
Atrazine Causes Autophagy- and Apoptosis-Related Neurodegenerative Effects in Dopaminergic Neurons in the Rat Nigrostriatal Dopaminergic System

Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
2.1.1. Body Weight and Physical Appearance
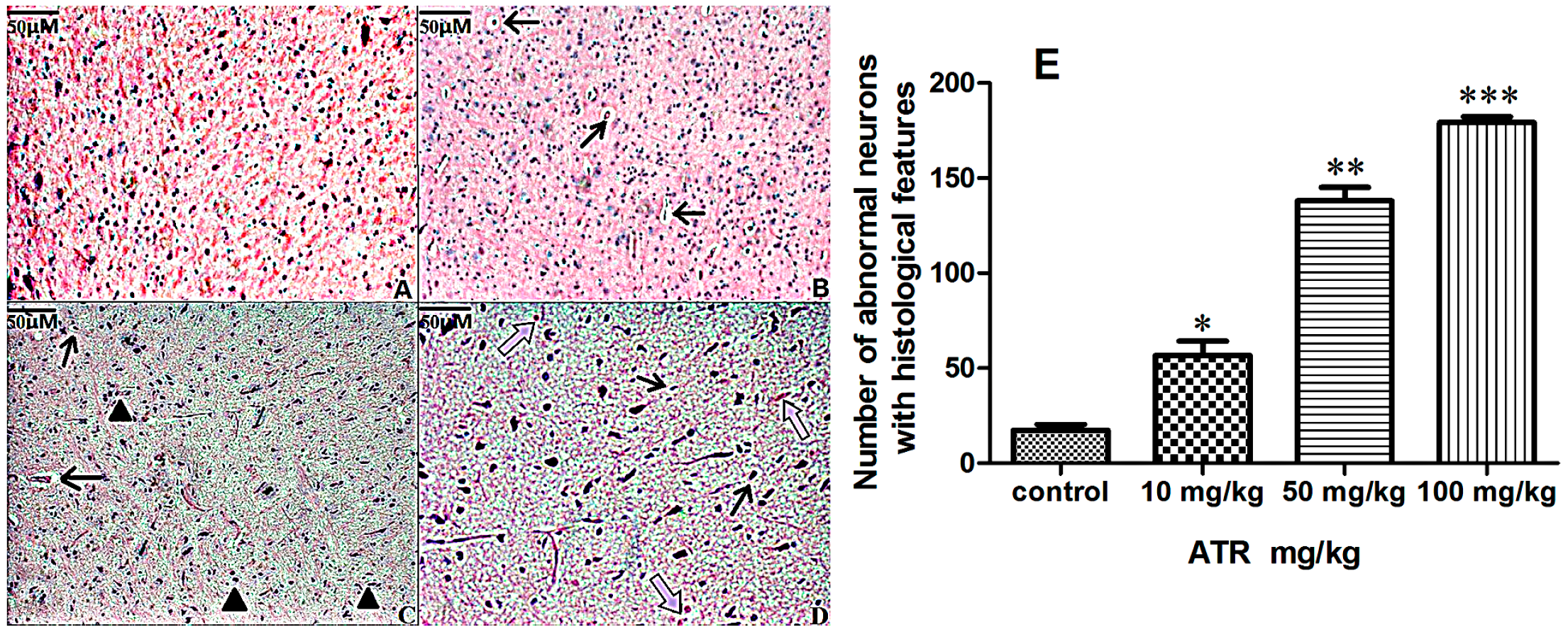
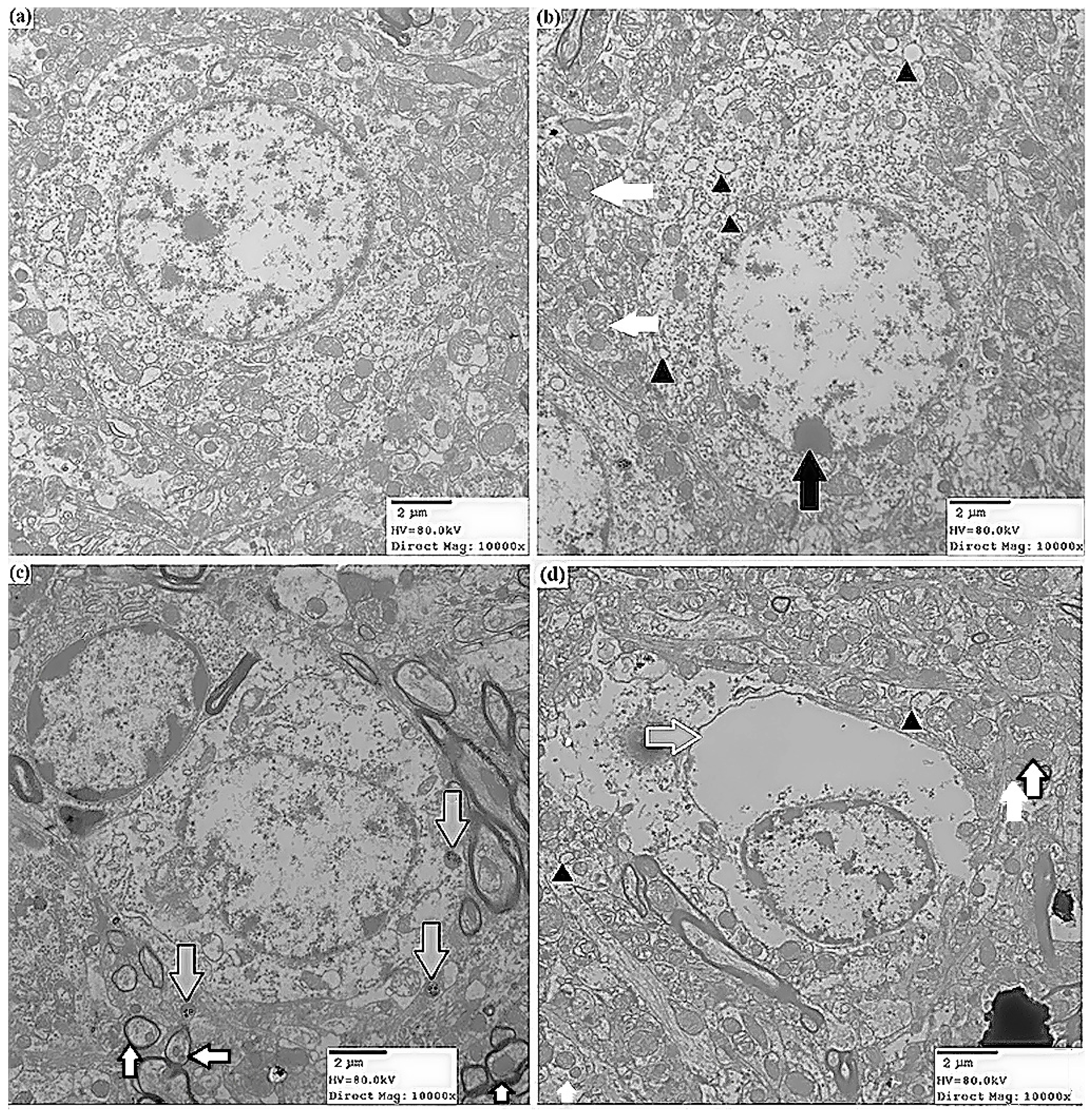
2.1.2. Effects of ATR on Histopathology
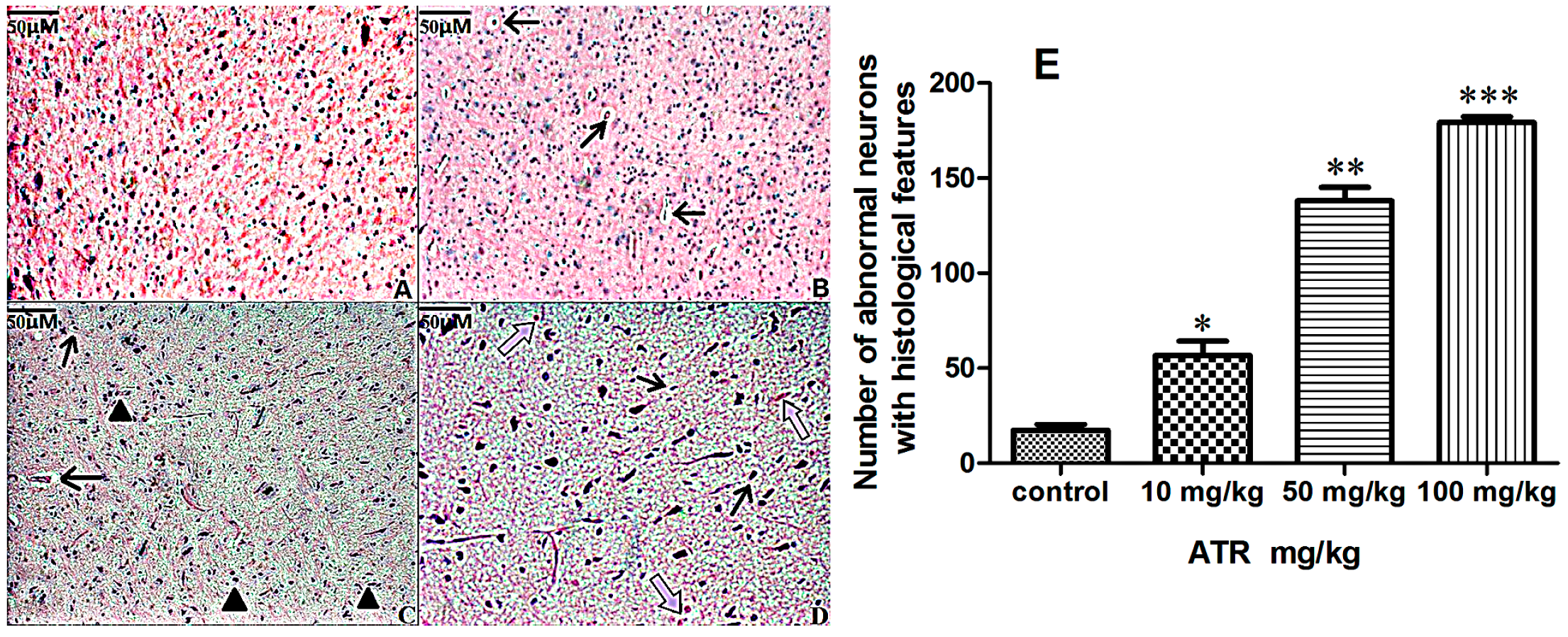
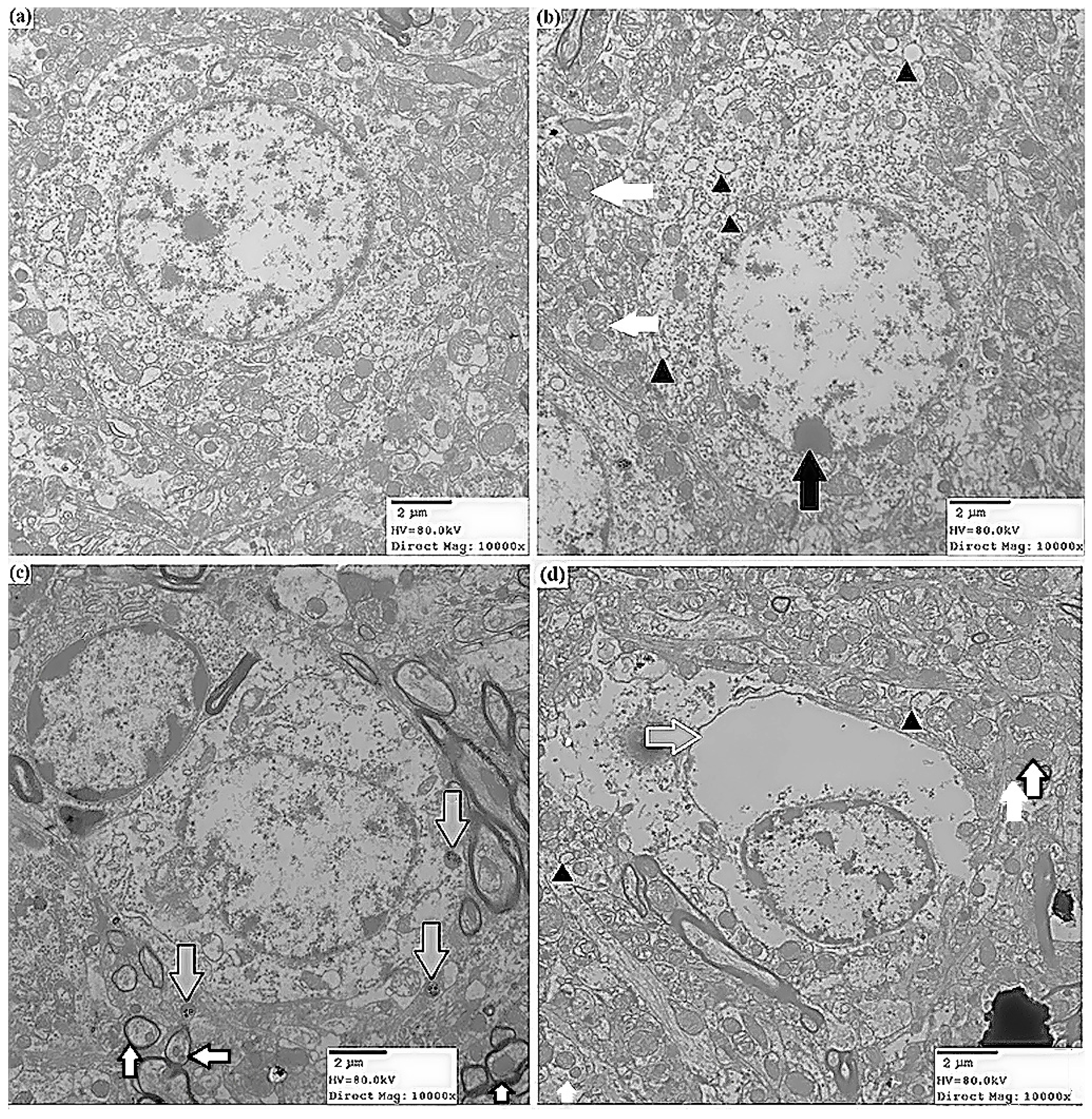
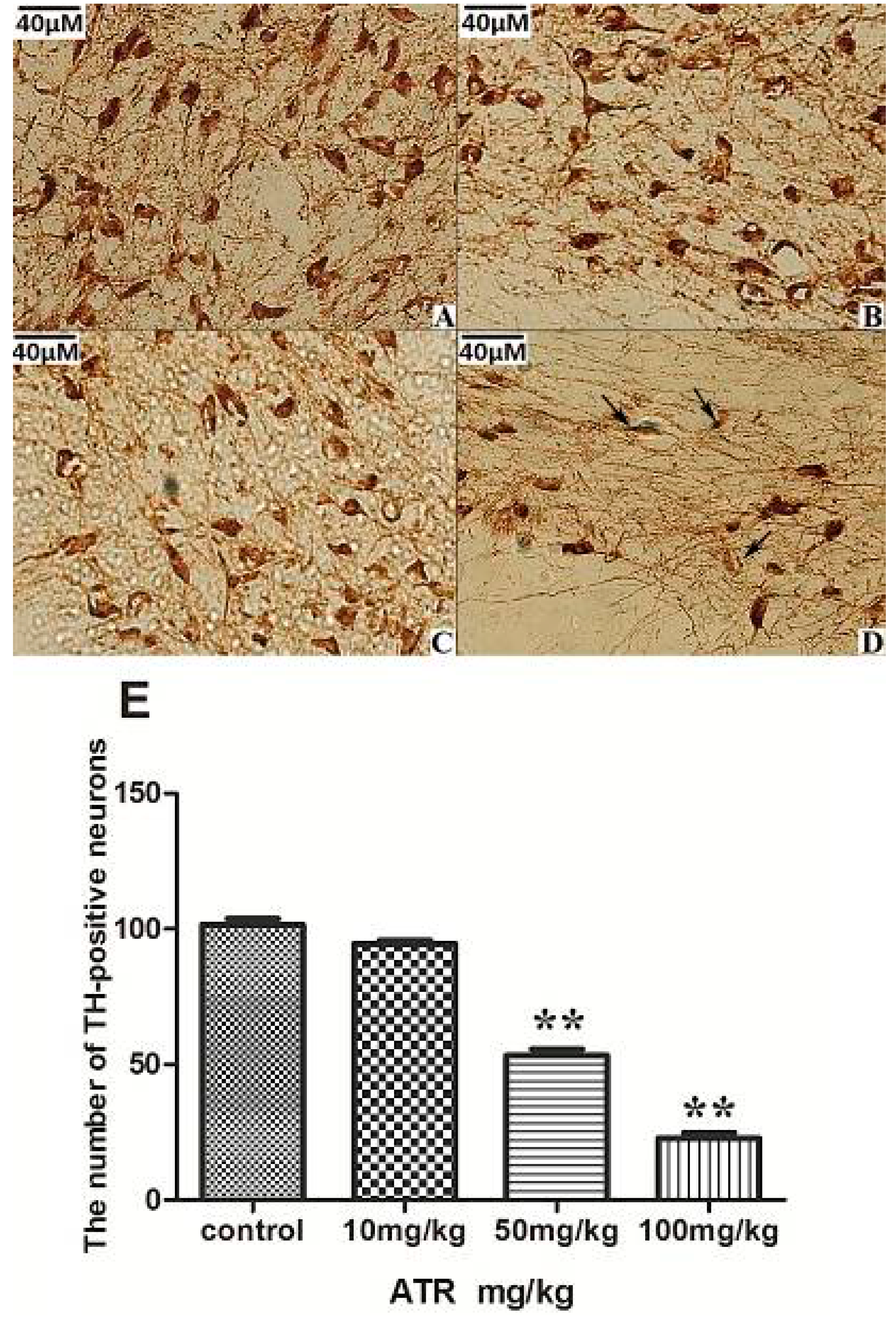
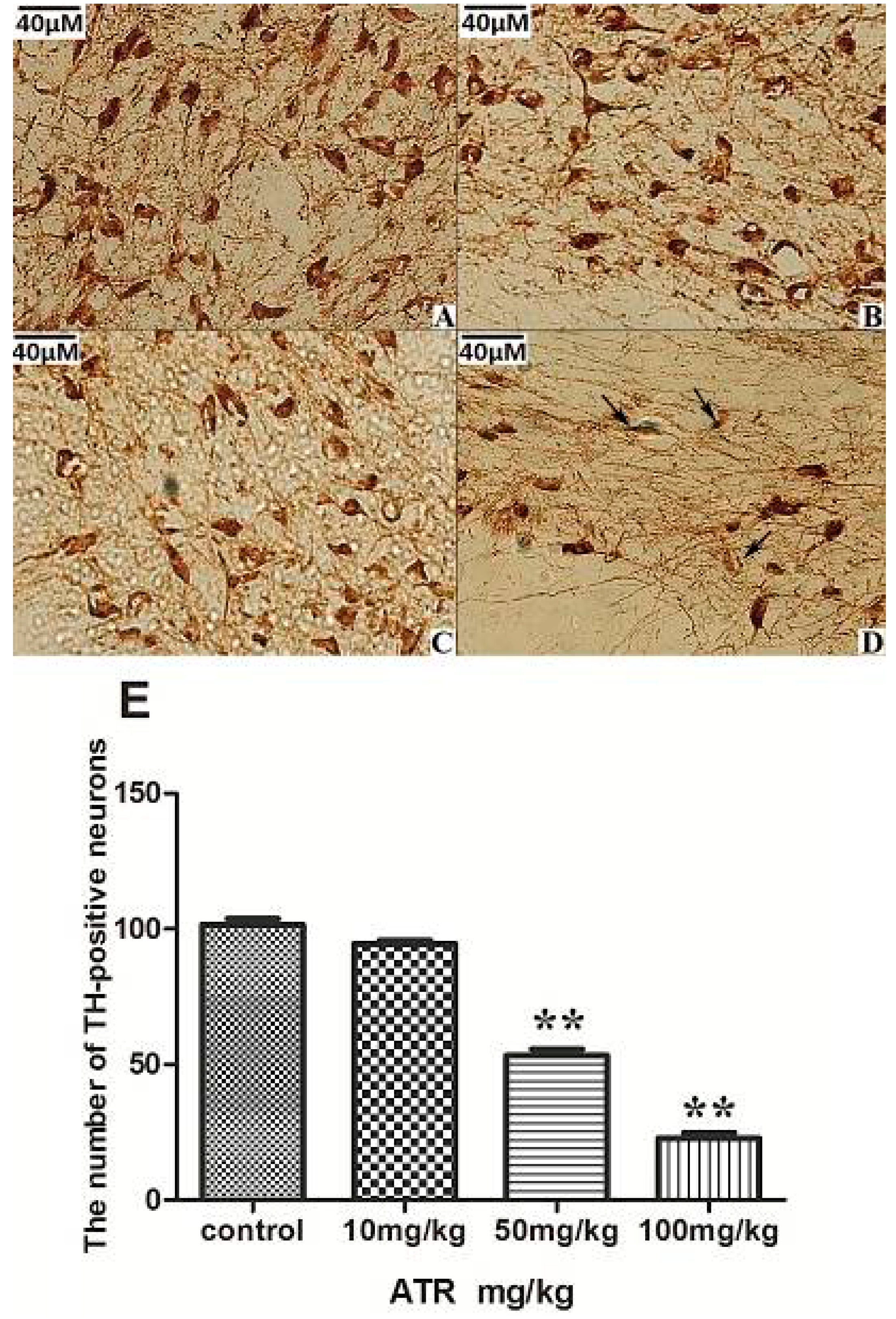
2.1.3. ATR Reduces TH-Positive Neurons in Rat Ventral Midbrain
2.1.4. ATR Alters the mRNA Levels of Several Autophagy- and Apoptosis-Related Genes
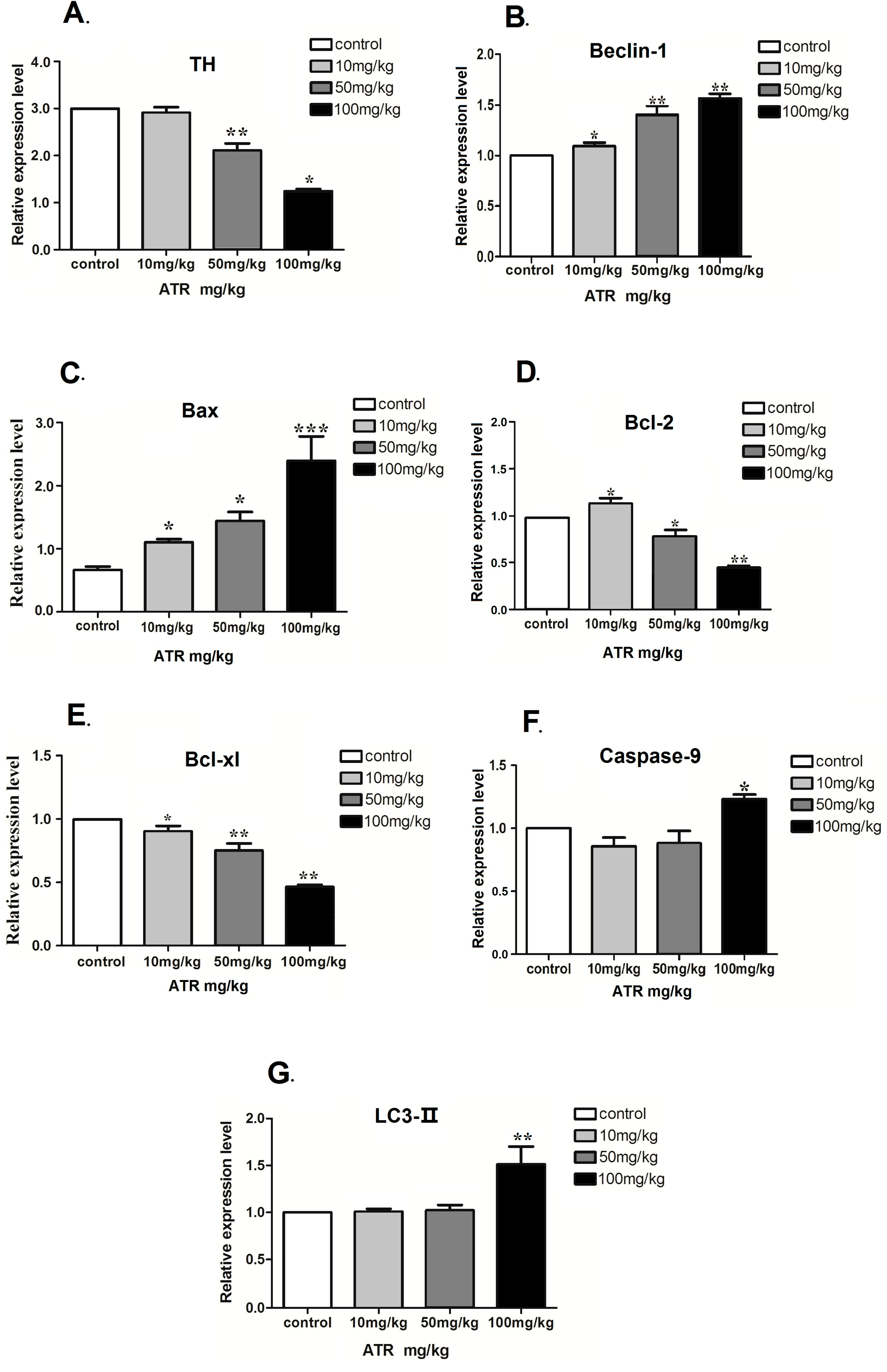
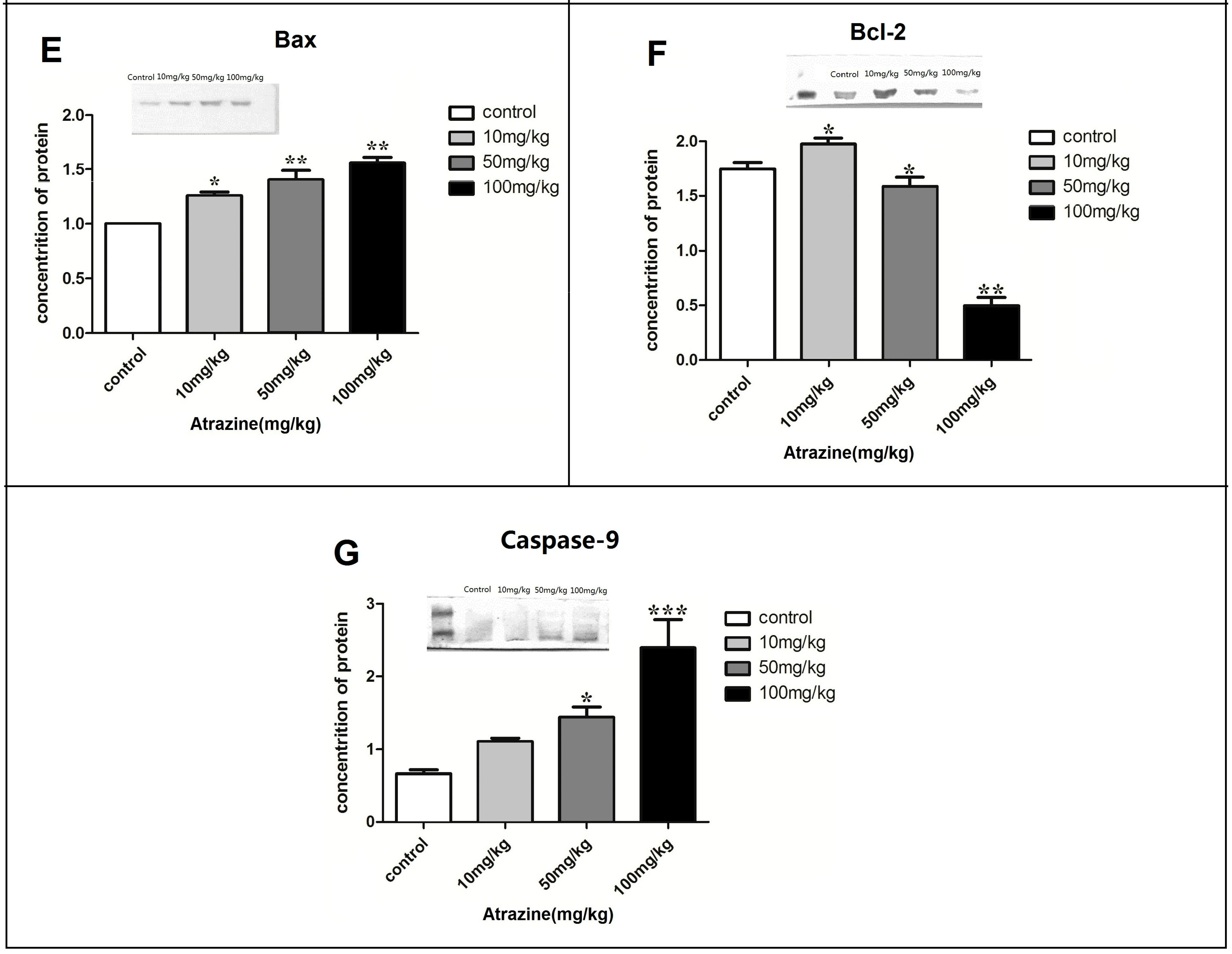
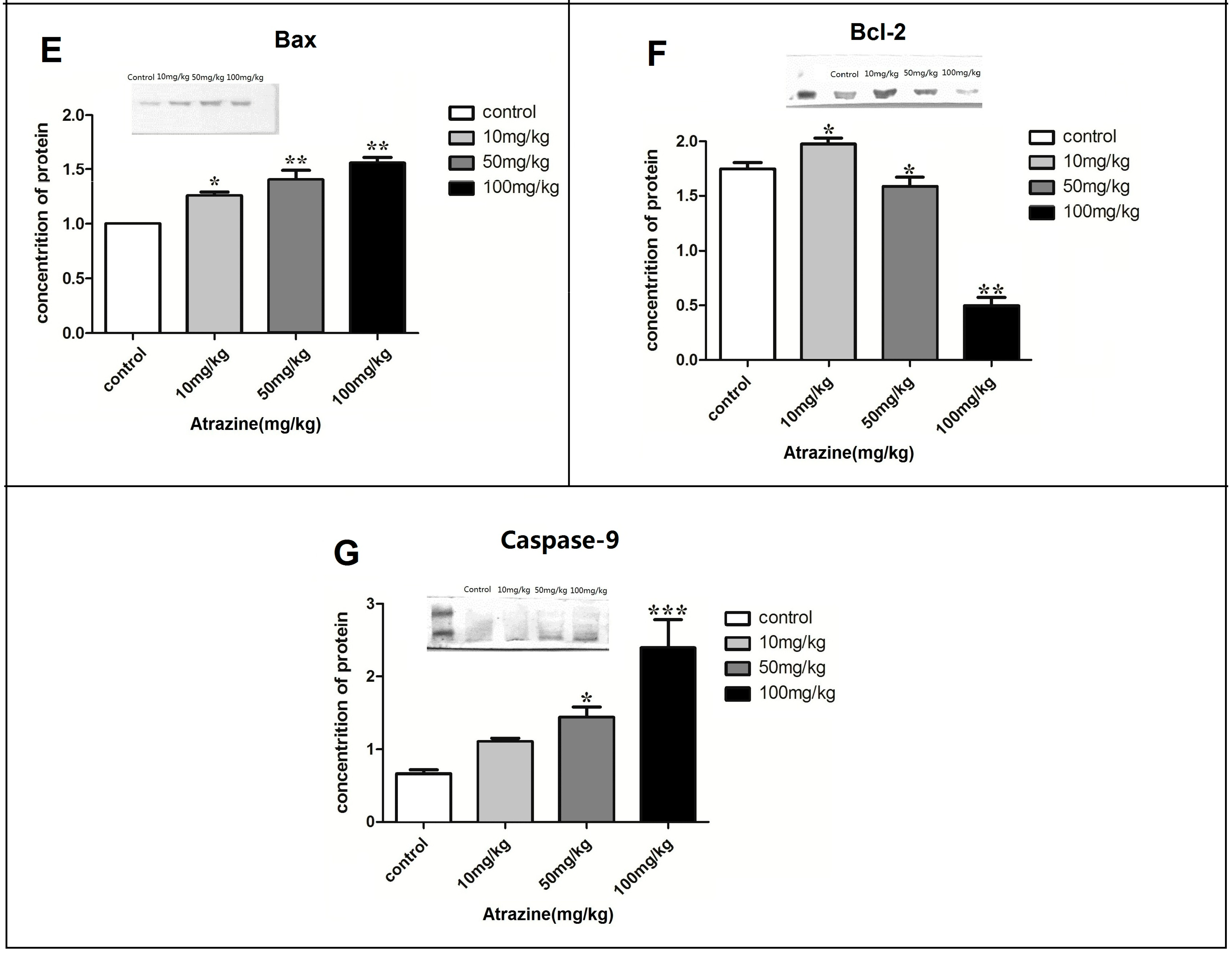
2.1.5. Effect of ATR on the Amounts of Autophagy- and Apoptosis- Related Proteins

2.2. Discussion
3. Experimental Section
3.1. Ethics Statement
3.2. Chemicals and Reagents
3.3. Animals and Treatments
3.4. Histopathological Evaluation
3.4.1. Light Microscopy
3.4.2. Transmission Electron Microscopy
3.4.3. Immunohistochemical Assays and Neuronal Counting
3.4.4. RNA Isolation and Reverse Transcription
3.4.5. Quantitative Real-Time PCR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer (5′→3′) | Product Size (bp) |
|---|---|---|
| β-actin | Forward: TGTTGGCATAGAGGTCTTTACGGReverse: TGGGTATGGAATCCTGTGGCA | 90 |
| Beclin-1 | Forward: GATGGTGTCTCTCGCAGATTCReverse: CTGTGCATTCCTCACAGAGTG | 138 |
| Bcl-xl | Forward: GCCACCTACCTGAATGACCACCReverse: TGAGCCCAGCAGAACTACACCA | 174 |
| Bcl-2 | Forward: GGCATCCCAGCCTCCGTTAT”Reverse: GTGTGTGGGGAGCGTCAATA | 121 |
| Caspase-9 | Forward: TCTACAAGGCAAGCCCAAGCReverse: CGTCCATCTGGTCATCTATTCC | 169 |
| TH | Forward: GTTCATCDDACGGCGACAGAReverse: ATGCCATATCATCGTCAGTTCCAC | 152 |
| LC3-II | Forward: AGAGCGAGAGAGATGAAGACGGReverse: TTGCCTTGGTAGGGGCTTAACA | 125 |
3.4.6. Western Blotting
3.4.7. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Still Poisoning the Well: Atrazine Continues to Contaminate Surface Water and Drinking Water in the United States. Available online: http://adams.vtls.com:9020/vital/access/manager/Repository/kcpl:9699 (accessed on 8 June 2015).
- Decision Documents for Atrazine. Available online: http://www.epa.gov/opp00001/chem_search/reg_actions/reregistration/red_PC-080803_1-Apr-06.pdf (accessed on 8 June 2015).
- Fawcett, R.S.; Huxley, F.C. Twenty years of university corn yield data with and without atrazine. North Cent. Weed Sci. Soc. Proc. 2008, 63, 137. [Google Scholar]
- Basic Information about Atrazine in Drinking Water. Available online: http://water.epa.gov/drink/contaminants/basicinformation/atrazine.cfm (accessed on 8 June 2015).
- USDA National Agricultural Statistics Service. Available online: http://quickstats.nass.usda.gov/?sectordesc=ENVIRONMENTAL (accessed on 8 June 2015).
- Guzzella, L.; Pozzoni, F.; Giuliano, G. Herbicide contamination of surficial groundwater in Northern Italy. Environ. Pollut. 2006, 142, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Brodkin, M.A.; Madhoun, H.; Rameswaran, M.; Vatnick, I. Atrazine is an immune disruptor in adult northern leopard frogs (Rana pipiens). Environ. Toxicol. Chem. 2007, 26, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Solomon, K.R.; Carr, J.A.; du Preez, L.H.; Giesy, J.P.; Kendall, R.J.; Smith, E.E.; van der Kraak, G.J. Effects of atrazine on fish, amphibians, and aquatic reptiles: A critical review. Crit. Rev. Toxicol. 2008, 38, 721–772. [Google Scholar] [CrossRef] [PubMed]
- Campos-Pereira, F.D.; Oliveira, C.A.; Pigoso, A.A.; Silva-Zacarin, E.C.M.; Barbieri, R.; Spatti, E.F.; Marin-Morales, M.A.; Severi-Aguiar, G.D.C. Early cytotoxic and genotoxic effects of atrazine on Wistar rat liver: A morphological, immunohistochemical, biochemical, and molecular study. Ecotoxicol. Environ. Saf. 2012, 78, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Foradori, C.D.; Zimmerman, A.D.; Hinds, L.R.; Zuloaga, K.L.; Breckenridge, C.B.; Handa, R.J. Atrazine inhibits pulsatile gonadotropin-releasing hormone (GnRH) release without altering GnRH messenger RNA or protein levels in the female rat. Biol. Reprod. 2013, 88. [Google Scholar] [CrossRef] [PubMed]
- Rayner, J.; Fenton, S. Atrazine: An environmental endocrine disruptor that alters mammary gland development and tumor susceptibility. In Environment and Breast Cancer; Russo, J., Ed.; Springer: New York, NY, USA, 2011; pp. 167–183. [Google Scholar]
- Nasseri, S.; Dehghani, M.; Amin, S.; Naddafi, K.; Zamanian, Z. Fate of atrazine in the agricultural soil of corn fields in Fars province of Iran. Iran. J. Environ. Health Sci. Eng. 2009, 6, 223–232. [Google Scholar]
- Bakke, B.; de Roos, A.J.; Barr, D.B.; Stewart, P.A.; Blair, A.; Freeman, L.B.; Lynch, C.F.; Allen, R.H.; Alavanja, M.C.R.; Vermeulen, R. Exposure to atrazine and selected non-persistent pesticides among corn farmers during a growing season. J. Expos. Sci. Environ. Epidemiol. 2008, 19, 544–554. [Google Scholar] [CrossRef] [PubMed]
- Hines, C.J.; Deddens, J.A.; Lu, C.; Fenske, R.; Striley, C.A. Mixed-effect models for evaluating multiple measures of atrazine exposure among custom applicators. J. Occup. Environ. Hyg. 2006, 3, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Priyadarshi, A.; Khuder, S.A.; Schaub, E.A.; Shrivastava, S. A meta-analysis of Parkinson’s disease and exposure to pesticides. Neurotoxicology 2000, 21, 435–440. [Google Scholar] [PubMed]
- Brown, T.P.; Rumsby, P.C.; Capleton, A.C.; Rushton, L.; Levy, L.S. Pesticides and Parkinson’s disease—Is there a link? Environ. Health Perspect. 2006, 114, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Cooper, R.L.; Stoker, T.E.; Tyrey, L.; Goldman, J.M.; McElroy, W.K. Atrazine disrupts the hypothalamic control of pituitary-ovarian function. Toxicol. Sci. 2000, 53, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Mokhtar, M.; Sharifi, E.; Soltani, A. The effects of atrazine on levels of pituitary-testis hormones in adult male rat. Egypt. Acad. J. Biol. Sci. 2010, 2, 53–60. [Google Scholar]
- Foradori, C.D.; Hinds, L.R.; Quihuis, A.M.; Lacagnina, A.F.; Breckenridge, C.B.; Handa, R.J. The differential effect of atrazine on luteinizing hormone release in adrenalectomized adult female wistar rats. Biol. Reprod. 2011, 85, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Cooper, R.L.; Stoker, T.E.; Goldman, J.M.; Parrish, M.B.; Tyrey, L. Effect of atrazine on ovarian function in the rat. Reprod. Toxicol. 1996, 10, 257–264. [Google Scholar] [CrossRef]
- Coban, A.; Filipov, N. Dopaminergic toxicity associated with oral exposure to the herbicide atrazine in juvenile male C57BL/6 mice. J. Neurochem. 2007, 100, 1177–1187. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, V.M.; Limón-Pacheco, J.H.; Mendoza-Trejo, M.S.; González-Gallardo, A.; Hernández-Plata, I.; Giordano, M. Repeated exposure to the herbicide atrazine alters locomotor activity and the nigrostriatal dopaminergic system of the albino rat. Neurotoxicology 2013, 34, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-M.; Shao, J.-Z.; Xiang, L.-X.; Chen, X.-Y. Cytotoxic effects and apoptosis induction of atrazine in a grass carp (Ctenopharyngodon idellus) cell line. Environ. Toxicol. 2006, 21, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, M.; Gao, S.; Ren, R.; Zheng, J.; Zhang, Y. Atrazine-induced apoptosis of splenocytes in BALB/C mice. BMC Med. 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Abarikwu, S.O.; Farombi, E.O.; Kashyap, M.P.; Pant, A.B. Kolaviron protects apoptotic cell death in PC12 cells exposed to Atrazine. Free Radic. Res. 2011, 45, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Bardullas, U.; Giordano, M.; Rodríguez, V.M. Chronic atrazine exposure causes disruption of the spontaneous locomotor activity and alters the striatal dopaminergic system of the male Sprague–Dawley rat. Neurotoxicol. Teratol. 2011, 33, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Filipov, N.M.; Stewart, M.A.; Carr, R.L.; Sistrunk, S.C. Dopaminergic toxicity of the herbicide atrazine in rat striatal slices. Toxicology 2007, 232, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, A.; Dunnett, S.B. Dopamine neuron systems in the brain: An update. Trends Neurosci. 2007, 30, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.M.; Filipov, N.M. Alteration of dopamine uptake into rat striatal vesicles and synaptosomes caused by an in vitro exposure to atrazine and some of its metabolites. Toxicology 2008, 248, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Ashby, J.; Tinwell, H.; Stevens, J.; Pastoor, T.; Breckenridge, C.B. The effects of atrazine on the sexual maturation of female rats. Regul. Toxicol. Pharmacol. 2002, 35, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Hayes, T.B.; Anderson, L.L.; Beasley, V.R.; de Solla, S.R.; Iguchi, T.; Ingraham, H.; Kestemont, P.; Kniewald, J.; Kniewald, Z.; Langlois, V.S.; et al. Demasculinization and feminization of male gonads by atrazine: Consistent effects across vertebrate classes. J. Steroid Biochem. Mol. Biol. 2011, 127, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.R.; Faldladdin, N.; Rand, A.D.; Pelzer, D.; Schrunk, E.M.; Dhanwada, K.R. Atrazine exposure leads to altered growth of HepG2 cells. Toxicol. Vitro 2011, 25, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Dodd, C.A.; Filipov, N.M. Differentiation state-dependent effects of in vitro exposure to atrazine or its metabolite diaminochlorotriazine in a dopaminergic cell line. Life Sci. 2013, 92, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Bardullas, U.; Giordano, M.; Rodríguez, V.M. Atrazine is primarily responsible for the toxicity of long-term exposure to a combination of atrazine and inorganic arsenic in the nigrostriatal system of the albino rat. Neurotoxicol. Teratol. 2013, 40, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Manger, P.; Fau-Genedani, S.; Genedani, S.; Fau-Agnati, L.; Agnati, L. The nigrostriatal DA pathway and Parkinson’s disease. J. Neural. Transm. Suppl. 2006, 70, 71–83. [Google Scholar] [PubMed]
- Lapointe, N.; st-Hilaire, M.; Martinoli, M.G.; Blanchet, J.; Gould, P.; Rouillard, C.; Cicchetti, F. Rotenone induces non-specific central nervous system and systemic toxicity. FASEB J. 2004, 18, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Das, P.C.; McElroy, W.K.; Cooper, R.L. Potential mechanisms responsible for chlorotriazine-induced alterations in catecholamines in pheochromocytoma (PC12) cells. Life Sci. 2003, 73, 3123–3138. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.X.; Fan, X.N.; Wang, H.M.; Wang, G.; Chen, S.D. Glyphosate induced cell death through apoptotic and autophagic mechanisms. Neurotoxicol. Teratol. 2012, 34, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; C, T.; Zhao, F.; Yao, T.; Chen, Y.; Liu, X.; Luo, W.; Chen, J. The role of α-synuclein and tau hyperphosphorylation-mediated autophagy and apoptosis in lead-induced learning and memory injury. Int. J. Biol. Sci. 2012, 8, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-H.; Lee, H.J.; Kang, B.; Yu, K.-N.; Minai-Tehrani, A.; Lee, S.; Kim, S.U.; Cho, M.-H. Methylmercury induces caspase-dependent apoptosis and autophagy in human neural stem cells. J. Toxicol. Sci. 2013, 38, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef] [PubMed]
- Gazzaniga, P.; Gradilone, A.; Vercillo, R.; Gandini, O.; Silvestri, I.; Napolitano, M.; Albonici, L.; Vincenzoni, A.; Gallucci, M.; Frati, L.; et al. Bcl-2/bax mRNA expression ratio as prognostic factor in low-grade urinary bladder cancer. Int. J. Cancer 1996, 69, 100–104. [Google Scholar] [CrossRef]
- Kurz, T.; Eaton, J.W.; Brunk, U.T. Redox activity within the lysosomal compartment: Implications for aging and apoptosis. Antioxid. Redox Signal. 2010, 13, 511–523. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Levine, B. The Beclin 1 interactome. Curr. Opin. Cell Biol. 2010, 22, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Apoptosome: The cellular engine for the activation of caspase-9. Structure 2002, 10, 285–288. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl2 antiapoptotic proteins inhibit beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yang, Y.; Xing, D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2011, 278, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Arsov, I.; Li, X.; Matthews, G.; Coradin, J.; Hartmann, B.; Simon, A.K.; Sealfon, S.C.; Yue, Z. BAC-mediated transgenic expression of fluorescent autophagic protein Beclin 1 reveals a role for Beclin 1 in lymphocyte development. Cell Death Differ. 2008, 15, 1385–1395. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-H.; Xu, L.; Duan, Z.-L.; Zeng, L.-Q.; Yan, N.-H.; Peng, Z.-L. Beclin 1-mediated macroautophagy involves regulation of caspase-9 expression in cervical cancer HeLa cells. Gynecol. Oncol. 2007, 107, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; de Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Furuya, D.; Tsuji, N.; Yagihashi, A.; Watanabe, N. Beclin 1 augmented cis-diamminedichloroplatinum induced apoptosis via enhancing caspase-9 activity. Exp. Cell Res. 2005, 307, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Rubinsztein, D.C. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: An effect rescued by Bcl-xL. Cell Death Differ. 2010, 17, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Breckenridge, C.B.; Charles Eldridge, J.; Stevens, J.T.; Simpkins, J.W. Chapter 79-Symmetrical triazine herbicides: A review of regulatory toxicity endpoints. In Hayes’ Handbook of Pesticide Toxicology, 3rd ed.; Krieger, R., Ed.; Academic Press: New York, NY, USA, 2010; pp. 1711–1723. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, X.-Y.; Li, J.-N.; Wu, Y.-P.; Zhang, B.; Li, B.-X. Atrazine Causes Autophagy- and Apoptosis-Related Neurodegenerative Effects in Dopaminergic Neurons in the Rat Nigrostriatal Dopaminergic System. Int. J. Mol. Sci. 2015, 16, 13490-13506. https://doi.org/10.3390/ijms160613490
Song X-Y, Li J-N, Wu Y-P, Zhang B, Li B-X. Atrazine Causes Autophagy- and Apoptosis-Related Neurodegenerative Effects in Dopaminergic Neurons in the Rat Nigrostriatal Dopaminergic System. International Journal of Molecular Sciences. 2015; 16(6):13490-13506. https://doi.org/10.3390/ijms160613490
Chicago/Turabian StyleSong, Xiao-Yao, Jia-Nan Li, Yan-Ping Wu, Bo Zhang, and Bai-Xiang Li. 2015. "Atrazine Causes Autophagy- and Apoptosis-Related Neurodegenerative Effects in Dopaminergic Neurons in the Rat Nigrostriatal Dopaminergic System" International Journal of Molecular Sciences 16, no. 6: 13490-13506. https://doi.org/10.3390/ijms160613490