
MD Simulations of tRNA and Aminoacyl-tRNA Synthetases: Dynamics, Folding, Binding, and Allostery

Abstract
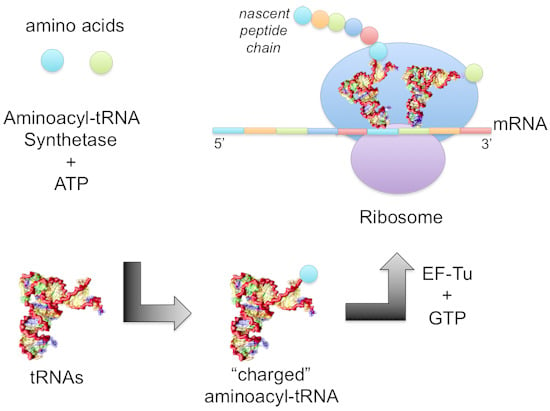
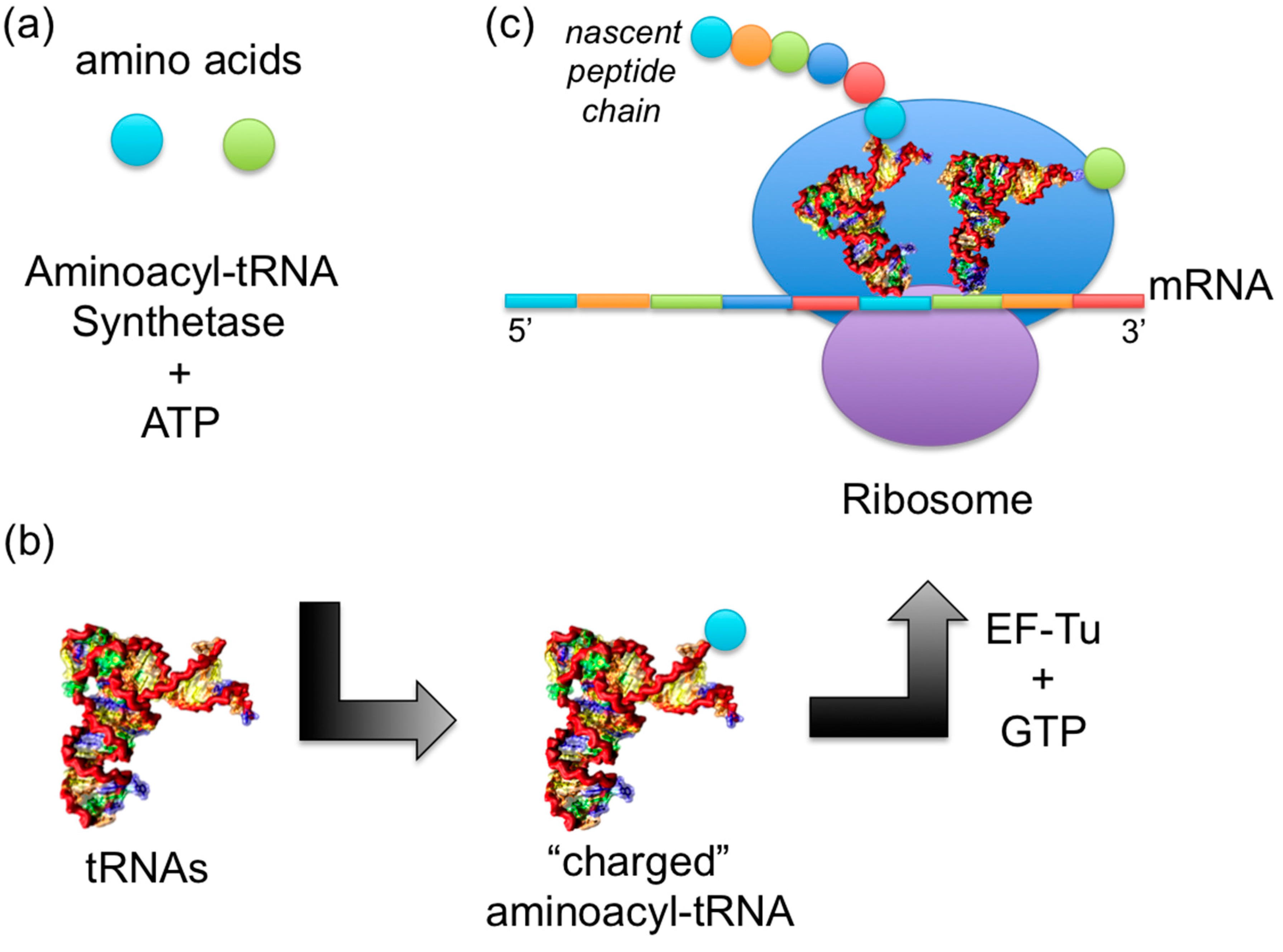
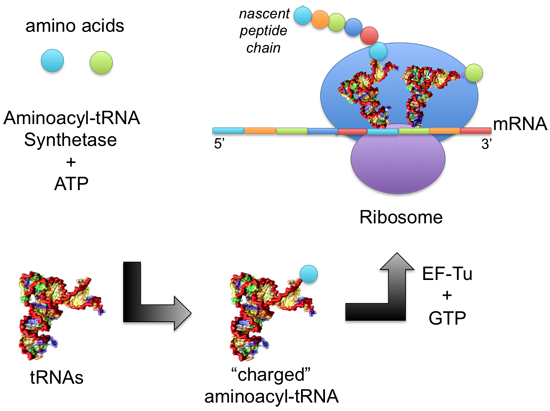
:
1. Introduction
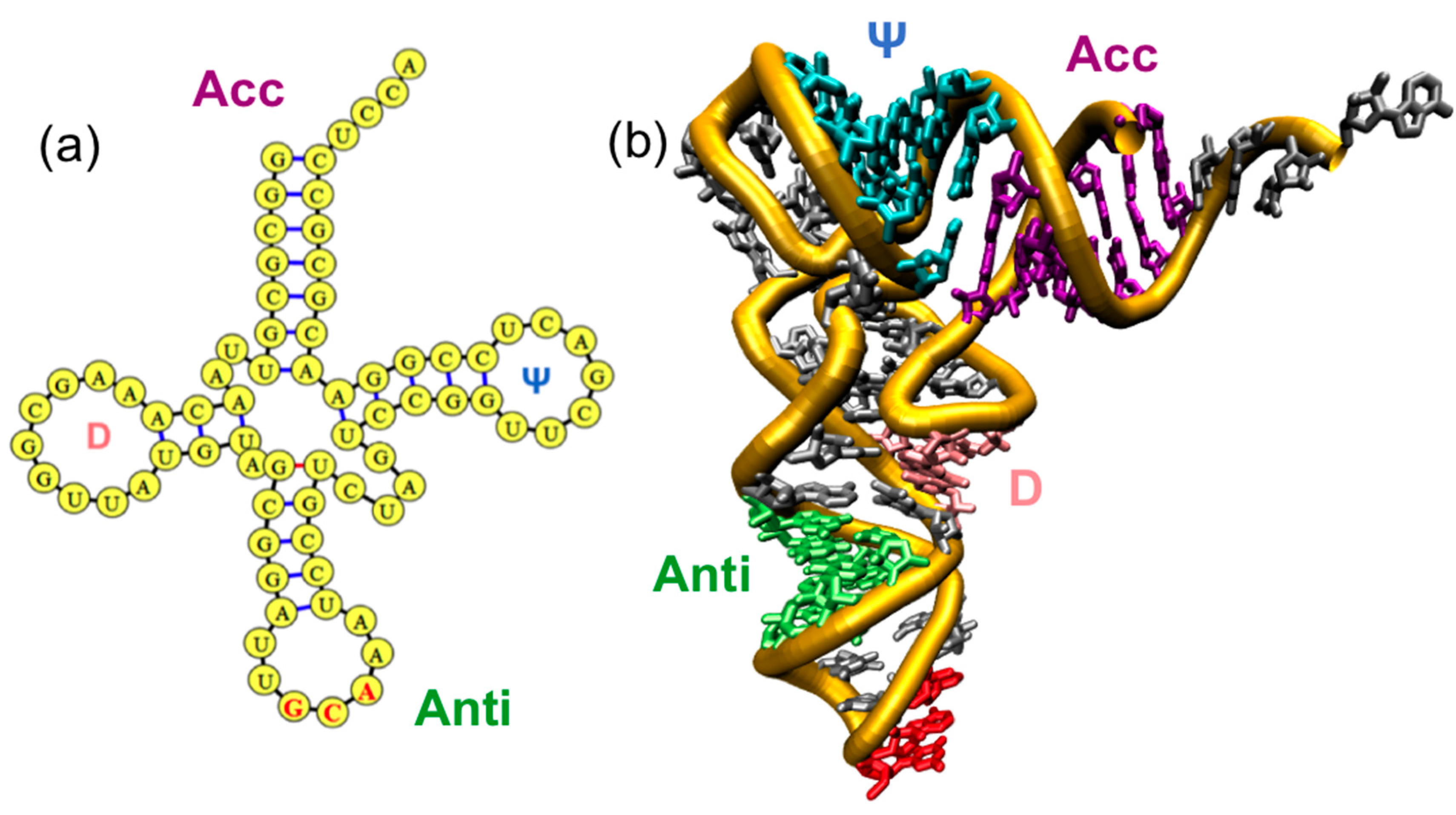
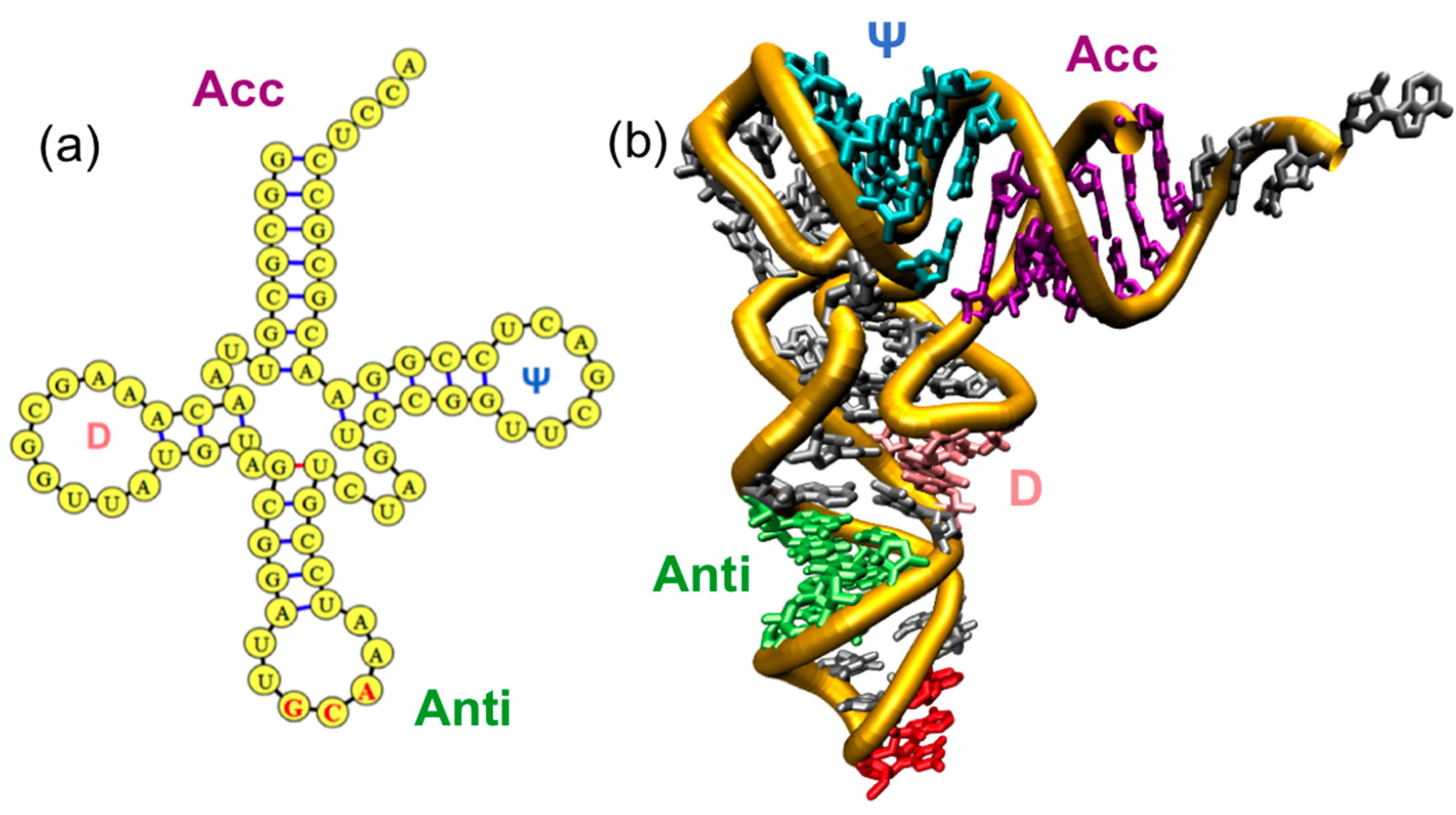
1.1. Overview of Family of tRNAs and AARSs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class I | Class II | |
|---|---|---|
| Group a | ArgRS (α) | GlyRS (α2) |
| CysRS (α/α2) | HisRS (α2) | |
| IleRS (α) | SerRS (α2) | |
| LeuRS (α) | ThrRS (α2) | |
| LysRS * (α/α2) | ||
| MetRS (α/α2) | ||
| ValRS (α) | ||
| Group b | GlnRS (α) | AsnRS (α2) |
| GluRS (α) | AspRS (α2) | |
| LysRS * (α2/(α2)2) | ||
| Group c | TrpRS (α2) | AlaRS (α, α2, α4) |
| TyrRS (α) | GlyRS (α2β2) | |
| PheRS (α, α2β2) |
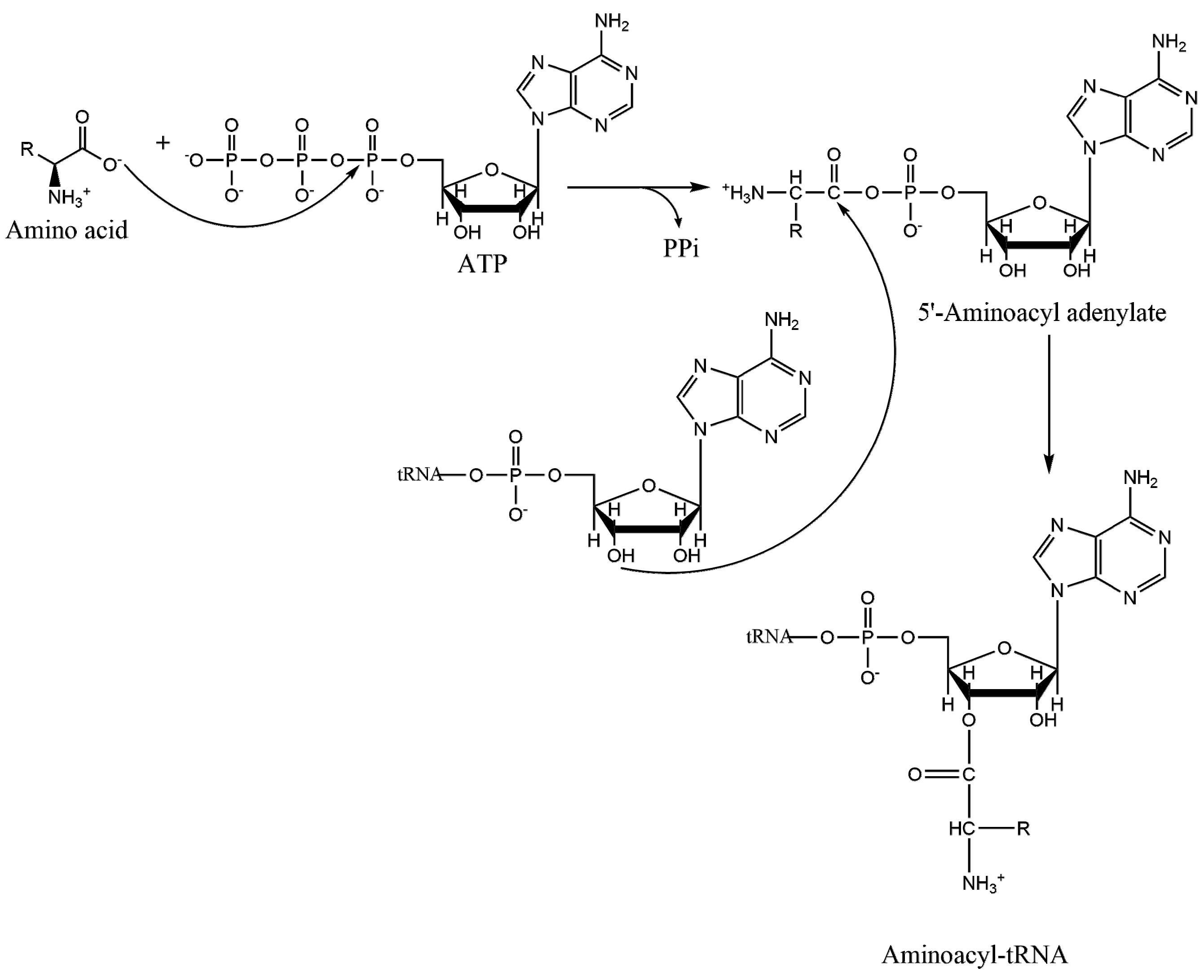
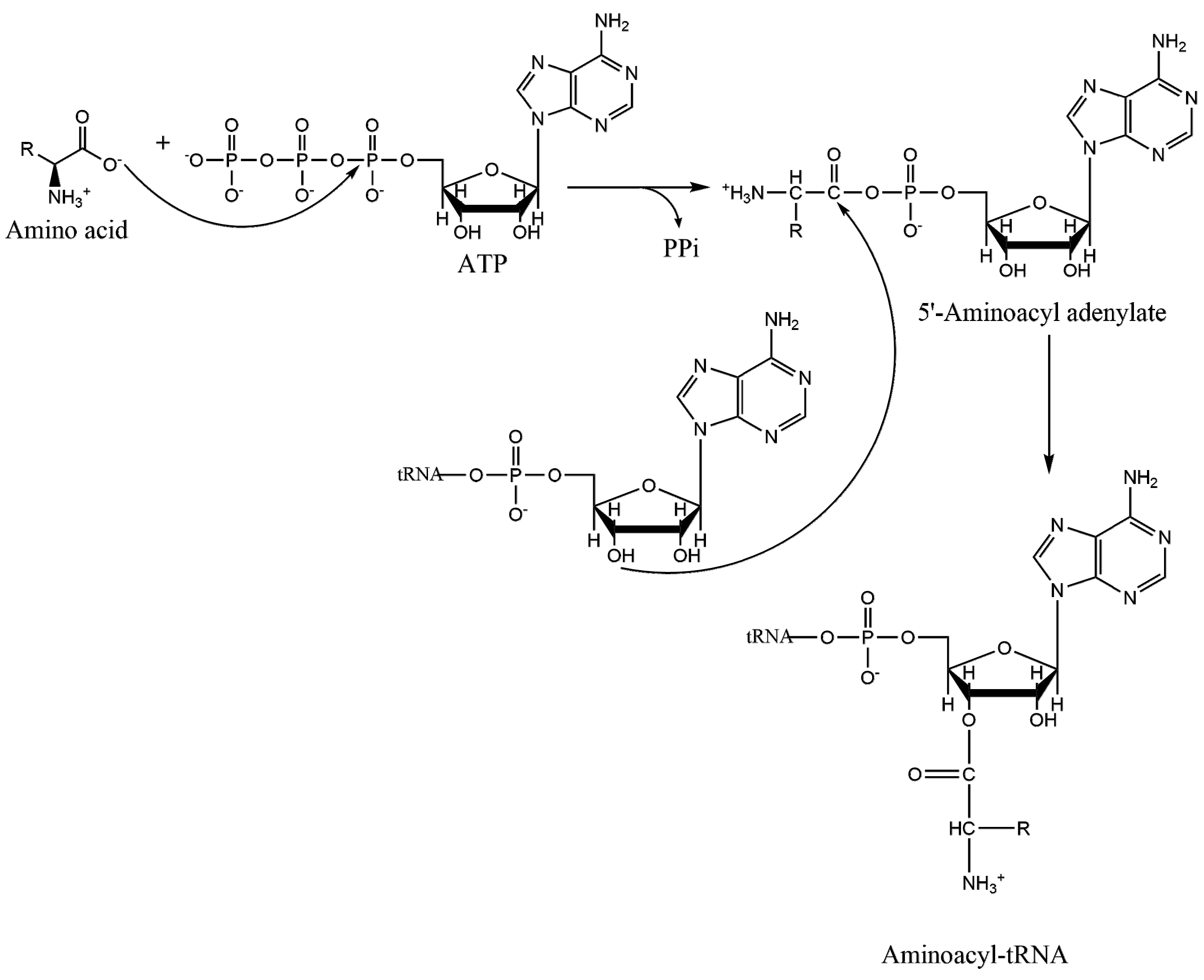
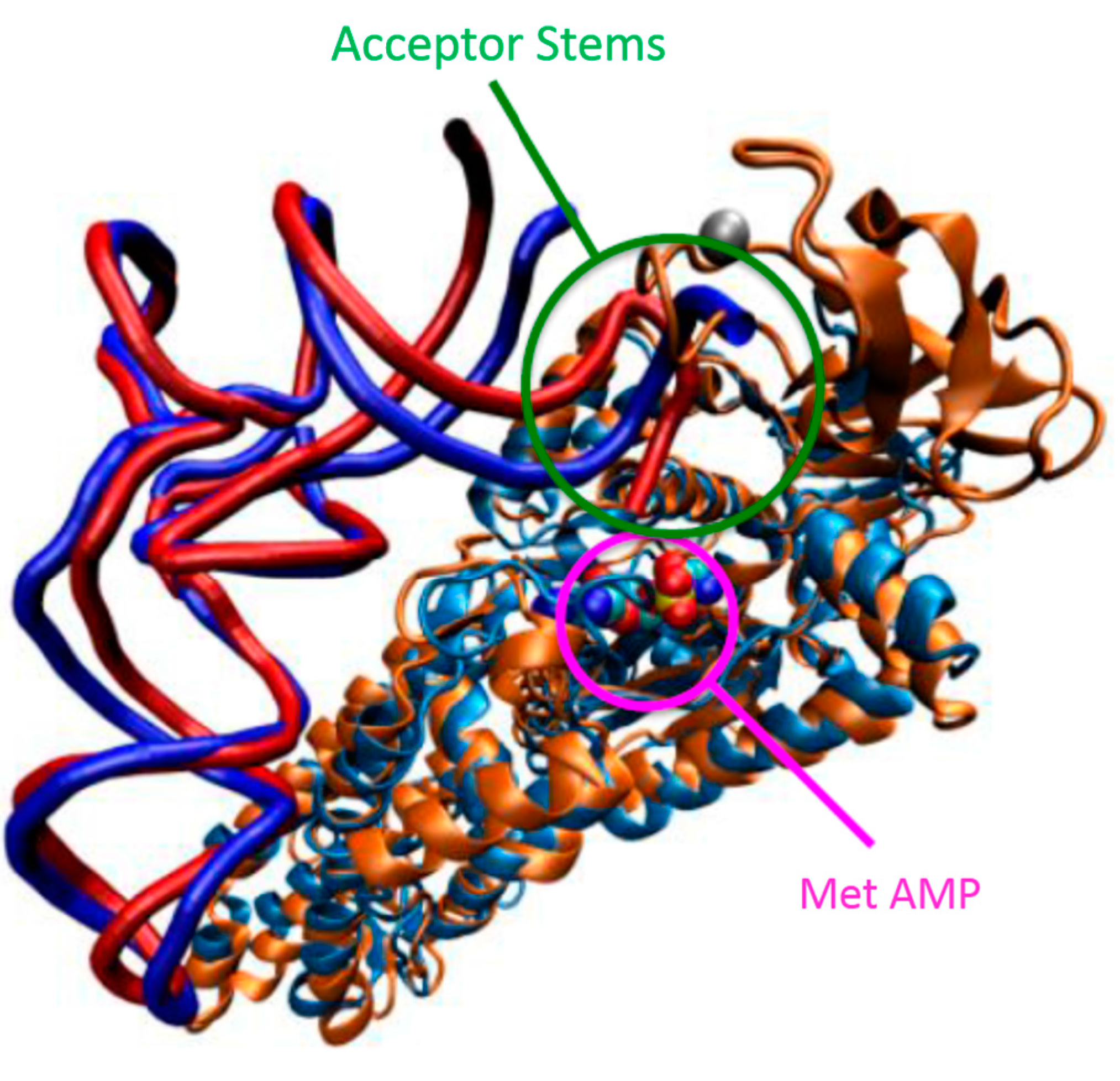
1.2. Mechanism of Aminoacylation Catalytic Reaction
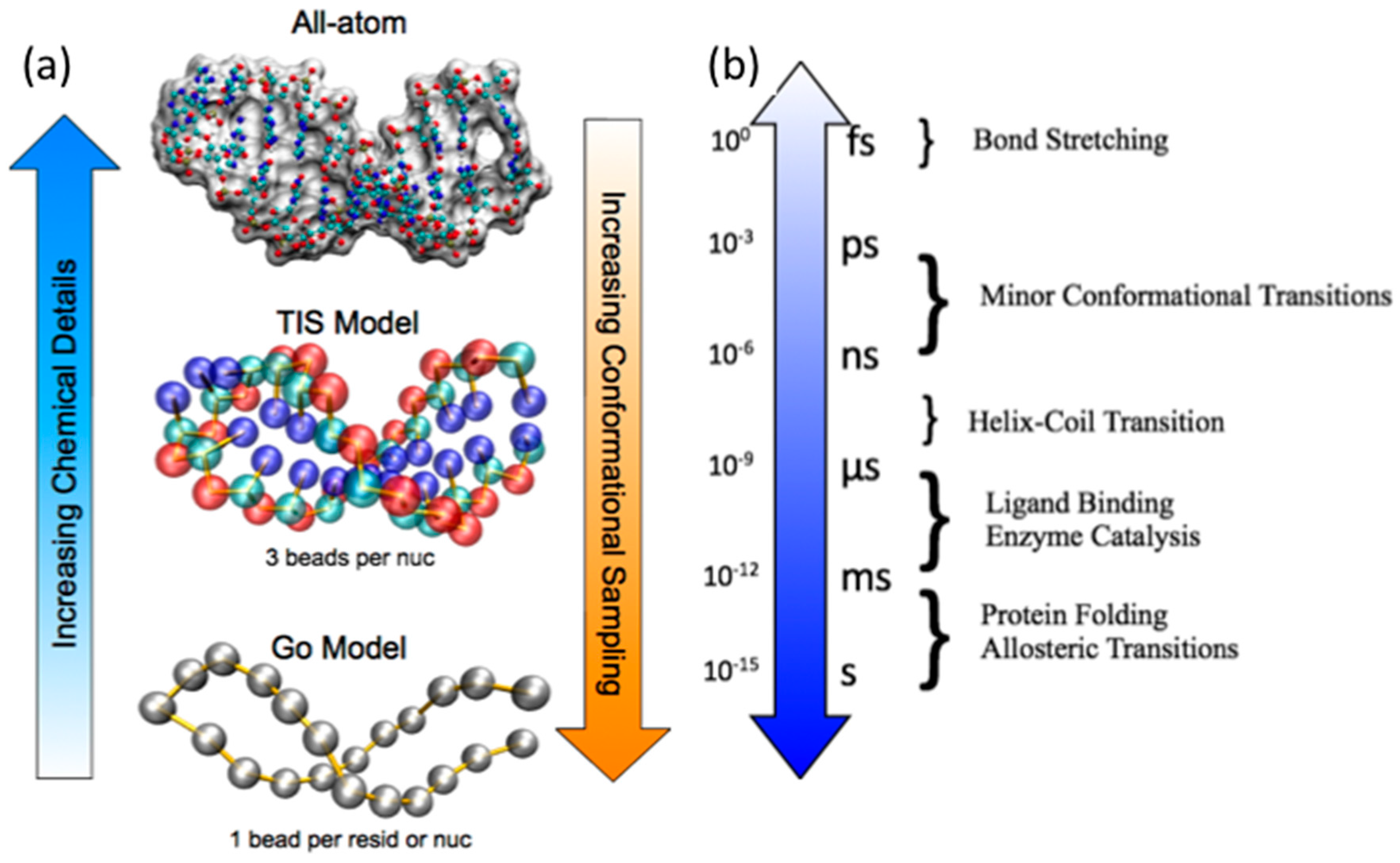
2. Biomolecular MD Simulations Approaches
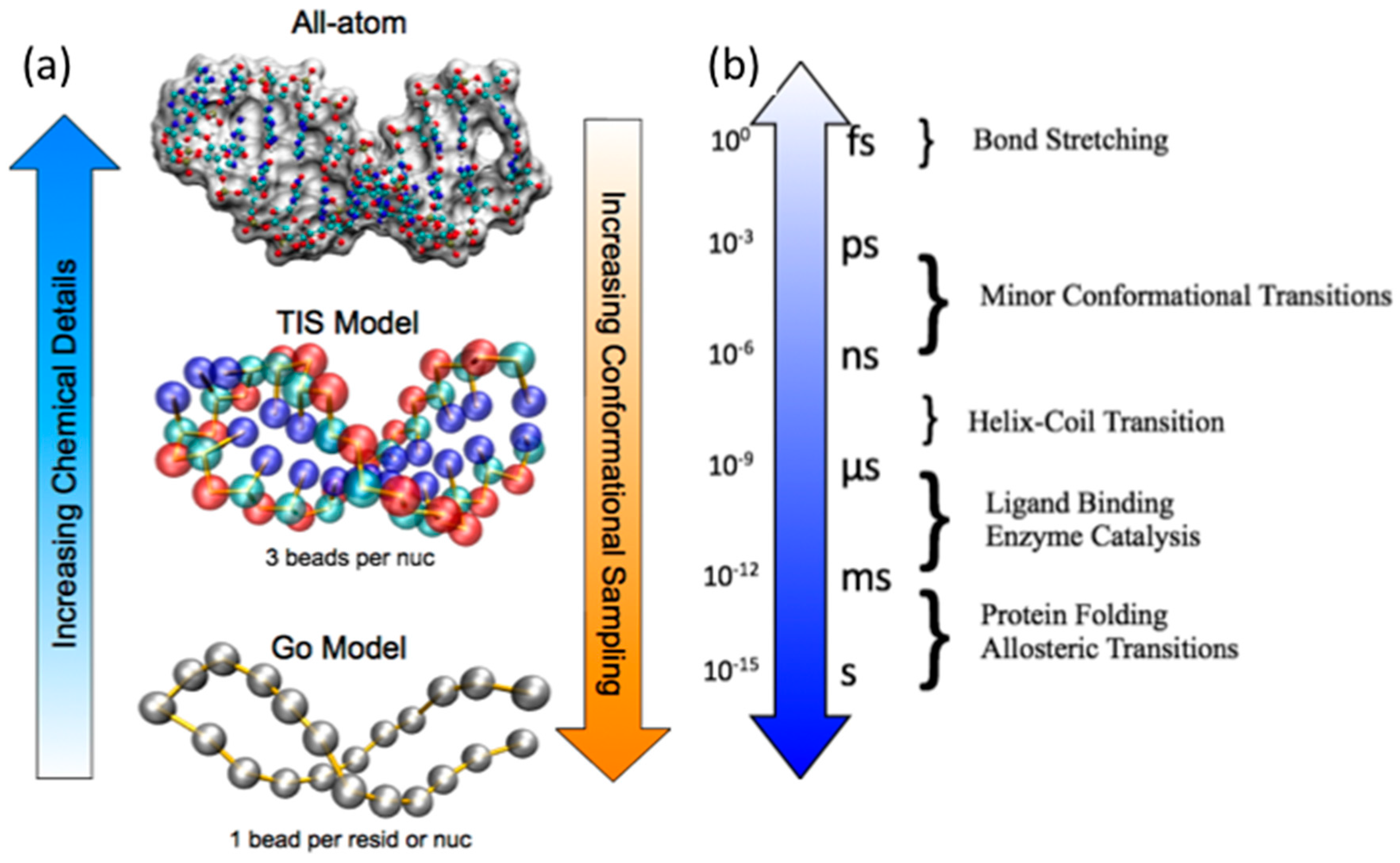
2.1. Atomistic Empirical Force Field MD Simulations
2.1.1. Addressing Computational Challenges
2.1.2. Improving Chemical Details
2.2. Coarse-Grained Native Structure Based MD Simulations
2.3. Coarse-Grained Topological Constraint Based RNA MD Simulations
3. tRNA Dynamics
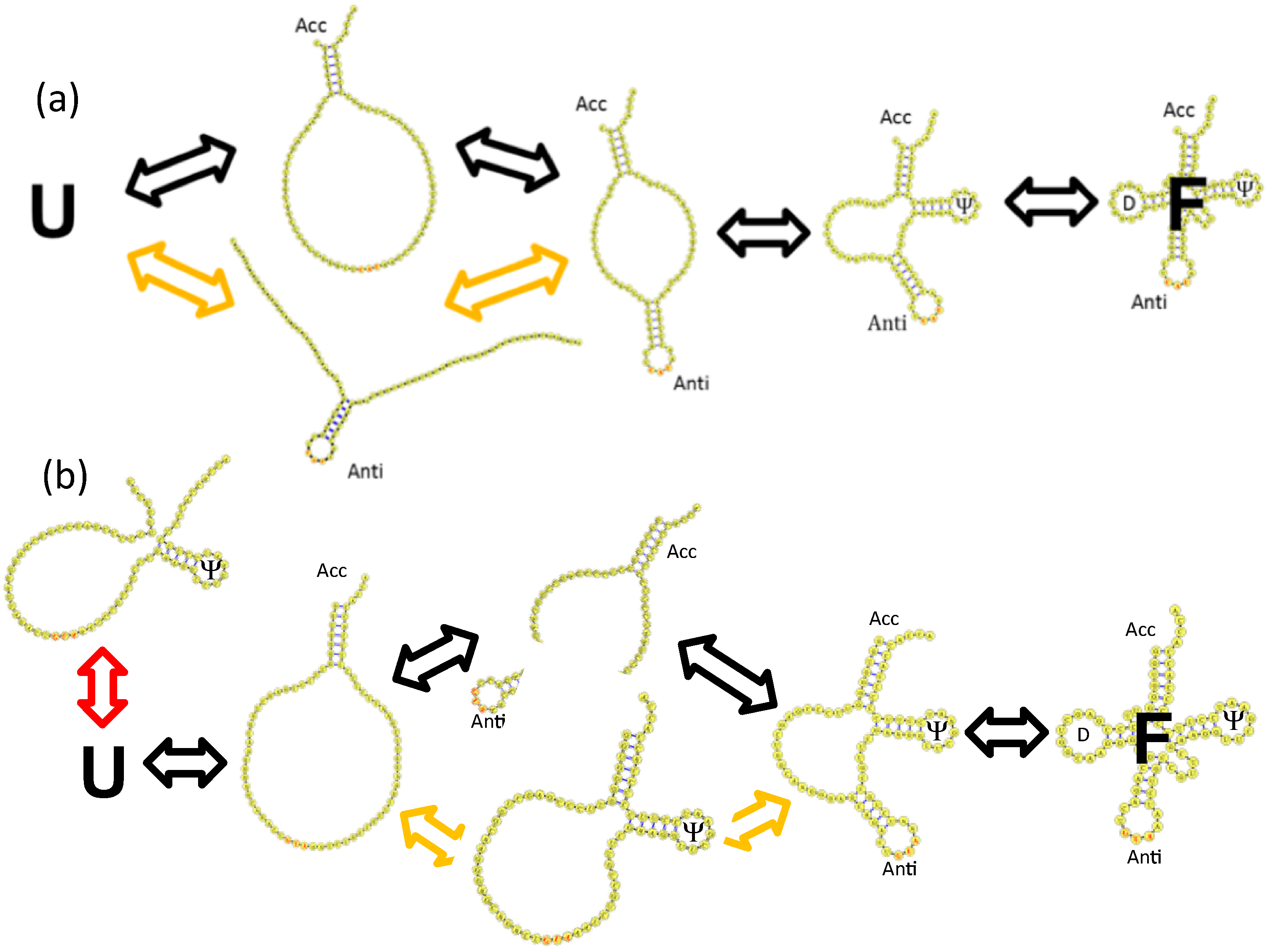
4. tRNA Folding Mechanisms
4.1. Brief Overview of Classical and Recent tRNA Folding Experiments
4.2. Base Stacking Interactions Are a Main Determinant of Parallel tRNA Folding Mechanisms
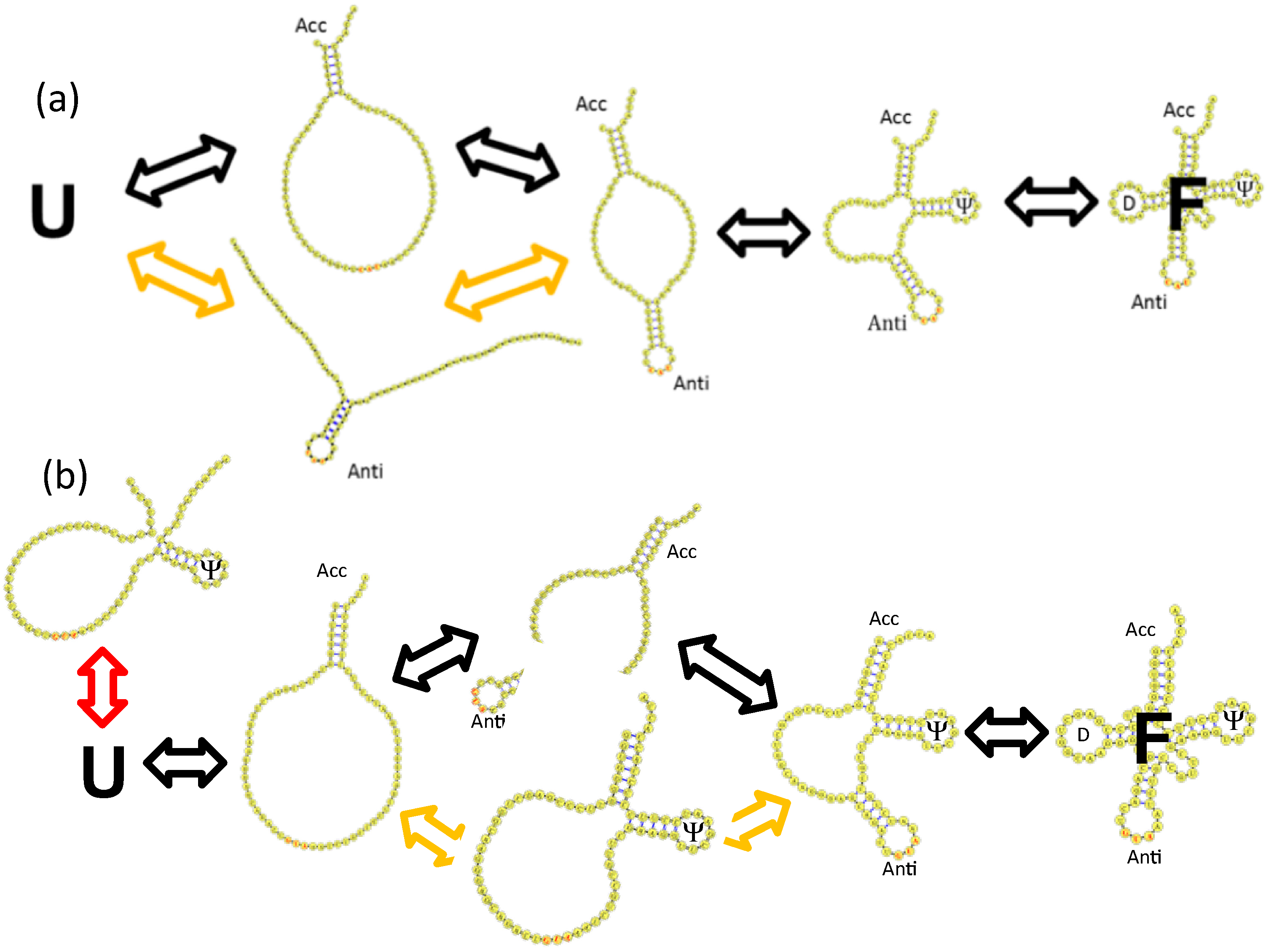
4.3. Backtracking Mechanisms Partitions Fast vs. Slow Folding
4.4. Topological Constraints in tRNA Tertiary Structure
5. MD Simulations of Aminoacyl-tRNA Synthetases
5.1. Brief Overview of MD Simulations of AARSs
| AARS | Ligands | Starting Structure(s) | Time | Reference |
|---|---|---|---|---|
| CysRS | +tRNACys:Cys-AMP (modeled) +Cys-AMP (modeled) | 1LI5 and models | 10 ns | Ghosh et al., JBC, 2011 [109] |
| GlnRS | +tRNAGln (modeled) | 4H3S and models | 70 ns | Grant et al., JMB, 2013 [110] |
| GlnRS | +tRNAGln | 1GTR, 1EXD and models | 6.5 ns | Yamasaki et al., Biophys. J, 2007 [111] |
| GluRS | +tRNAGlu:Glutamol-AMP | 1N78 | 20 ns | Pyrkosz et al., JMB, 2010 [112] |
| GluRS | +tRNAGlu:Glu-AMP | 1N78 | 20 ns | Sethi et al., PNAS, 2009 [113] |
| LeuRS | CP domains from 3PZ0, 3PZ6 | 20 ns | Liu et al., Biochem. J, 2011 [114] | |
| LeuRS | +tRNALeu:Leu-AMP (modeled) | 1WZ2, 2V0C | 20 ns | Sethi et al., PNAS, 2009 [113] |
| LeuRS | 1H3N | 55 ns | Strom et al., J. Mol. Model., 2014 [115] | |
| LeuRS | +Val-tRNALeu (modeled) | 2BYT, 10BC and models | 1 ns | Hagiwara et al., FEBS, 2009 [116] |
| MetRS | tRNAMet:Met-AMP | 2CSX, 2CT8 and models | 10 ns | Ghosh et al., PNAS, 2007 [117] |
| MetRS | 1QQT | 12 ns | Budiman et al., Proteins, 2007 [118] | |
| MetRS | +Met, +ATP, +Met-AMP, +tRNA:MetAMP (modeled) | 1QQT, 1F4L, 1PFY and models | 10 ns | Ghosh et al., Biochem., 2008 [119] |
| MetRS | 1QQT | 30 ns | Strom et al., J. Mol. Model., 2014 [115] | |
| TrpRS | +Trp-AMP, +tRNATrp:Trp-AMP (modeled) | 2DR2, 1R6U and models | 5 ns | Bhattacharyya et al., Proteins, 2008 [120] |
| TrpRS | + ATP, + Trp, +ATP:Trp, +ATP:Mg, +ATP:Trp:Mg | 1MAW, 1MB2, 1MAU, 1M83, 1I6L | 5 ns | Kapustina et al., JMB, 2006 [121] |
| TyrRS | +Tyr, +ATP, +Tyr-AMP, +inhibitor | 1JIL, 4TS1, 1H3E, 3TS1, 1I6K and models | 12 ns | Li et al., Eur. Biophys. J., 2008 [122] |
| TyrRS | Tyr | 4TS1 | 540 ps | Lau et al., JMB, 1994 [108] |
| TyrRS | +Tyr, +Tyr:ATP, +Tyr-AMP | 2JAN, 1X8X, 1H3E, 1VBM and models | 100 ns | Mykuliak et al., Eur. Biophys. J., 2014 [123] |
| TyrRS | Assembled N and C domains from 1N3L and 1NTG | 100 ns | Savytskyi et al., J. Mol. Recognit., 2013 [124] | |
| ValRS | +tRNAVal:Val-AMP, +tRNAVal:ThrAMP (modeled) | 1GAX and models | 10 ns | Li et al., J. Mol. Model., 2011 [125] |
| ValRS | +editing substrates (modeled) | 1WK9 (CP domain), 1GAX (ValRS + tRNA) and models | 2 ns for full 5 ns for CP | Bharatham et al., Biophys. Chem., 2009 [126] |
| AARS | Ligands | Starting Structure(s) | Time | Reference |
|---|---|---|---|---|
| AspRS | +Asp:ATP (modeled), + Asn:ATP (modeled) | 1IL2, 1COA and models | 500 ps | Thompson et al., Chem. Bio. Chem., 2006 [127] |
| AspRS | +Asp:ATP (modeled), +Asn:ATP (modeled) | 1IL2, 1COA and models | 0.5–3 ns | Thompson et al., JBC, 2006 [128] |
| AspRS | - | 1ASZ and models | 5 ns | Ul-Haq et al., J. Mol. Graph. Model., 2010 [129] |
| AspRS | +Asp, +Asn (modeled) | 1C0Z | 300 ps | Archontis et al., JMB, 2001 [130] |
| AsnRS | +Asn-AMP, +Asp-AMP (modeled) | T. thermophilus AsnRS | 4 ns | Polydorides et al., Proteins, 2011 [131] |
| HisRS | +His-AMP, +His (modeled), +HisOH (modeled) | 1KMM, 1KMN and modeled variants | 600 ps | Arnez et al., Proteins, 1998 [132] |
| LysRS (LysU) | +Lys:AMPPCP | 1E22, dimer modeled | 1 ns | Hughes et al., BMC Struct. Biol., 2003 [133] |
| LysRS (LysU) | +Lys:AMPPCP | 1E22, dimer modeled | 520 ps | Hughes et al., Proteins, 2006 [134] |
| ProRS | +Pro-AMP (modeled) | 2J3M | 30 ns | Strom et al., J. Mol. Model., 2014 [115] |
| ProRS | - | 2J3M and modeled variants | 12 ns | Sanford et al., Biochemistry, 2012 [135] |
| SerRS | +tRNASer | 3W3S and modeled dimer | 2 ns | Dutta et al., J. Phys. Chem. B, 2015 [136] |
| ThrRS | +tRNAThr:Thr-AMP (modeled) | 1QF6 | 15 ns | Bushnell et al., J. Phys. Chem. B, 2012 [137] |
5.2. Flexibility and Allosteric Communication Networks in AARSs
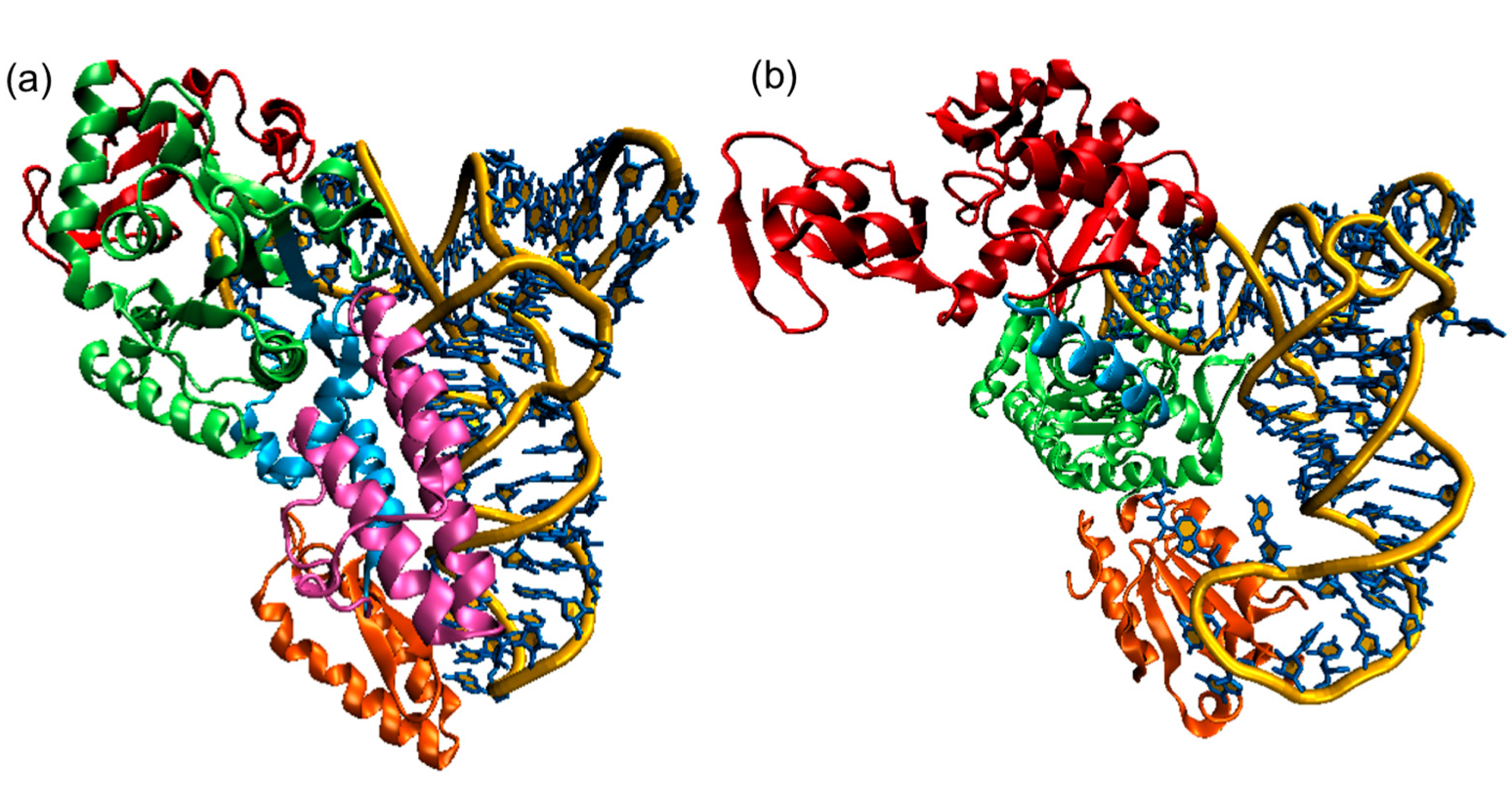
5.3. Conformational Flexibility of tRNA upon Binding to AARSs
5.4. Catalytic Aminoacylation Mechanisms of AARSs
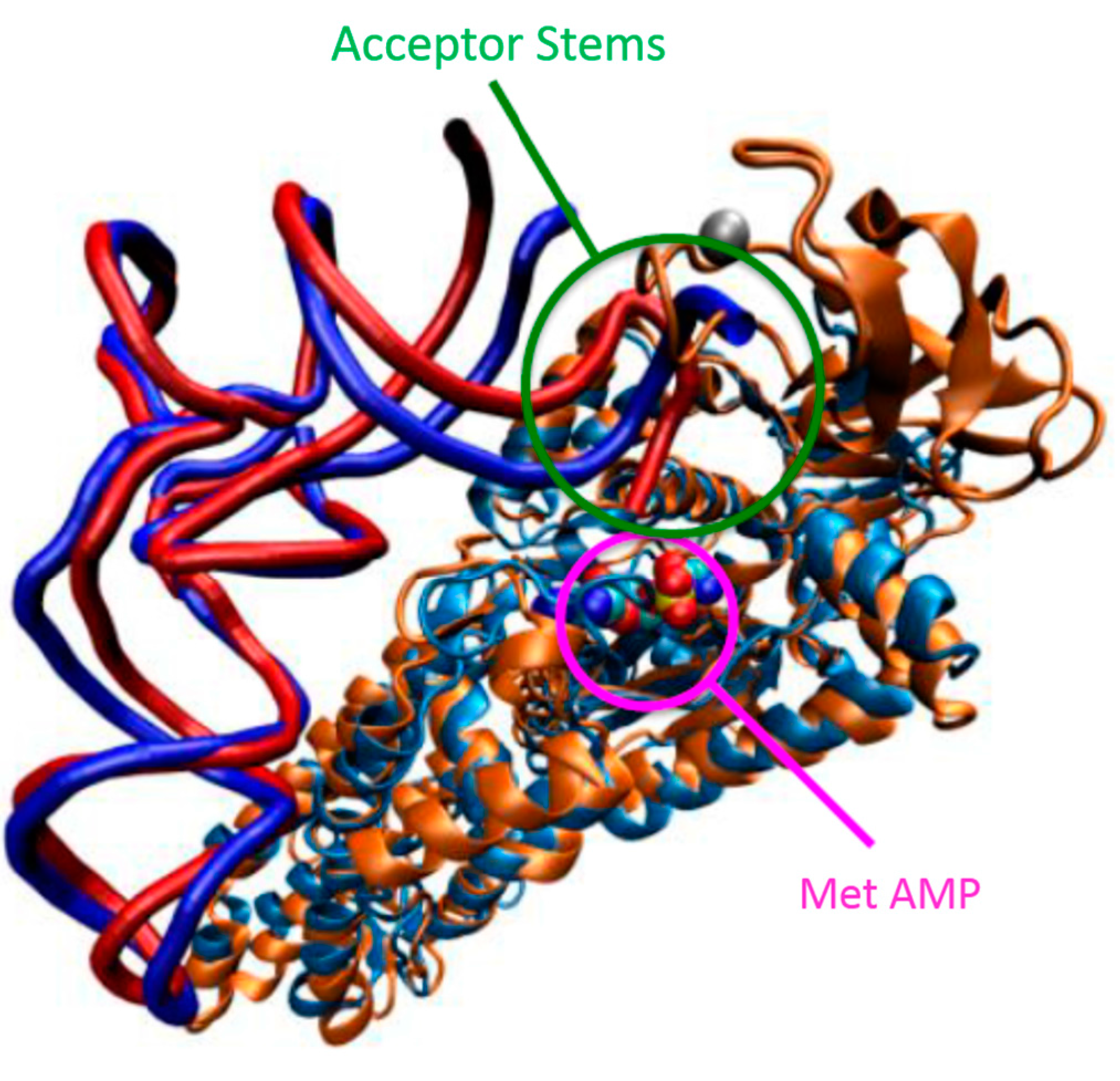
5.5. Functional and Structural Roles of Editing Domains
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Crick, F.H. On protein synthesis. Symp. Soc. Exp. Biol. 1958, 12, 138–163. [Google Scholar] [PubMed]
- Hoagland, M.B.; Keller, E.B.; Zamecnik, P.C. Enzymatic carboxyl activation of amino acids. J. Biol. Chem. 1956, 218, 345–358. [Google Scholar] [PubMed]
- Robertus, J.D.; Ladner, J.E.; Finch, J.T.; Rhodes, D.; Brown, R.S.; Clark, B.F.C.; Klug, A. Structure of yeast phenylalanine tRNA at 3 Å resolution. Nature 1974, 250, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Moore, P.B. The crystal structure of yeast phenylalanine tRNA at 1.93 Å resolution: A classic structure revisited. RNA 2000, 6, 1091–1105. [Google Scholar] [CrossRef] [PubMed]
- Westhof, E.; Auffinger, P. Transfer RNA structure. eLS 2012. [Google Scholar]
- Wuite, J.; Davies, B.I.; Go, M.J.; Lambers, J.C.; Jackson, D.; Mellows, G.; Tasker, T.C. Pseudomonic acid, a new antibiotic for topical therapy. J. Am. Acad. Dermatol. 1985, 12, 1026–1031. [Google Scholar] [CrossRef]
- Park, S.G.; Schimmel, P.; Kim, S. Aminoacyl tRNA synthetases and their connections to disease. Proc. Natl. Acad. Sci. USA 2008, 105, 11043–11049. [Google Scholar] [CrossRef] [PubMed]
- Perona, J.J.; Hadd, A. Structural diversity and protein engineering of the aminoacyl-tRNA synthetases. Biochemistry (Mosc.) 2012, 51, 8705–8729. [Google Scholar] [CrossRef] [PubMed]
- Alexander, R.W.; Schimmel, P. Domain-domain communication in aminoacyl-tRNA synthetases. Prog. Nucl. Acid Res. Mol. Biol. 2001, 69, 317–349. [Google Scholar]
- Nussinov, R. The significance of the 2013 Nobel Prize in chemistry and the challenges ahead. PLoS Comput. Biol. 2014, 10, e1003423. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.C.; Roux, B. Eppur Si Muove! The 2013 Nobel Prize in chemistry. Structure 2013, 21, 2102–2105. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press: New York City, NY, USA, 1989. [Google Scholar]
- Eaton, W.A.; Munoz, V.; Hagen, S.J.; Jas, G.S.; Lapidus, L.J.; Henry, E.R.; Hofrichter, J. Fast kinetics and mechanisms in protein folding. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 327–359. [Google Scholar] [CrossRef] [PubMed]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Wolynes, P.G.; Onuchic, J.N.; Thirumalai, D. Navigating the folding routes. Science 1995, 267, 1619–1620. [Google Scholar] [CrossRef] [PubMed]
- Shakhnovich, E. Protein folding thermodynamics and dynamics: Where physics, chemistry, and biology meet. Chem. Rev. 2006, 106, 1559–1588. [Google Scholar] [CrossRef] [PubMed]
- Shea, J.E.; Brooks, C.L. From folding theories to folding proteins: A review and assessment of simulation studies of protein folding and unfolding. Annu. Rev. Phys. Chem. 2001, 52, 499–535. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Pérez, A.; Marchán, I.; Svozil, D.; Sponer, J.; Cheatham, T.E., III; Laughton, C.A.; Orozco, M. Refinement of the AMBER force field for nucleic acids: Improving the description of α/γ conformers. Biophys. J. 2007, 92, 3817–3829. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; van Gunsteren, W.F. Refinement of the application of the GROMOS 54A7 force field to β-peptides. J. Comput. Chem. 2013, 34, 2796–2805. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Perilla, J.R.; Yufenyuy, E.L.; Meng, X.; Chen, B.; Ning, J.; Ahn, J.; Gronenborn, A.M.; Schulten, K.; Aiken, C.; et al. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 2013, 497, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Voelz, V.A.; Bowman, G.R.; Beauchamp, K.; Pande, V.S. Molecular simulation of ab initio protein folding for a millisecond folder NTL9(1–39). J. Am. Chem. Soc. 2010, 132, 1526–1528. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Zhang, D.; McCammon, J.A. Computation of electrostatic forces between solvated molecules determined by the Poisson-Boltzmann equation using a boundary element method. J. Chem. Phys. 2005, 122, 214102. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Salsbury, F.R.; Olson, M.A. An efficient hybrid explicit/implicit solvent method for biomolecular simulations. J. Comput. Chem. 2004, 25, 1967–1978. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.; Case, D.A.; Bashford, D. Effective Born radii in the generalized Born approximation: The importance of being perfect. J. Comput. Chem. 2002, 23, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Roux, B.; Simonson, T. Implicit solvent models. Biophys. Chem. 1999, 78, 1–20. [Google Scholar] [CrossRef]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. [Google Scholar] [CrossRef] [PubMed]
- Kirk, D.B.; Hwu, W.W. Programming Massively Parallel Processors: A Hands-on Approach, 1st ed.; Morgan Kaufmann: Burlington, MA, USA, 2010. [Google Scholar]
- Sanders, J.; Kandrot, E. CUDA by Example: An Introduction to General-Purpose GPU Programming, 1st ed.; Addison-Wesley Professional: Upper Saddle River, NJ, USA, 2010. [Google Scholar]
- Stone, J.E.; Phillips, J.C.; Freddolino, P.L.; Hardy, D.J.; Trabuco, L.G.; Schulten, K. Accelerating molecular modeling applications with graphics processors. J. Comput. Chem. 2007, 28, 2618–2640. [Google Scholar] [CrossRef] [PubMed]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Schmid, N.; Christ, C.D.; Christen, M.; Eichenberger, A.P.; van Gunsteren, W.F. Architecture, implementation and parallelisation of the GROMOS software for biomolecular simulation. Comput. Phys. Commun. 2012, 183, 890–903. [Google Scholar] [CrossRef]
- Anderson, J.A.; Glotzer, S.C. The development and expansion of HOOMD-blue through six years of GPU proliferation. Comput. Phys. 2013. [Google Scholar]
- Brown, W.M.; Wang, P.; Plimpton, S.J.; Tharrington, A.N. Implementing molecular dynamics on hybrid high performance computers—Short range forces. Comput. Phys. Commun. 2011, 182, 898–911. [Google Scholar] [CrossRef]
- Eastman, P.; Friedrichs, M.S.; Chodera, J.D.; Radmer, R.J.; Bruns, C.M.; Ku, J.P.; Beauchamp, K.A.; Lane, T.J.; Wang, L.-P.; Shukla, D.; et al. OpenMM 4: A reusable, extensible, hardware independent library for high performance molecular simulation. J. Chem. Theory Comput. 2013, 9, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Zhmurov, A.; Dima, R.I.; Kholodov, Y.; Barsegov, V. Sop-GPU: Accelerating biomolecular simulations in the centisecond timescale using graphics processors. Proteins Struct. Funct. Bioinform. 2010, 78, 2984–2999. [Google Scholar] [CrossRef] [PubMed]
- Leuchter, J.D.; Green, A.T.; Gilyard, J.; Rambarat, C.G.; Cho, S.S. Coarse-grained and atomistic MD simulations of RNA and DNA folding. Isr. J. Chem. 2014, 54, 1152–1164. [Google Scholar] [CrossRef]
- Lopes, P.E.M.; Huang, J.; Shim, J.; Luo, Y.; Li, H.; Roux, B.; MacKerell, A.D. Polarizable force field for peptides and proteins based on the classical drude oscillator. J. Chem. Theory Comput. 2013, 9, 5430–5449. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.M.; Anisimov, V.M.; MacKerell, A.D. Development of CHARMM polarizable force field for nucleic acid bases based on the classical drude oscillator model. J. Phys. Chem. B 2011, 115, 580–596. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Hardy, D.J.; Phillips, J.C.; MacKerell, A.D.; Schulten, K.; Roux, B. High-performance scalable molecular dynamics simulations of a polarizable force field based on classical drude oscillators in NAMD. J. Phys. Chem. Lett. 2011, 2, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Lee, M.S.; Salsbury, F.R.; Brooks, C.L. Constant-pH molecular dynamics using continuous titration coordinates. Proteins Struct. Funct. Bioinform. 2004, 56, 738–752. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Morrow, B.H.; Shi, C.; Shen, J.K. Recent development and application of constant pH molecular dynamics. Mol. Simul. 2014, 40, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.A.; Wang, Y.; Shi, C.; Pastoor, K.J.; Nguyen, B.-L.; Xia, K.; Shen, J.K. Toward accurate prediction of pKa values for internal protein residues: The importance of conformational relaxation and desolvation energy. Proteins Struct. Funct. Bioinform. 2011, 79, 3364–3373. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.B.; Hulbert, B.S.; Zhou, H.; Brooks, C.L. Constant pH molecular dynamics of proteins in explicit solvent with proton tautomerism. Proteins Struct. Funct. Bioinform. 2014, 82, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Levinthal, C. Mossbauer spectroscopy in biological systems. In Proceedings of a meeting held at Allerton House; Debrunner, P., Tsibris, J.C.M., Munck, E., Eds.; University of Illinois Press: Urbana, IL, USA, 1969. [Google Scholar]
- Doudna, J.A.; Cech, T.R. The chemical repertoire of natural ribozymes. Nature 2002, 418, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Prasanth, K.V.; Spector, D.L. Eukaryotic regulatory RNAs: An answer to the “genome complexity” conundrum. Genes Dev. 2007, 21, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Storz, G. An expanding universe of noncoding RNAs. Science 2002, 296, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.S.; Pincus, D.L.; Thirumalai, D. Assembly mechanisms of RNA pseudoknots are determined by the stabilities of constituent secondary structures. Proc. Natl. Acad. Sci. USA 2009, 106, 17349–17354. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Ge, H.W.; Cho, S.S. Sequence-dependent base-stacking stabilities guide tRNA folding energy landscapes. J. Phys. Chem. B 2013, 117, 12943–12952. [Google Scholar] [CrossRef] [PubMed]
- Pincus, D.L.; Cho, S.S.; Hyeon, C.; Thirumalai, D. Minimal models for proteins and RNA from folding to function. Prog. Mol. Biol. Transl. Sci. 2008, 84, 203–250. [Google Scholar] [PubMed]
- Hills, R.D.; Brooks, C.L. Insights from coarse-grained Gō models for protein folding and dynamics. Int. J. Mol. Sci. 2009, 10, 889–905. [Google Scholar] [CrossRef] [PubMed]
- Clementi, C.; Nymeyer, H.; Onuchic, J.N. Topological and energetic factors: What determines the structural details of the transition state ensemble and “en-route” intermediates for protein folding? An investigation for small globular proteins. J. Mol. Biol. 2000, 298, 937–953. [Google Scholar] [CrossRef] [PubMed]
- Chavez, L.L.; Onuchic, J.N.; Clementi, C. Quantifying the roughness on the free energy landscape: Entropic bottlenecks and protein folding rates. J. Am. Chem. Soc. 2004, 126, 8426–8432. [Google Scholar] [CrossRef] [PubMed]
- Levy, Y.; Cho, S.S.; Onuchic, J.N.; Wolynes, P.G. A survey of flexible protein binding mechanisms and their transition states using native topology based energy landscapes. J. Mol. Biol. 2005, 346, 1121–1145. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, C.; Thirumalai, D. Mechanical unfolding of RNA hairpins. Proc. Natl. Acad. Sci. USA 2005, 102, 6789–6794. [Google Scholar] [CrossRef] [PubMed]
- Biyun, S.; Cho, S.S.; Thirumalai, D. Folding of human telomerase RNA pseudoknot using ion-jump and temperature-quench simulations. J. Am. Chem. Soc. 2011, 133, 20634–20643. [Google Scholar] [CrossRef] [PubMed]
- Koculi, E.; Cho, S.S.; Desai, R.; Thirumalai, D.; Woodson, S.A. Folding path of P5abc RNA involves direct coupling of secondary and tertiary structures. Nucleic Acids Res. 2012, 40, 8011–8020. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, R.; Velmurugu, Y.; Kuznetsov, S.V.; Ansari, A. Fast folding of RNA pseudoknots initiated by laser temperature-jump. J. Am. Chem. Soc. 2011, 133, 18767–18774. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shakhnovich, E.I. Constructing, verifying, and dissecting the folding transition state of chymotrypsin inhibitor 2 with all-atom simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13014–13018. [Google Scholar] [CrossRef] [PubMed]
- Whitford, P.C.; Noel, J.K.; Gosavi, S.; Schug, A.; Sanbonmatsu, K.Y.; Onuchic, J.N. An all-atom structure-based potential for proteins: Bridging minimal models with all-atom empirical forcefields. Proteins Struct. Funct. Bioinform. 2009, 75, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Karanicolas, J.; Brooks, C.L. The origins of asymmetry in the folding transition states of protein L and protein G. Protein Sci. 2002, 11, 2351–2361. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.S.; Levy, Y.; Wolynes, P.G. Quantitative criteria for native energetic heterogeneity influences in the prediction of protein folding kinetics. Proc. Natl. Acad. Sci. USA 2009, 106, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Cheung, M.S.; García, A.E.; Onuchic, J.N. Protein folding mediated by solvation: Water expulsion and formation of the hydrophobic core occur after the structural collapse. Proc. Natl. Acad. Sci. USA 2002, 99, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Mustoe, A.M.; Al-Hashimi, H.M.; Brooks, C.L. Coarse grained models reveal essential contributions of topological constraints to the conformational free energy of RNA bulges. J. Phys. Chem. B 2014, 118, 2615–2627. [Google Scholar] [CrossRef] [PubMed]
- Crothers, D.M.; Cole, P.E.; Hilbers, C.W.; Shulman, R.G. The molecular mechanism of thermal unfolding of Escherichia coli formylmethionine transfer RNA. J. Mol. Biol. 1974, 87, 63–88. [Google Scholar] [CrossRef]
- Wu, M.; Tinoco, I. RNA folding causes secondary structure rearrangement. Proc. Natl. Acad. Sci. USA 1998, 95, 11555–11560. [Google Scholar] [CrossRef] [PubMed]
- Andersen, A.A.; Collins, R.A. Intramolecular secondary structure rearrangement by the kissing interaction of the Neurospora VS ribozyme. Proc. Natl. Acad. Sci. USA 2001, 98, 7730–7735. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.B.; Lipfert, J.; Bai, Y.; Pande, V.S.; Doniach, S.; Herschlag, D. Do conformational biases of simple helical junctions influence RNA folding stability and specificity? RNA 2009, 15, 2195–2205. [Google Scholar] [CrossRef] [PubMed]
- Bailor, M.H.; Sun, X.; Al-Hashimi, H.M. Topology links RNA secondary structure with global conformation, dynamics, and adaptation. Science 2010, 327, 202–206. [Google Scholar] [CrossRef] [PubMed]
- McCammon, J.A.; Harvey, S.C. Dynamics of Proteins and Nucleic Acids; Cambridge University Press: Cambridge, UK, 1988. [Google Scholar]
- Auffinger, P.; Westhof, E. Simulations of the molecular dynamics of nucleic acids. Curr. Opin. Struct. Biol. 1998, 8, 227–236. [Google Scholar] [CrossRef]
- Stadlbauer, P.; Trantírek, L.; CheathamI, T.E., II; Koča, J.; Šponer, J. Triplex intermediates in folding of human telomeric quadruplexes probed by microsecond-scale molecular dynamics simulations. Biochimie 2014, 105, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, P.; Westhof, E. RNA hydration: Three nanoseconds of multiple molecular dynamics simulations of the solvated tRNAAsp anticodon hairpin1. J. Mol. Biol. 1997, 269, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.H.; Briber, R.M.; Damjanovic, A.; Thirumalai, D.; Woodson, S.A.; Sokolov, A.P. Dynamics of tRNA at different levels of hydration. Biophys. J. 2009, 96, 2755–2762. [Google Scholar] [CrossRef] [PubMed]
- Cole, P.E.; Yang, S.K.; Crothers, D.M. Conformational changes of transfer ribonucleic acid. Equilibrium phase diagrams. Biochemistry (Mosc.) 1972, 11, 4358–4368. [Google Scholar] [CrossRef]
- Yang, S.K.; Crothers, D.M. Conformational changes of transfer ribonucleic acid. Comparison of the early melting transition of two tyrosine-specific transfer ribonucleic acids. Biochemistry (Mosc.) 1972, 11, 4375–4381. [Google Scholar] [CrossRef]
- Hilbers, C.W.; Shulman, R.G.; Kim, S.H. High resolution NMR study of the melting of tRNAYeastPhe. Biochem. Biophys. Res. Commun. 1973, 55, 953–960. [Google Scholar] [CrossRef]
- Treiber, D.K.; Williamson, J.R. Exposing the kinetic traps in RNA folding. Curr. Opin. Struct. Biol. 1999, 9, 339–345. [Google Scholar] [CrossRef]
- Shelton, V.M.; Sosnick, T.R.; Pan, T. Altering the intermediate in the equilibrium folding of unmodified yeast tRNAPhe with monovalent and divalent cations. Biochemistry (Mosc.) 2001, 40, 3629–3638. [Google Scholar] [CrossRef]
- Serebrov, V.; Clarke, R.J.; Gross, H.J.; Kisselev, L. Mg2+-induced tRNA folding. Biochemistry (Mosc.) 2001, 40, 6688–6698. [Google Scholar] [CrossRef]
- Bhaskaran, H.; Rodriguez-Hernandez, A.; Perona, J.J. Kinetics of tRNA folding monitored by aminoacylation. RNA 2012, 18, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Nobles, K.N.; Yarian, C.S.; Liu, G.; Guenther, R.H.; Agris, P.F. Highly conserved modified nucleosides influence Mg2+-dependent tRNA folding. Nucleic Acids Res. 2002, 30, 4751–4760. [Google Scholar] [CrossRef] [PubMed]
- Aduri, R.; Psciuk, B.T.; Saro, P.; Taniga, H.; Schlegel, H.B.; SantaLucia, J. AMBER force field parameters for the naturally occurring modified nucleosides in RNA. J. Chem. Theory Comput. 2007, 3, 1464–1475. [Google Scholar] [CrossRef]
- Sorin, E.J.; Nakatani, B.J.; Rhee, Y.M.; Jayachandran, G.; Vishal, V.; Pande, V.S. Does native state topology determine the RNA folding mechanism? J. Mol. Biol. 2004, 337, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Sharma, S.; Chalasani, P.; Demidov, V.V.; Broude, N.E.; Dokholyan, N.V. Ab initio RNA folding by discrete molecular dynamics: From structure prediction to folding mechanisms. RNA 2008, 14, 1164–1173. [Google Scholar] [CrossRef] [PubMed]
- Gosavi, S.; Chavez, L.L.; Jennings, P.A.; Onuchic, J.N. Topological frustration and the folding of interleukin-1β. J. Mol. Biol. 2006, 357, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Gosavi, S.; Whitford, P.C.; Jennings, P.A.; Onuchic, J.N. Extracting function from a β-trefoil folding motif. Proc. Natl. Acad. Sci. USA 2008, 105, 10384–10389. [Google Scholar] [CrossRef] [PubMed]
- Hills, R.D., Jr.; Brooks, C.L., III. Coevolution of function and the folding landscape: Correlation with density of native contacts. Biophys. J. 2008, 95, L57–L59. [Google Scholar] [CrossRef] [PubMed]
- Nobrega, R.P.; Arora, K.; Kathuria, S.V.; Graceffa, R.; Barrea, R.A.; Guo, L.; Chakravarthy, S.; Bilsel, O.; Irving, T.C.; Brooks, C.L.; et al. Modulation of frustration in folding by sequence permutation. Proc. Natl. Acad. Sci. USA 2014, 111, 10562–10567. [Google Scholar] [CrossRef] [PubMed]
- Mustoe, A.M.; Brooks, C.L.; Al-Hashimi, H.M. Topological constraints are major determinants of tRNA tertiary structure and dynamics and provide basis for tertiary folding cooperativity. Nucleic Acids Res. 2014, 42, 11792–11804. [Google Scholar] [CrossRef] [PubMed]
- Mustoe, A.M.; Liu, X.; Lin, P.J.; Al-Hashimi, H.M.; Fierke, C.A.; Brooks, C.L. Noncanonical secondary structure stabilizes mitochondrial tRNASer (UCN) by reducing the entropic cost of tertiary folding. J. Am. Chem. Soc. 2015, 137, 3592–3599. [Google Scholar] [CrossRef] [PubMed]
- Lau, F.T.K.; Karplus, M. Molecular recognition in proteins: Simulation analysis of substrate binding by a tyrosyl-tRNA synthetase mutant. J. Mol. Biol. 1994, 236, 1049–1066. [Google Scholar] [CrossRef]
- Ghosh, A.; Sakaguchi, R.; Liu, C.; Vishveshwara, S.; Hou, Y.-M. Allosteric communication in cysteinyl tRNA synthetase: A network of direct and indirect readout. J. Biol. Chem. 2011, 286, 37721–37731. [Google Scholar] [CrossRef] [PubMed]
- Grant, T.D.; Luft, J.R.; Wolfley, J.R.; Snell, M.E.; Tsuruta, H.; Corretore, S.; Quartley, E.; Phizicky, E.M.; Grayhack, E.J.; Snell, E.H. The structure of yeast glutaminyl-tRNA synthetase and modeling of its interaction with tRNA. J. Mol. Biol. 2013, 425, 2480–2493. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Nakamura, S.; Terada, T.; Shimizu, K. Mechanism of the difference in the binding affinity of E. coli tRNAGln to glutaminyl-tRNA synthetase caused by noninterface nucleotides in variable loop. Biophys. J. 2007, 92, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Black Pyrkosz, A.; Eargle, J.; Sethi, A.; Luthey-Schulten, Z. Exit strategies for charged tRNA from GluRS. J. Mol. Biol. 2010, 397, 1350–1371. [Google Scholar] [CrossRef] [PubMed]
- Sethi, A.; Eargle, J.; Black, A.A.; Luthey-Schulten, Z. Dynamical networks in tRNA:protein complexes. Proc. Natl. Acad. Sci. USA 2009, 106, 6620–6625. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Tan, M.; Du, D.; Xu, B.; Eriani, G.; Wang, E. Peripheral insertion modulates the editing activity of the isolated CP1 domain of leucyl-tRNA synthetase. Biochem. J. 2011, 440, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Strom, A.M.; Fehling, S.C.; Bhattacharyya, S.; Hati, S. Probing the global and local dynamics of aminoacyl-tRNA synthetases using all-atom and coarse-grained simulations. J. Mol. Model. 2014, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, Y.; Nureki, O.; Tateno, M. Identification of the nucleophilic factors and the productive complex for the editing reaction by leucyl-tRNA synthetase. FEBS Lett. 2009, 583, 1901–1908. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Vishveshwara, S. A study of communication pathways in methionyl-tRNA synthetase by molecular dynamics simulations and structure network analysis. Proc. Natl. Acad. Sci. USA 2007, 104, 15711–15716. [Google Scholar] [CrossRef] [PubMed]
- Budiman, M.E.; Knaggs, M.H.; Fetrow, J.S.; Alexander, R.W. Using molecular dynamics to map interaction networks in an aminoacyl-tRNA synthetase. Proteins Struct. Funct. Bioinform. 2007, 68, 670–689. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, G.; Pelka, H.; Schulman, L.H. Identification of the tRNA anticodon recognition site of Escherichia coli methionyl-tRNA synthetase. Biochemistry (Mosc.) 1990, 29, 2220–2225. [Google Scholar] [CrossRef]
- Bhattacharyya, M.; Ghosh, A.; Hansia, P.; Vishveshwara, S. Allostery and conformational free energy changes in human tryptophanyl-tRNA synthetase from essential dynamics and structure networks. Proteins Struct. Funct. Bioinform. 2010, 78, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Kapustina, M.; Carter, C.W., Jr. Computational studies of tryptophanyl-tRNA synthetase: Activation of ATP by induced-fit. J. Mol. Biol. 2006, 362, 1159–1180. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Froeyen, M.; Herdewijn, P. Comparative structural dynamics of Tyrosyl-tRNA synthetase complexed with different substrates explored by molecular dynamics. Eur. Biophys. J. 2008, 38, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Mykuliak, V.V.; Dragan, A.I.; Kornelyuk, A.I. Structural states of the flexible catalytic loop of M. tuberculosis tyrosyl-tRNA synthetase in different enzyme-substrate complexes. Eur. Biophys. J. EBJ 2014, 43, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Savytskyi, O.V.; Yesylevskyy, S.O.; Kornelyuk, A.I. Asymmetric structure and domain binding interfaces of human tyrosyl-tRNA synthetase studied by molecular dynamics simulations. J. Mol. Recognit. 2013, 26, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yu, L.; Huang, Q. Molecular trigger for pre-transfer editing pathway in Valyl-tRNA synthetase: A molecular dynamics simulation study. J. Mol. Model. 2010, 17, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Bharatham, N.; Bharatham, K.; Lee, Y.; Woo Lee, K. Molecular dynamics simulation study of valyl-tRNA synthetase with its pre- and post-transfer editing substrates. Biophys. Chem. 2009, 143, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.; Plateau, P.; Simonson, T. Free-Energy simulations and experiments reveal long-range electrostatic interactions and substrate-assisted specificity in an aminoacyl-tRNA synthetase. ChemBioChem 2006, 7, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.; Simonson, T. Molecular dynamics simulations show that bound Mg2+ contributes to amino acid and aminoacyl adenylate binding specificity in aspartyl-tRNA synthetase through long range electrostatic interactions. J. Biol. Chem. 2006, 281, 23792–23803. [Google Scholar] [CrossRef] [PubMed]
- Ul-Haq, Z.; Khan, W.; Zarina, S.; Sattar, R.; Moin, S.T. Template-based structure prediction and molecular dynamics simulation study of two mammalian aspartyl-tRNA synthetases. J. Mol. Graph. Model. 2010, 28, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Archontis, G.; Simonson, T.; Karplus, M. Binding free energies and free energy components from molecular dynamics and Poisson-Boltzmann calculations. Application to amino acid recognition by aspartyl-tRNA synthetase1. J. Mol. Biol. 2001, 306, 307–327. [Google Scholar] [CrossRef] [PubMed]
- Polydorides, S.; Amara, N.; Aubard, C.; Plateau, P.; Simonson, T.; Archontis, G. Computational protein design with a generalized born solvent model: Application to asparaginyl-tRNA synthetase. Proteins Struct. Funct. Bioinform. 2011, 79, 3448–3468. [Google Scholar] [CrossRef] [PubMed]
- Arnez, J.G.; Flanagan, K.; Moras, D.; Simonson, T. Engineering an Mg2+ site to replace a structurally conserved arginine in the catalytic center of histidyl-tRNA synthetase by computer experiments. Proteins Struct. Funct. Bioinform. 1998, 32, 362–380. [Google Scholar] [CrossRef]
- Hughes, S.J.; Tanner, J.A.; Hindley, A.D.; Miller, A.D.; Gould, I.R. Functional asymmetry in the lysyl-tRNA synthetase explored by molecular dynamics, free energy calculations and experiment. BMC Struct. Biol. 2003, 3. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.J.; Tanner, J.A.; Miller, A.D.; Gould, I.R. Molecular dynamics simulations of LysRS: An asymmetric state. Proteins Struct. Funct. Bioinform. 2006, 62, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Sanford, B.; Cao, B.; Johnson, J.M.; Zimmerman, K.; Strom, A.M.; Mueller, R.M.; Bhattacharyya, S.; Musier-Forsyth, K.; Hati, S. Role of coupled dynamics in the catalytic activity of prokaryotic-like prolyl-tRNA synthetases. Biochemistry (Mosc.) 2012, 51, 2146–2156. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Nandi, N. Dynamics of the active sites of dimeric seryl tRNA synthetase from methanopyrus kandleri. J. Phys. Chem. B 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, E.A.C.; Huang, W.; Llano, J.; Gauld, J.W. Molecular dynamics investigation into substrate binding and identity of the catalytic base in the mechanism of threonyl-tRNA synthetase. J. Phys. Chem. B 2012, 116, 5205–5212. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.J.; Nyborg, J.; Reid, B.R.; Blow, D.M. The crystal structure of tyrosyl-transfer RNA synthetase at 2.7 Å resolution. J. Mol. Biol. 1976, 105, 577–586. [Google Scholar] [CrossRef]
- Blow, D.M. Flexibility and rigidity in protein crystals. Ciba Found. Symp. 1977, 55–61. [Google Scholar]
- Williamson, J.R. Induced fit in RNA–protein recognition. Nat. Struct. Mol. Biol. 2000, 7, 834–837. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.; Panvert, M.; Blanquet, S.; Mechulam, Y. Transition state stabilization by the “high” motif of class I aminoacyl-tRNA synthetases: The case of Escherichia coli methionyl-tRNA synthetase. Nucleic Acids Res. 1995, 23, 4793–4798. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Ogiso, Y.; Nakama, T.; Fukai, S.; Nureki, O. Structural basis for anticodon recognition by methionyl-tRNA synthetase. Nat. Struct. Mol. Biol. 2005, 12, 931–932. [Google Scholar] [CrossRef] [PubMed]
- Yesylevskyy, S.O.; Savytskyi, O.V.; Odynets, K.A.; Kornelyuk, A.I. Interdomain compactization in human tyrosyl-tRNA synthetase studied by the hierarchical rotations technique. Biophys. Chem. 2011, 154, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, K.; Schimmel, P. Two distinct cytokines released from a human aminoacyl-tRNA synthetase. Science 1999, 284, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Meinnel, T.; Mechulam, Y.; Corre, D.L.; Panvert, M.; Blanquet, S.; Fayat, G. Selection of suppressor methionyl-tRNA synthetases: Mapping the tRNA anticodon binding site. Proc. Natl. Acad. Sci. USA 1991, 88, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Rould, M.A.; Perona, J.J.; Soll, D.; Steitz, T.A. Structure of E. coli glutaminyl-tRNA synthetase complexed with tRNAGln and ATP at 2.8 A resolution. Science 1989, 246, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Nureki, O.; Shimada, A.; Vassylyev, D.G.; Yokoyama, S. Structural basis for anticodon recognition by discriminating glutamyl-tRNA synthetase. Nat. Struct. Mol. Biol. 2001, 8, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Hauenstein, S.; Zhang, C.-M.; Hou, Y.-M.; Perona, J.J. Shape-selective RNA recognition by cysteinyl-tRNA synthetase. Nat. Struct. Mol. Biol. 2004, 11, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Ibba, M.; Söll, D. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 2000, 69, 617–650. [Google Scholar] [CrossRef] [PubMed]
- Delagoutte, B.; Moras, D.; Cavarelli, J. tRNA aminoacylation by arginyl-tRNA synthetase: Induced conformations during substrates binding. EMBO J. 2000, 19, 5599–5610. [Google Scholar] [CrossRef] [PubMed]
- Minajigi, A.; Francklyn, C.S. RNA-assisted catalysis in a protein enzyme: The 2′-hydroxyl of tRNAThr A76 promotes aminoacylation by threonyl-tRNA synthetase. Proc. Natl. Acad. Sci. USA 2008, 105, 17748–17753. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Gauld, J.W. Substrate-assisted catalysis in the aminoacyl transfer mechanism of histidyl-tRNA synthetase: A density functional theory study. J. Phys. Chem. B 2008, 112, 16874–16882. [Google Scholar] [CrossRef] [PubMed]
- Cramer, F.; Englisch, U.; Freist, W.; Sternbach, H. Aminoacylation of tRNAs as critical step of protein biosynthesis. Biochimie 1991, 73, 1027–1035. [Google Scholar] [CrossRef]
- Mascarenhas, A.P.; An, S.; Rosen, A.E.; Martinis, S.A.; Musier-Forsyth, K. Fidelity mechanisms of the aminoacyl-tRNA synthetases. In Protein Engineering; Köhrer, C., RajBhandary, U.L., Eds.; Nucleic Acids and Molecular Biology; Springer: Berlin, Germany; Heidelberg, Germany, 2009; pp. 155–203. [Google Scholar]
- Pauling, L. The Probability of Errors in the Process of Synthesis of Protein Molecules; Birkhauser: Basel, Switzerland, 1957. [Google Scholar]
- Fersht, A.R. Editing mechanisms in protein synthesis. Rejection of valine by the isoleucyl-tRNA synthetase. Biochemistry (Mosc.) 1977, 16, 1025–1030. [Google Scholar] [CrossRef]
- Martinis, S.A.; Boniecki, M.T. The balance between pre- and post-transfer editing in tRNA synthetases. FEBS Lett. 2010, 584, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Tukalo, M.; Yaremchuk, A.; Fukunaga, R.; Yokoyama, S.; Cusack, S. The crystal structure of leucyl-tRNA synthetase complexed with tRNALeu in the post-transfer–editing conformation. Nat. Struct. Mol. Biol. 2005, 12, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Francklyn, C.S. DNA polymerases and aminoacyl-tRNA synthetases: Shared mechanisms for ensuring the fidelity of gene expression. Biochemistry (Mosc.) 2008, 47, 11695–11703. [Google Scholar] [CrossRef] [PubMed]
- Palencia, A.; Crépin, T.; Vu, M.T.; Lincecum, T.L., Jr.; Martinis, S.A.; Cusack, S. Structural dynamics of the aminoacylation and proofreading functional cycle of bacterial leucyl-tRNA synthetase. Nat. Struct. Mol. Biol. 2012, 19, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Perona, J.J.; Gruic-Sovulj, I. Synthetic and editing mechanisms of aminoacyl-tRNA synthetases. Top. Curr. Chem. 2014, 344, 1–41. [Google Scholar] [PubMed]
- Cvetesic, N.; Perona, J.J.; Gruic-Sovulj, I. Kinetic partitioning between synthetic and editing pathways in class I aminoacyl-tRNA synthetases occurs at both pre-transfer and post-transfer hydrolytic steps. J. Biol. Chem. 2012, 287, 25381–25394. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Macnamara, L.M.; Leuchter, J.D.; Alexander, R.W.; Cho, S.S. MD Simulations of tRNA and Aminoacyl-tRNA Synthetases: Dynamics, Folding, Binding, and Allostery. Int. J. Mol. Sci. 2015, 16, 15872-15902. https://doi.org/10.3390/ijms160715872
Li R, Macnamara LM, Leuchter JD, Alexander RW, Cho SS. MD Simulations of tRNA and Aminoacyl-tRNA Synthetases: Dynamics, Folding, Binding, and Allostery. International Journal of Molecular Sciences. 2015; 16(7):15872-15902. https://doi.org/10.3390/ijms160715872
Chicago/Turabian StyleLi, Rongzhong, Lindsay M. Macnamara, Jessica D. Leuchter, Rebecca W. Alexander, and Samuel S. Cho. 2015. "MD Simulations of tRNA and Aminoacyl-tRNA Synthetases: Dynamics, Folding, Binding, and Allostery" International Journal of Molecular Sciences 16, no. 7: 15872-15902. https://doi.org/10.3390/ijms160715872
APA StyleLi, R., Macnamara, L. M., Leuchter, J. D., Alexander, R. W., & Cho, S. S. (2015). MD Simulations of tRNA and Aminoacyl-tRNA Synthetases: Dynamics, Folding, Binding, and Allostery. International Journal of Molecular Sciences, 16(7), 15872-15902. https://doi.org/10.3390/ijms160715872