
Regioselective Alcoholysis of Silychristin Acetates Catalyzed by Lipases †

 ,
, 
Abstract
:
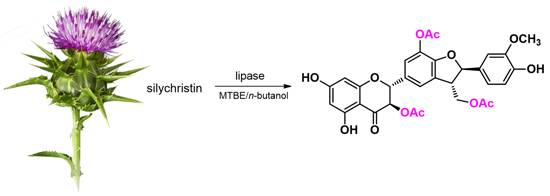
1. Introduction
2. Results and Discussion
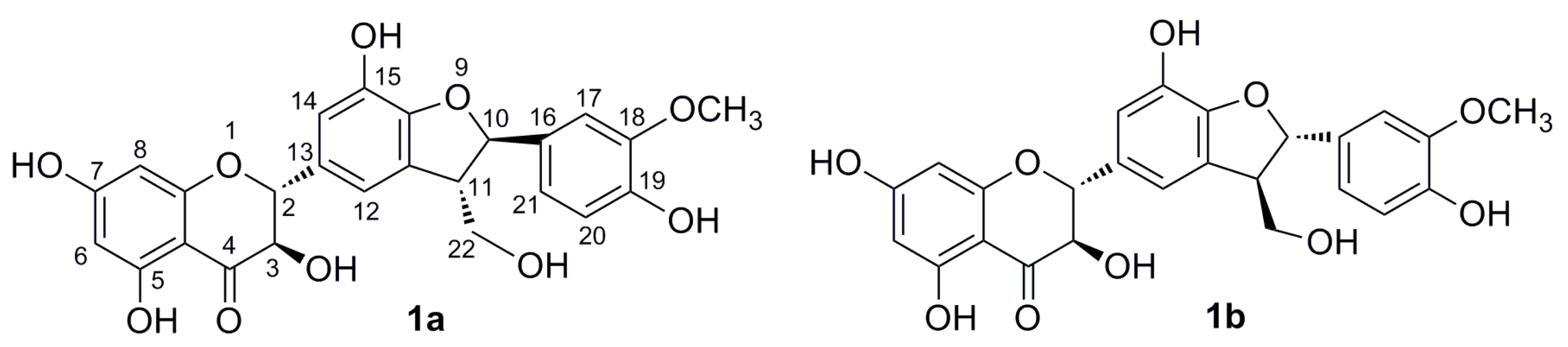
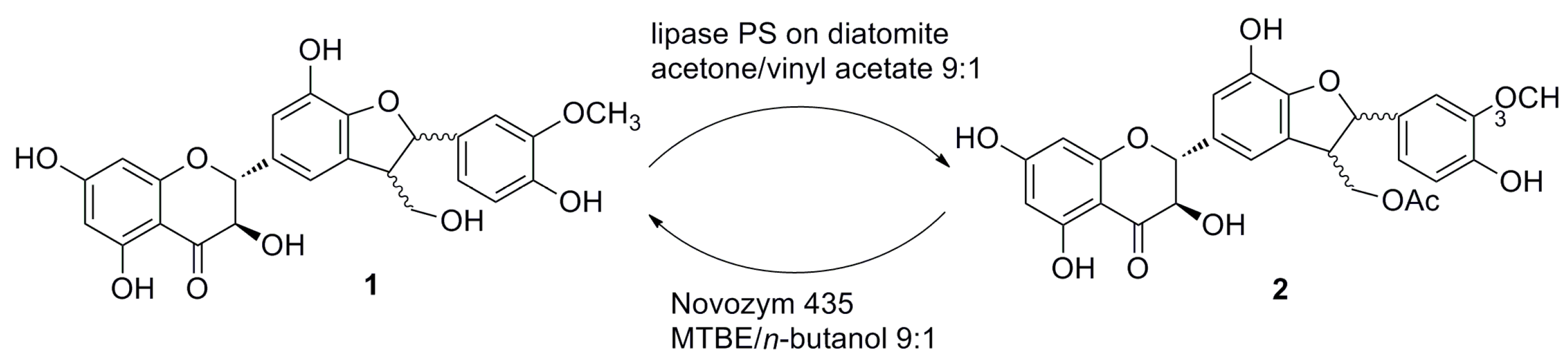
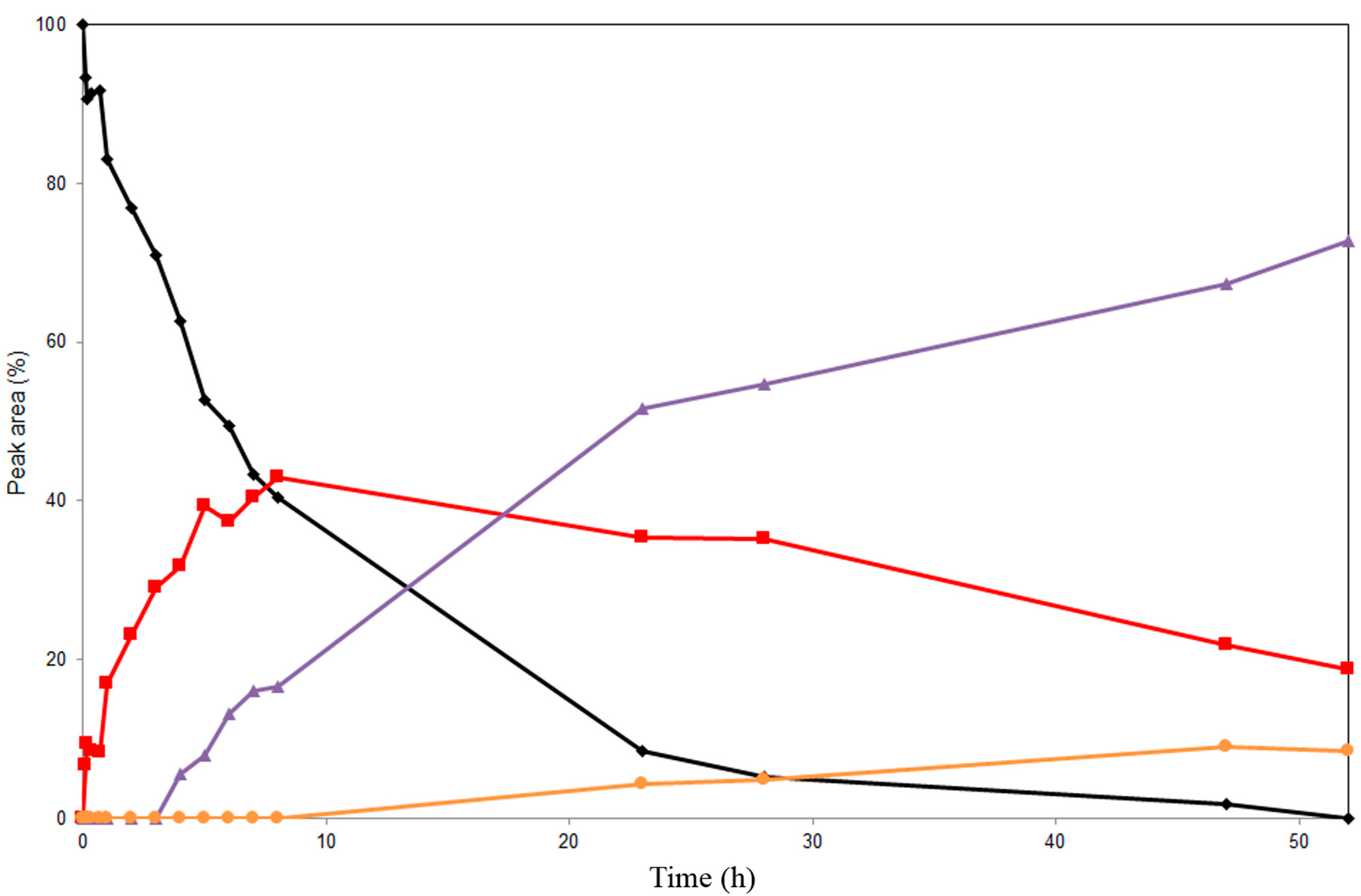
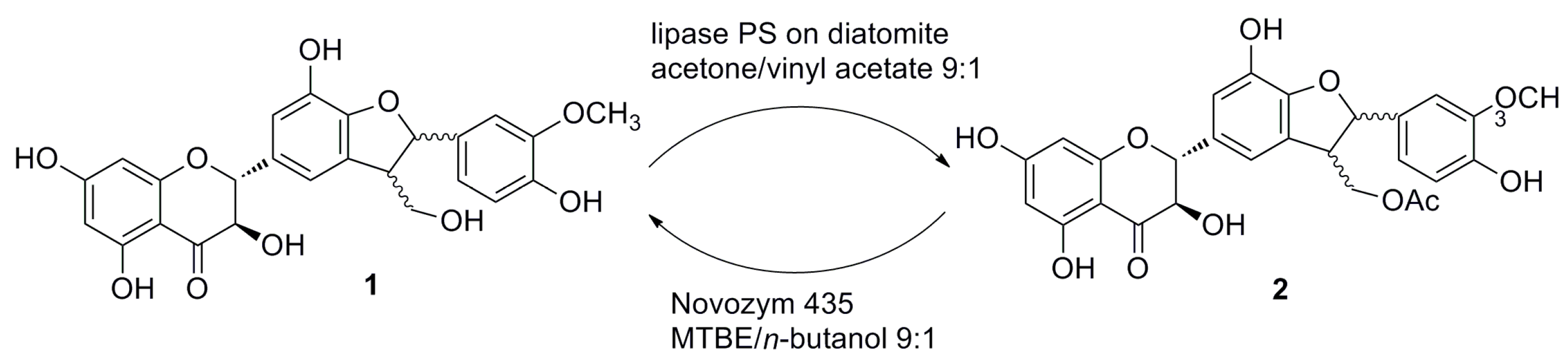
2.1. Enzymatic Acetylation of Silychristin (1)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Source | Provider | Acetylation a | Alcoholysis of 22-Acetate (2) b | Alcoholysis of Peracetate (3) c |
|---|---|---|---|---|---|
| Lipase AK | Pseudomonas sp. | Amano | NR d | NR | NR |
| Lipase PS immobilized on celite | Pseudomonas cepacia | Amano | ++ | NR | ++ |
| Lipase PS immobilized on diatomite | Pseudomonas cepacia | Sigma | ++ | NR | ++ |
| PPL | porcine pancreas | Sigma | NR | NR | NR |
| Novozym 435 | Candida antarctica (Lip. B) | Novozymes | + | ++ | ++ |
| Lipase G | Penicillium camemberti | Amano | NR | NR | NR |
| Lipase CE | Humicola lanuginosa | Amano | NR | NR | + |
| Lipase A | Aspergillus niger | Amano | NR | NR | + |
| Lipase F-AP15 | Rhizopus oryzae | Amano | NR | NR | NR |
| Lipase CV | Chromobacterium viscosum | Amano | NR | NR | NR |
| Lipase CR | Candida rugosa | Amano | NR | NR | + |
| Lipozyme | Rhizomucor miehei | Amano | + | + | + |
| Lipolase | Thermomyces lanuginosa | Amano | + | + | + |

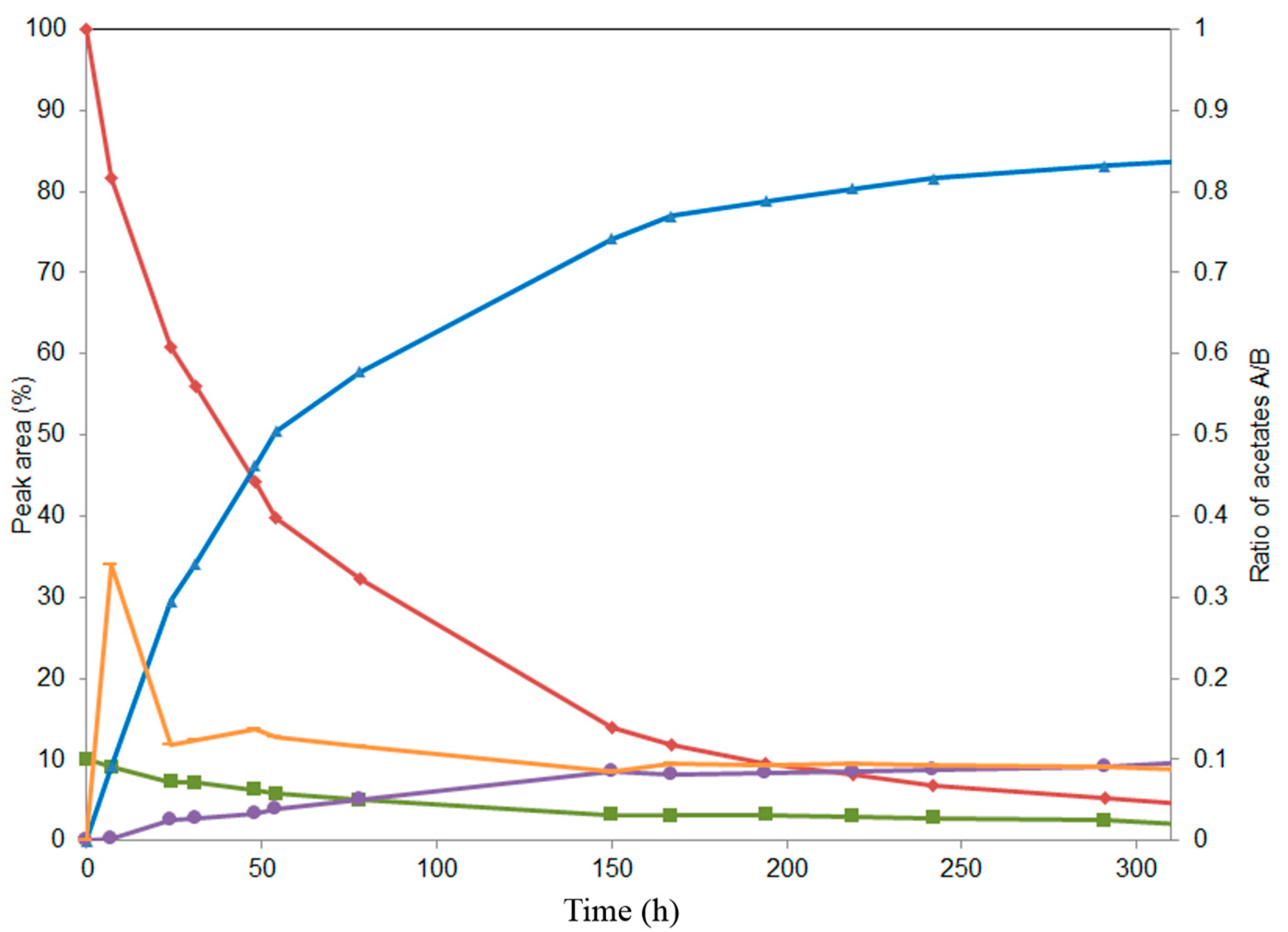
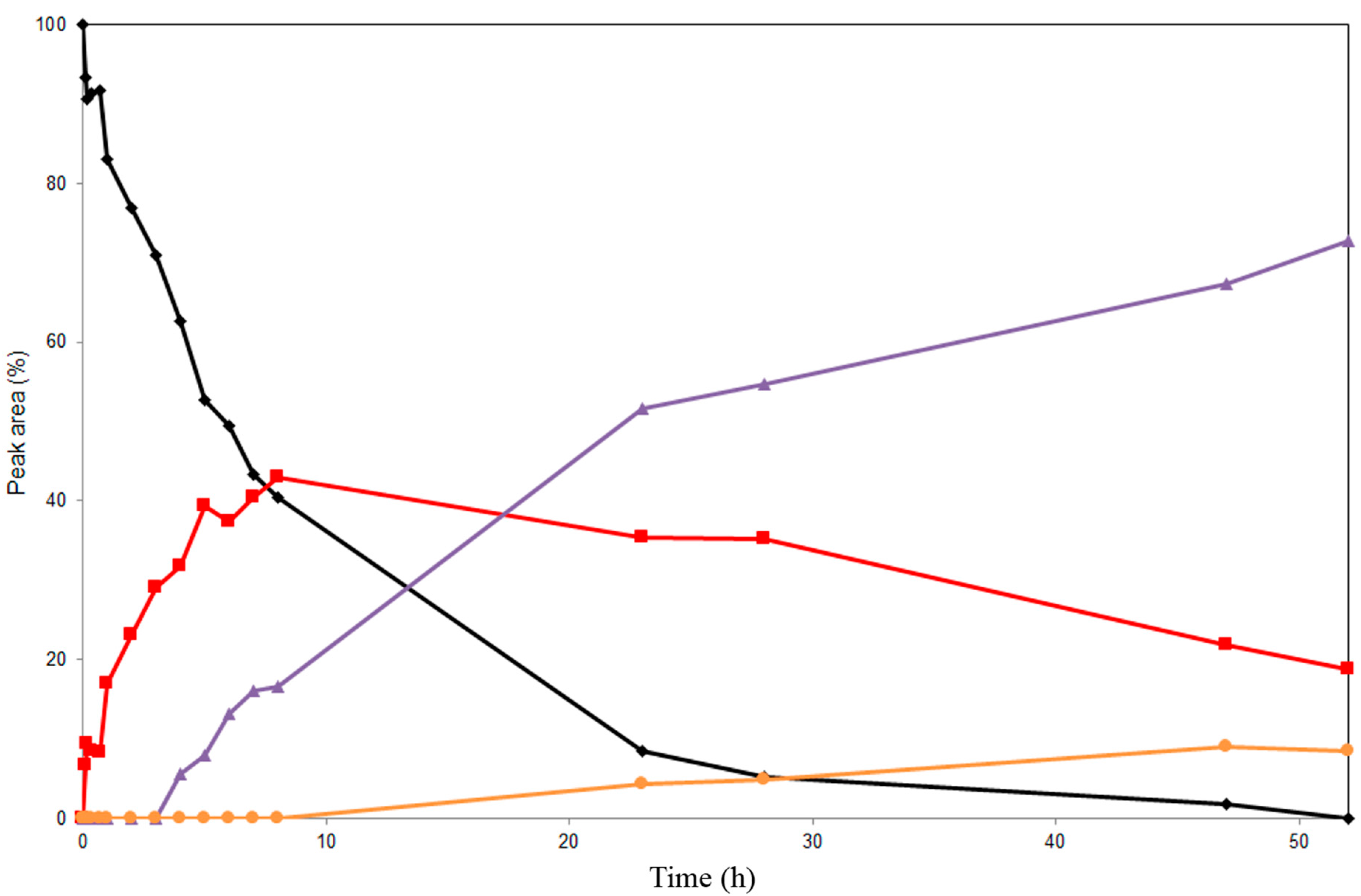
2.2. Alcoholysis of 22-O-Acetyl Silychristin (2)
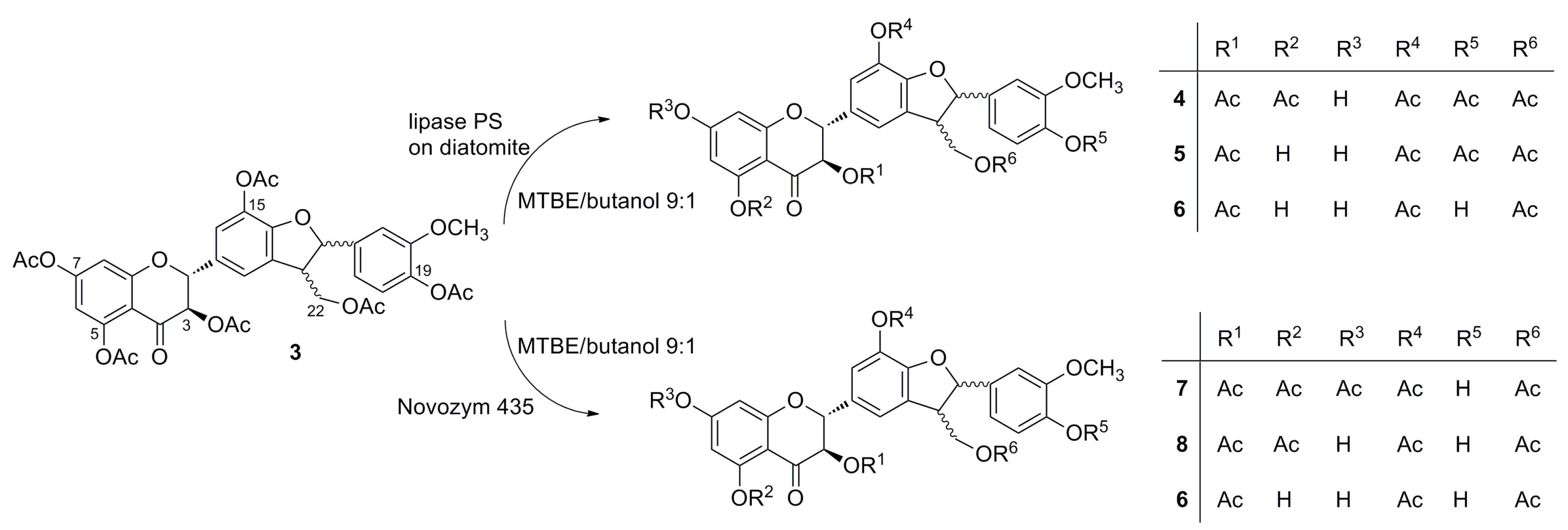
2.3. Screening of Lipases for the Selective Alcoholysis of 3,5,7,15,19,22-Hexa-O-acetyl silychristin (3)
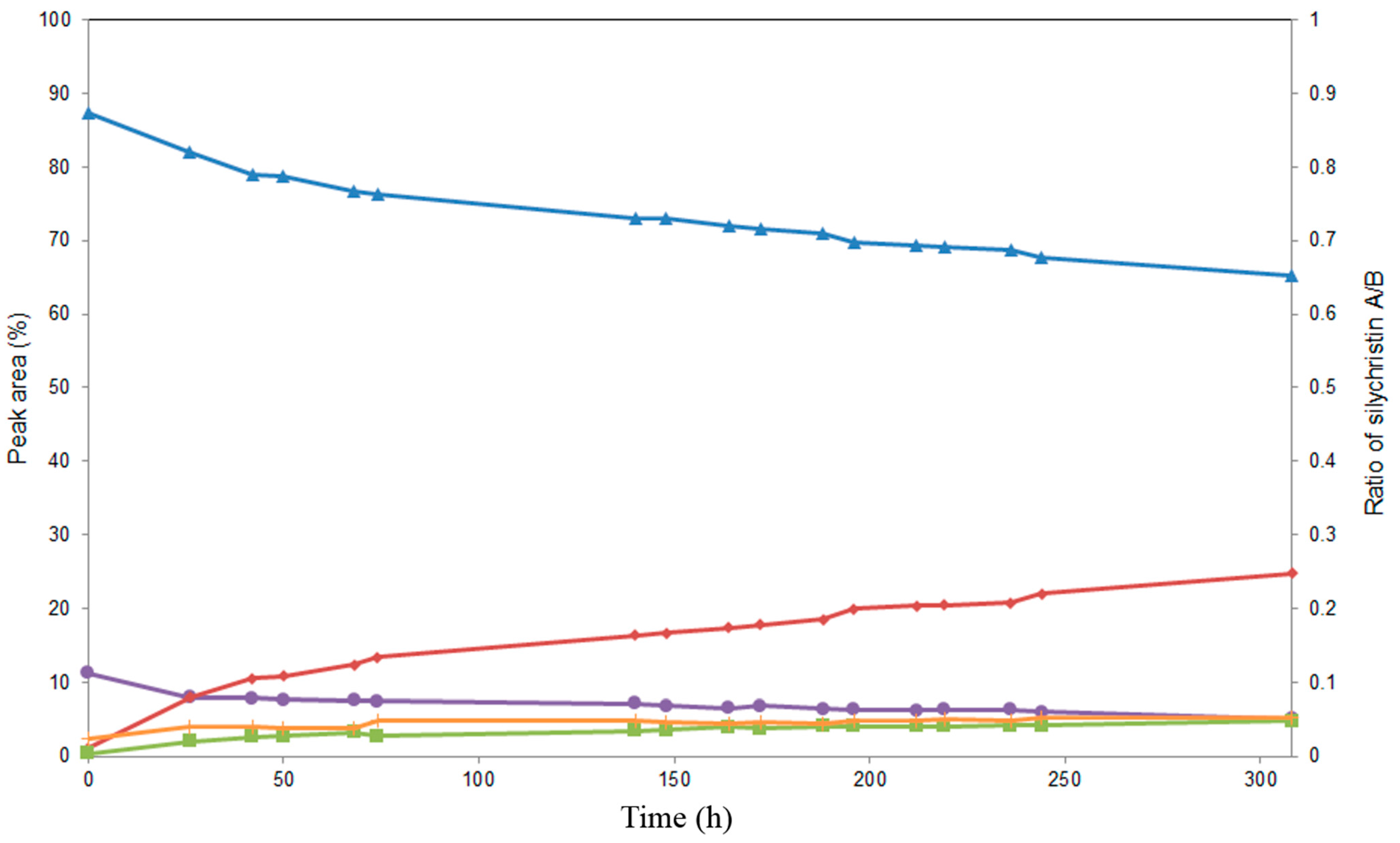
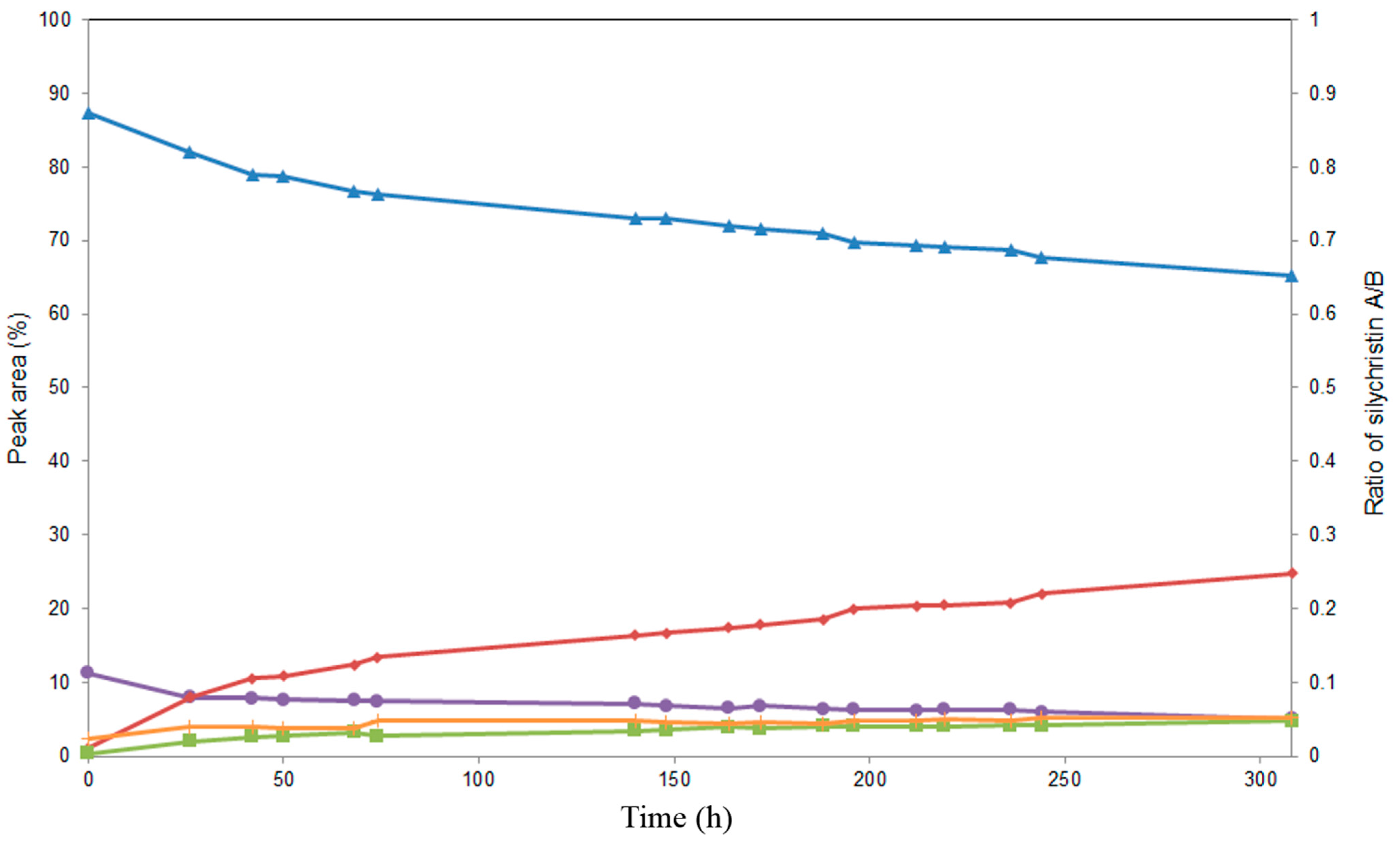
2.4. Alcoholysis of 3,5,7,15,19,22-Hexa-O-acetyl silychristin (3)
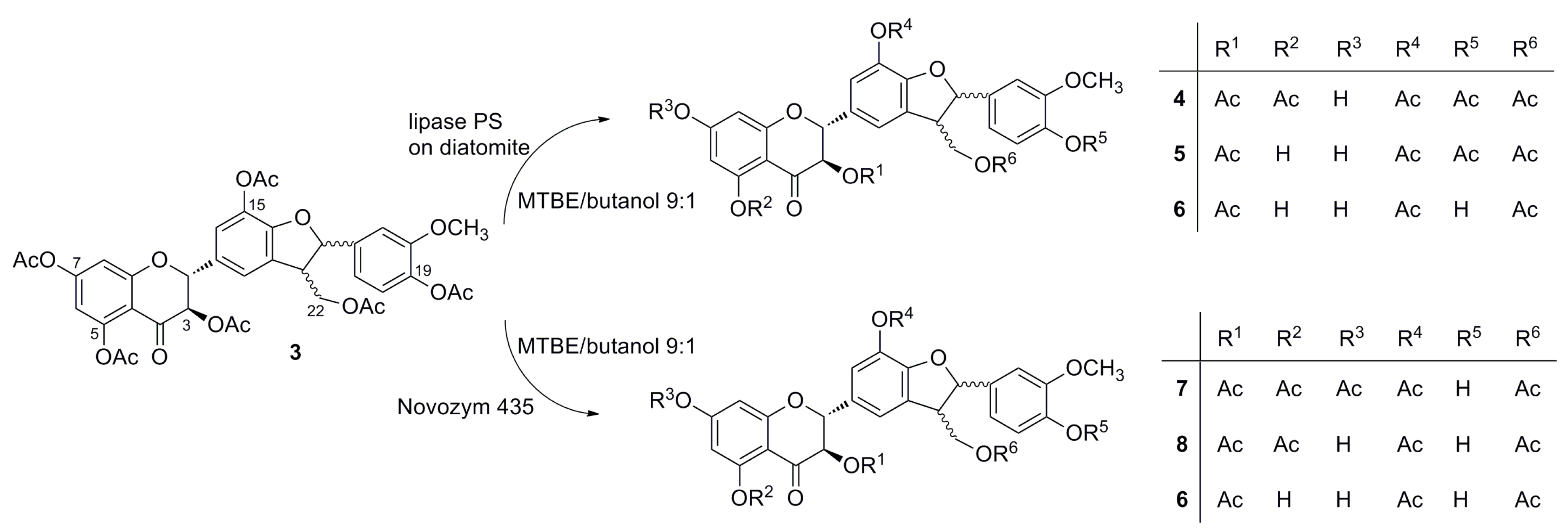

3. Experimental Section
3.1. Chemicals and Reagents
3.2. General Methods
3.3. HPLC
3.4. Chemistry
3.4.1. Screening of Enzymatic Acetylation of Silychristin
3.4.2. Enzymatic Synthesis of 22-O-Acetyl-silychristin (2)
3.4.3. Enzymatic Alcoholysis of 22-O-Acetyl-silychristin (2)
3.4.4. Preparation of 3,5,7,15,19,22-Hexa-O-acetyl-silychristin (3)
3.4.5. Screening of Enzymatic Alcoholysis of 3,5,7,15,19,22-Hexa-O-acetyl-silychristin (3)
3.4.6. Preparative Alcoholysis of 3,5,7,15,19,22-Hexa-O-acetyl silychristin (3) Catalyzed by Lipase PS on Diatomite
3.4.7. Preparation of 3,5,15,19,22-Penta-O-acetyl-silychristin (4) Catalyzed by Lipase PS on Diatomite
3.4.8. Preparative Alcoholysis of 3,5,7,15,19,22-Hexa-O-acetyl silychristin (3) catalyzed by Novozym 435
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gažák, R.; Walterová, D.; Křen, V. Silybin and silymarin—New and emerging applications in medicine. Curr. Med. Chem. 2007, 14, 315–338. [Google Scholar] [PubMed]
- Biedermann, D.; Vavříková, E.; Cvak, L.; Křen, V. Chemistry of silybin. Nat. Prod. Rep. 2014, 31, 1138–1157. [Google Scholar] [CrossRef] [PubMed]
- Křenek, K.; Marhol, P.; Peikerová, Z.; Křen, V.; Biedermann, D. Preparatory separation of the silymarin flavonolignans by Sephadex LH-20 gel. Food Res. Int. 2014, 65, 115–120. [Google Scholar] [CrossRef]
- Wagner, H.; Seligmann, O.; Seitz, M.; Abraham, D.; Sonnenbichler, J. Silydianin and silychristin, two isomeric silymarins from Silybum marianum L. Gaertn. (milk thistle). Z. Naturforsch B 1976, 31, 876–884. (In German) [Google Scholar] [CrossRef]
- Pelter, A.; Hansel, R.; Kaloga, M. The structure of silychristin. Tetrahedron Lett. 1977, 4547–4548. [Google Scholar] [CrossRef]
- Smith, W.A.; Lauren, D.R.; Burgess, E.J.; Perry, N.B.; Martin, R.J. A silychristin isomer and variation of flavonolignan levels in milk thistle (Silybum marianum) fruits. Planta Med. 2005, 71, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Šeršeň, F.; Vencel, T.; Annus, J. Silymarin and its components scavenge phenylglyoxylic ketyl radicals. Fitoterapia 2006, 77, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.L.; Li, D.N.; Qiao, J.Q.; Lian, H.Z.; Wang, S.K. Determination of silymarin flavonoids by HPLC and LC-MS and investigation of extraction rate of silymarin in Silybum marianum fruits by boiling water. Asian J. Chem. 2009, 21, 63–74. [Google Scholar]
- Chebil, L.; Humeau, C.; Falcimaigne, A.; Engasser, J.M.; Ghoul, M. Enzymatic acylation of flavonoids. Process Biochem. 2006, 41, 2237–2251. [Google Scholar] [CrossRef]
- Gavezzotti, P.; Vavříková, E.; Valentová, K.; Fronza, G.; Kudanga, T.; Kuzma, M.; Riva, S.; Biedermann, D.; Křen, V. Enzymatic oxidative dimerization of silymarin flavonolignans. J. Mol. Catal. B 2014, 109, 24–30. [Google Scholar] [CrossRef]
- Vavříková, E.; Vacek, J.; Valentová, K.; Marhol, P.; Ulrichová, J.; Kuzma, M.; Křen, V. Chemo-enzymatic synthesis of silybin and 2,3-dehydrosilybin dimers. Molecules 2014, 19, 4115–4134. [Google Scholar] [CrossRef] [PubMed]
- Monti, D.; Gažák, R.; Marhol, P.; Biedermann, D.; Purchartová, K.; Fedrigo, M.; Riva, S.; Křen, V. Enzymatic kinetic resolution of silybin diastereoisomers. J. Nat. Prod. 2010, 73, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Gažák, R.; Marhol, P.; Purchartová, K.; Monti, D.; Biedermann, D.; Riva, S.; Cvak, L.; Křen, V. Large-scale separation of silybin diastereoisomers using lipases. Process Biochem. 2010, 45, 1657–1663. [Google Scholar] [CrossRef]
- Purchartová, K.; Marhol, P.; Gažák, R.; Monti, D.; Riva, S.; Kuzma, M.; Křen, V. Regioselective alcoholysis of silybin A and B acetates with lipases. J. Mol. Catal. B 2011, 71, 119–123. [Google Scholar] [CrossRef]
- Lambusta, D.; Nicolosi, G.; Patti, A.; Sanfilippo, C. Application of lipase catalysis in organic solvents for selective protection-deprotection of bioactive compounds. J. Mol. Catal. B 2003, 22, 271–277. [Google Scholar] [CrossRef]
- D’Antona, N.; Lambusta, D.; Nicolosi, G.; Bovicelli, P. Preparation of regioprotected morins by lipase-catalysed transesterification. J. Mol. Catal. B 2008, 52, 78–81. [Google Scholar] [CrossRef]
- Natoli, M.; Nicolosi, G.; Piattelli, M. Regioselective alcoholysis of flavonoid acetates with lipase in an organic solvent. J. Org. Chem. 1992, 57, 5776–5778. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vavříková, E.; Gavezzotti, P.; Purchartová, K.; Fuksová, K.; Biedermann, D.; Kuzma, M.; Riva, S.; Křen, V. Regioselective Alcoholysis of Silychristin Acetates Catalyzed by Lipases. Int. J. Mol. Sci. 2015, 16, 11983-11995. https://doi.org/10.3390/ijms160611983
Vavříková E, Gavezzotti P, Purchartová K, Fuksová K, Biedermann D, Kuzma M, Riva S, Křen V. Regioselective Alcoholysis of Silychristin Acetates Catalyzed by Lipases. International Journal of Molecular Sciences. 2015; 16(6):11983-11995. https://doi.org/10.3390/ijms160611983
Chicago/Turabian StyleVavříková, Eva, Paolo Gavezzotti, Kateřina Purchartová, Kateřina Fuksová, David Biedermann, Marek Kuzma, Sergio Riva, and Vladimír Křen. 2015. "Regioselective Alcoholysis of Silychristin Acetates Catalyzed by Lipases" International Journal of Molecular Sciences 16, no. 6: 11983-11995. https://doi.org/10.3390/ijms160611983