
A Series of New Ligustrazine-Triterpenes Derivatives as Anti-Tumor Agents: Design, Synthesis, and Biological Evaluation

 , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
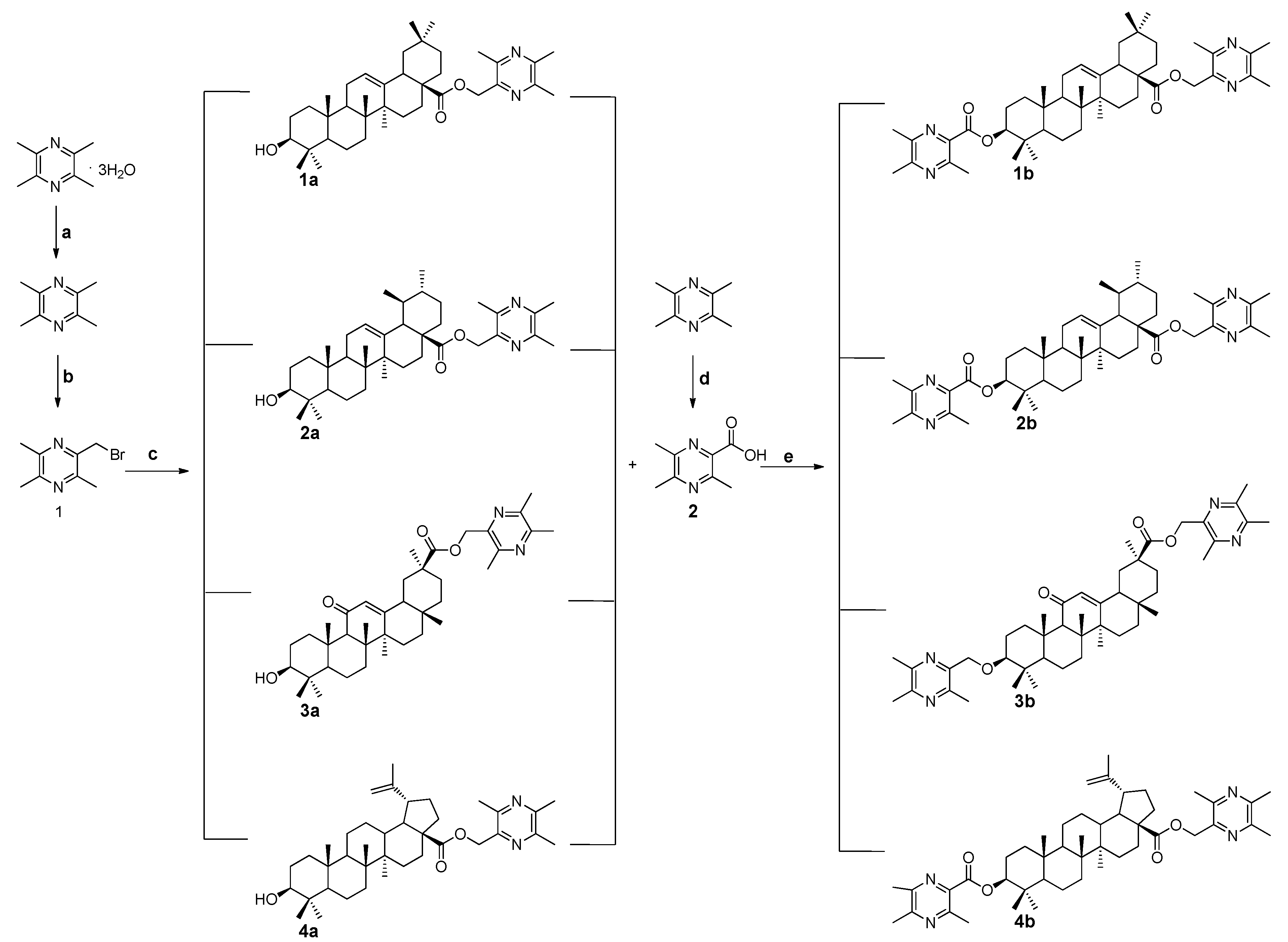
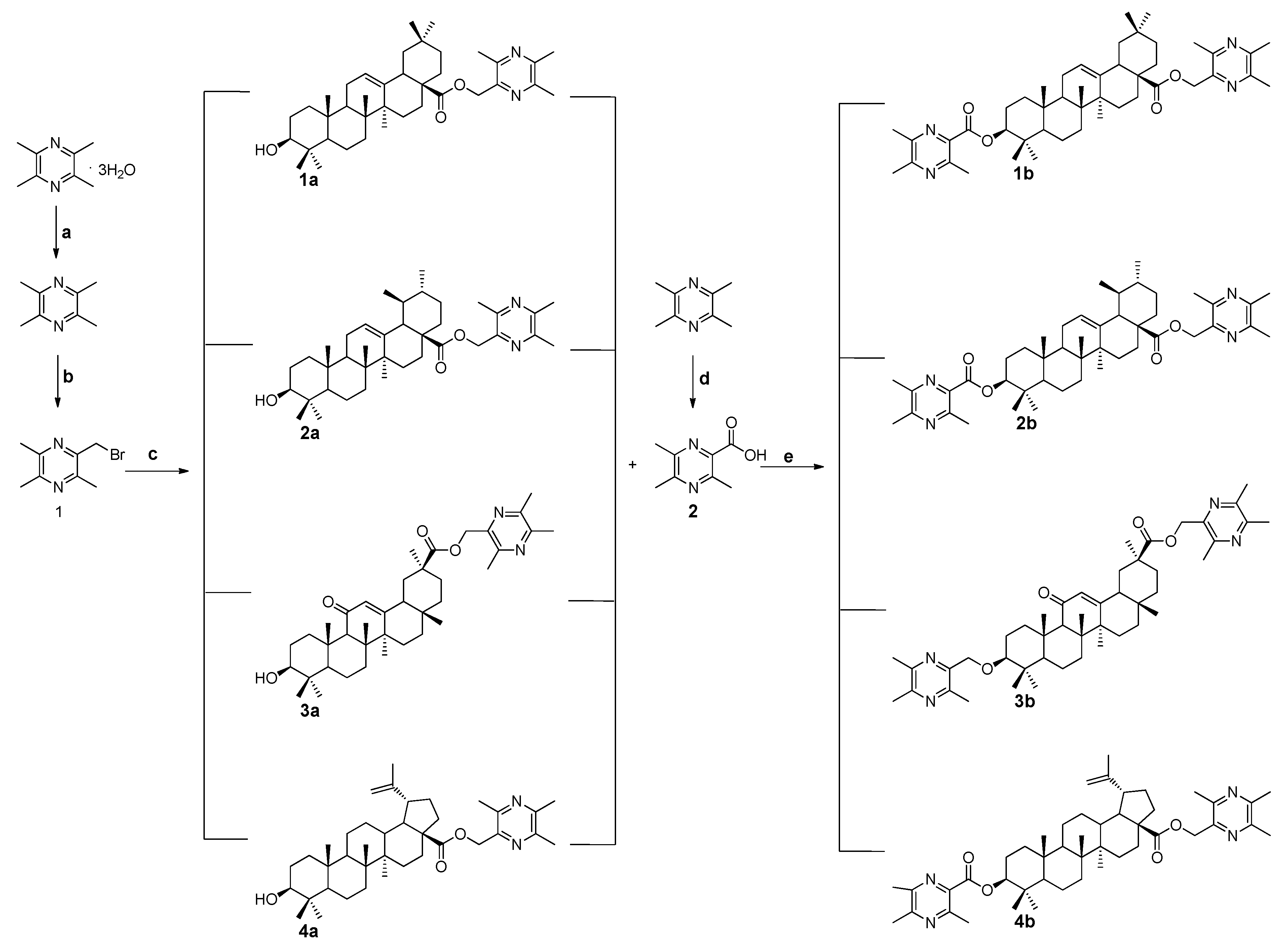
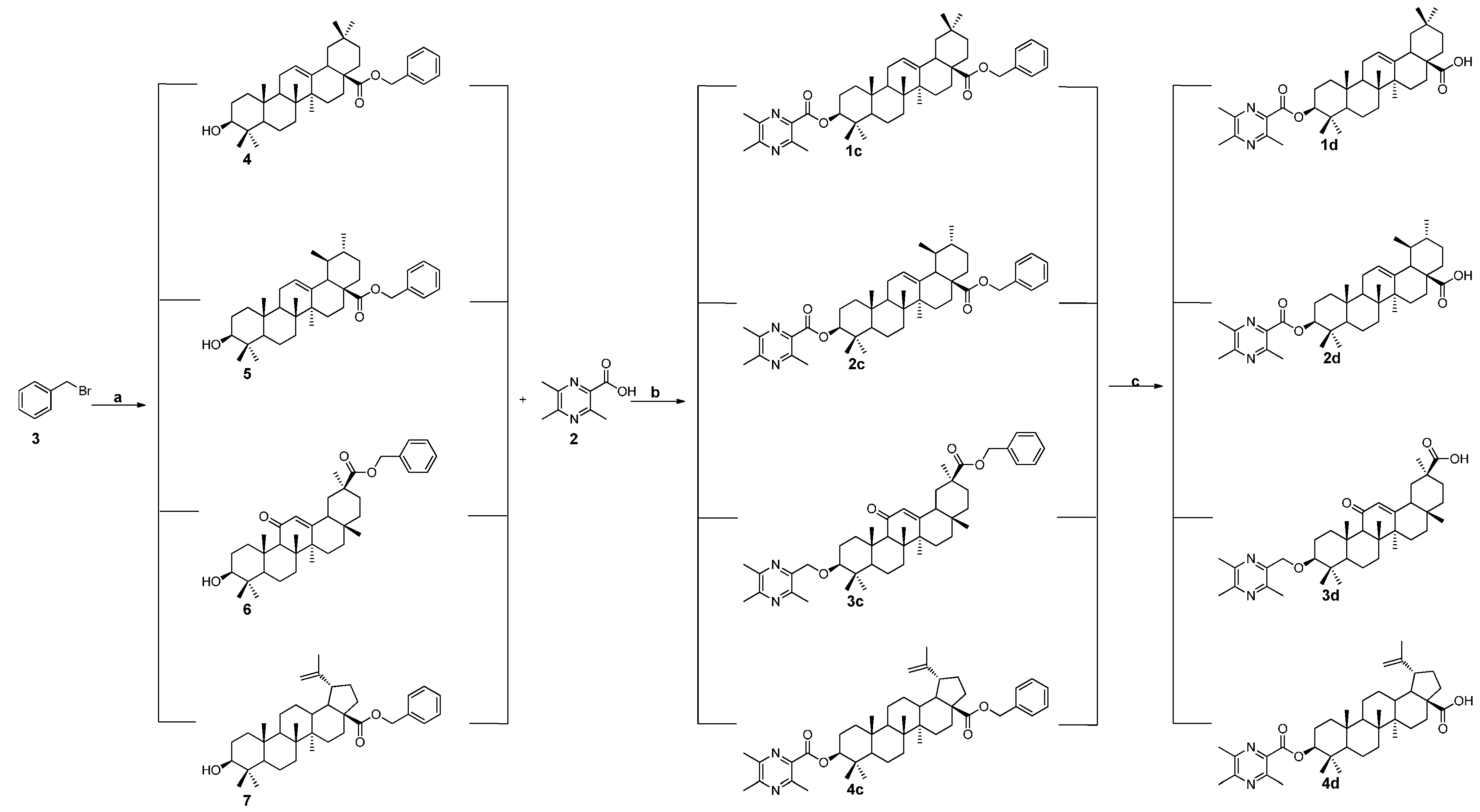
2.1. Chemistry
2.2. Cytotoxicity Assay

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) | ClogP | |||||
|---|---|---|---|---|---|---|---|
| Bel-7402 | HT-29 | HepG2 | MCF-7 | Hela | MDCK | ||
| TMP, OA, UA, GA, BA | >20 a | >20 | >20 | >20 | >20 | ND b | NC c |
| 1a | 10.24 | 13.22 | 14.85 | 15.17 | 14.07 | >20 | 9.72 |
| 1b | >20 | >20 | 12.09 | 10.55 | >20 | >20 | 11.26 |
| 1c | >20 | >20 | >20 | >20 | >20 | ND | 12.32 |
| 1d | 9.28 | 8.43 | 7.94 | 5.69 | 4.37 | 16.39 | 10.16 |
| 2a | 8.01 | 17.61 | >20 | >20 | 19.96 | >20 | 9.72 |
| 2b | >20 | >20 | >20 | >20 | >20 | ND | 11.26 |
| 2c | >20 | >20 | >20 | >20 | >20 | ND | 12.32 |
| 2d | 10.14 | 9.70 | >20 | 9.49 | 9.66 | >20 | 10.16 |
| 3a | >20 | 19.87 | >20 | >20 | 11.17 | >20 | 7.57 |
| 3b | >20 | >20 | >20 | >20 | >20 | >20 | 9.08 |
| 3c | >20 | >20 | 9.06 | 7.58 | 17.51 | >20 | 10.14 |
| 3d | >20 | >20 | >20 | >20 | >20 | ND | 7.99 |
| 4a | 4.19 | 5.23 | 4.48 | 4.23 | 4.34 | >20 | 9.57 |
| 4b | >20 | >20 | 13.20 | 12.87 | >20 | >20 | 11.11 |
| 4c | >20 | >20 | >20 | >20 | >20 | ND | 12.17 |
| 4d | 13.35 | 8.38 | 7.42 | 6.22 | 12.15 | >20 | 10.01 |
| DDP | 5.91 | 5.28 | 4.57 | 5.17 | 6.22 | 14.61 | NC |
2.3. Computer Simulation of ClogP
2.4. Preliminary Investigation of the Cytotoxicity Selectivity and the Apoptosis-Inducing Effect of 4a
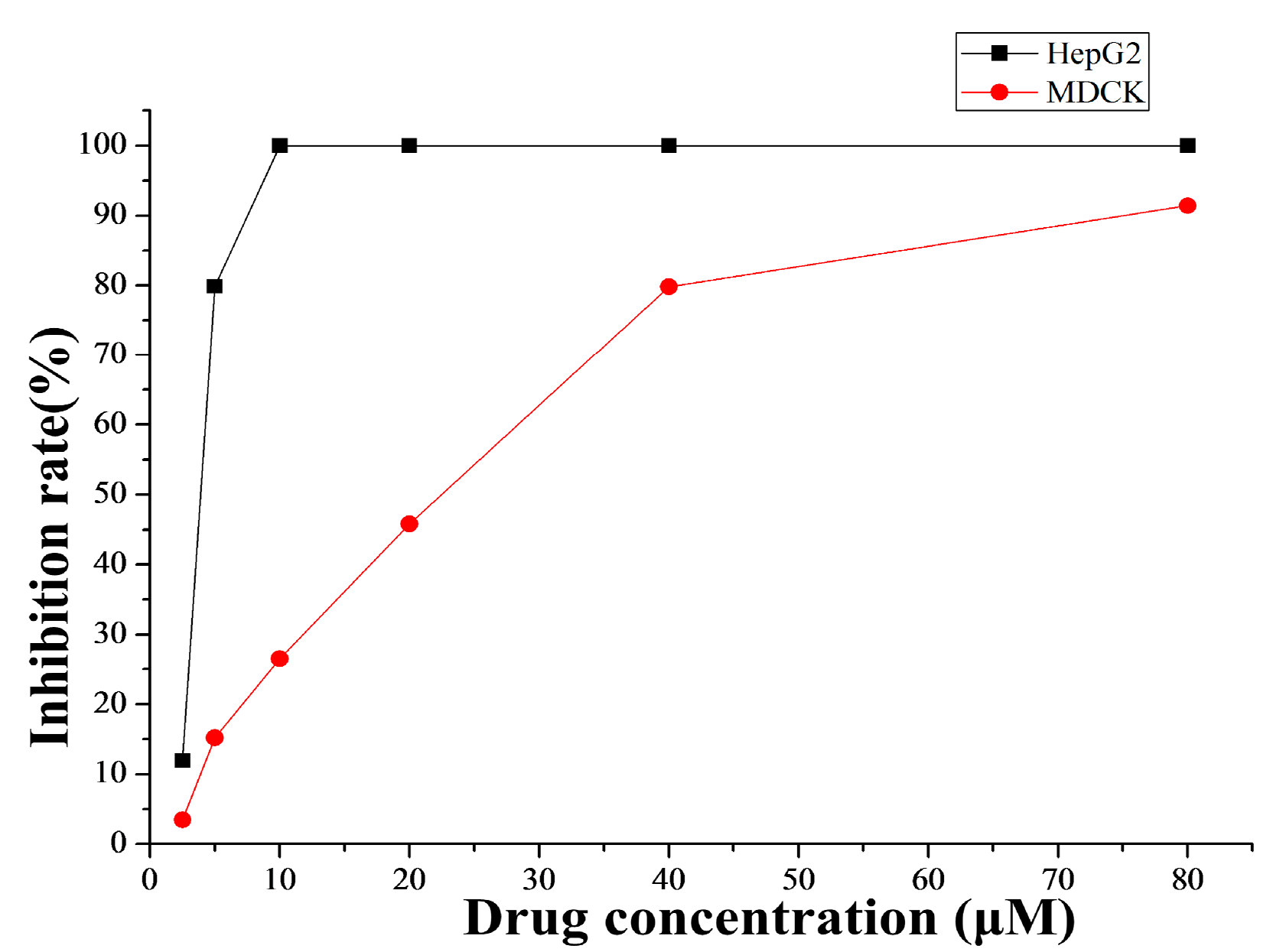
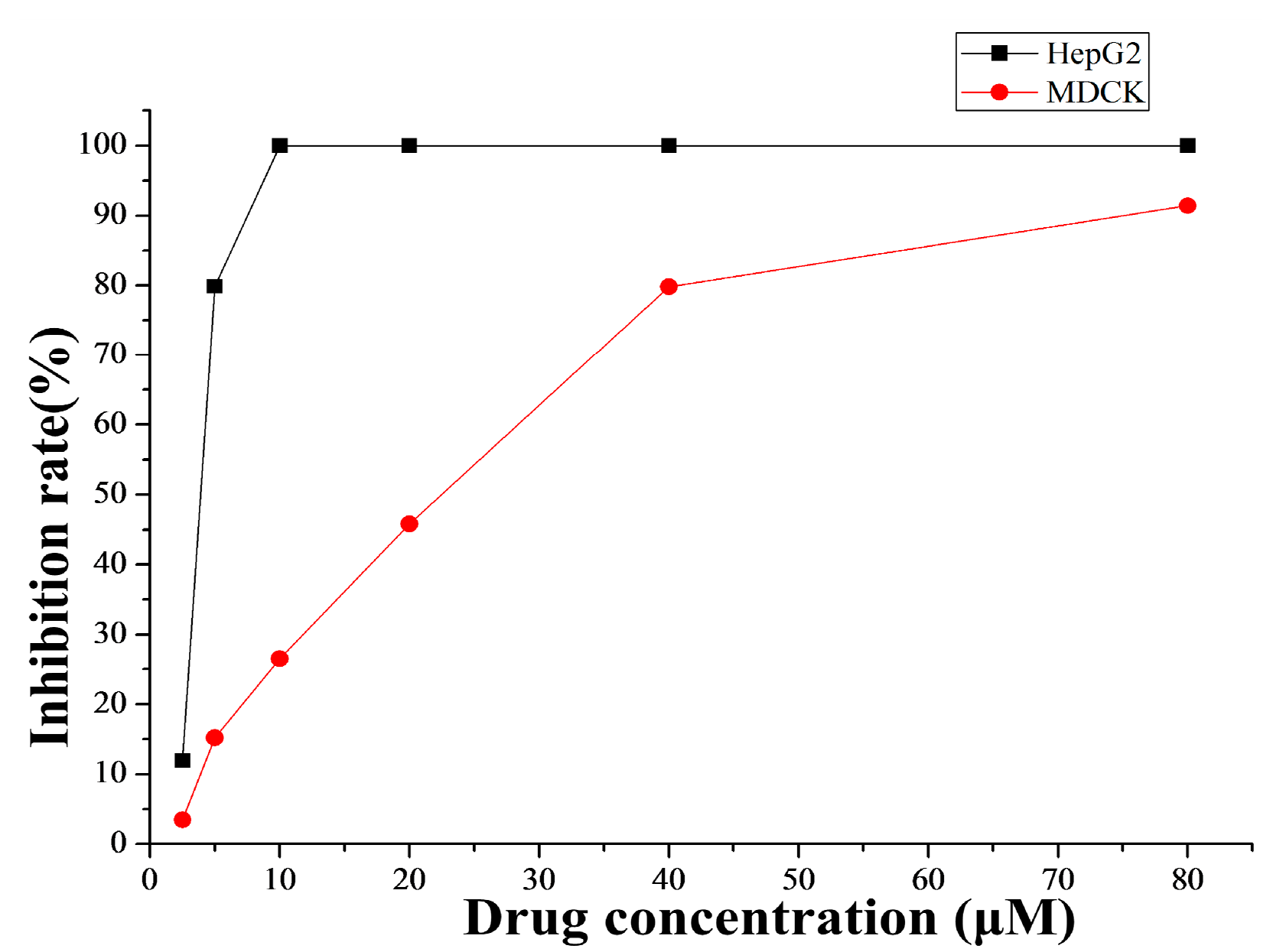
2.4.1. Cytotoxicity Selectivity
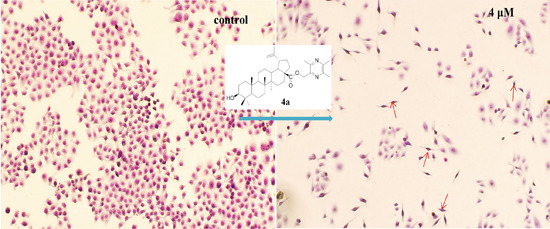
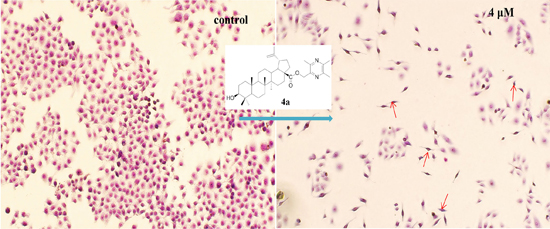
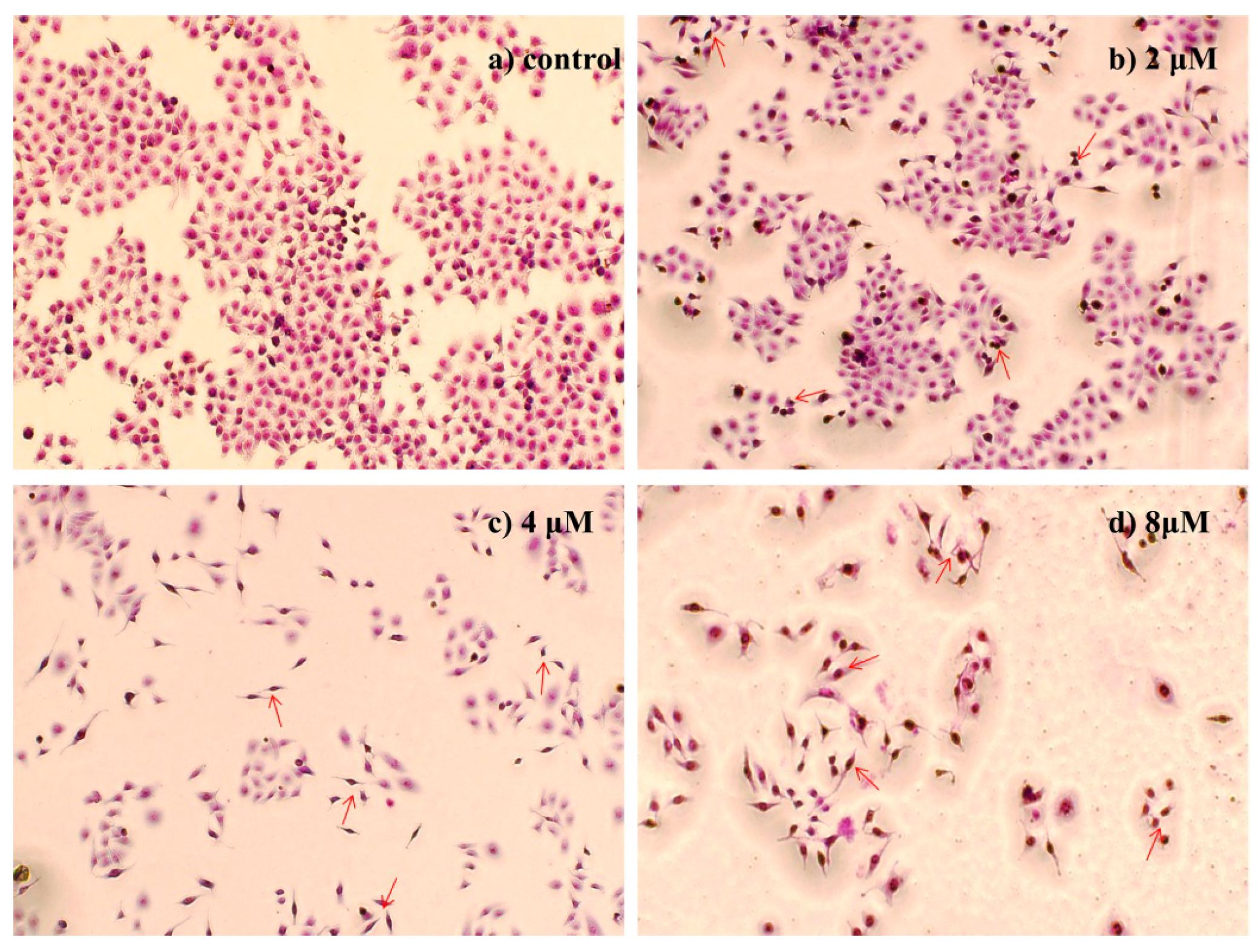
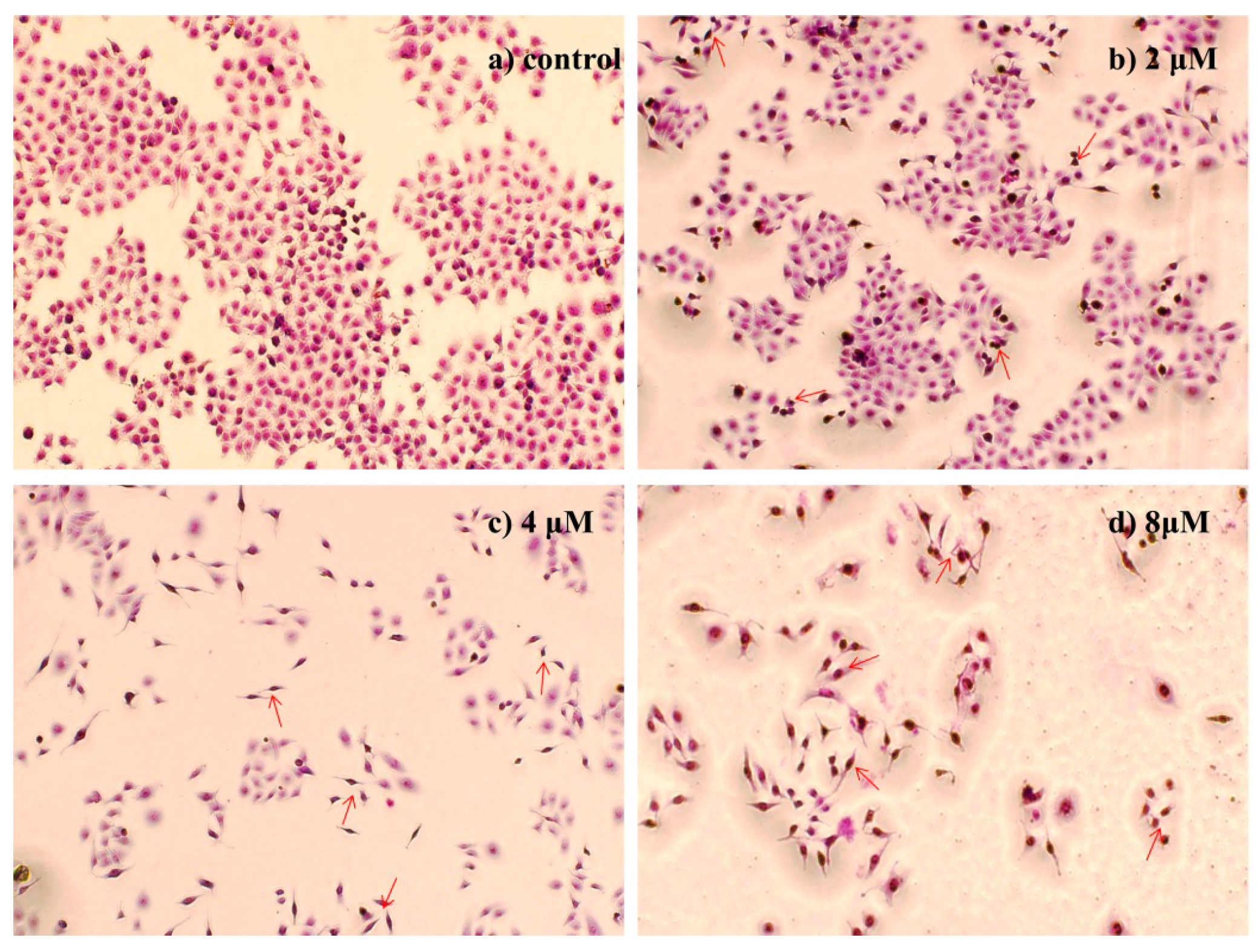
2.4.2. Morphological Observation of HepG2 Cells Using Giemsa Staining
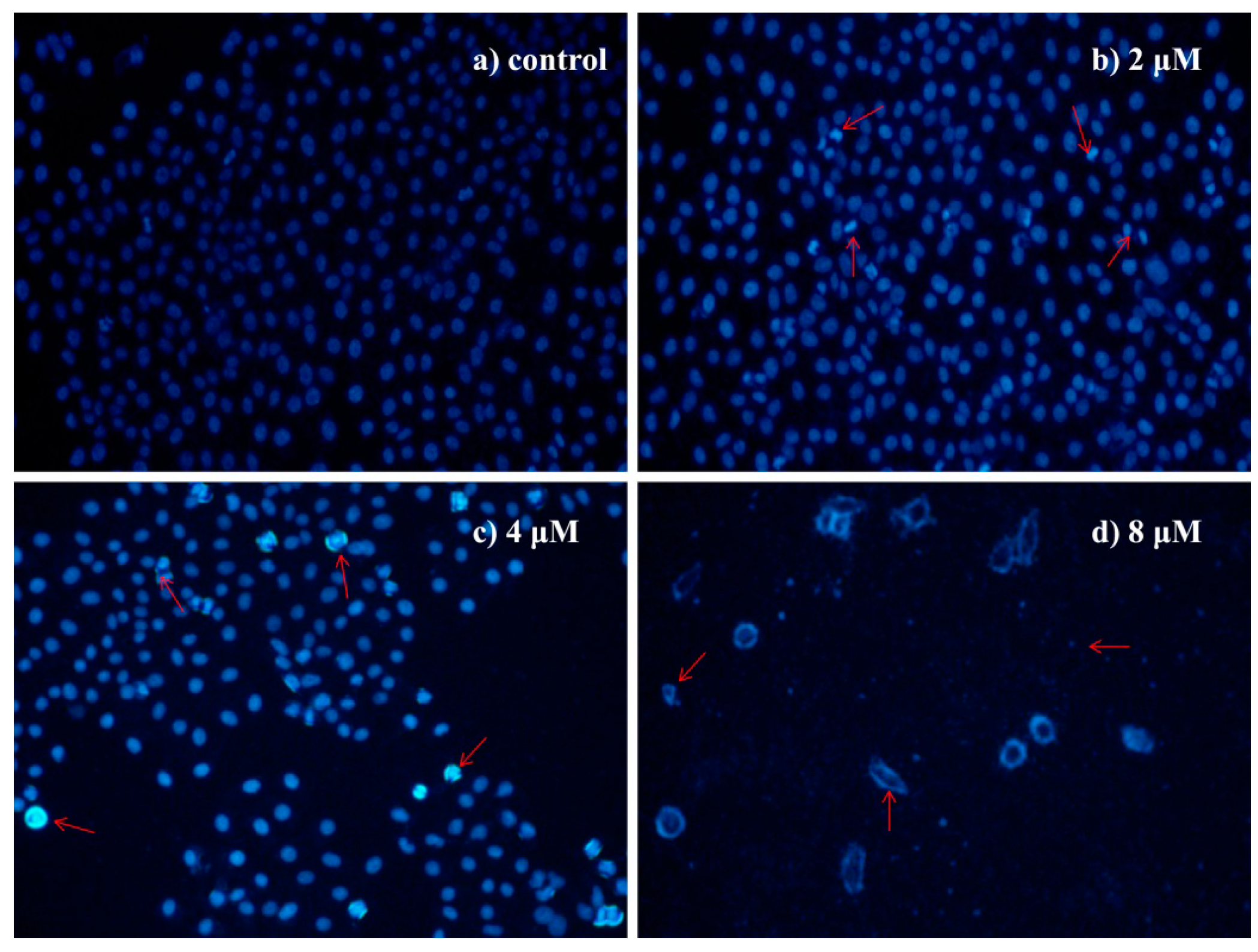
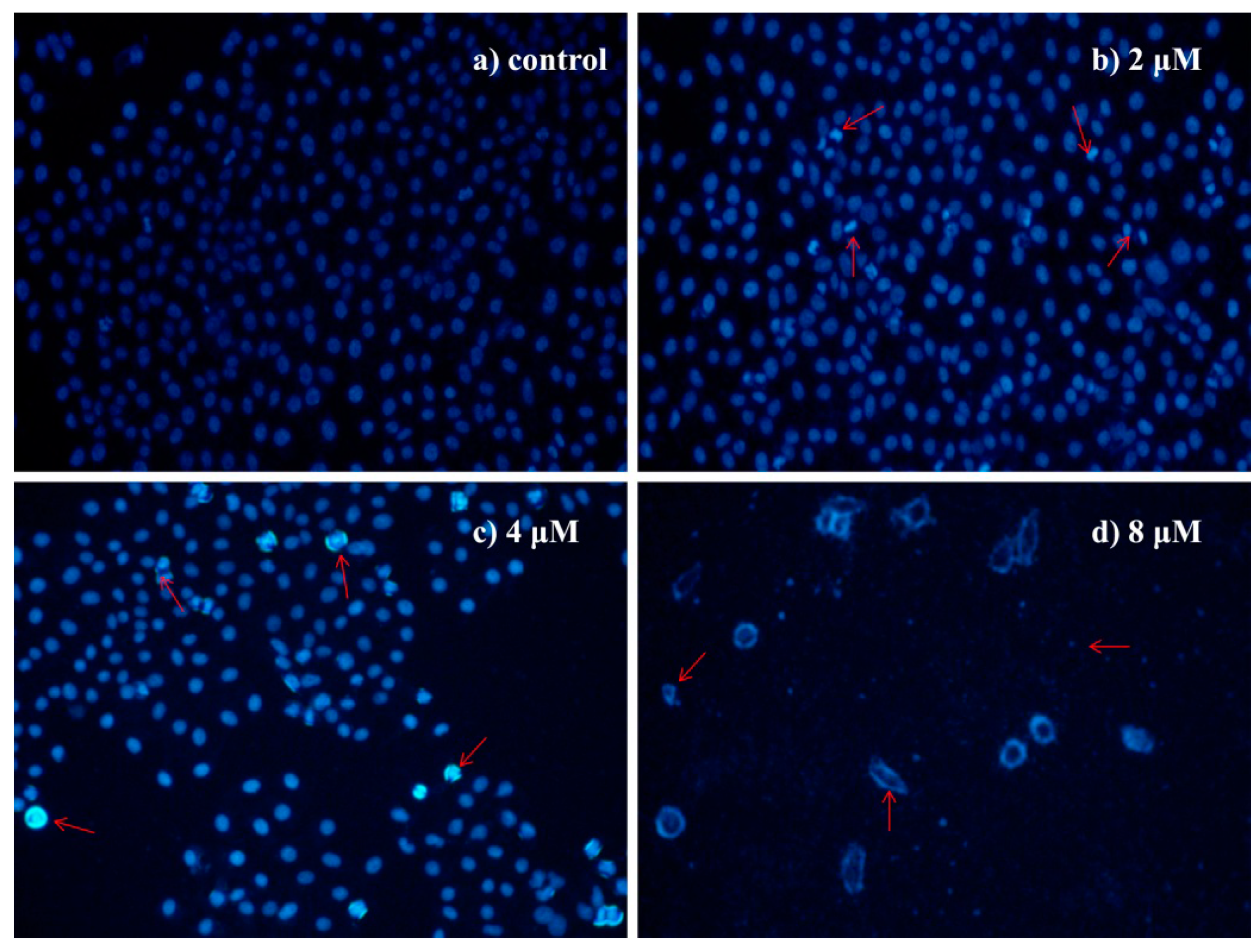
2.4.3. Morphological Observation of HepG2 Cells Using DAPI Staining
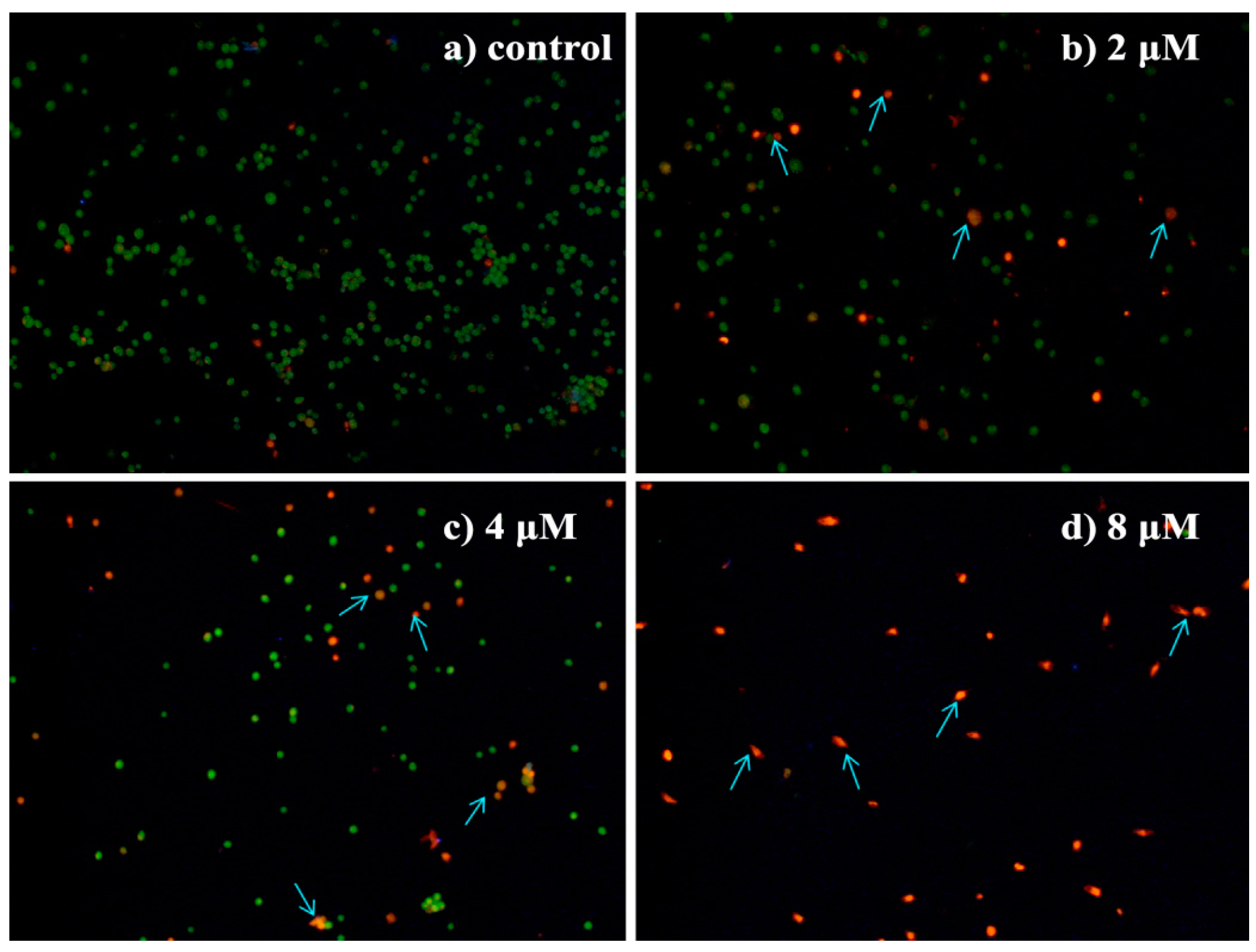
2.4.4. Morphological Observation of HepG2 Cells Using Acridine Orange/Ethidium Bromide (AO/EB) Double Staining
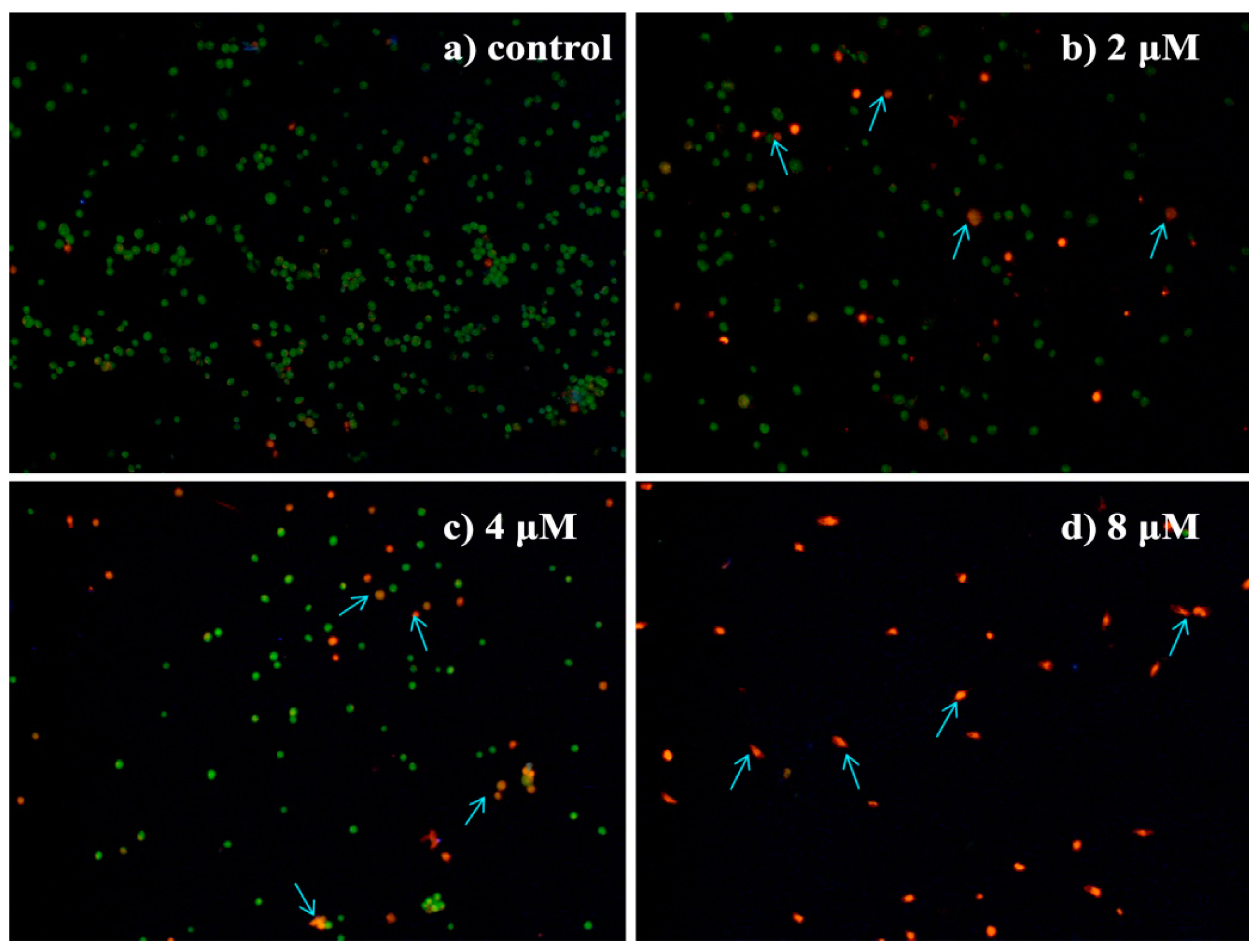
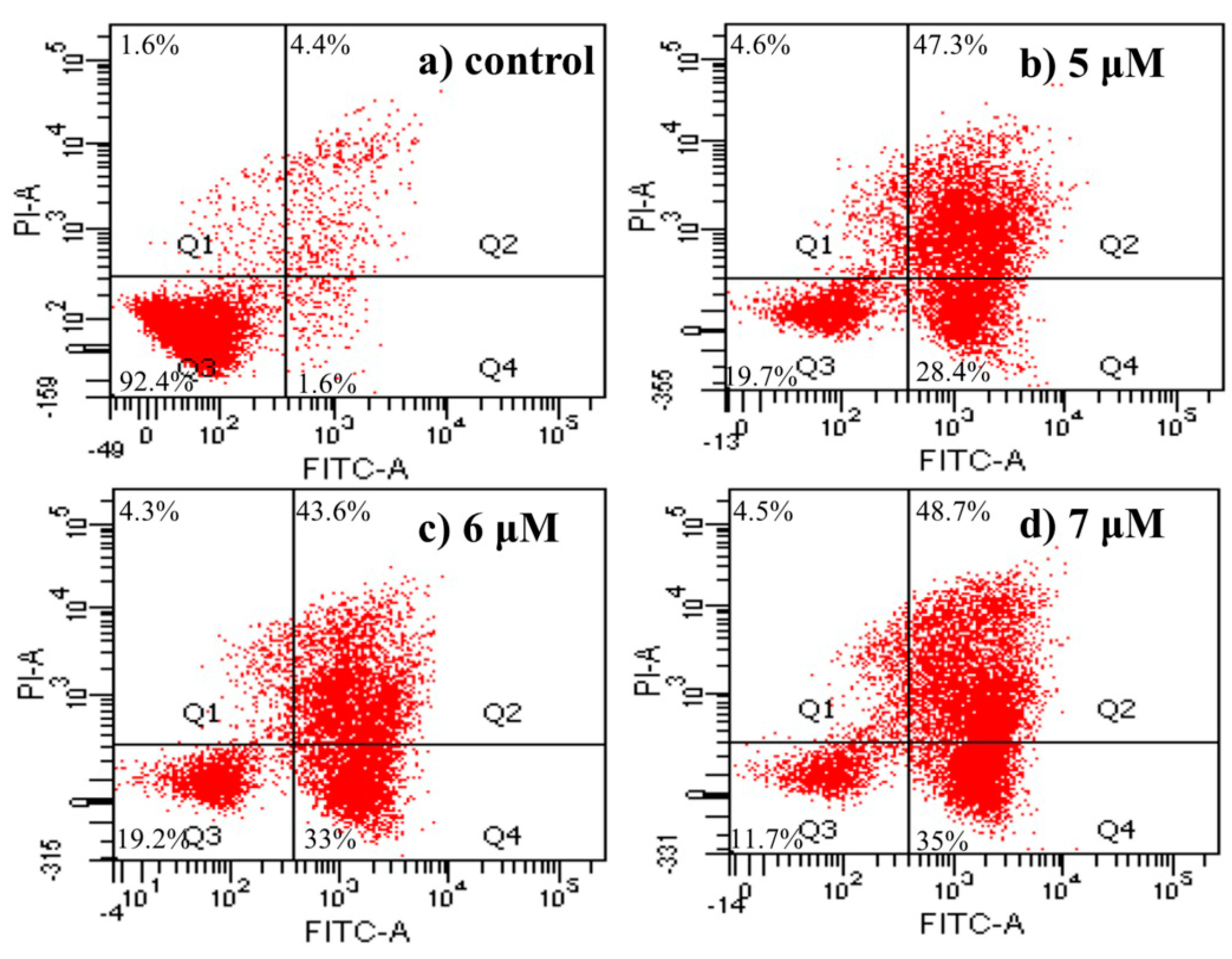
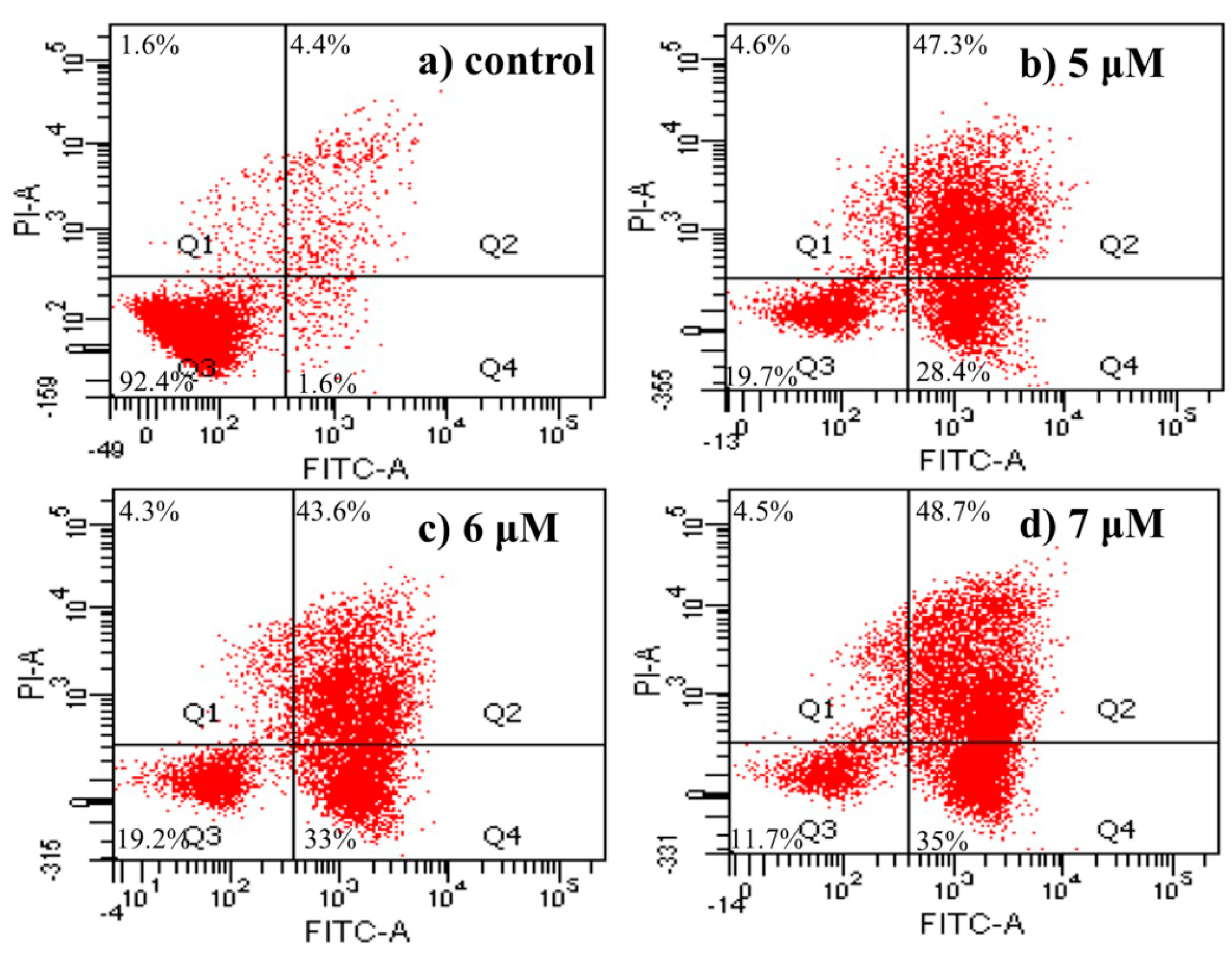
2.4.5. Apoptosis Analysis by Flow Cytometric Using Annexin V-FITC/Propidium Iodide (PI) Staining
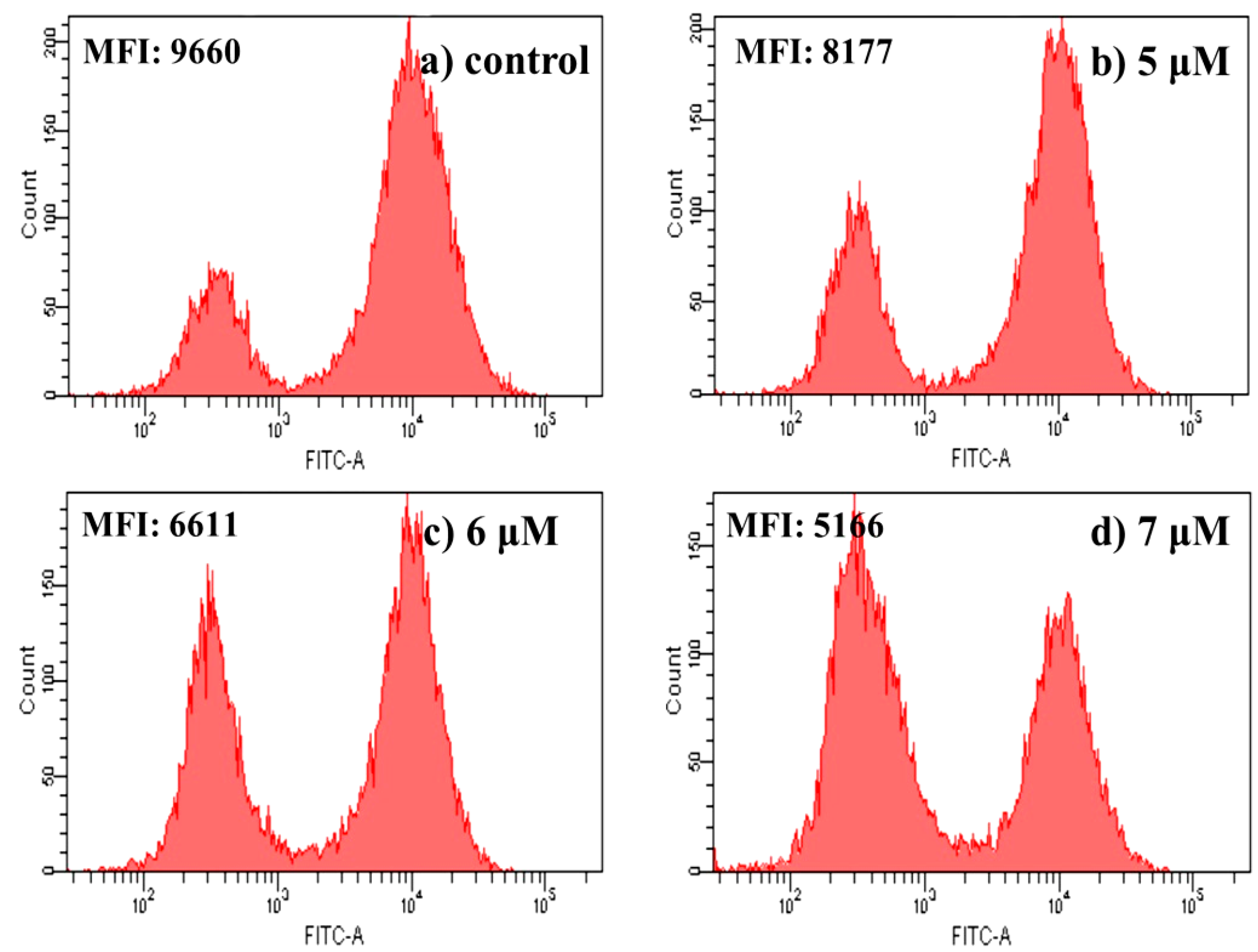
2.4.6. Measurement of Mitochondrial Membrane Potential

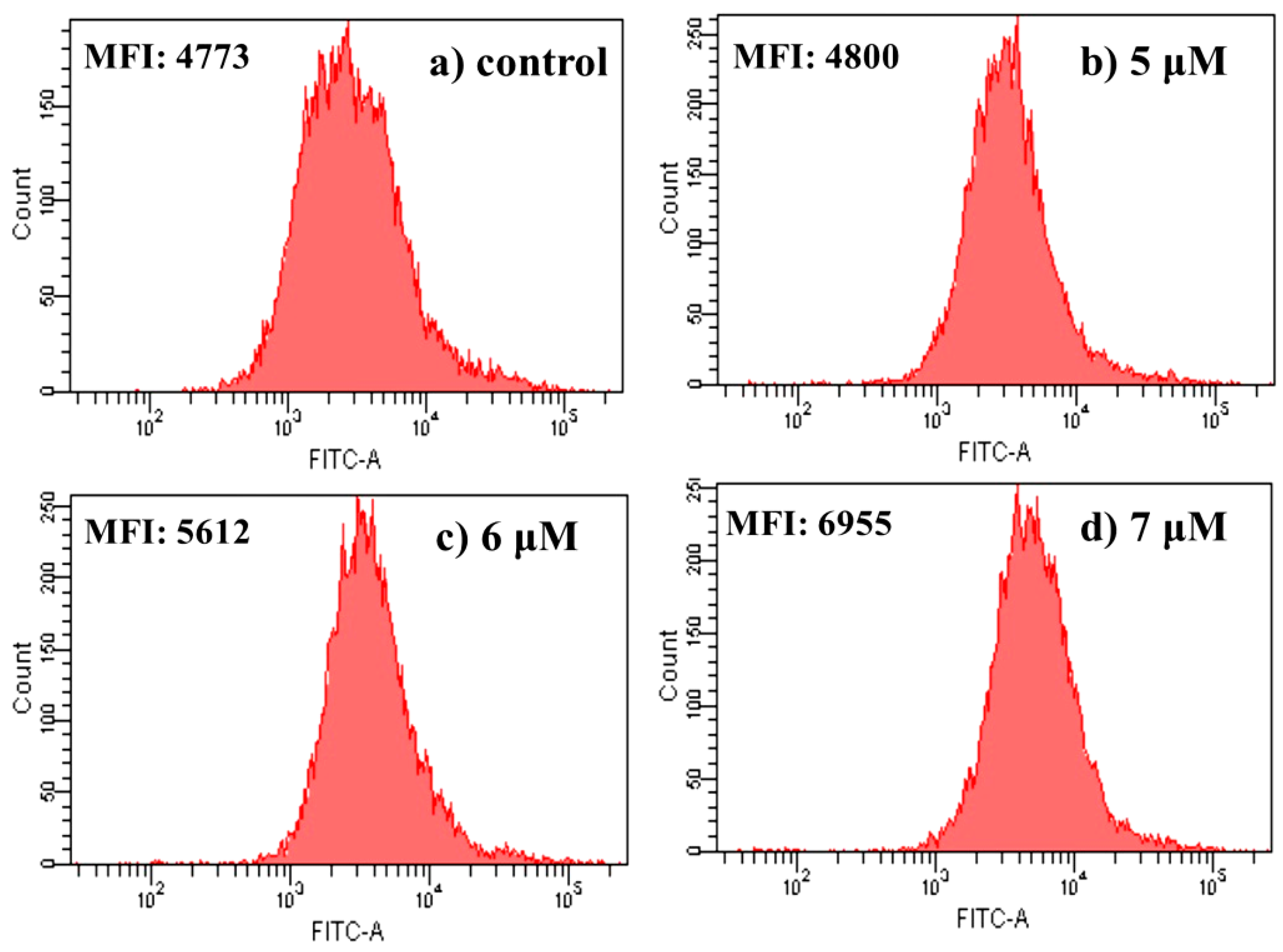
2.4.7. Intracellular Free Ca2+ Detection

3. Experimental Section
3.1. Chemistry
3.1.1. General Procedure for the Preparation of Ligustrazine-Triterpenes Derivatives 1a–4a
3.1.2. General Procedure for the Preparation of Ligustrazine-Triterpenes Derivatives 1b–4b
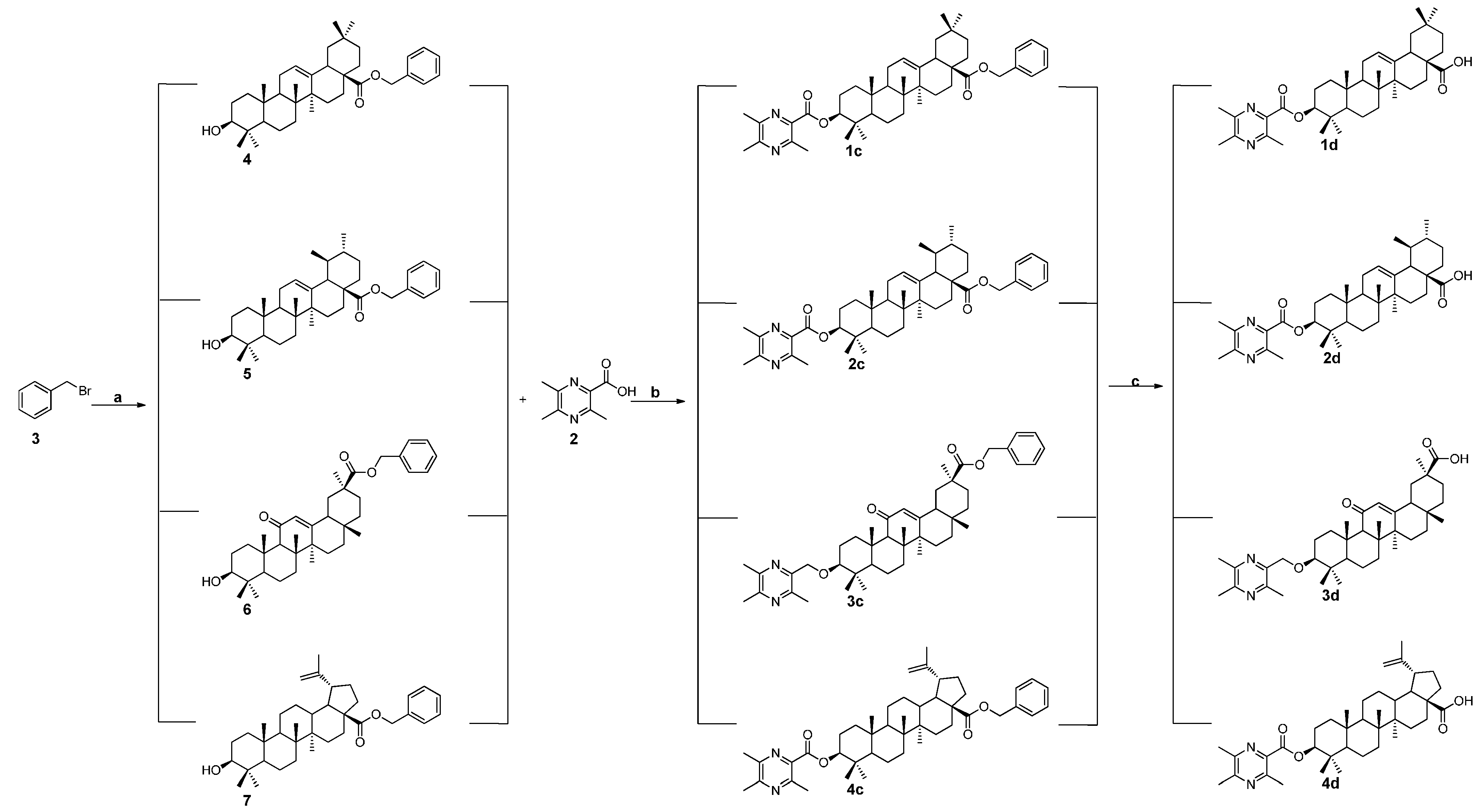
3.1.3. General Procedure for the Preparation of the Intermediates 4, 5, 6, and 7
3.1.4. General Procedure for the Preparation of Ligustrazine-triterpenes Derivatives 1c–4c
3.1.5. General Procedure for the Preparation of Ligustrazine-Triterpenes Derivatives 1d–4d
3.2. Bio-Evaluation Methods
3.2.1. Cell Culture
3.2.2. Cytotoxicity Assay
3.2.3. Morphological Observation of HepG2 Cells Using Giemsa Staining
3.2.4. Morphological Observation of HepG2 Cells Using DAPI Staining
3.2.5. Morphological Observation of HepG2 Cells Using AO/EB Double Staining
3.2.6. Apoptosis Analysis by FCM Using Annexin V-FITC/PI Staining
3.2.7. Mitochondrial Membrane Potential (ΔΨm) Analysis
3.2.8. Intracellular Free Ca2+ Detection
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M.; Snader, K.M. Natural products as sources of new drugs over the period 1981–2002. J. Nat. Prod. 2003, 66, 1022–1037. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bai, H.; Zhang, X.D.; Liu, J.Z.; Cao, P.P.; Liao, N.; Zhang, W.; Wang, Z.; Hai, C.X. Inhibitory effect of oleanolic acid on hepatocellular carcinoma via ERK-p53-mediated cell cycle arrest and mitochondrial-dependent apoptosis. Carcinogenesis 2013, 34, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Wang, E.; Kumar, N.; Glickman, R.D. Ursolic acid differentially modulates apoptosis in skin melanoma and retinal pigment epithelial cells exposed to UV-VIS broadband radiation. Apoptosis 2014, 19, 816–828. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Ko, H.S.; Sohn, E.J.; Kim, B.; Kim, J.H.; Kim, H.J.; Kim, C.; Kim, J.E.; Kim, S.H. Inhibition of protein kinase C alpha/beta II and activation of c-Jun NH2-terminal kinase mediate glycyrrhetinic acid induced apoptosis in non-small cell lung cancer NCI-H460 cells. Bioorg. Med. Chem. Lett. 2014, 24, 1188–1191. [Google Scholar] [CrossRef] [PubMed]
- Jonnalagadda, S.C.; Corsello, M.A.; Sleet, C.E. Betulin-betulinic acid natural product based analogs as anti-cancer agents. Anticancer Agents Med. Chem. 2013, 13, 1477–1499. [Google Scholar] [CrossRef] [PubMed]
- Mullauer, F.B.; Kessler, J.H.; Medema, J.P. Betulinic acid, a natural compound with potent anticancer effects. Anticancer Drugs 2010, 21, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Csuk, R.; Schwarz, S.; Kluge, R.; Strohl, D. Improvement of the cytotoxicity and tumor selectivity of glycyrrhetinic acid by derivatization with bifunctional aminoacids. Arch. Pharm. 2011, 344, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Salomatina, O.V.; Markov, A.V.; Logashenko, E.B.; Korchagina, D.V.; Zenkova, M.A.; Salakhutdinov, N.F.; Vlassov, V.V.; Tolstikov, G.A. Synthesis of novel 2-cyano substituted glycyrrhetinic acid derivatives as inhibitors of cancer cells growth and NO production in LPS-activated J-774 cells. Bioorg. Med. Chem. 2014, 22, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, S.; Siewert, B.; Xavier, N.M.; Jesus, A.R.; Rauter, A.P.; Csuk, R. A “natural” approach: Synthesis and cytoxicity of monodesmosidic glycyrrhetinic acid glycosides. Eur. J. Med. Chem. 2014, 72, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Dai, X.; Kumar, A.P.; Tan, B.K.H.; Sethi, G.; Bishayee, A. Oleanolic acid and its synthetic derivatives for the prevention and therapy of cancer. Cancer Lett. 2014, 346, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Wang, M.; Gou, S.H.; Liu, X.Y.; Zhang, H.; Cao, F. Combination of amino acid/dipeptide with nitric oxide donating oleanolic acid derivatives as PepT1 targeting antitumor prodrugs. J. Med. Chem. 2014, 57, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Ashour, A.; El-Sharkawy, S.; Amer, M.; Bar, F.A.; Katakura, Y.; Miyamoto, T.; Toyota, N.; Tran, B.H.; Kondo, R.; Shimizu, K. Rational design and synthesis of topoisomerase I and II inhibitors based on oleanolic acid moiety for new anti-cancer drugs. Bioorg. Med. Chem. 2014, 22, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.G.; Su, C.H.; Yang, L.D.; Liu, J.; Chen, Z.F. Synthesis of oleanolic acid dimers linked at C-28 and evaluation of anti-tumor activity. Eur. J. Med. Chem. 2015, 89, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.; Dar, B.A.; Majeed, R.; Hamid, A.; Bhat, B.A. Synthesis and biological evaluation of ursolic acid-triazolyl derivatives as potential anti-cancer agents. Eur. J. Med. Chem. 2013, 66, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Parida, P.K.; Sau, A.; Ghosh, T.; Jana, K.; Biswas, K.; Raha, S.; Misra, A.K. Synthesis and evaluation of triazole linked glycosylated 18 beta-glycyrrhetinic acid derivatives as anticancer agents. Bioorg. Med. Chem. Lett. 2014, 24, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.L.; She, G.M.; Yang, Y.N.; Li, Q.; Zhang, H.G.; Liu, J.; Cao, Y.Q.; Xu, X.; Lei, H.M. Synthesis and biological evaluation of new ligustrazine derivatives as anti-tumor agents. Molecules 2012, 17, 4972–4985. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.L.; Cheng, Y.T.; Xu, K.; An, Y.W.; Wang, W.; Li, Q.S.; Han, Q.J.; Li, Q.; Zhang, H.G.; Lei, H.M. Synthesis and antitumor evaluation of one novel tetramethylpyrazine-rhein derivative. Asian J. Chem. 2013, 25, 4885–4888. [Google Scholar]
- Wang, P.L.; Zhang, H.G.; Chu, F.H.; Xu, X.; Lin, J.X.; Chen, C.X.; Li, G.L.; Cheng, Y.T.; Wang, L.; Li, Q.; et al. Synthesis and protective effect of new ligustrazine-benzoic acid derivatives against CoCl2-induced neurotoxicity in differentiated PC12 Cells. Molecules 2013, 18, 13027–13042. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.L.; Zhang, Y.Z.; Xu, K.; Li, Q.; Zhang, H.G.; Guo, J.; Pang, D.; Cheng, Y.T.; Lei, H.M. A new ligustrazine derivative - pharmacokinetic evaluation and antitumor activity by suppression of NF- kB/p65 and COX-2 expression in S180 mice. Pharmazie 2013, 68, 782–789. [Google Scholar] [PubMed]
- Hou, P.; Ni, J.; Cao, S.L.; Lei, H.M.; Cai, Z.J.; Zhang, T.; Yu, F.; Tan, Q.Z. Preparation and evaluation of solid dispersions of a new antitumor compound based on early-stage preparation discovery concept. AAPS PharmSciTech. 2013, 14, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Cao, S.L.; Ni, J.; Zhang, T.; Cai, Z.J.; Liu, J.J.; Wang, Y.; Wang, P.L.; Lei, H.M.; Liu, Y. In-vitro and in vivo comparison of T-OA microemulsions and solid dispersions based on EPDC. Drug Dev. Ind. Pharm. 2015, 41, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Kwon, T.H.; Lee, B.; Chung, S.H.; Kim, D.H.; Lee, Y.S. Synthesis and NO production inhibitory activities of ursolic acid and oleanolic acid derivatives. B. Korean Chem. Soc. 2009, 30, 119–123. [Google Scholar]
- Yamashita, T.; Dohta, Y.; Nakamura, T.; Fukami, T. High-speed solubility screening assay using ultra-performance liquid chromatography/mass spectrometry in drug discovery. J. Chromatogr. A 2008, 1182, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Dohta, Y.; Yamashita, T.; Horiike, S.; Nakamura, T.; Fukami, T. A system for LogD screening of 96-well plates using a water-plug aspiration/injection method combined with high-performance liquid chromatography-mass spectrometry. Anal. Chem. 2007, 79, 8312–8315. [Google Scholar] [CrossRef] [PubMed]
- Alelyunas, Y.W.; Pelosi-Kilby, L.; Turcotte, P.; Kary, M.B.; Spreena, R.C. A high throughput dried DMSO LogD lipophilicity measurement based on 96-well shake-flask and atmospheric pressure photoionization mass spectrometry detection. J. Chromatogr. A 2010, 1217, 1950–1955. [Google Scholar] [CrossRef] [PubMed]
- Saggiomo, V.; Otto, S.; Marques, I.; Félix, V.; Torroba, T.; Quesada, R. The role of lipophilicity in transmembrane anion transport. Chem. Commun. 2012, 48, 5274–5276. [Google Scholar]
- Csuk, R.; Schwarz, S.; Siewert, B.; Kluge, R.; Strohl, D. Synthesis and cytotoxic activity of methyl glycyrrhetinate esterified with amino acids. Z. Naturforsch. 2012, 67, 731–746. [Google Scholar] [CrossRef]
- Vijayan, P.; Viswanathamurthi, P.; Silambarasan, V.; Velmurugan, D.; Velmurugan, K.; Nandhakumar, R.; Butcher, R.J.; Silambarasan, T.; Dhandapani, R. Dissymmetric thiosemicarbazone ligands containing substituted aldehyde arm and their ruthenium(II) carbonyl complexes with PPh3/AsPh3 as ancillary ligands: Synthesis, structural characterization, DNA/BSA interaction and in vitro anticancer activity. J. Organomet. Chem. 2014, 768, 163–177. [Google Scholar] [CrossRef]
- Mohankumar, K.; Pajaniradje, S.; Sridharan, S.; Singh, V.K.; Ronsard, L.; Banerjea, A.C.; Benson, C.S.; Coumar, M.S.; Rajagopalan, R. Mechanism of apoptotic induction in human breast cancer cell, MCF-7, by an analog of curcumin in comparison with curcumin—An in vitro and in silico approach. Chem. Biol. Interact. 2014, 210, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, J.S.; Qin, Q.Z.; Jiang, Y.; Yang, G.T.; Rao, K.M.; Wang, Q.; Xiong, W.; Yuan, J. Mono-2-ethylhexyl phthalate induced loss of mitochondrial membrane potential and activation of Caspase3 in HepG2 cells. Environ. Toxicol. Pharmacol. 2012, 33, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.X.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.; Puthli, A.; Balakrishnan, S.; Sapra, B.K.; Mishra, K.P. Betulinic acid-induced cytotoxicity in human breast tumor cell lines MCF-7 and T47D and its modification by tocopherol. Cancer Investig. 2014, 32, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.K.; Ho, J.C.K.; Cheung, F.W.K.; Liu, B.P.L.; Ye, W.C.; Che, C.T. Apoptotic activity of betulinic acid derivatives on murine melanoma B16 cell line. Eur. J. Pharmacol. 2004, 498, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Ruan, Y.X.; Chen, Q.; Li, S.P.; Wang, Q.L.; Cai, J.Y. Curcumin induced HepG2 cell apoptosis-associated mitochondrial membrane potential and intracellular free Ca2+ concentration. Eur. J. Pharmacol. 2011, 650, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.H.; Gong, L.; Li, X.F.; Ye, L.; Wang, B.; Liu, J.; Qiu, J.Y.; Jiao, H.D.; Zhang, W.D.; Chen, J.Z.; Wang, J.P. Infrasound increases intracellular calcium concentration and induces apoptosis in hippocampi of adult rats. Mol. Med. Rep. 2012, 5, 73–77. [Google Scholar] [PubMed]
- Li, Z.Y.; Yu, F.; Cui, L.; Zhan, P.; Wang, S.X.; Shen, Y.M.; Liu, X.Y. Ligustrazine derivatives. Part 6: Design, synthesis and evaluation of novel ligustrazinyl acylguanidine derivatives as potential cardiovascular agents. Med. Chem. 2012, 8, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Sakalar, C.; Yuruk, M.; Kaya, T.; Aytekin, M.; Kuk, S.; Canatan, H. Pronounced transcriptional regulation of apoptotic and TNF-NF-kappa-B signaling genes during the course of thymoquinone mediated apoptosis in HeLa cells. Mol. Cell. Biochem. 2013, 383, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Nakkala, J.R.; Mata, R.; Gupta, A.K.; Sadras, S.R. Biological activities of green silver nanoparticles synthesized with Acorous calamus rhizome extract. Eur. J. Med. Chem. 2014, 85, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, S.; Csuk, R. Synthesis and antitumour activity of glycyrrhetinic acid derivatives. Bioorg. Med. Chem. 2010, 18, 7458–7474. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.; Pinheiro, R.O.; Dutra, P.M.L.; Santos, R.F.; Cunha-Junior, E.F.; Torres-Santos, E.C.; da Silva, A.J.M.; Costa, P.R.R.; Da-Silva, S.A.G. Pterocarpanquinone LQB-118 induces apoptosis in leishmania (Viannia) braziliensis and controls lesions in infected hamsters. PLoS ONE 2014, 9, 109672. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, B.; Chu, F.; Zhang, Y.; Wang, X.; Li, Q.; Liu, W.; Xu, X.; Xing, Y.; Chen, J.; Wang, P.; et al. A Series of New Ligustrazine-Triterpenes Derivatives as Anti-Tumor Agents: Design, Synthesis, and Biological Evaluation. Int. J. Mol. Sci. 2015, 16, 21035-21055. https://doi.org/10.3390/ijms160921035
Xu B, Chu F, Zhang Y, Wang X, Li Q, Liu W, Xu X, Xing Y, Chen J, Wang P, et al. A Series of New Ligustrazine-Triterpenes Derivatives as Anti-Tumor Agents: Design, Synthesis, and Biological Evaluation. International Journal of Molecular Sciences. 2015; 16(9):21035-21055. https://doi.org/10.3390/ijms160921035
Chicago/Turabian StyleXu, Bing, Fuhao Chu, Yuzhong Zhang, Xiaobo Wang, Qiang Li, Wei Liu, Xin Xu, Yanyi Xing, Jing Chen, Penglong Wang, and et al. 2015. "A Series of New Ligustrazine-Triterpenes Derivatives as Anti-Tumor Agents: Design, Synthesis, and Biological Evaluation" International Journal of Molecular Sciences 16, no. 9: 21035-21055. https://doi.org/10.3390/ijms160921035