cancer and immune cells. Biochim. Biophys. Acta 2013, 1831, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Hinson, D.D.; Chambliss, K.L.; Toth, M.J.; Tanaka, R.D.; Gibson, K.M. Post-translational regulation of mevalonate kinase by intermediates of the cholesterol and nonsterol isoprene biosynthetic pathways. J. Lipid Res. 1997, 38, 2216–2223. [Google Scholar] [PubMed]
- Ikeda, U.; Shimada, K. Pleiotropic effects of statins on the vascular tissue. Curr. Drug Targets Cardiovasc. Haematol. Disord. 2001, 1, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.; DeBose-Boyd, R.A. Control of cholesterol synthesis through regulated ER-associated degradation of HMG CoA reductase. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Goldstein, J.L.; Brown, M.S. Multivalent control of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Mevalonate-derived product inhibits translation of mRNA and accelerates degradation of enzyme. J. Biol. Chem. 1988, 263, 8929–8937. [Google Scholar] [PubMed]
- Hampton, R.; Dimster-Denk, D.; Rine, J. The biology of HMG-CoA reductase: The pros of contra-regulation. Trends Biochem. Sci. 1996, 21, 140–145. [Google Scholar] [CrossRef]
- Crisby, M. Modulation of the inflammatory process by statins. Drugs Today 2003, 39, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. Progress in understanding the LDL receptor and HMG-CoA reductase, two membrane proteins that regulate the plasma cholesterol. J. Lipid Res. 1984, 25, 1450–1461. [Google Scholar] [PubMed]
- Jira, P. Cholesterol metabolism deficiency. Handb. Clin. Neurol. 2013, 113, 1845–1850. [Google Scholar] [PubMed]
- Marcuzzi, A.; Decorti, G.; Pontillo, A.; Ventura, A.; Tommasini, A. Decreased cholesterol levels reflect a consumption of anti-inflammatory isoprenoids associated with an impaired control of inflammation in a mouse model of mevalonate kinase deficiency. Inflamm. Res. 2010, 59, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Herman, G.E. Disorders of cholesterol biosynthesis: Prototypic metabolic malformation syndromes. Hum. Mol. Genet. 2003, 12, R75–R88. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzi, A.; Pontillo, A.; De Leo, L.; Tommasini, A.; Decorti, G.; Not, T.; Ventura, A. Natural isoprenoids are able to reduce inflammation in a mouse model of mevalonate kinase deficiency. Pediatr. Res. 2008, 64, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Mulders-Manders, C.M.; Simon, A. Hyper-IgD syndrome/mevalonate kinase deficiency: What is new? Semin. Immunopathol. 2015, 37, 371–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varret, M.; Abifadel, M.; Rabès, J.P.; Boileau, C. Genetic heterogeneity of autosomal dominant hypercholesterolemia. Clin. Genet. 2008, 73, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Van der Meer, J.W.; Vossen, J.M.; Radl, J.; van Nieuwkoop, J.A.; Meyer, C.J.; Lobatto, S.; van Furth, R. Hyperimmunoglobulinaemia D and periodic fever: A new syndrome. Lancet 1984, 1, 1087–1090. [Google Scholar] [CrossRef]
- Drenth, J.P.; van der Meer, J.W. Hereditary periodic fever. N. Engl. J. Med. 2001, 345, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Opitz, J.M.; Penchaszadeh, V.B.; Holt, M.C.; Spano, L.M. Smith-Lemli-Opitz (RSH) syndrome bibliography. Am. J. Med. Genet. 1987, 28, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Tint, G.S.; Irons, M.; Elias, E.R.; Batta, A.K.; Frieden, R.; Chen, T.S.; Salen, G. Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N. Engl. J. Med. 1994, 330, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.I. Inborn errors of cholesterol biosynthesis. Adv. Pediatr. 2000, 47, 1–53. [Google Scholar] [PubMed]
- Platt, F.M.; Wassif, C.; Colaco, A.; Dardis, A.; Lloyd-Evans, E.; Bembi, B.; Porter, F.D. Disorders of cholesterol metabolism and their unanticipated convergent mechanisms of disease. Annu. Rev. Genom. Hum. Genet. 2014, 15, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Derry, J.M.; Gormally, E.; Means, G.D.; Zhao, W.; Meindl, A.; Kelley, R.I.; Boyd, Y.; Herman, G.E. Mutations in a delta 8-Δ7 sterol isomerase in the tattered mouse and X-linked dominant chondrodysplasia punctata. Nat. Genet. 1999, 22, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Falek, A.; Heath, C.W., Jr.; Ebbin, A.J.; McLean, W.R. Unilateral limb and skin deformities with congenital heart disease in two siblings: A lethal syndrome. J. Pediatr. 1968, 73, 910–913. [Google Scholar] [CrossRef]
- Happle, R.; Koch, H.; Lenz, W. The CHILD syndrome. Congenital hemidysplasia with ichthyosiform erythroderma and limb defects. Eur. J. Pediatr. 1980, 134, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, C.R.; Rimoin, D.L.; Gruber, H.E.; DeSa, D.J.; Reed, M.; Lachman, R.S. A new autosomal recessive lethal chondrodystrophy with congenital hydrops. Am. J. Med. Genet. 1988, 29, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, N.; Corso, G.; Rossi, M.; Ferrari, P.; Balli, F.; Rivasi, F.; Annunziata, I.; Ballabio, A.; Russo, A.D.; Andria, G.; et al. Lathosterolosis, a novel multiple-malformation/mental retardation syndrome due to deficiency of 3β-hydroxysteroid-Δ5-desaturase. Am. J. Hum. Genet. 2002, 71, 952–958. [Google Scholar] [CrossRef] [PubMed]
- FitzPatrick, D.R.; Keeling, J.W.; Evans, M.J.; Kan, A.E.; Bell, J.E.; Porteous, M.E.; Mills, K.; Winter, R.M.; Clayton, P.T. Clinical phenotype of desmosterolosis. Am. J. Med. Genet. 1998, 75, 145–152. [Google Scholar] [CrossRef]
- Waterham, H.R.; Koster, J.; Romeijn, G.J.; Hennekam, R.C.; Vreken, P.; Andersson, H.C.; FitzPatrick, D.R.; Kelley, R.I.; Wanders, R.J. Mutations in the 3β-hydroxysterol Δ24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis. Am. J. Hum. Genet. 2001, 69, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M. Emerging inflammasome effector mechanisms. Nat. Rev. Immunol. 2011, 11, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Masters, S.L. Specific inflammasomes in complex diseases. Clin. Immunol. 2013, 147, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Faustin, B.; Lartigue, L.; Bruey, J.M.; Luciano, F.; Sergienko, E.; Bailly-Maitre, B.; Volkmann, N.; Hanein, D.; Rouiller, I.; Reed, J.C. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol. Cell 2007, 25, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Minkiewicz, J.; de Rivero Vaccari, J.P.; Keane, R.W. Human astrocytes express a novel NLRP2 inflammasome. Glia 2013, 61, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, E.; Campbell, M.; Doyle, S.L. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: Current perspectives. J. Inflamm. Res. 2015, 8, 15–27. [Google Scholar] [PubMed]
- Anand, P.K.; Malireddi, R.K.; Lukens, J.R.; Vogel, P.; Bertin, J.; Lamkanfi, M.; Kanneganti, T.D. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 2012, 488, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Dorfleutner, A.; Bryan, N.B.; Yun, C.; Radian, A.D.; de Almeida, L.; Rojanasakul, Y.; Stehlik, C. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 2012, 36, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Tuncer, S.; Fiorillo, M.T.; Sorrentino, R. The multifaceted nature of NLRP12. J. Leukoc. Biol. 2014, 96, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Vance, R.E. The NAIP/NLRC4 inflammasomes. Curr. Opin. Immunol. 2015, 32, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Haneklaus, M.; O’Neill, L.A. NLRP3 at the interface of metabolism and inflammation. Immunol. Rev. 2015, 265, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, F.; Bartok, E.; Rieger, A.; Franchi, L.; Nunez, G.; Hornung, V. Cutting edge: Reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 2011, 187, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 8, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Cuellar, E.; Tsuchiya, K.; Hara, H.; Fang, R.; Sakai, S.; Kawamura, I.; Akira, S.; Mitsuyama, M. Cutting edge: Nitric oxide inhibits the NLRP3 inflammasome. J. Immunol. 2012, 189, 5113–5117. [Google Scholar] [CrossRef] [PubMed]
- Caito, S.W.; Aschner, M. Mitochondrial redox dysfunction and environmental exposures. Antioxid. Redox Signal. 2015. [Google Scholar] [CrossRef] [PubMed]
- Sorriento, D.; Pascale, A.V.; Finelli, R.; Carillo, A.L.; Annunziata, R.; Trimarco, B.; Iaccarino, G. Targeting mitochondria as therapeutic strategy for metabolic disorders. Sci. World J. 2014, 2014, 604685. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Ungvari, Z. Role of mitochondrial oxidative stress in hypertension. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1417–H1427. [Google Scholar] [CrossRef] [PubMed]
- Münzel, T.; Gori, T.; Bruno, R.M.; Taddei, S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart J. 2010, 22, 2741–2748. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.X. Mitochondrial dysfunction, oxidative stress and diabetic cardiovascular disorders. Cardiovasc. Hematol. Disord. Drug Targets 2012, 12, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.R.; Cheng, K.K.; Figg, N.; Gorenne, I.; Mahmoudi, M.; Griffin, J.; Vidal-Puig, A.; Logan, A.; Murphy, M.P.; Bennett, M. DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ. Res. 2010, 107, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Vercesi, A.E.; Castilho, R.F.; Kowaltowski, A.J.; Oliveira, H.C. Mitochondrial energy metabolism and redox state in dyslipidemias. IUBMB Life 2007, 59, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Clendening, J.W.; Pandyra, A.; Boutros, P.C.; el Ghamrasni, S.; Khosravi, F.; Trentin, G.A.; Martirosyan, A.; Hakem, A.; Hakem, R.; Jurisica, I.; et al. Dysregulation of the mevalonate pathway promotes transformation. Proc. Natl. Acad. Sci. USA 2010, 107, 15051–15056. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Dysregulation of cholesterol balance in the brain: Contribution to neurodegenerative diseases. Dis. Model. Mech. 2012, 6, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Van der Burgh, R.; Boes, M. Mitochondria in autoinflammation: Cause, mediator or bystander? Trends Endocrinol. Metab. 2015, 26, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Venditti, P.; di Stefano, L.; di Meo, S. Mitochondrial metabolism of reactive oxygen species. Mitochondrion 2013, 13, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, G.; Benedict, M.A.; Hu, Y.; Inohara, N. Caspases: The proteases of the apoptotic pathway. Oncogene 1998, 17, 3237–3245. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Tricarico, P.M.; Marcuzzi, A.; Piscianz, E.; Monasta, L.; Crovella, S.; Kleiner, G. Mevalonate kinase deficiency and neuroinflammation: Balance between apoptosis and pyroptosis. Int. J. Mol. Sci. 2013, 14, 23274–23288. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzi, A.; Tricarico, P.M.; Piscianz, E.; Kleiner, G.; Vecchi Brumatti, L.; Crovella, S. Lovastatin induces apoptosis through the mitochondrial pathway in an undifferentiated SH-SY5Y neuroblastoma cell line. Cell Death Dis. 2013, 4, e585. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Yang, Y.; Ming, M.; Liu, B. Mitochondrial ROS generation for regulation of autophagic pathways in cancer. Biochem. Biophys. Res. Commun. 2011, 414, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Klionsky, D.J. Mitochondria removal by autophagy. Autophagy 2011, 7, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M. Keeping the immune system in check: a role for mitophagy. Immunol. Cell Biol. 2015, 93, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Braschi, E.; McBride, H.M. Mitochondria and the culture of the Borg: Understanding the integration of mitochondrial function within the reticulum, the cell, and the organism. Bioessays 2010, 32, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Pinton, P. Mitophagy and mitochondrial balance. Methods Mol. Biol. 2015, 1241, 181–194. [Google Scholar] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Ren, G.; Steiner, R.D.; Merkens, L.; Roullet, J.B.; Korade, Z.; DiMuzio, P.J.; Tulenko, T.N. Elevated autophagy and mitochondrial dysfunction in the Smith-Lemli-Opitz Syndrome. Mol. Genet. Metab. Rep. 2014, 1, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Van der Burgh, R.; Pervolaraki, K.; Turkenburg, M.; Waterham, H.R.; Frenkel, J.; Boes, M. Unprenylated RhoA contributes to IL-1β hypersecretion in mevalonate kinase deficiency model through stimulation of Rac1 activity. J. Biol. Chem. 2014, 289, 27757–27765. [Google Scholar] [CrossRef] [PubMed]
- Van der Burgh, R.; Nijhuis, L.; Pervolaraki, K.; Compeer, E.B.; Jongeneel, L.H.; van Gijn, M.; Coffer, P.J.; Murphy, M.P.; Mastroberardino, P.G.; Frenkel, J.; et al. Defects in mitochondrial clearance predispose human monocytes to interleukin-1β hypersecretion. J. Biol. Chem. 2014, 289, 5000–5012. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzi, A.; Crovella, S.; Monasta, L.; Vecchi Brumatti, L.; Gattorno, M.; Frenkel, J. Mevalonate kinase deficiency: Disclosing the role of mevalonate pathway modulation in inflammation. Curr. Pharm. Des. 2012, 18, 5746–5752. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.; Feely, J. Pharmacokinetic-pharmacodynamic drug interactions with HMG-CoA reductase inhibitors. Clin. Pharmacokinet. 2002, 41, 343–370. [Google Scholar] [CrossRef] [PubMed]
- Endo, A. The discovery and development of HMG-CoA reductase inhibitors. J. Lipid Res. 1992, 33, 1569–1582. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S; Yeganeh, B.; Stelmack, G.L.; Kashani, H.H.; Sharma, P.; Cunnington, R.; Rattan, S.; Bathe, K.; Klonisch, T.; Dixon, I.M.; et al. Apoptosis, autophagy and ER stress in mevalonate cascade inhibition-induced cell death of human atrial fibroblasts. Cell Death Dis. 2012, 3, e330. [Google Scholar] [CrossRef]
- Stone, N.J. Current drug treatments for lipid management. Manag. Care 2002, 9S, 4–9. [Google Scholar]
- Bandeali, S.J.; Daye, J.; Virani, S.S. Novel therapies for treating familial hypercholesterolemia. Curr. Atheroscler. Rep. 2014, 16, 382. [Google Scholar] [CrossRef] [PubMed]
- Fordyce, C.B.; Roe, M.T.; Ahmad, T.; Libby, P.; Borer, J.S.; Hiatt, W.R.; Bristow, M.R.; Packer, M.; Wasserman, S.M.; Braunstein, N.; et al. Cardiovascular drug development: Is it dead or just hibernating? J. Am. Coll. Cardiol. 2015, 65, 1567–1582. [Google Scholar] [CrossRef] [PubMed]
- Šimić, I.; Reiner, Ž. Adverse effects of statins—Myths and reality. Curr. Pharm. Des. 2015, 21, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Catapano, A.L. Statin intolerance: Diagnosis and remedies. Curr. Cardiol. Rep. 2015, 17, 27. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulou, M.; Corsini, A.; Roden, M. The role of mitochondria in statin-induced myopathy. Eur. J. Clin. Investig. 2015, 45, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Izadpanah, R.; Schächtele, D.J.; Pfnür, A.B.; Lin, D.; Slakey, D.P.; Kadowitz, P.J.; Alt, E.U. The impact of statins on biological characteristics of stem cells provides a novel explanation for their pleotropic beneficial and adverse clinical effects. Am. J. Physiol. Cell Physiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Luckman, S.P.; Hughes, D.E.; Coxon, F.P.; Graham, R.; Russell, G.; Rogers, M.J. Nitrogen-containing bisphosphonates inhibit the mevalonate pathway and prevent post-translational prenylation of GTP-binding proteins, including Ras. J. Bone Miner. Res. 1998, 13, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.J. New insights into the molecular mechanisms of action of bisphosphonates. Curr. Pharm. Des. 2003, 9, 2643–2658. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.G. Bisphosphonates: the first 40 years. Bone 2011, 1, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Merrella, M.A.; Wakchourea, S.; Lehenkarib, P.P.; Harrisa, K.W.; Selander, K.S. Inhibition of the mevalonate pathway and activation of p38 MAP kinase are independently regulated by nitrogen-containing bisphosphonates in breast cancer cells. Eur. J. Pharmacol. 2007, 570, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Frith, J.C.; Monkkonen, J.; Blackburn, G.M.; Russell, R.G.; Rogers, M.J. Clodronate and liposome-encapsulated clodronate are metabolized to a toxic ATP analog, adenosine 5′-(β,γ-dichloromethylene) triphosphate, by mammalian cells in vitro. J. Bone Miner. Res. 1997, 12, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Lehenkari, P.P.; Kellinsalmi, M.; Näpänkangas, J.P.; Ylitalo, K.V.; Mönkkönen, J.; Rogers, M.J.; Azhayev, A.; Väänänen, H.K.; Hassinen, I.E. Further insight into mechanism of action of clodronate: Inhibition of mitochondrial ADP/ATP translocase by a nonhydrolyzable, adenine-containing metabolite. Mol. Pharmacol. 2002, 5, 1255–1262. [Google Scholar] [CrossRef]
- Diel, I.J.; Bergner, R.; Grötz, K.A. Adverse effects of bisphosphonates: Current issues. J. Support. Oncol. 2007, 5, 475–482. [Google Scholar] [PubMed]
- Chang, J.T.; Green, L.; Beitz, J. Renal failure with the use of zoledronic acid. N. Engl. J. Med. 2003, 349, 1676–1678. [Google Scholar] [PubMed]
- Bergner, R.; Diel, I.J.; Henrich, D.; Hoffmann, M.; Uppenkamp, M. Differences in nephrotoxicity of intravenous bisphosphonates for the treatment of malignancy-related bone disease. Onkologie 2006, 29, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Pecherstorfer, M.; Jilch, R.; Sauty, A.; Horn, E.; Keck, A.V.; Zimmer-Roth, I.; Thiebaud, D. Effect of first treatment with aminobisphosphonates pamidronate and ibandronate on circulating lymphocyte subpopulations. J. Bone Miner. Res. 2000, 15, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Thiébaud, D.; Sauty, A.; Burckhardt, P.; Leuenberger, P.; Sitzler, L.; Green, J.R.; Kandra, A.; Zieschang, J.; Ibarra de Palacios, P. An in vitro and in vivo study of cytokines in the acute-phase response associated with bisphosphonates. Calcif. Tissue Int. 1997, 61, 386–392. [Google Scholar] [PubMed]
- Rosen, L.S.; Gordon, D.; Tchekmedyian, N.S.; Yanagihara, R.; Hirsh, V.; Krzakowski, M.; Pawlicki, M.; de Souza, P.; Zheng, M.; Urbanowitz, G.; et al. Long-term efficacy and safety of zoledronic acid in the treatment of skeletal metastases in patients with nonsmall cell lung carcinoma and other solid tumors: A randomized, Phase III, double-blind, placebo-controlled trial. Cancer 2004, 100, 2613–2621. [Google Scholar] [CrossRef] [PubMed]
- Dicuonzo, G.; Vincenzi, B.; Santini, D.; Avvisati, G.; Rocci, L.; Battistoni, F.; Gavasci, M.; Borzomati, D.; Coppola, R.; Tonini, G. Fever after zoledronic acid administration is due to increase in TNF-α and IL-6. J. Interferon Cytokine Res. 2003, 23, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.; Rogers, M.J. Statins prevent bisphosphonate induced γ, Δ-T-cell proliferation and activation in vitro. J. Bone Miner. Res. 2004, 19, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.K. The gastrointestinal tolerability and safety of oral bisphosphonates. Expert Opin. Drug Saf. 2002, 1, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Migliorati, C.A.; Casiglia, J.; Epstein, J.; Jacobsen, P.L.; Siegel, M.A.; Woo, S.B. Managing the care of patients with bisphosphonate-associated osteonecrosis: An American academy of oral medicine position paper. J. Am. Dent. Assoc. 2005, 136, 1658–1668. [Google Scholar]
- Elsayed, R.K.; Evans, J.D. Emerging lipid-lowering drugs: Squalene synthase inhibitors. Expert Opin. Emerg. Drugs 2008, 13, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K. Squalene synthase inhibitor lapaquistat acetate: Could anything be better than statins? Circulation 2011, 123, 1925–1928. [Google Scholar] [CrossRef] [PubMed]
- Mo, H.; Elson, C.E. Studies of the isoprenoid-mediated inhibition of mevalonate synthesis applied to cancer chemotherapy and chemoprevention. Exp. Biol. Med. 2004, 229, 567–585. [Google Scholar]
- Marcuzzi, A.; de Leo, L.; Decorti, G.; Crovella, S.; Tommasini, A.; Pontillo, A. The farnesyltransferase inhibitors tipifarnib and lonafarnib inhibit cytokines secretion in a cellular model of mevalonate kinase deficiency. Pediatr. Res. 2011, 70, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Olszewska, A.; Szewczyk, A. Mitochondria as a pharmacological target: Magnum overview. IUBMB Life 2013, 65, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Cochemé, H.M.; Kelso, G.F.; James, A.M.; Ross, M.F.; Trnka, J.; Mahendiran, T.; Asin-Cayuela, J.; Blaikie, F.H.; Manas, A.R.; Porteous, C.M.; et al. Mitochondrial targeting of quinones: Therapeutic implications. Mitochondrion 2007, S94–S102. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, A.; Kotamraju, S.; Kalivendi, S.V.; Matsunaga, T.; Shang, T.; Keszler, A.; Joseph, J.; Kalyanaraman, B. Supplementation of endothelial cells with mitochondria-targeted antioxidants inhibit peroxide-induced mitochondrial iron uptake, oxidative damage, and apoptosis. J. Biol. Chem. 2004, 279, 37575–37587. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Porteous, C.M.; Coulter, C.V.; Murphy, M.P. Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 1999, 263, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Mitochondrial oxidative damage in aging and Alzheimer’s disease: Implications for mitochondrially targeted antioxidant therapeutics. J. Biomed. Biotechnol. 2006, 3, 31372. [Google Scholar] [CrossRef] [PubMed]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef] [PubMed]
- Hwang, P.M.; Bunz, F.; Yu, J.; Rago, C.; Chan, T.A.; Murphy, M.P.; Kelso, G.F.; Smith, R.A.; Kinzler, K.W.; Vogelstein, B. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat. Med. 2001, 7, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Saretzki, G.; Murphy, M.P.; von Zglinicki, T. MitoQ counteracts telomere shortening and elongates lifespan of fibroblasts under mild oxidative stress. Aging Cell 2003, 2, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.; Schafer, C.; Ewald, N.; Piper, H.M.; Noll, T. Role of redox signaling in the autonomous proliferative response of endothelial cells to hypoxia. Circ. Res. 2003, 92, 1010–1015. [Google Scholar] [CrossRef] [PubMed]
- Barhoumi, R.; Faske, J.; Liu, X.; Tjalkens, R.B. Manganese potentiates lipopolysaccharide-induced expression of NOS2 in C6 glioma cells through mitochondrial-dependent activation of nuclear factor κB. Brain Res. Mol. Brain Res. 2004, 122, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Kalivendi, S.V.; Konorev, E.A.; Cunningham, S.; Vanamala, S.K.; Kaji, E.H.; Joseph, J.; Kalyanaraman, B. Doxorubicin activates nuclear factor of activated T-lymphocytes and Fas ligand transcription: Role of mitochondrial reactive oxygen species and calcium. Biochem. J. 2005, 389, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.; Verkaart, S.; Visch, H.J.; van der Westhuizen, F.H.; Murphy, M.P.; van den Heuvel, L.W.; Smeitink, J.A.; Willems, P.H. Inhibition of complex I of the electron transport chain causes view the MathML sourceO2—Mediated mitochondrial outgrowth. Am. J. Physiol. Cell Physiol. 2005, 288, C1440–C1450. [Google Scholar] [CrossRef] [PubMed]
- Pletjushkina, O.Y.; Fetisova, E.K.; Lyamzaev, K.G.; Ivanova, O.Y.; Domnina, L.V.; Vyssokikh, M.Y.; Pustovidko, A.V.; Vasiliev, J.M.; Murphy, M.P.; Chernyak, B.V.; et al. Long-distance apoptotic killing of cells is mediated by hydrogen peroxide in a mitochondrial ROS-dependent fashion. Cell Death Differ. 2005, 12, 1442–1444. [Google Scholar] [CrossRef] [PubMed]
- Siler-Marsiglio, K.I.; Pan, Q.; Paiva, M.; Madorsky, I.; Khurana, N.C.; Heaton, M.B. Mitochondrially targeted vitamin E and vitamin E mitigate ethanol-mediated effects on cerebellar granule cell antioxidant defense systems. Brain Res. 2005, 1052, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.F.; Kelso, G.F.; Blaikie, F.H.; James, A.M.; Cocheme, H.M.; Filipovska, A.; da Ros, T.; Hurd, T.R.; Smith, R.A.; Murphy, M.P. Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochemistry 2005, 70, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Crane, F.L. Hydroquinone dehydrogenases. Annu. Rev. Biochem. 1977, 46, 439–469. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Serbinova, E.A.; Stoyanovsky, D.A.; Khwaja, S.; Packer, L. Assay of ubiquinones and ubiquinols as antioxidants. Methods Enzymol. 1994, 234, 343–354. [Google Scholar] [PubMed]
- Maguire, J.J.; Wilson, D.S.; Packer, L. Mitochondrial electron transport-linked tocopheroxyl radical reduction. J. Biol. Chem. 1989, 264, 21462–21465. [Google Scholar] [PubMed]
- Ernster, L.; Forsmark, P.; Nordenbrand, K. The mode of action of lipid-soluble antioxidants in biological membranes: Relationship between the effects of ubiquinol and vitamin E as inhibitors of lipid peroxidation in submitochondrial particles. BioFactors 1992, 3, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Ingold, K.U.; Bowry, V.W.; Stocker, R.; Walling, C. Autoxidation of lipids and antioxidation by α-tocopherol and ubiquinol in homogeneous solution and in aqueous dispersions of lipids: Unrecognized consequences of lipid particle size as exemplified by oxidation of human low density lipoprotein. Proc. Natl. Acad. Sci. USA 1993, 90, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Jauslin, M.L.; Meier, T.; Smith, R.A.; Murphy, M.P. Mitochondria targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003, 17, 1972–1974. [Google Scholar] [CrossRef] [PubMed]
- Bedogni, B.; Pani, G.; Colavitti, R.; Riccio, A.; Borrello, S.; Murphy, M.; Smith, R.; Eboli, M.L.; Galeotti, T. Redox regulation of cAMP-responsive element-binding protein and induction of manganous superoxide dismutase in nerve growth factordependent cell survival. J. Biol. Chem. 2003, 278, 16510–16519. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; di Venosa, N.; Federici, A.; Ruggiero, F.M. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ. Res. 2004, 94, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar] [CrossRef] [PubMed]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia reperfusion injury. FASEB J. 2005, 19, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.B.; Yang, S.K.; Zhou, Q.Y.; Pan, P.; Zhang, H.; Liu, F.; Xu, X.Q. Mitochondria-targeted peptides prevent on contrast-induced acute kidney injury in the rats with hypercholesterolemia. Ren. Fail. 2013, 35, 1124–1129. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 2014, 1, 3–6. [Google Scholar] [CrossRef] [PubMed]
- McCormack, W.J.; Parker, A.E.; O’Neill, L.A. Toll-like receptors and NOD-like receptors in rheumatic diseases. Arthritis Res. Ther. 2009, 11, 243. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [PubMed]
- Carta, S.; Penco, F.; Lavieri, R.; Martini, A.; Dinarello, C.A.; Gattorno, M.; Rubartelli, A. Cell stress increases ATP release in NLRP3 inflammasome-mediated autoinflammatory diseases, resulting in cytokine imbalance. Proc. Natl. Acad. Sci. USA 2015, 112, 2835–2840. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Sun, Y.; Liu, W.; Wu, X.; Guo, L.; Cai, P.; Wu, X.; Wu, X.; Shen, Y.; Shu, Y.; et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy 2014, 10, 972–985. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Mueller, J.L.; Vitari, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell. Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Chojnacki, J.; Toldo, S.; Mezzaroma, E.; Tranchida, N.; Rose, S.W.; Federici, M.; Van Tassell, B.W.; Zhang, S.; Abbate, A. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J. Cardiovasc. Pharmacol. 2014, 63, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.C.; Chen, C.W.; Chen, J.C.; Lin, W.W. HMG-CoA reductase inhibitors inhibit inducible nitric oxide synthase gene expression in macrophages. J. Biomed. Sci. 2003, 10, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Novelli, G.; D’Apice, M.R. Protein farnesylation and disease. J. Inherit. Metab. Dis. 2012, 35, 917–926. [Google Scholar] [CrossRef] [PubMed]
- McTaggart, S.J. Isoprenylated proteins. Cell. Mol. Life Sci. 2006, 63, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Rikitake, Y.; Liao, J.K. Rho GTPases, statins, and nitric oxide. Circ. Res. 2005, 97, 1232–1235. [Google Scholar] [CrossRef] [PubMed]
- Simon, A. Cholesterol metabolism and immunity. N. Engl. J. Med. 2014, 371, 1933–1935. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcuzzi, A.; Piscianz, E.; Loganes, C.; Vecchi Brumatti, L.; Knowles, A.; Bilel, S.; Tommasini, A.; Bortul, R.; Zweyer, M. Innovative Target Therapies Are Able to Block the Inflammation Associated with Dysfunction of the Cholesterol Biosynthesis Pathway. Int. J. Mol. Sci. 2016, 17, 47. https://doi.org/10.3390/ijms17010047
Marcuzzi A, Piscianz E, Loganes C, Vecchi Brumatti L, Knowles A, Bilel S, Tommasini A, Bortul R, Zweyer M. Innovative Target Therapies Are Able to Block the Inflammation Associated with Dysfunction of the Cholesterol Biosynthesis Pathway. International Journal of Molecular Sciences. 2016; 17(1):47. https://doi.org/10.3390/ijms17010047
Chicago/Turabian StyleMarcuzzi, Annalisa, Elisa Piscianz, Claudia Loganes, Liza Vecchi Brumatti, Alessandra Knowles, Sabrine Bilel, Alberto Tommasini, Roberta Bortul, and Marina Zweyer. 2016. "Innovative Target Therapies Are Able to Block the Inflammation Associated with Dysfunction of the Cholesterol Biosynthesis Pathway" International Journal of Molecular Sciences 17, no. 1: 47. https://doi.org/10.3390/ijms17010047