
Identification of Genes Putatively Involved in Chitin Metabolism and Insecticide Detoxification in the Rice Leaf Folder (Cnaphalocrocis medinalis) Larvae through Transcriptomic Analysis

Abstract
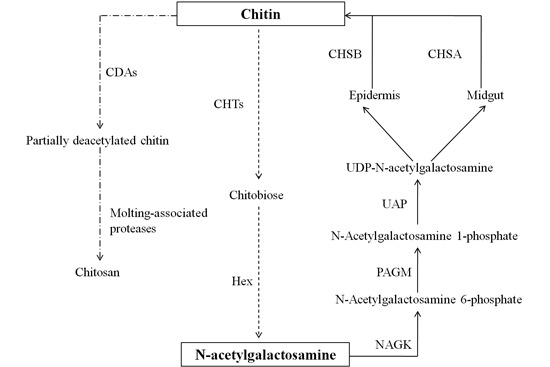
:
1. Introduction
2. Results
2.1. Illumina Sequencing and Assembly
2.2. Open Reading Frame Prediction and Global Gene Expression
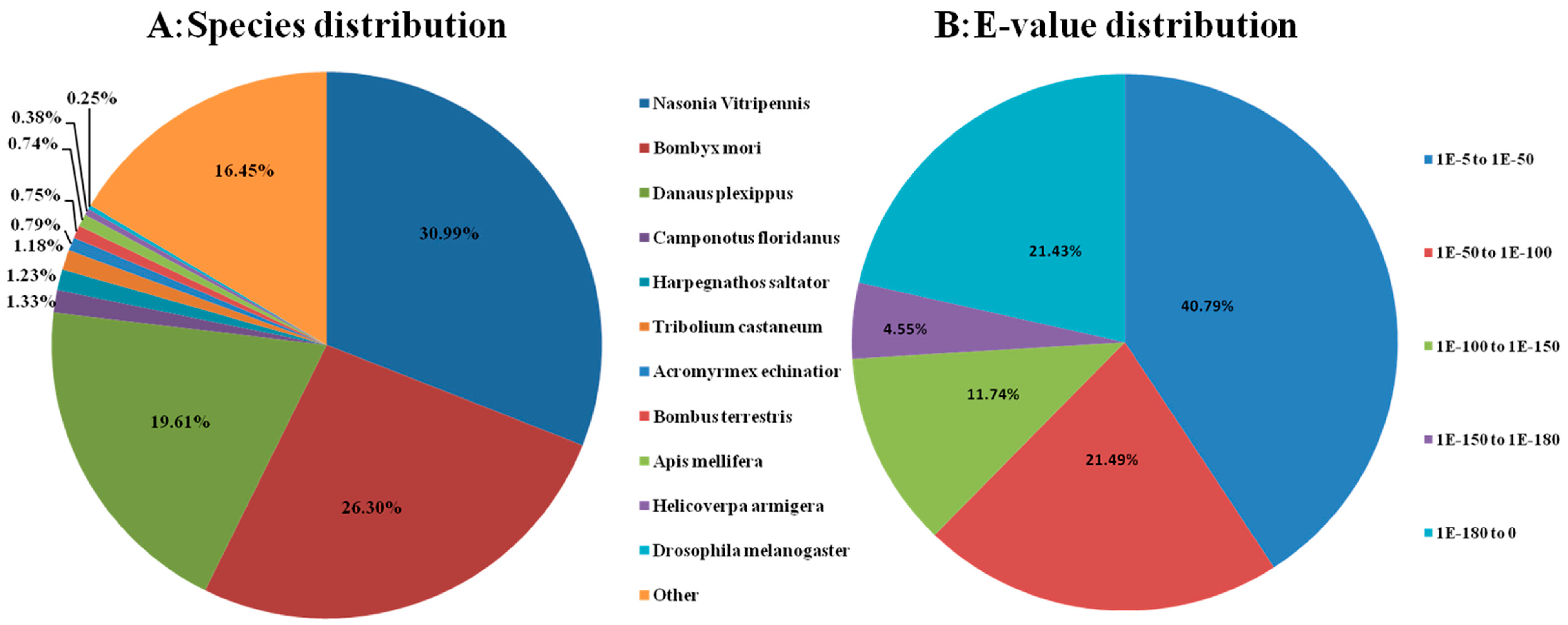
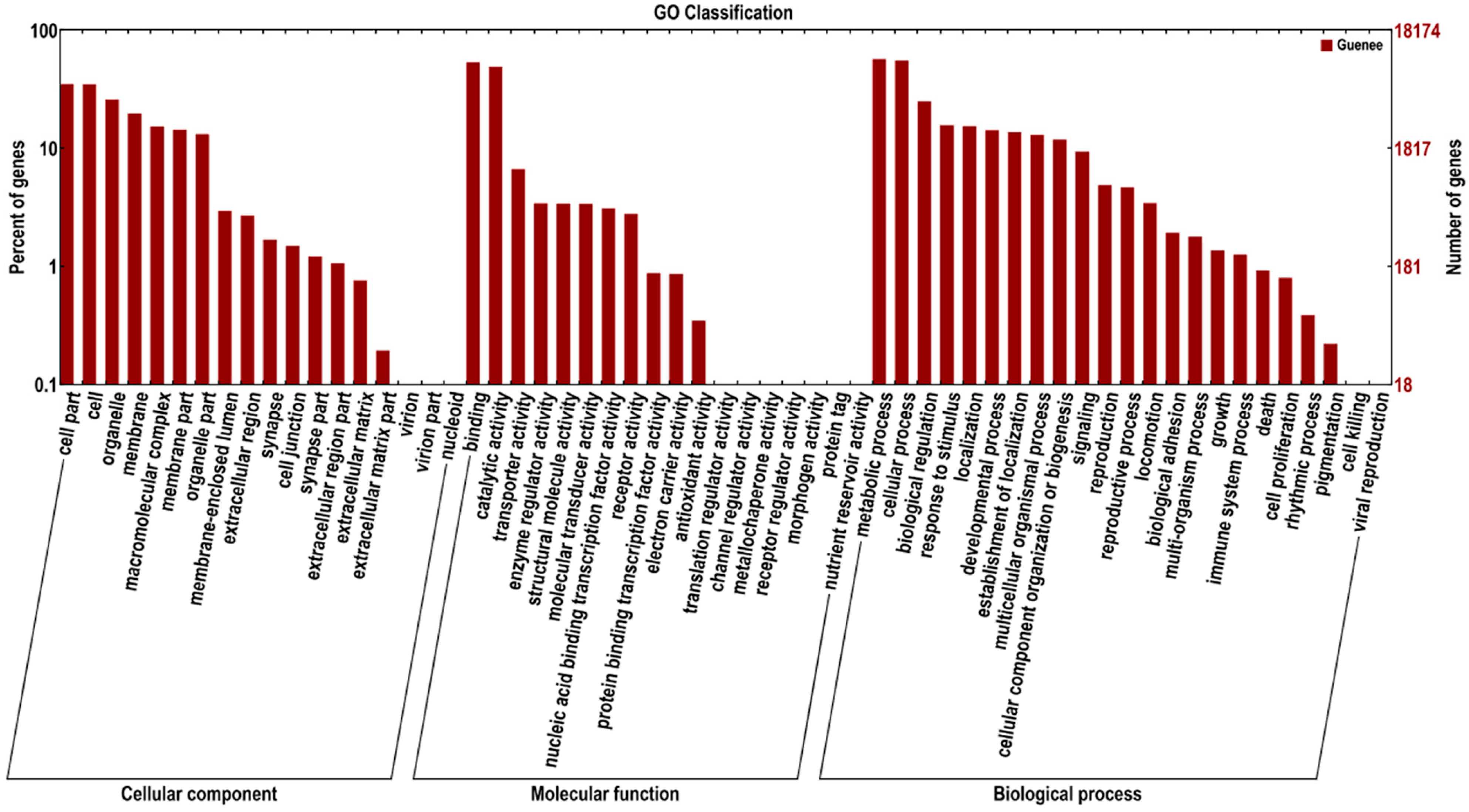
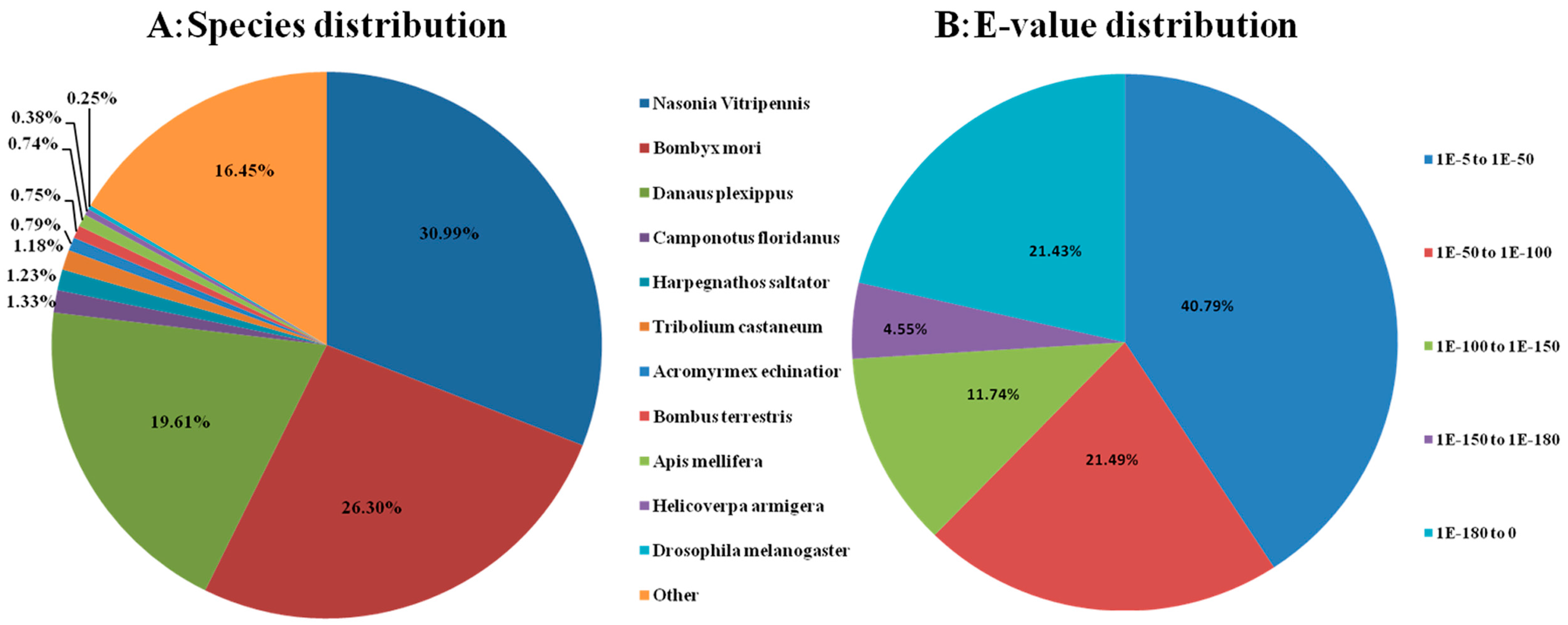
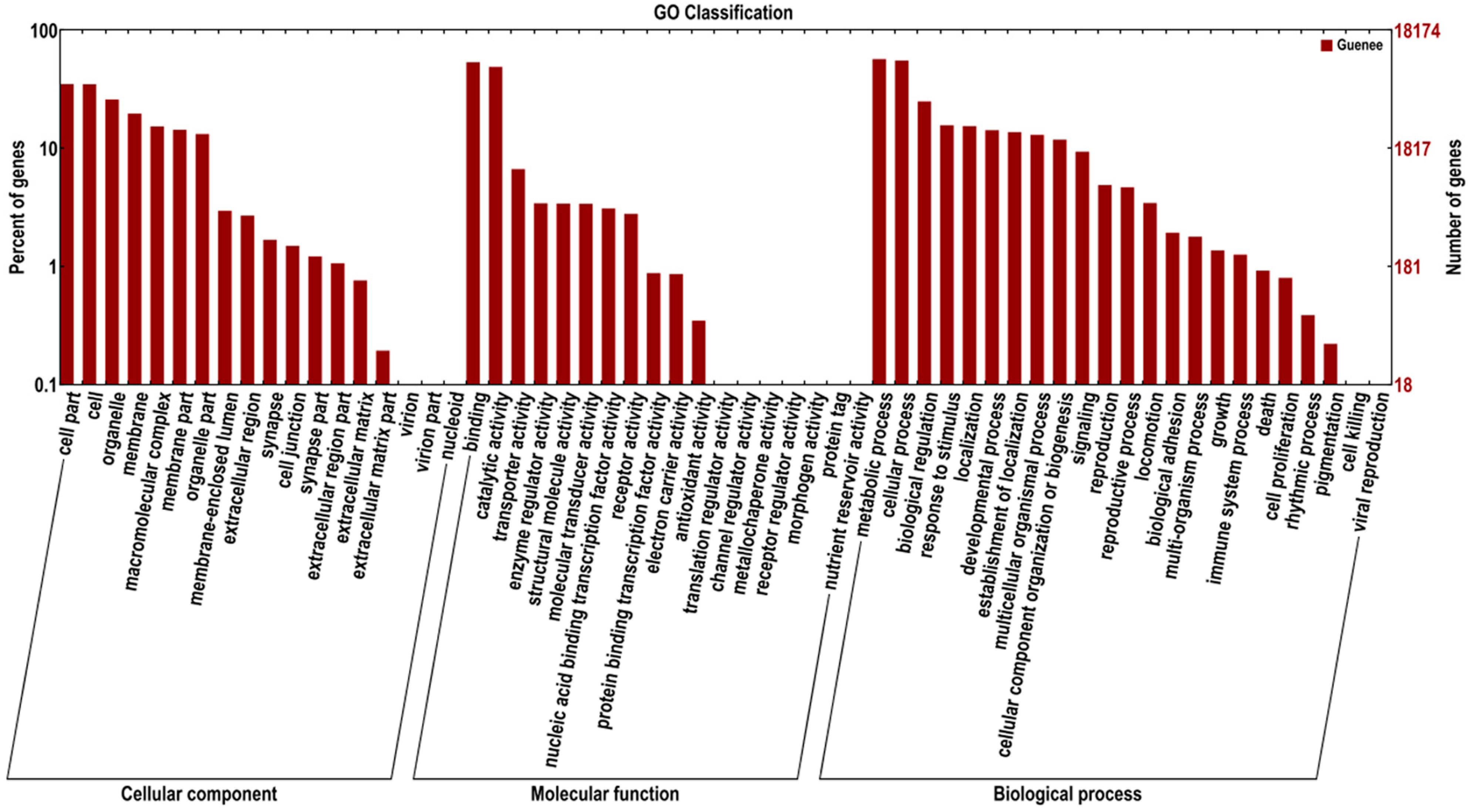
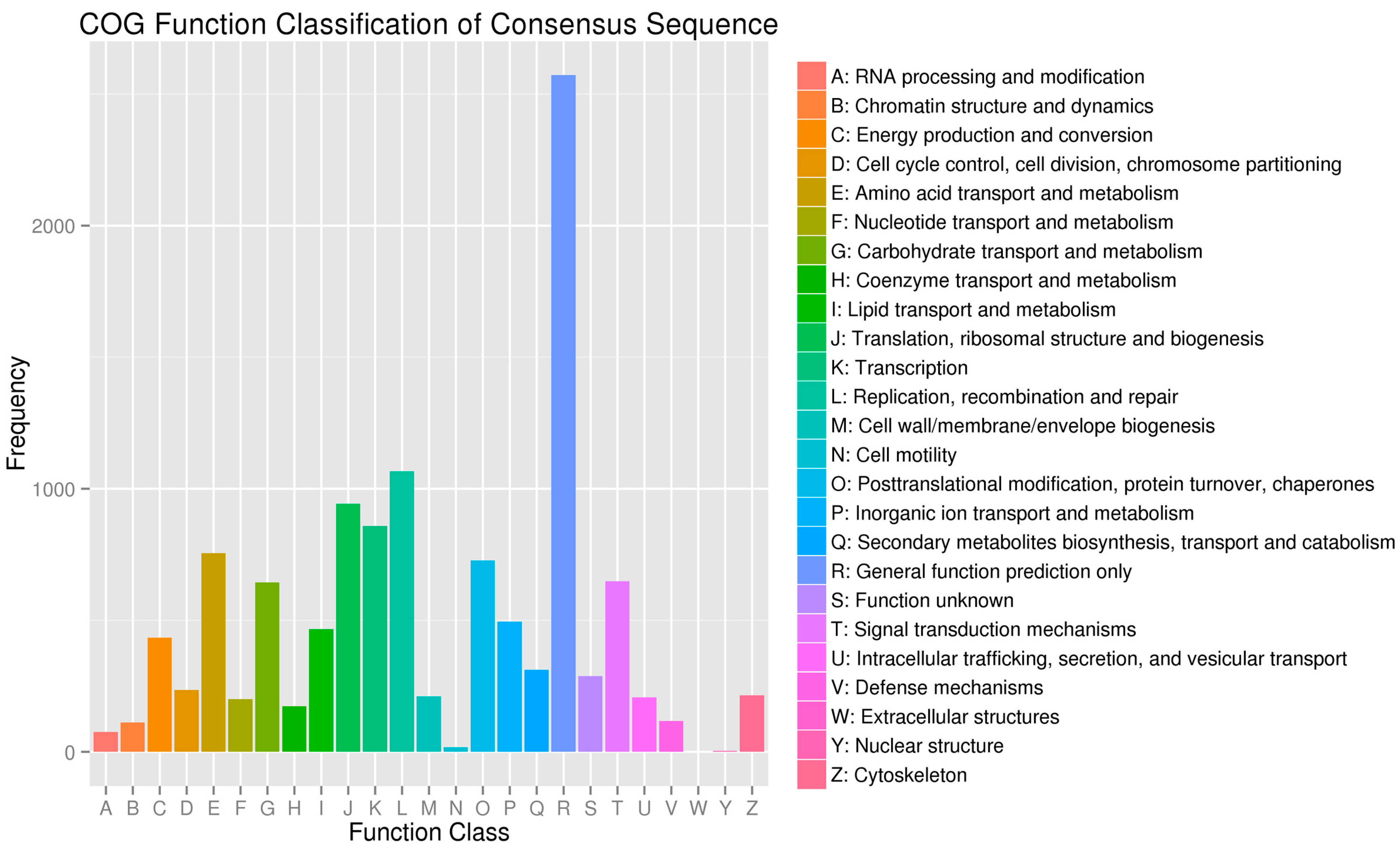
2.3. Sequence Annotation and Classification

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Annotated Databases | Annotated Number | 300 ≤ Length < 1000 bp | Length ≥ 1000 bp |
|---|---|---|---|
| COG_Annotation | 8669 | 3410 | 4372 |
| GO_Annotation | 18,176 | 7955 | 7174 |
| KEGG_Annotation | 10,043 | 4217 | 4482 |
| Swissprot_Annotation | 24,246 | 10,627 | 10,539 |
| nr_Annotation | 31,810 | 14,722 | 11,261 |
| All_Annotated | 32,035 | 14,834 | 11,270 |
| Repeat Motif | Number | Percentage (%) |
|---|---|---|
| Single-nucleotide | ||
| A/T | 4803 | |
| C/G | 247 | |
| Total | 5050 | 48.59 |
| Di-nucleotide | ||
| AC/GT | 372 | |
| AG/CT | 775 | |
| AT/AT | 535 | |
| CG/CG | 144 | |
| Total | 1826 | 17.57 |
| Tri-nucleotide | ||
| AAC/GTT/AAG/CTT/AAT/ATT | 757 | |
| ACC/GGT/ACG/CGT/ACT/AGT | 800 | |
| AGC/CTG/AGG/CCT/ATC/ATG/CCG/CGG | 1851 | |
| Total | 3408 | 36.60 |
| Tetra-nucleotide | ||
| AAAC/GTTT/AAAG/CTTT/AAAT/ATTT/AACC/GGTT | 54 | |
| AACG/CGTT/AAGC/CTTG/AAGG/CCTT/AATC/ATTG | 11 | |
| AATG/ATTC/ACAG/CTGT/ACAT/ATGT/ACCG/CGGT | 13 | |
| ACCT/AGGT/ACGC/CGTG/ACGG/CCGT/ACTC/AGTG | 10 | |
| ACTG/AGTC/AGAAGCG/CGCTT/ATCT/AGCC/CTGG/ | 6 | |
| AGGC/CCTG/AGGG/CCCT/ATCC/ATCG/ATCG/ATCG | 7 | |
| Total | 101 | 0.97 |
| Penta-nucleotide | ||
| AAAAC/GTTTT/AAAAT/ATTTT | 3 | |
| AAGTG/ACTTC/AATTC/AATTG | 2 | |
| AGGCG/CCTCG | 1 | |
| Total | 6 | 0.058 |
| Hexa- nucleotide | ||
| ACCATG/ATGGTC | 1 | |
| ACCGCC/CGGTGG | 1 | |
| AGCCGC/CGGCTG | 1 | |
| Total | 3 | 0.029 |
2.4. Simple Sequence Repeats (SSR) Discovery
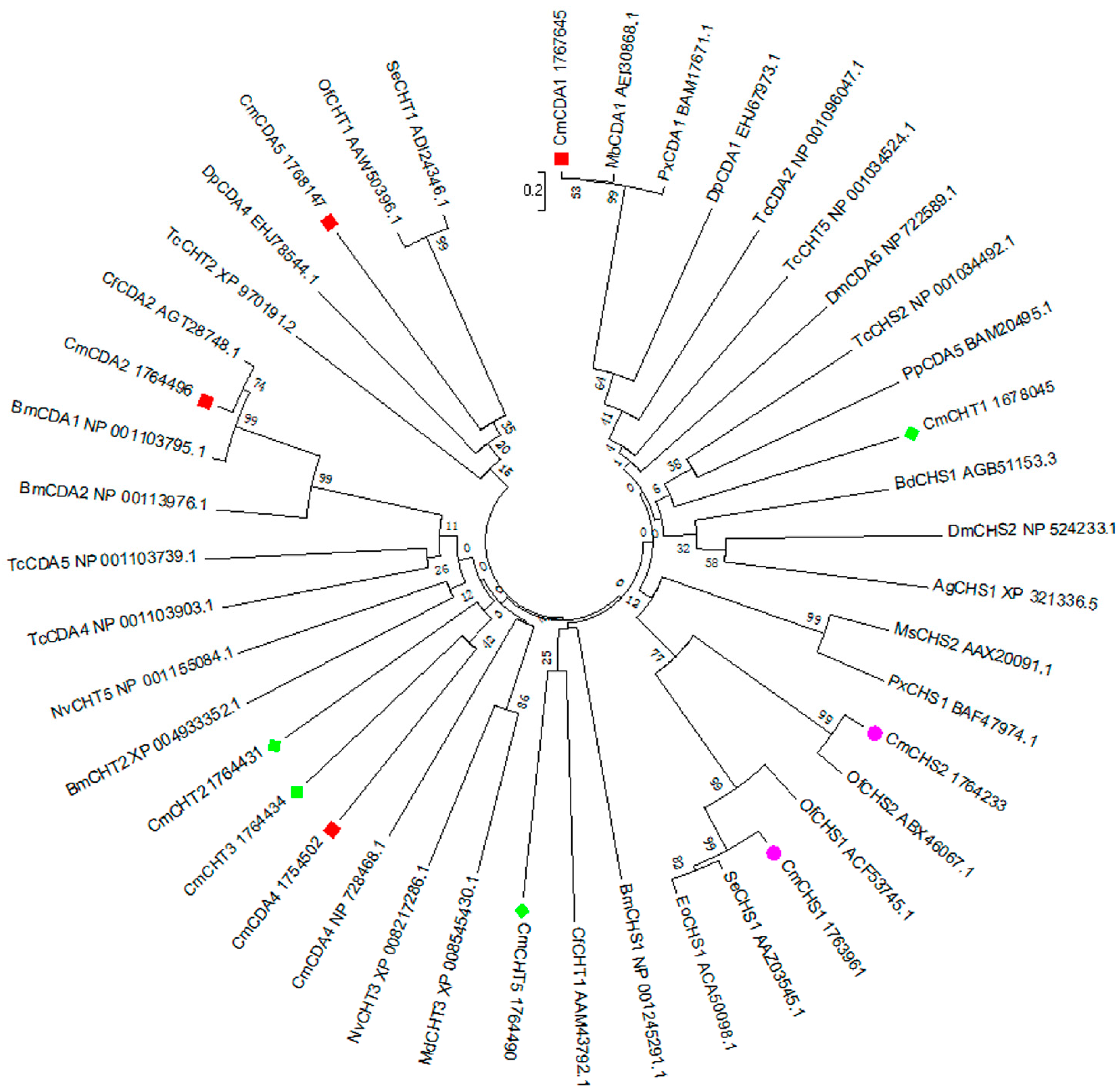
2.5. Transcripts Encoding Chitin Metabolism Enzymes
| Gene Name | GeneBank Accession No | Length (bp) | Function (Ontology) |
|---|---|---|---|
| Chitin synthase 1 (CHS1) | KP000843 | 4868 | chitin biosynthetic process |
| Chitin synthase 2 (CHS2) | KP000844 | 4651 | |
| N-Phosphoacetylglucosamine mutase (PAGM) | KP000845 | 1934 | carbohydrate metabolic process phosphoacetylglucosamine mutase activity, UDP-N-acetylglucosamine biosynthetic process |
| UDP-N-acetylglucosamine pyrophosphorylase | KP000846 | 1173 (partial) | transferase activity, transferring glycosyl groups, UMP salvage catalytic activity |
| N-Acetyl-d-glucosamine kinase(NAGK) | KP000849 | 733 (partial) | N-acetylglucosamine kinase activity |
| Chitinase 1 | KF897513 | 2039 | chitin catabolic process, carbohydrate metabolic process, chitinase activity chitin binding, carbohydrate metabolic process, extracellular region, cuticle chitin catabolic process, chitinase activity chitin catabolic process, carbohydrate metabolic process, imaginal disc development, extracellular region, chitinase activity chitin catabolic process, carbohydrate metabolic process, chitinase activity β-N-acetylhexosaminidase activity peptidoglycan-based cell wall biogenesis peptidoglycan turnover, chitin catabolic process, chitin deacetylase activity |
| Chitinase 2 | KP000847 | 1897 | |
| Chitinase 3 | KP000848 | 4311 | |
| Chitinase 5 | KP000850 | 2105 (partial) | |
| Chitinase 7 | none | none | |
| β-l-N-Acetylhexosaminidase | KP000851 | 1377 (partial) | |
| Chitin deacetylase 1 | KP000854 | 2020 | |
| Chitin deacetylase 2 | KP000852 | 2158 | |
| Chitin deacetylase 4 | KP000853 | 1537 | |
| Chitin deacetylase 5 | 1768147 | 765 (partial) | |
| Chitin deacetylase 6 | 1787955 | 349 (partial) |
| Gene Name | Primer Sequence | Length of Product (bp) |
|---|---|---|
| Reverse Transcription Quantitative PCR (RT-qPCR) | ||
| CmCHS1 | F: CTGGAACAGCAACGGAAACT | 190 |
| R: TCCGTTGTTGATGAGCCAGA | ||
| CmCHT1 | F: TGCTGGCAACAACAACTACAATC | 231 |
| R: ATCTCCACGCTCTTAGGGTCTT | ||
| CmCDA1 | F: AAGAAATGGCGGGTATGAGGGTG | 142 |
| R: AGTGGTGGATTAGACAAAGGTGCG | ||
| CmActin | F: GAGCGTGGTTACTCATTCA | 283 |
| R: TGTCAACATCGCACTTCA | ||
| CmCYPA792 | F: TTGATGCGGATGTTGACG | 164 |
| R: ATGCCCTTTGGAGTTGGA | ||
| CmGST | F: AACTGTATTCGGCTTTATCC | 234 |
| R: ACACACTTGCTGCCTTTTCC | ||
| PCR for cDNA Sequencing | ||
| CmCDA1 | F1: GTCTACGGTTGCGTTGCTCC | 911 |
| R1: GAGCAACGCAACCGTAGACGACTGG | ||
| F2: CATCATCGTTGTGCGTGACAGAGTG | 970 | |
| R2: ACGACAGCACAATCACCGCACCTT | ||
| F3: GCCGTCCTCGGAGCAGAAACAGTCA | 830 | |
| R3: CCAACAACGA CGAATATCTTCCAGG | ||
| CmCHT1 | F1: TGTCCTCGATGCCGTACCTGCCCAC | 1300 |
| R1: AAGGGGTGCTGACCGCTGCTGTGCCGCT | ||
| F2: CACGCCGGGGCGGTACACCGCC | 300 | |
| R2: TGGGAAGACAAGGGCTGTCCAACCAACA | ||
| CmCHSA | F1: ACTATGTGGCACGAAACGAA | 354 |
| R1: TAGTACATGTACAT | ||
| F2: CATTTGAAAGATAAGGC | 580 | |
| R2: ACCAACATRAGRAADAT | ||
| F3: CGCCTTACATCGCTTACC | 1258 | |
| R3: ACBARACCRATNGGYTCC | ||

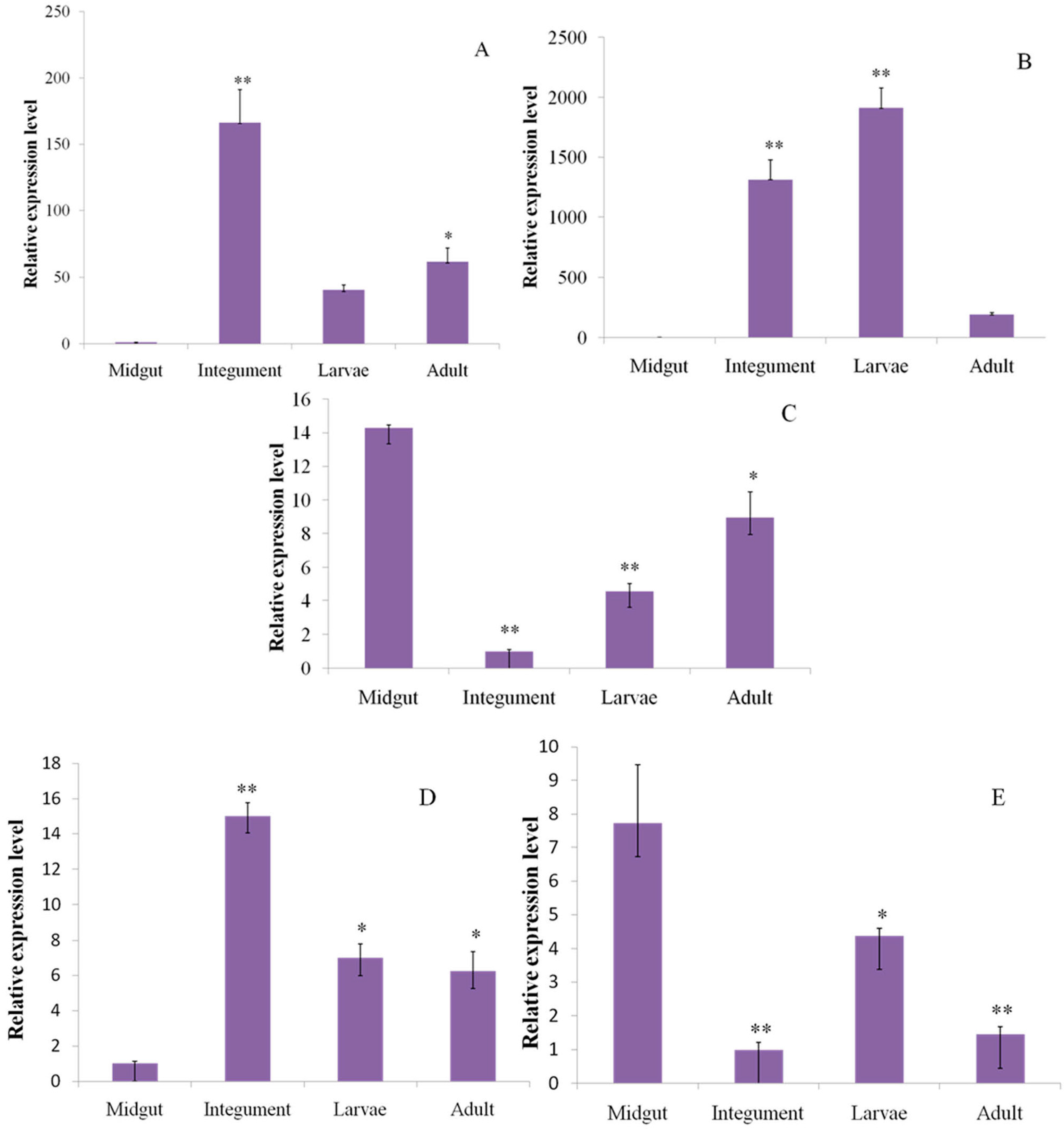
2.6. Reverse Transcription Quantitative PCR (RT-qPCR) Analysis

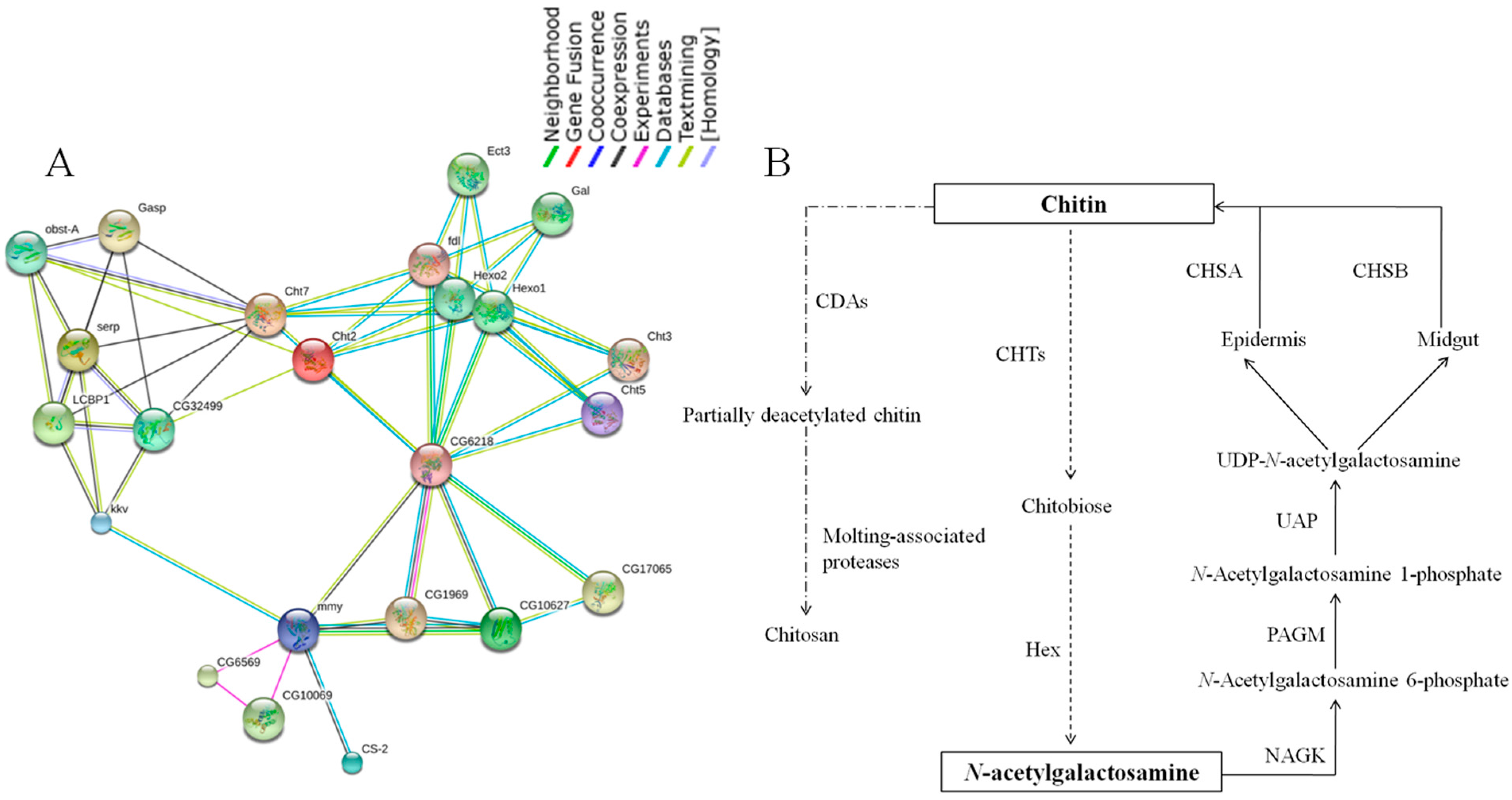
2.7. Chitin Metabolism Enzyme Networks of C. medinalis
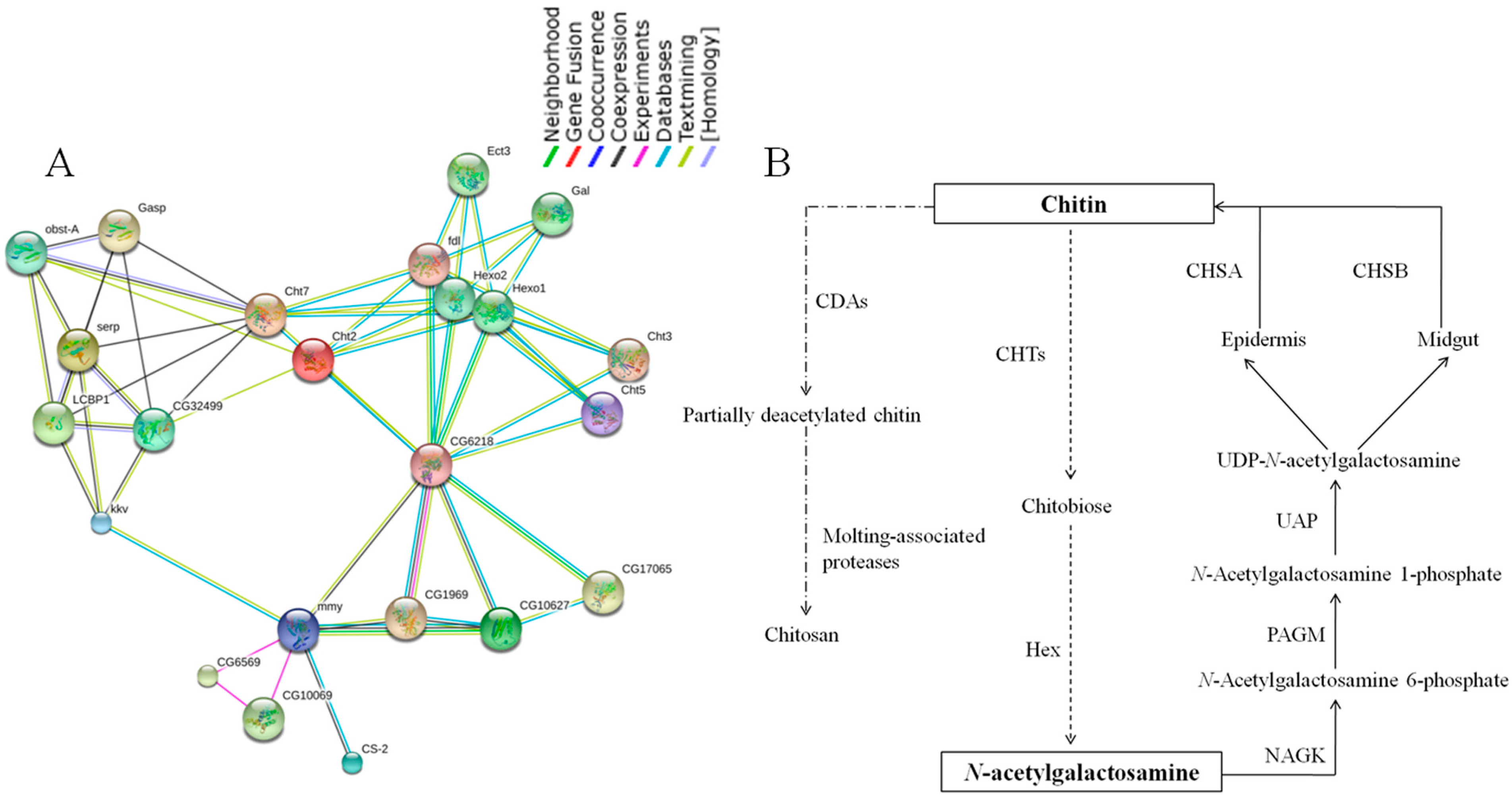
2.8. Identification of Insecticide Resistance Related Genes and RT-qPCR Validation
3. Experimental Section
3.1. Insect Sample Preparation
3.2. Isolation of Total RNA and cDNA Library Construction
3.3. Sequence Data Analysis and Assembly
3.4. Functional Annotation and Classification
3.5. Identification of Insecticide Resistance Genes and Chitin Metabolism Enzymes
3.6. SSR Detection
3.7. The Predicted Functional Association Networks
3.8. cDNA Cloning of Chitin Genes and Sequence Analysis
3.9. RT-qPCR Analysis
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zheng, X.S.; Ren, X.B.; Su, J.Y. Insecticide susceptibility of Cnaphalocrocis medinalis (Lepidoptera: Pyralidae) in China. J. Econ. Entomol. 2011, 104, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Pathak, M.D.; Khan, Z.R. Insect Pests of Ric; International Rice Research Institute: Manila, Philippines, 1994. [Google Scholar]
- Bautista, R.C.; Heinrichs, E.A.; Rejesus, R.S. Economic injury levels for the rice leaffolder Cnaphalocrocis medinalis (Lepidoptera: Pyralidae): Insect infestation and artificial leaf removal. Environ. Entomol. 1984, 13, 439–443. [Google Scholar] [CrossRef]
- Huang, J.K.; Hu, R.F.; Pray, C.; Qiao, F.B.; Rozelle, S. Biotechnology as an alternative to chemical pesticides: A case study of Bt cotton in China. Agric. Econ. 2003, 29, 55–67. [Google Scholar] [CrossRef]
- Mumuni, A.; Shepard, B.M.; Mitchel, P.L. Parasitism and predation on eggs of Leptoglossus phyllopus (L.) (Hemiptera: Coreidae) in cowpea: Impact of endosulfan sprays. J. Agric. Urban Entomol. 2001, 18, 105–115. [Google Scholar]
- Zhang, S.K.; Ren, X.B.; Wang, Y.C.; Su, J.Y. Resistance in Cnaphalocrocis medinalis (Lepidoptera: Pyralidae) to new chemistry insecticides. J. Econ. Entomol. 2014, 2, 483–879. [Google Scholar] [CrossRef]
- Sun, X.; Zhou, W.; Liu, H.; Zhang, A.J.; Ai, C.R.; Zhou, S.S.; Zhou, C.X.; Wang, M.Q. Transgenic Bt rice does not challenge host preference of the target pest of rice leaffolder, Cnaphalocrocis medinalis (Lepidoptera: Pyralidae). PLoS ONE 2013, 8, e79032. [Google Scholar] [CrossRef] [PubMed]
- Chai, H.N.; Du, Y.Z.; Zhai, B.P. Characterization of the complete mitochondrial genomes of Cnaphalocrocis medinalis and Chilo suppressalis (Lepidoptera: Pyralidae). Int. J. Biol. Sci. 2012, 8, 561–579. [Google Scholar] [CrossRef] [PubMed]
- An, B.G.; Deng, X.L.; Shi, H.R.; Ding, M.; Lan, J.; Yang, J.; Li, S.Y. Development and characterization of microsatellite markers for rice leaffolder, Cnaphalocrocis medinalis (Guenee) and cross-species amplification in other Pyralididae. Mol. Biol. Rep. 2014, 41, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Li, S.W.; Yang, H.; Liu, Y.F.; Liao, Q.R.; Du, J.; Jin, D.C. Transcriptome and gene expression analysis of the rice leaf folder, Cnaphalocrosis medinalis. PLoS ONE 2012, 7, e47401. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Watanabe, T.; Yui, T.; Sugiyama, J.J. The directionality of chitin biosynthesis: A revisit. Biochem. J. 2003, 374, 775–760. [Google Scholar] [CrossRef] [PubMed]
- Merzendorfer, H.; Zimoch, L. Chitin metabolism in insects: Structure, function and regulation of chitin synthases and chitinases. J. Exp. Biol. 2003, 206, 4393–4412. [Google Scholar] [CrossRef] [PubMed]
- Babiker, M.A.; Abdel, B.; Daizo, K. A genomic clone for a chitinase gene from the silkworm, Bombyx mori: Structural organization identifies functional motifs. Insect Biochem. Mol. Biol. 2001, 31, 497–508. [Google Scholar]
- Ranson, H.; Claudianos, C.; Ortelli, F.; Abgrall, C.; Hemingway, J.; Sharakhova, M.V.; Collins, F.H.; Feyereisen, R. Evolution of supergene families associated with insecticide resistance. Science 2002, 298, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.H.; Yoon, K.S.; Clark, J.M.; Lee, S.H. A point mutation in a glutamate-gated chloride channel confers abamectin resistance in the two-spotted spider mite, Tetranychus urticae Koch. Insect Mol. Biol. 2010, 19, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Khajehali, J.; Leeuwen, T.V.; Grispou, M.; Morou, E.; Alout, H.; Weill, M.; Tirry, L.; Vontas, J.; Tsagkarakou, A. Acetycholinesterase point mutations in european strains of Tetranychus urticae (Acari: Tetrany-chidae) resistant to organophosphates. Pest Manag. Sci. 2010, 66, 220–228. [Google Scholar] [PubMed]
- Morrow, C.S.; Smitherman, P.K.; Diah, S.K.; Schneider, E.; Townsend, A.J. Coordinated action of glutathione S-transferases (GSTs) and multidrug resistance protein 1 (MRP1) in antineoplastic drug detoxification. J. Biol. Chem. 2002, 277, 2000–2005. [Google Scholar]
- Ranson, H.; Rossiter, L.; Ortelli, F.; Jensen, B.; Wang, X.L.; Roth, C.W.; Collins, F.H.; Hemingway, J. Identification of a novel class of insect glutathione S-transferases involved in resistance to DDT in the malaria vector Anopheles gambiae. Biochem. J. 2001, 359, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Fonnum, F.; Sterri, S.H.; Aas, P.; Johnsen, H. Carboxylesterases, importance for detoxification of organophosphorus anticholinesterases and trichothecenes. Fundam. Appl. Toxicol. 1985, 5, S29–S38. [Google Scholar] [CrossRef]
- Yu, Q.Y.; Lu, C.; Li, W.L.; Xiang, Z.H.; Zhang, Z. Annotation and expression of carboxylesterases in the silkworm Bombyx mori. BMC Genom. 2009, 10, 553. [Google Scholar] [CrossRef] [PubMed]
- Feyereisen, R. Insect P450 enzymes. Annu. Rev. Entomol. 1999, 44, 507–533. [Google Scholar] [CrossRef] [PubMed]
- Marguiles, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.J.; Chen, Z.T.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, B.T.; Marguerat, S.; Watt, S.; Schubert, F.; Wood, V.; Goodhead, I.; Penkett, C.J.; Rogers, J.; Bahler, J. Dynamic repertoire of a eukaryotic transcriptome surveryed at single-nucletotide resolution. Nature 2008, 453, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Novaes, E.; Drost, D.R.; Farmerie, W.G.; Pappas, G.J.; Grattapaglia, D.; Sederoff, R.R.; Kirst, M. High-throughput gene and SNP discovery in eucalyplus grandis, an unclaracterized genome. BMC Genom. 2008, 9, 312. [Google Scholar] [CrossRef] [PubMed]
- Hou, R.; Bao, Z.M.; Wang, S.; Su, H.L.; Li, Y.; Du, H.X.; Hu, X.L.; Wang, S. Transcriptome sequencing and de novo analysis for Yesso scallop (Patinopecten yessoensis) using 454 GS FLX. PLoS ONE 2011, 6, e21560. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.Y.; Zhao, C.Q.; Wu, G. Progress in molecular mechanisms of insect resistance to in secticides. Entomol. J. East China 2007, 16, 136–140. [Google Scholar]
- Li, R.Q.; Zhu, H.M.; Ruan, J.; Qian, W.B.; Fang, X.D.; Shi, Z.B.; Li, S.T.; Shan, G.; Kristiansen, K.; Li, S.G. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.; Zahid, S.; Wan, Y.Y.; Forster, J.; Karim, A.A.; Nawabi, A.M.; Azhar, A.; Rahman, M.A.; Ahmed, N. Protein expression profiling of nuclear membrane protein reveals potential biomarker of human hepatocellular carcinoma. Clin. Proteom. 2013, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Park, R.D.; Muzzarelli, R. Chitin deacetylases: Properties and applications. Mar. Drugs 2010, 8, 24–46. [Google Scholar] [CrossRef] [PubMed]
- Peneff, C.; Ferrari, P.; Charrier, V.; Taburet, Y.; Monnier, C.; Zamboni, V.; Winter, J.; Harnois, M.; Fassy, F.; Bourne, Y. Crystal structures of two human pyrophosphorylase isoforms in complexes with UDPGlc(Gal)NAc: Role of the alternatively spliced insert in the enzyme oligomeric assembly and active site architecture. EMBO J. 2001, 20, 6191–6202. [Google Scholar] [CrossRef] [PubMed]
- Marschall, H.U.; Matern, H.; Wietholtz, H.; Egestad, B.; Matern, S.; Sjovall, J. Bile acid N-acetylglucosaminidation in vivo and in vitro evidence for a selective conjugation reaction of 7 β-hydroxylated bile acids in humans. J. Clin. Investig. 1992, 89, 1981–1987. [Google Scholar] [CrossRef] [PubMed]
- Eisenhaber, B.; Maurer-Stroh, S.; Novatchkova, M.; Schneider, G.; Eisenhaber, F. Enzymes and auxiliary factors for GPI lipid anchor biosynthesis and posttranslational transfer to proteins. Bioessays 2003, 25, 367–385. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E. Chitin synthesis and inhibition: A revisit. Pest Manag. Sci. 2001, 57, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Emboss Getorf. Available online: http://emboss.sourceforge.net/apps/cvs/emboss/apps/getorf.html (accessed on 10 June 2014).
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Gramene Ssrtool. Available online: http://archire.gramene.org/db/markers/ssrtool (accessed on 25 Auguest 2014).
- EMBL-EBI ClustaIW2. Available online: http://www.ebi.ac.uk/Tools/msa/clustalw2/ (accessed on 25 August 2014).
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Merzendorfer, H. Insect chitin synthases: A review. J. Comp. Physiol. B 2006, 176, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Arakane, Y.; Beeman, R.W.; Kramer, K.J.; Muthukrishnan, S. Functional specialization among insect chitinase family genes revealed by RNA interference. Proc. Natl. Acad. Sci. USA 2008, 105, 6650–6655. [Google Scholar] [CrossRef] [PubMed]
- Zimoch, L.; Merzendorfer, H. Immunolocalization of chitin synthase in the tobacco hornworm. Cell Tissue Res. 2002, 308, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, P.M.; Deleury, E.; Davies, G.J.; Henrissat, B. An evolving hierarchical family classification for glycosyltransferases. J. Mol. Biol. 2003, 328, 307–317. [Google Scholar] [CrossRef]
- George, E.L.; Ober, M.B.; Emerson, C.P. Functional domains of the Drosophila melanogaster muscle myosin heavy-chain gene are encoded by alternatively spliced exons. Mol. Cell. Biol. 1989, 9, 2957–2974. [Google Scholar] [PubMed]
- Dixit, R.; Arakane, Y.; Specht, C.A.; Richard, C.; Kramer, K.J.; Beeman, R.W.; Muthukrishnan, S. Domain organization and phylogenetic analysis of proteins from the chitin deacetylase gene family of Tribolium castaneum and three other species of insects. Insect Biochem. Mol. Biol. 2008, 38, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Pan, P.L.; Ye, Y.X.; Yu, B.; Zhang, C.X. Chitin deacetylase family genes in the brown planthopper, Nilaparvata lugens (Hemiptera: Delphacidae). Insect Mol. Biol. 2014, 6, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, J.Z.; Park, Y.; Zhu, K.Y. Identification and characterization of two chitin synthase genes in African malaria mosquito, Anopheles gamniae. Insect Biochem. Mol. Biol. 2012, 42, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Reese, J.; Christenson, M.K.; Leng, N.; Saha, S.; Cantarel, B.; Lindeberg, M.; Tamborindeguy, C.; MacCarthy, J.; Weaver, D.; Trease, A.J. Characterization of the Asian citrus psyllid transcriptome. J. Genom. 2014, 2, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.S.; Chung, P.G.; Murugan, K. Effect of botanical insecticides and bacterial toxins on the gut enzyme of the rice leaf folder Cnaphalocrocis medinalis. Phytoparasitica 2004, 32, 433–443. [Google Scholar] [CrossRef]
- Nathan, S.S.; Chung, P.G.; Murugan, K. Effect of biopesticides applied separately or together on nutritional indices of the rice leaf folder Cnaphalocrocis medinalis. Entomology 2002, 33, 187–195. [Google Scholar]
- Shah, M.A.; Muhammad, A.A.; Sanaullah, C.; Shaheen, I. Population trends and chemical control of rice leaf folder, Cnaphalocrocis medinalis on rice crop. Int. J. Agric. Biol. 2003 2003, 4, 615–617. [Google Scholar]
- Berge, J.B.; Feyereisen, R.; Amichot, M. Cytochrome P450 monooxygenases and insecticide resistance in insect. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1998, 353, 1701–1705. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Baudry, J.; Berenbaum, M.R.; Schuler, M.A. Structural and functional divergence of insect CYP6B proteins: From specialist to generalist cytochrome P450. Proc. Natl. Acad. Sci. USA 2004, 9, 2939–2944. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.A. Protease inhibitors in plants: Genes for improving defenses against insects and pathogens. Annu. Rev. Phytopathol. 1990, 28, 425–449. [Google Scholar] [CrossRef]
- Kellenberger, C.; Boudier, C.; Bermudez, I.; Bieth, J.G.; Luu, B.; Hietter, H. Serine protease inhibition by insect peptides containing a cysteine knot and a triple-stranded β-sheet. J. Biol. Chem. 1995, 43, 25514–25519. [Google Scholar] [CrossRef]
- Enayati, A.A.; Ranson, H.; Hemingway, J. Insect glutathione transferases and insecticide resistance. Insect Mol. Biol. 2005, 14, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Snchuler, M.A.; Berenbaum, M.R. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Annu. Rev. Entomol. 2007, 52, 231–253. [Google Scholar] [CrossRef] [PubMed]
- Fabrick, J.A.; Pei, J.X.; Hull, J.J.; Yool, A.J. Molecular and functional characterization of multiple aquaporin water channel proteins from the western tarnished plant bug, Lygus hesperus. Insect Mol. Biol. 2014, 45, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Gorman, M.J.; Paskewitz, S.M. Serine proteases as mediators of mosquito immune responses. Insect Mol. Biol. 2001, 31, 257–262. [Google Scholar] [CrossRef]
- Qin, L.G.; Xia, H.C.; Shi, H.F.; Zhou, Y.J.; Chen, L.; Yao, Q.; Liu, X.Y.; Feng, F.; Yuan, Y.; Chen, K.P. Comparative proteomic analysis reveals that caspase-1 and serine protease may be involved in silkworm resistance to Bombyx mori nuclear polyhedrosis virus. J. Proteom. 2012, 75, 3630–3638. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.Y.; Qiao, G.R.; Jiang, J.; Yang, H.Q.; Xie, L.H.; Xie, J.Z.; Zhuo, R.Y. Transcriptome sequencing and de novo analysis for Ma Bamboo (Dendrocalamus latiflorus Munro) using the Illumina platform. PLoS ONE 2012, 7, e46766. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Graner, A.; Sorrells, M. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–45. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, H.-Z.; Wen, D.-F.; Wang, W.-L.; Geng, L.; Zhang, Y.; Xu, J.-P. Identification of Genes Putatively Involved in Chitin Metabolism and Insecticide Detoxification in the Rice Leaf Folder (Cnaphalocrocis medinalis) Larvae through Transcriptomic Analysis. Int. J. Mol. Sci. 2015, 16, 21873-21896. https://doi.org/10.3390/ijms160921873
Yu H-Z, Wen D-F, Wang W-L, Geng L, Zhang Y, Xu J-P. Identification of Genes Putatively Involved in Chitin Metabolism and Insecticide Detoxification in the Rice Leaf Folder (Cnaphalocrocis medinalis) Larvae through Transcriptomic Analysis. International Journal of Molecular Sciences. 2015; 16(9):21873-21896. https://doi.org/10.3390/ijms160921873
Chicago/Turabian StyleYu, Hai-Zhong, De-Fu Wen, Wan-Lin Wang, Lei Geng, Yan Zhang, and Jia-Ping Xu. 2015. "Identification of Genes Putatively Involved in Chitin Metabolism and Insecticide Detoxification in the Rice Leaf Folder (Cnaphalocrocis medinalis) Larvae through Transcriptomic Analysis" International Journal of Molecular Sciences 16, no. 9: 21873-21896. https://doi.org/10.3390/ijms160921873