
Detection of Selection Signatures on the X Chromosome in Three Sheep Breeds

 and
and
Abstract
:
1. Introduction
2. Results
2.1. Markers and Core Haplotypes
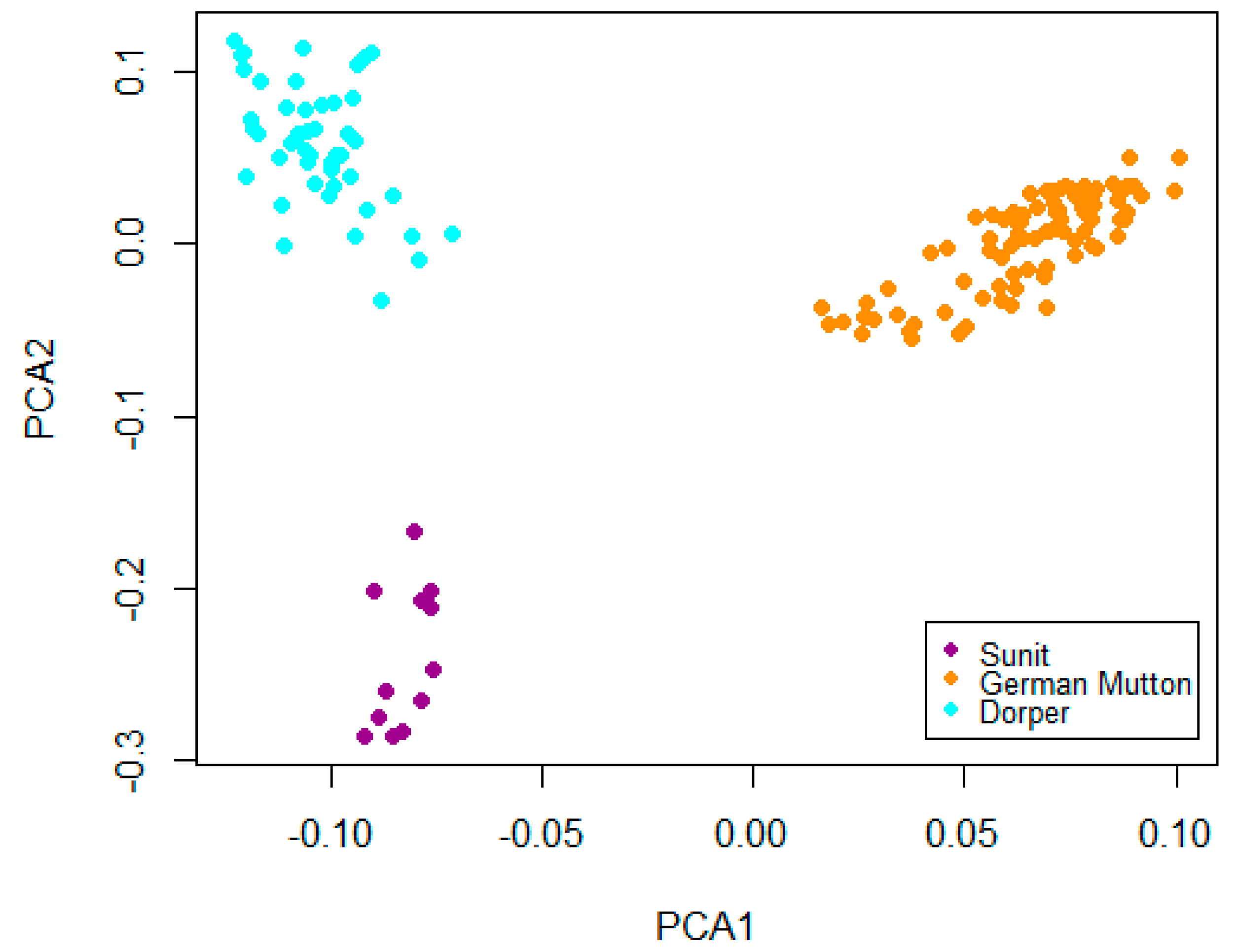
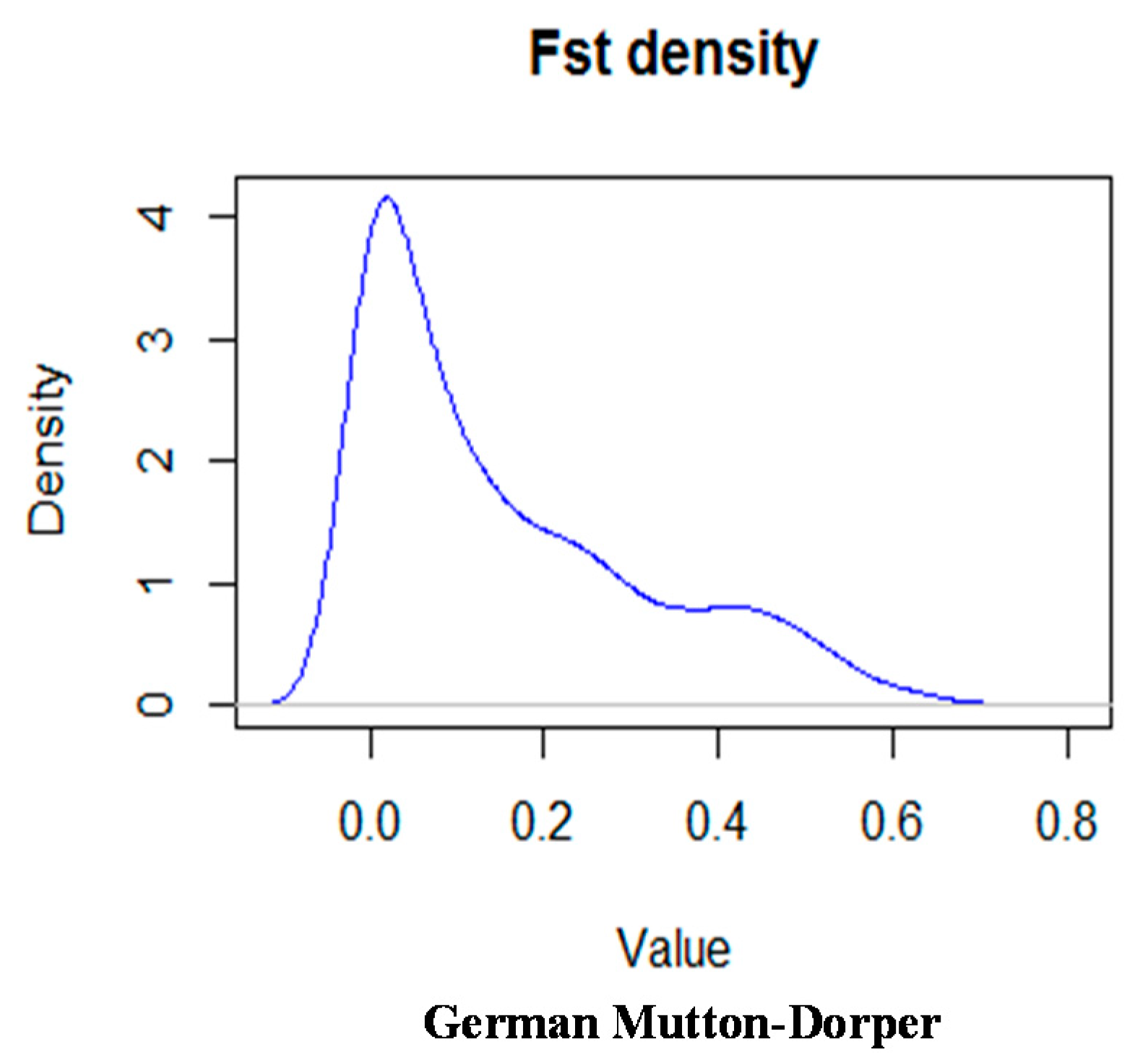
2.2. Empirical Distribution of Test Statistics

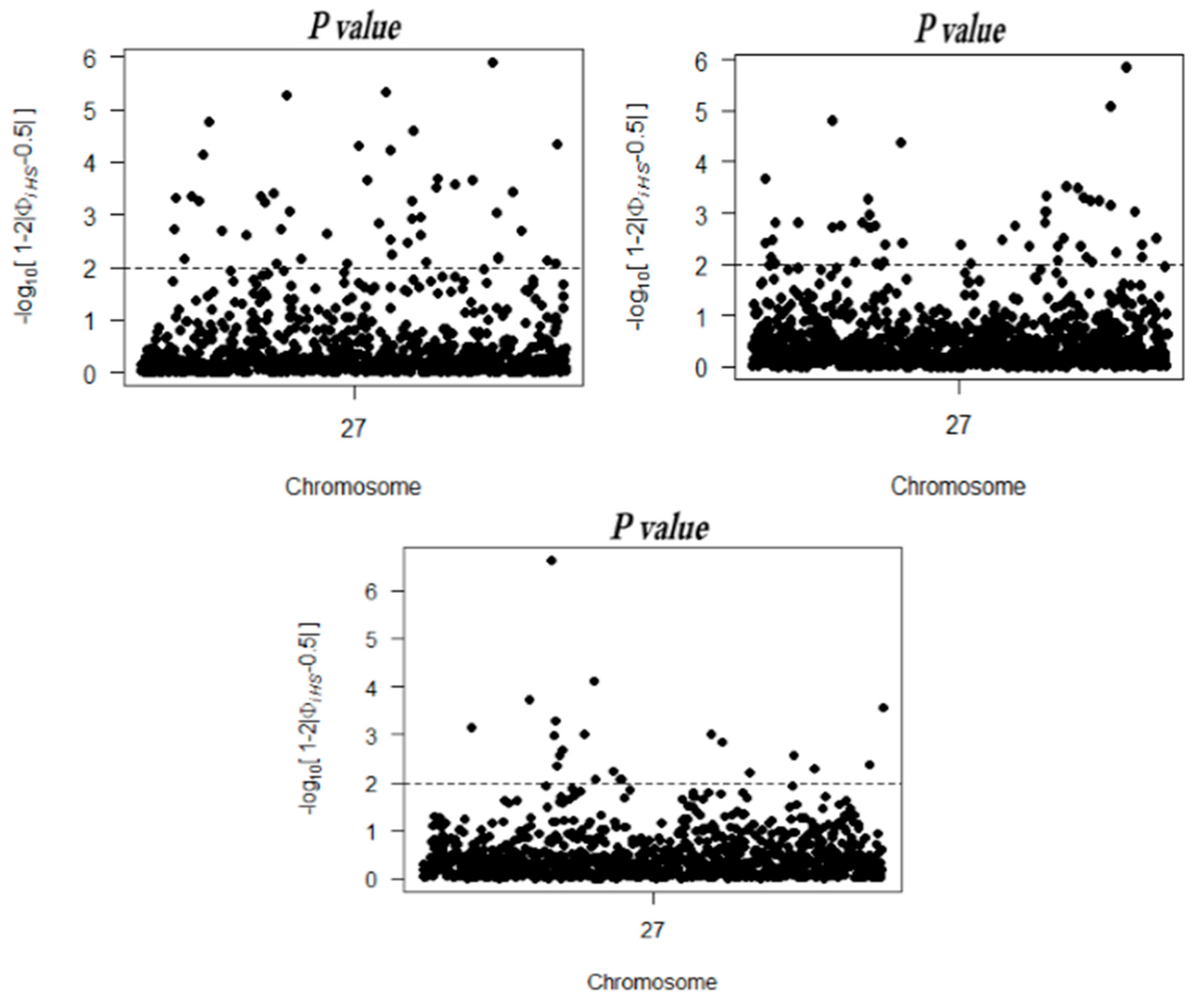
2.3. Identification of Recent Selection Signatures on the X Chromosome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | iHS | FST | |||||
|---|---|---|---|---|---|---|---|
| G M | D | S | G M-D-S | G M-D | S-G M | S-D | |
| Number of SNPs | 51 | 21 | 46 | 12 | 12 | 12 | 12 |
| Number of regions | 35 | 17 | 37 | 10 | 9 | 11 | 9 |
| Average length (Mb) | 0.43 | 0.44 | 0.43 | 0.43 | 0.43 | 0.40 | 0.45 |
| Total length (Mb) | 14.78 | 7.59 | 15.93 | 4.35 | 3.89 | 4.44 | 40.3 |

2.4. Candidate Selection Regions
2.5. Identification of Candidate Genes in Selection Regions and Functional Analysis
| Breed | Region (Mb) | |iHS| | Gene |
|---|---|---|---|
| German Mutton | 122.16–122.56 | 5.85 | TCEAL4, TCEAL1, MORF4L2, GLRA4, PLP1, RAB9B, TMSB15B, CYSLTR1 |
| 118.98–119.38 | 5.07 | NXT2 | |
| 19.83–20.23 | 4.80 | - | |
| 40.51–40.91 | 4.39 | FAM192A | |
| 3.77–4.17 | 3.66 | - | |
| Dorper | 30.58–30.98 | 6.63 | LAMP1 |
| 41.36–41.76 | 4.12 | EFHC2 | |
| 23.67–24.07 | 3.72 | - | |
| 134.90–135.30 | 3.56 | PABPC5 | |
| 31.27–31.67 | 3.29 | - | |
| Sunit | 103.45–103.85 | 3.63 | TEX13A |
| 31.98–32.38 | 3.44 | TMEM47 | |
| 40.13–40.53 | 3.32 | - | |
| 116.80–117.2 | 3.30 | TRPC5, ALG13 | |
| 94.82–95.22 | 3.25 | - |
| Breed Pair | Position (Mb) | FST Value | Gene Name |
|---|---|---|---|
| German Mutton-Dorper-Sunit | 51.51–51.91 | 0.71 | SRSF10, DGKK, CCNB3 |
| 117.91–118.31 | 0.64 | TMEM164 | |
| 42.80–43.21 | 0.64 | ZXDB | |
| 86.91–87.31 | 0.63 | SLITRK2 | |
| 45.82–46.22 | 0.59 | MSN, MMD, VSIG4, HEPH | |
| 134.78–135.18 | 0.59 | PCDH11X, ZGC:112234, PABPC5 | |
| 125.43–125.83 | 0.58 | TIMM8A, BTK, RPL36A, GLA, HNRNPH2, ARMCX4, CSNK1A1, ARMCX1, ARMCX6, ARMCX3, ARMCX2 | |
| 117.84–118.24 | 0.58 | RGAG1, AMMECR1 | |
| 73.69–74.09 | 0.57 | CHM, DACH2 | |
| 77.77–78.17 | 0.57 | FAM58A, ATP2B3, BGN, HAUS7, TREX2, ZNF275, KIR3DL1 | |
| 125.15–125.55 | 0.57 | XKRX, ARL13A, TRMT2B, TMEM35, CENPI, DRP2, TAF7L | |
| 84.41–84.81 | 0.56 | SLITRK2, SLITRK4 | |
| German Mutton-Dorper | 86.65–87.30 | 0.71 | SLITRK4 |
| 45.82–46.22 | 0.67 | MSN, MMD, VSIG4, HEPH | |
| 125.15–125.55 | 0.64 | XKRX, ARL13A, TRMT2B, TMEM35, CENPI, DRP2, TAF7L | |
| 9.35–9.75 | 0.63 | - | |
| 4.85–5.25 | 0.61 | PNPLA4 | |
| 116.23–116.63 | 0.61 | - | |
| 37.26–37.66 | 0.61 | BCOR, DUT | |
| 17.56–17.96 | 0.60 | RPS6KA3 | |
| 117.84–118.24 | 0.59 | RGAG1, AMMECR1 | |
| Sunit-German Mutton | 117.91–118.31 | 0.97 | RGAG1, AMMECR1, TMEM164 |
| 134.78–135.18 | 0.95 | PCDH11X, ZGC:112234, PABPC5 | |
| 42.80–43.20 | 0.94 | ZXDB | |
| 51.17–51.61 | 0.93 | SHROOM4, SRSF10 | |
| 10.63–11.03 | 0.91 | EGFL6, RAD51L3 | |
| 52.36–52.76 | 0.91 | USP27X, SUMO2, PPP1R3F, FOXP3, CCDCC22, CACNA1F, SYP, PRICKLE3, PLP2, MAGIX, GPKOW, RPL36, WDR45, PRAF2 | |
| 79.89–80.29 | 0.90 | CD99L2, MTMR1, MTM1, MAMLD1 | |
| 20.82–21.22 | 0.89 | PRDX4, ACOT9, SAT1 | |
| 73.69–74.09 | 0.88 | CHM, DACH2 | |
| 117.91–118.31 | 0.88 | RGAG1, AMMECR1, TMEM164 | |
| Sunit-Dorper | 60.80–61.20 | 0.87 | OGT, ACRC, CXCR3, DMRTC2 |
| 74.29–74.69 | 0.87 | DACH2 | |
| 118.09–118.49 | 0.82 | AMMECR1, TMEM164 | |
| 80.30–80.76 | 0.82 | MAMLD1 | |
| 117.72–118.12 | 0.81 | CHRDL1, RGAG1 | |
| 81.49–81.89 | 0.81 | AFF2 | |
| 51.51–51.91 | 0.80 | SRSF10, DGKK, CCNB3 | |
| 112.66–113.06 | 0.79 | PLS3 | |
| 92.61–93.01 | 0.79 | - | |
| 17.08–17.48 | 0.77 | MAP3K15, RPL17, SH3KBP1, CXORF23 |
3. Discussion
4. Experimental Section
4.1. Experimental Animals and DNA Samples
4.2. Genotyping and Quality Control
4.3. Analyses Integrated Haplotype Score
4.4. Population Differentiation Index
4.5. Identifying the Region in the X Chromosome under Selection
4.6. Enrichment Analysis
4.7. Gene Annotation
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zeder, M.A. Domestication and early agriculture in the Mediterranean Basin: Origins, diffusion, and impact. Proc. Natl. Acad. Sci. USA 2008, 105, 11597–11604. [Google Scholar] [CrossRef] [PubMed]
- Chessa, B.; Pereira, F.; Arnaud, F.; Amorim, A.; Goyache, F.; Mainland, I.; Kao, R.R.; Pemberton, J.M.; Beraldi, D.; Stear, M.J. Revealing the history of sheep domestication using retrovirus integrations. Science 2009, 324, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Kijas, J.W.; Lenstra, J.A.; Hayes, B.; Boitard, S.; Neto, L.P.; Cristobal, M.S.; Servin, B.; McCulloch, R.; Whan, V.; Gietzen, K.; et al. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Wei, C.; Zhang, L.; Liu, J.; Wang, G.; Zeng, T.; Du, L. A genome scan of recent positive selection signatures in three sheep populations. J. Integr. Agric. 2015, 15. [Google Scholar] [CrossRef]
- Stainton, J.J.; Haley, C.S.; Charlesworth, B.; Kranis, A.; Watson, K.; Wiener, P. Detecting signatures of selection in nine distinct lines of broiler chickens. Anim. Genet. 2014, 46, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, H.; Zhang, Q.; Ding, X. Identification of Selection Footprints on the X Chromosome in Pig. PLoS ONE 2014, 9, e94911. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.; Lu, Z.H.; Megens, H.J.; Archibald, A.L.; Haley, C.; Jackson, I.J.; Groenen, M.A.; Crooijmans, R.P.; Ogden, R.; Wiener, P. Signatures of diversifying selection in European pig breeds. PLoS Genet. 2013, 9, e1003453. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y. Statistical methods for detecting natural selection from genomic data. Genes Genet. Syst. 2010, 85, 359–376. [Google Scholar] [CrossRef] [PubMed]
- Voight, B.F.; Kudaravalli, S.; Wen, X.; Pritchard, J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006, 4, e72. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Riebler, A.; Held, L.; Stephan, W. Bayesian variable selection for detecting adaptive genomic differences among populations. Genetics 2008, 178, 1817–1829. [Google Scholar] [CrossRef] [PubMed]
- Mcrae, K.M.; Mcewan, J.C.; Dodds, K.G.; Gemmell, N.J. Signatures of selection in sheep bred for resistance or susceptibility to gastrointestinal nematodes. BMC Geno 2014, 15, 637–637. [Google Scholar] [CrossRef] [PubMed]
- Rubina, C.J.; Megensb, H.-J.; Barrioa, A.M.; Maqboolc, K.; Sayyabc, S.; Schwochowc, D.; Wang, C.; Örjan, C.; Jerna, P.; Jørgensen, C.B.; et al. Strong signatures of selection in the domestic pig genome. Proc. Natl. Acad. Sci. USA 2012, 109. [Google Scholar] [CrossRef] [PubMed]
- Heyer, E.; Segurel, L. Looking for signatures of sex-specific demography and local adaptation on the X chromosome. Genome Biol. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- McVicker, G.; Gordon, D.; Davis, C.; Green, P. Widespread genomic signatures of natural selection in hominid evolution. PLoS Genet. 2009, 5, e1000471. [Google Scholar] [CrossRef] [PubMed]
- Moradi, M.H.; Nejati-Javaremi, A.; Moradi-Shahrbabak, M.; Dodds, K.G.; Mcewan, J.C. Genomic scan of selective sweeps in thin and fat tail sheep breeds for identifying of candidate regions associated with fat deposition. BMC Genet. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Disteche, C.M. Dosage compensation of the active X chromosome in mammals. Nat. Genet. 2006, 38, 47–53. [Google Scholar]
- Graves, J.A.M. Sex chromosome specialization and degeneration in mammals. Cell 2006, 124, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shen, M.; Li, H.; Gao, L.; Liang, Y.; Yang, J.; Liu, S.; Wang, X.; Gan, S. Detection and analysis of polymorphisms of 59571364 and 59912586 loci on X chromosome in fat-tail and thin-tail sheep flocks (In Chinses). Yi Chuan = Hereditas/Zhongguo Yi Chuan Xue Hui Bian Ji 2013, 35, 1384–1390. [Google Scholar] [CrossRef] [PubMed]
- Sabeti, P.C.; Patrick, V.; Ben, F.; Jason, L.; Elizabeth, H.; Chris, C.; Xiaohui, X.; Byrne, E.H.; Mccarroll, S.A.; Rachelle, G. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, S.; Li, F.; Pan, X.; Li, C.; Zhang, X.; Ma, Y.; La, Y.; Xi, R.; Li, T. Polymorphisms of the Ovine BMPR-IB, BMP-15 and FSHR and Their Associations with Litter Size in Two Chinese Indigenous Sheep Breeds. Int. J. Mol. Sci. 2015, 16, 11385–11397. [Google Scholar] [CrossRef] [PubMed]
- Doerks, T.; Copley, R.R.; Schultz, J.; Ponting, C.P.; Bork, P. Systematic identification of novel protein domain families associated with nuclear functions. Genome Res. 2002, 12, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Kelley, J.L.; Madeoy, J.; Calhoun, J.C.; Swanson, W.; Akey, J.M. Genomic signatures of positive selection in humans and the limits of outlier approaches. Genome Res. 2006, 16, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Kodama, G.; Baldi, P.; Moyzis, R.K. Global landscape of recent inferred Darwinian selection for Homo sapiens. Proc. Natl. Acad. Sci. USA 2006, 103, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Bailey, D.K.; Awad, T.; Liu, G.; Xing, G.; Cao, M.; Valmeekam, V.; Retief, J.; Matsuzaki, H.; Taub, M.; et al. A whole genome long-range haplotype (WGLRH) test for detecting imprints of positive selection in human populations. Bioinformatics 2006, 22, 2122–2128. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Thornton, K.R.; Stoneking, M. A new approach for using genome scans to detect recent positive selection in the human genome. PLoS Biol. 2007, 5, e171. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; McParland, S.; Kearney, F.; Du, L.; Berry, D.P. Detection of selection signatures in dairy and beef cattle using high-density genomic information. Genet. Sel. Evol. 2015, 47, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, S. The genetical structure of populations. Ann. Eugen. 1949, 15, 323–354. [Google Scholar] [CrossRef]
- McGill, R.; Tukey, J.W.; Larsen, W.A. Variations of box plots. Am. Stat. 1978, 32, 12–16. [Google Scholar]
- China National Commission of Animal Genetic Resources: 2012. Animal genetic resource in China-Sheep and Goats; China Agriculture Press: Beijing, China, 2012. [Google Scholar]
- Helmy, K.Y.; Katschke, K.J., Jr.; Gorgani, N.N.; Kljavin, N.M.; Elliott, J.M.; Diehl, L.; Scales, S.J.; Ghilardi, N.; van Lookeren Campagne, M. CRIg: A macrophage complement receptor required for phagocytosis of circulating pathogens. Cell 2006, 124, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chen, Y.; Kubota, F.; Pan, J.J.; Murakami, T. Expression of protocadherin-19 in the nervous system of the embryonic zebrafish. Int. J. Dev. Biol. 2010, 54. [Google Scholar] [CrossRef] [PubMed]
- Satterthwaite, A.B.; Li, Z.; Witte, O.N. Btk function in B cell development and response. Semi Immunol. 1998, 10, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Gauld, S.B.; Porto, J.M.D.; Cambier, J.C.B. Cell Antigen Receptor Signaling: Roles in Cell Development and Disease. Science 2002, 296, 1641–1642. [Google Scholar] [CrossRef] [PubMed]
- Demars, J.; Fabre, S.; Sarry, J.; Rossetti, R.; Gilbert, H.; Persani, L.; Tosser-Klopp, G.; Mulsant, P.; Nowak, Z.; Drobik, W. Genome-Wide Association Studies Identify Two Novel BMP15 Mutations Responsible for an Atypical Hyperprolificacy Phenotype in Sheep. PLoS Genet. 2013, 9, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Worley, K.; Carey, J.; Veitch, A.; Coltman, D.W. Detecting the signature of selection on immune genes in highly structured populations of wild sheep (Ovis dalli). Mol. Ecol. 2006, 15, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Kijas, J.W. Haplotype-based analysis of selective sweeps in sheep. Genome 2014, 57, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Amaral, A.J.; Ferretti, L.; Megens, H.-J.; Crooijmans, R.P.; Nie, H.; Ramos-Onsins, S.E.; Perez-Enciso, M.; Schook, L.B.; Groenen, M.A. Genome-wide footprints of pig domestication and selection revealed through massive parallel sequencing of pooled DNA. PLoS ONE 2011, 6, e14782. [Google Scholar] [CrossRef] [PubMed]
- Browning, B.L.; Browning, S.R. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am. J. Hum. Genet. 2009, 84, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Gautier, M.; Vitalis, R. Rehh: An R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics 2012, 28, 1176–1177. [Google Scholar] [CrossRef] [PubMed]
- MacEachern, S.; Hayes, B.; McEwan, J.; Goddard, M. An examination of positive selection and changing effective population size in Angus and Holstein cattle populations (Bos taurus) using a high density SNP genotyping platform and the contribution of ancient polymorphism to genomic diversity in Domestic cattle. BMC Genom. 2009, 10. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xie, M.; Chen, W.; Talbot, R.; Maddox, J.F.; Faraut, T.; Wu, C.; Muzny, D.M.; Li, Y.; Zhang, W. The sheep genome illuminates biology of the rumen and lipid metabolism. Science 2014, 344, 1168–1173. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, C.; Fan, H.; Yuan, Z.; Hu, S.; Zhang, L.; Wei, C.; Zhang, Q.; Zhao, F.; Du, L. Detection of Selection Signatures on the X Chromosome in Three Sheep Breeds. Int. J. Mol. Sci. 2015, 16, 20360-20374. https://doi.org/10.3390/ijms160920360
Zhu C, Fan H, Yuan Z, Hu S, Zhang L, Wei C, Zhang Q, Zhao F, Du L. Detection of Selection Signatures on the X Chromosome in Three Sheep Breeds. International Journal of Molecular Sciences. 2015; 16(9):20360-20374. https://doi.org/10.3390/ijms160920360
Chicago/Turabian StyleZhu, Caiye, Hongying Fan, Zehu Yuan, Shijin Hu, Li Zhang, Caihong Wei, Qin Zhang, Fuping Zhao, and Lixin Du. 2015. "Detection of Selection Signatures on the X Chromosome in Three Sheep Breeds" International Journal of Molecular Sciences 16, no. 9: 20360-20374. https://doi.org/10.3390/ijms160920360