
Serum Amyloid A Receptor Blockade and Incorporation into High-Density Lipoprotein Modulates Its Pro-Inflammatory and Pro-Thrombotic Activities on Vascular Endothelial Cells




Abstract
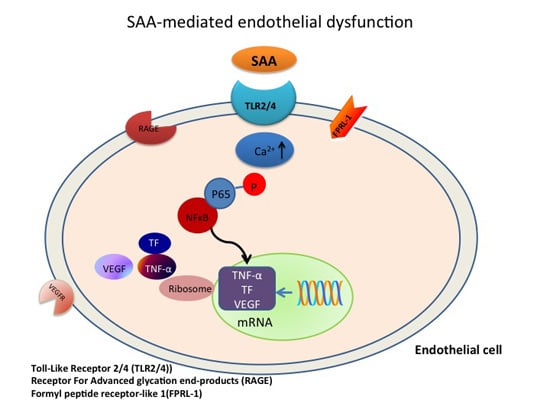
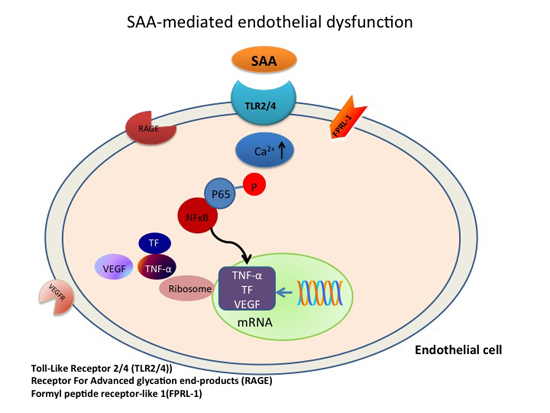
:
1. Introduction
2. Results
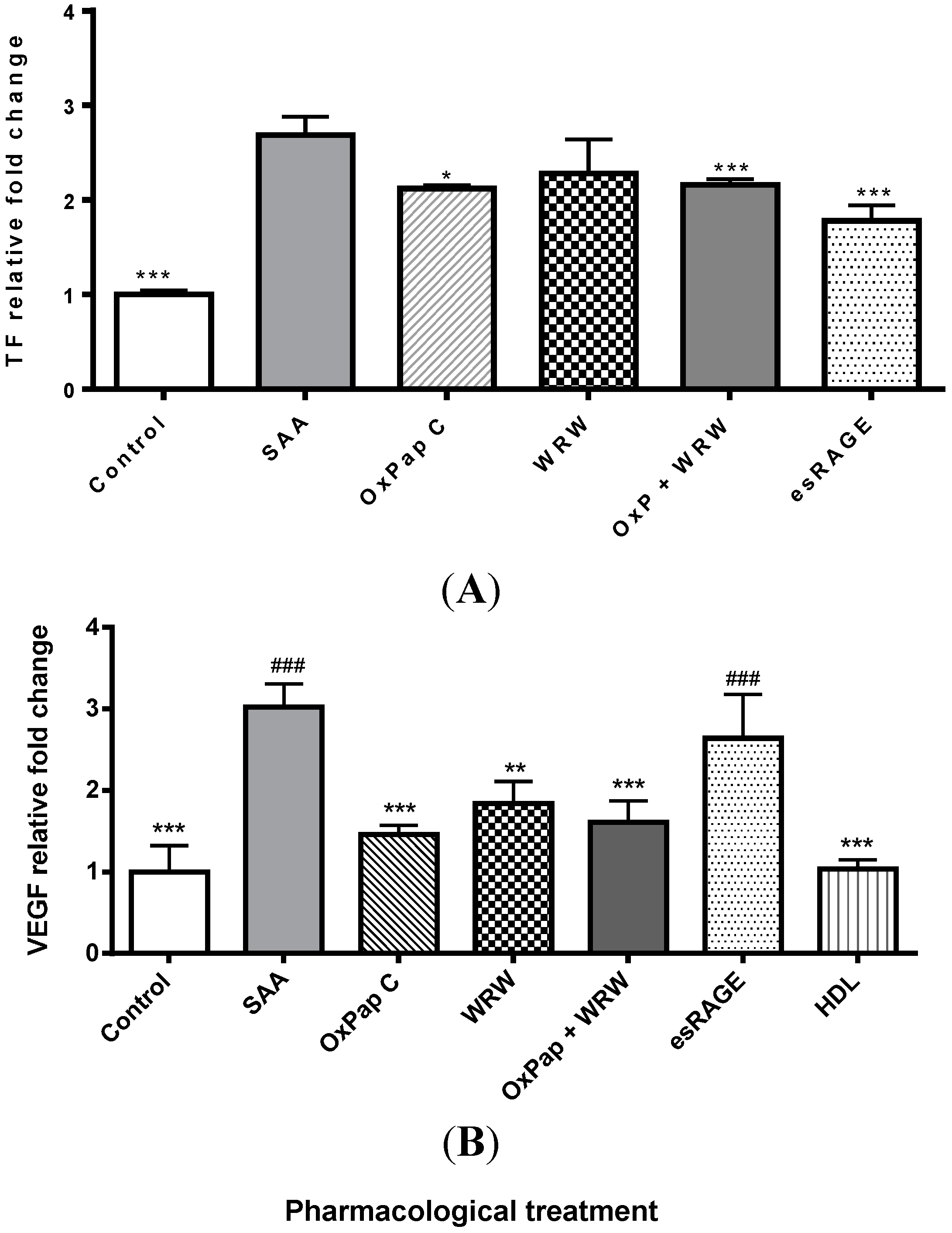
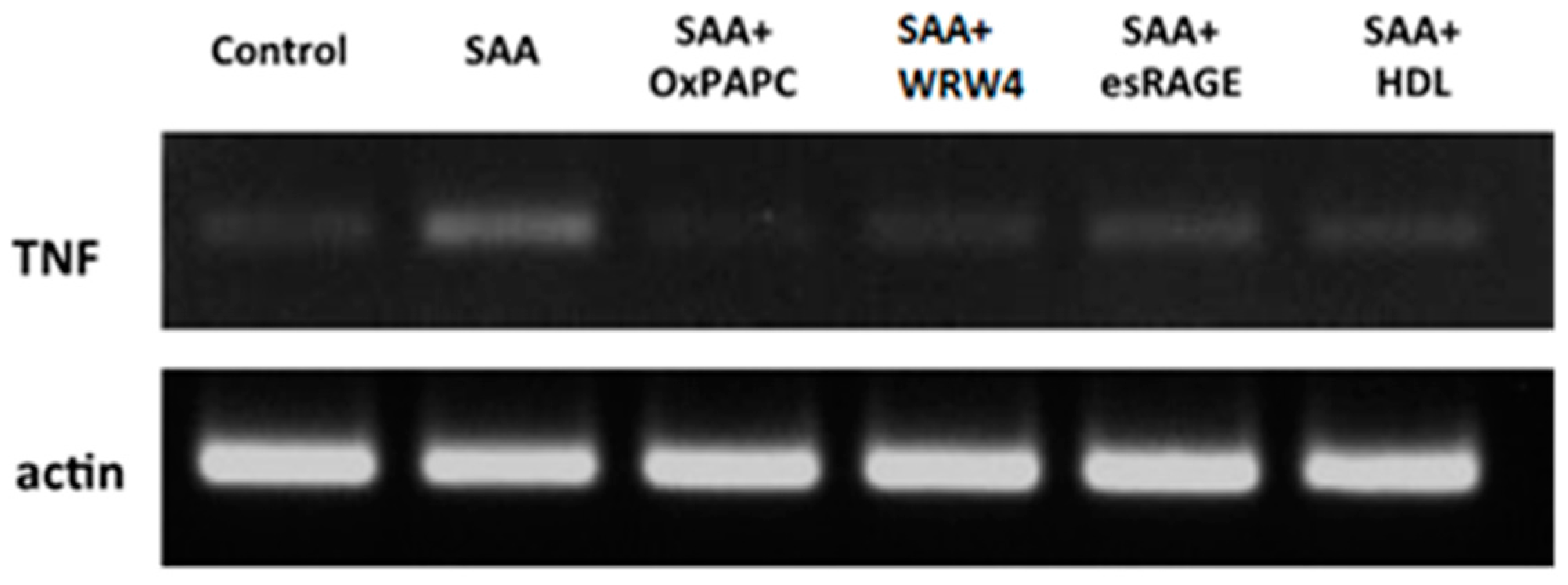
2.1. SAA Receptor Inhibitor/Antagonists and HDL Suppress SAA-Induced Pro-Atherogenic Gene Expression in Endothelial Cells

{kind=link}
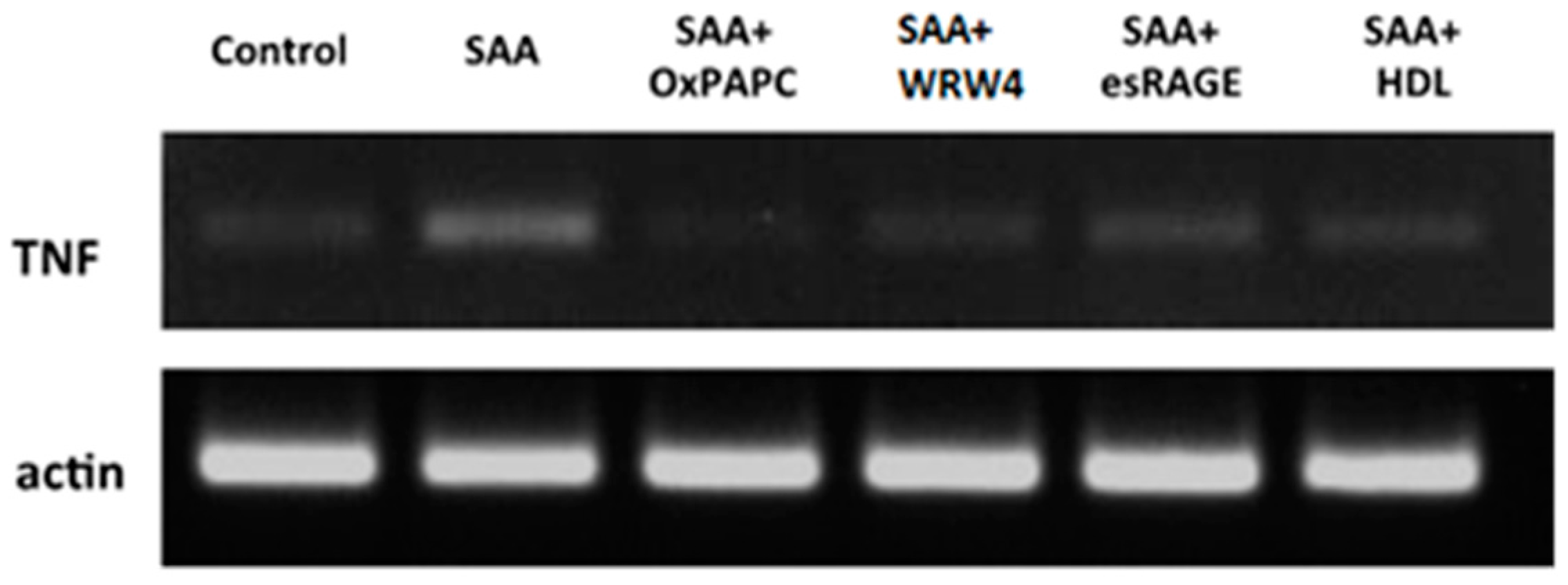{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | ||||||||
|---|---|---|---|---|---|---|---|---|
| Gene | Control | SAA | OxPap C (25 μg/mL) | OxPap C (45 μg/mL) | WRW4 | OxPap C + WRW4 | esRAGE | HDL |
| TF | 1 ± 0.02 | 4.4 ± 0.6 * | 3.4 ± 0.3 *,# | 3.2 ± 0.1 *,# | 3.9 ± 0.1 * | 3.3 ± 0.5 *,# | 3.1 ± 0.2 *,# | 0.9 ± 0.4 # |
| NFκB | 1 ± 0.02 | 3.4 ± 0.3 * | 2.2 ± 0.3 *,# | 2.0 ± 0.3 *,# | 2.4 ± 0.4 *,# | 1.9 ± 0.2 *,# | 3.3 ± 0.2 * | 1.4 ± 0.4 # |
| TNF-α | 1 ± 0.17 | 6.8 ± 0.6 * | 2.7 ± 0.4 *,# | 2.6 ± 0.9 *,# | 3.7 ± 0.7 *,# | 2.3 ± 0.3 *,# | 5.5 ± 1.2 * | 1.7 ± 0.3 # |
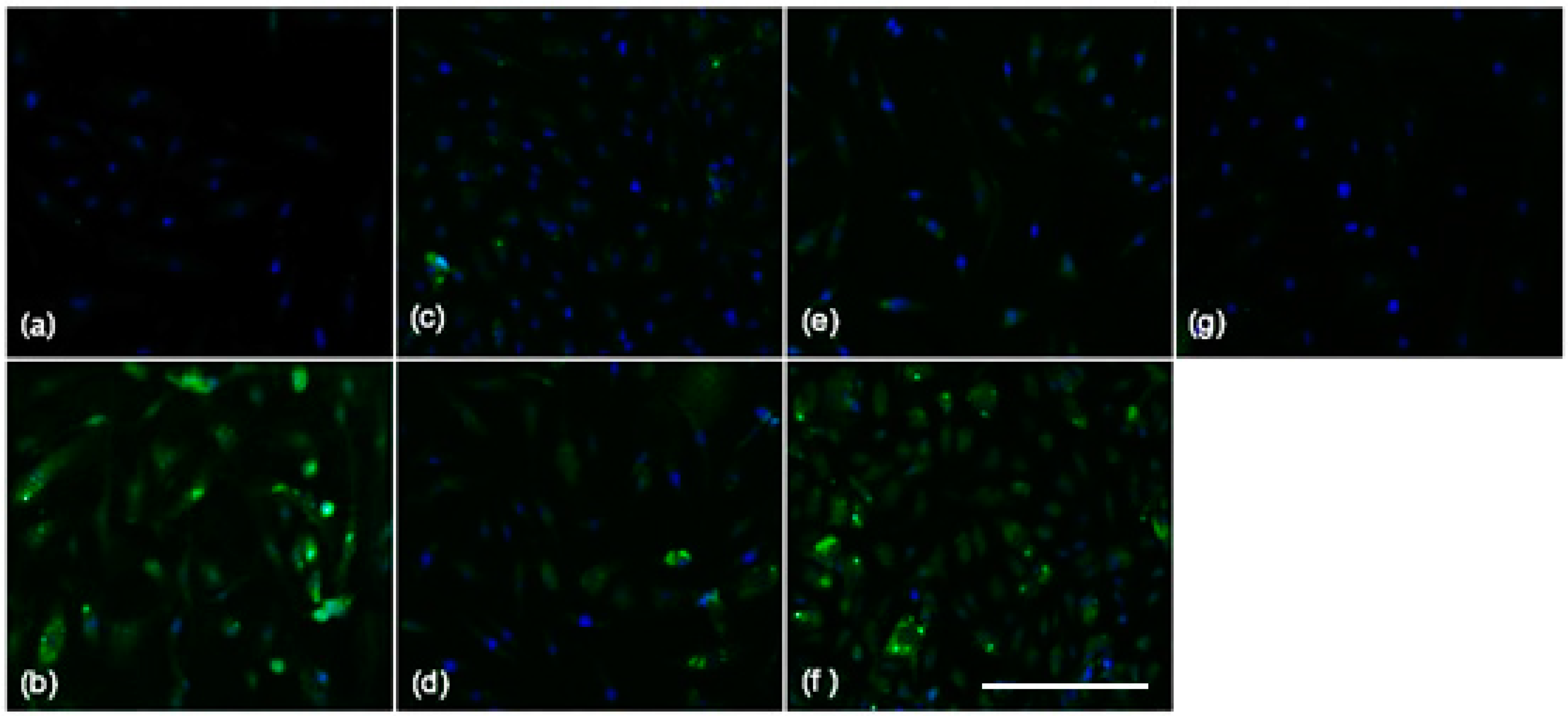
2.2. HDL Is a Chief Suppressor of SAA-Induced Pro-Atherogenic Protein Expression
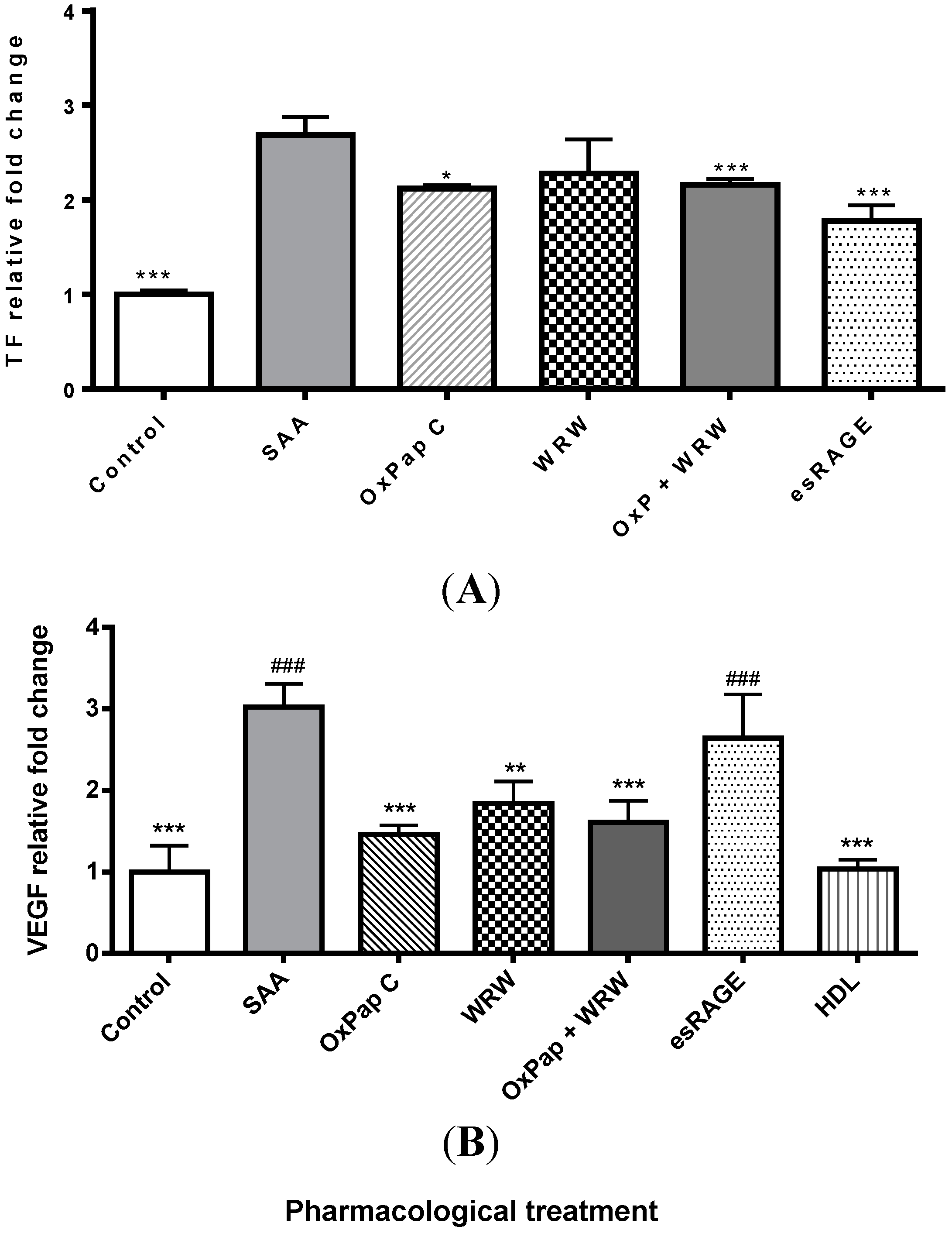
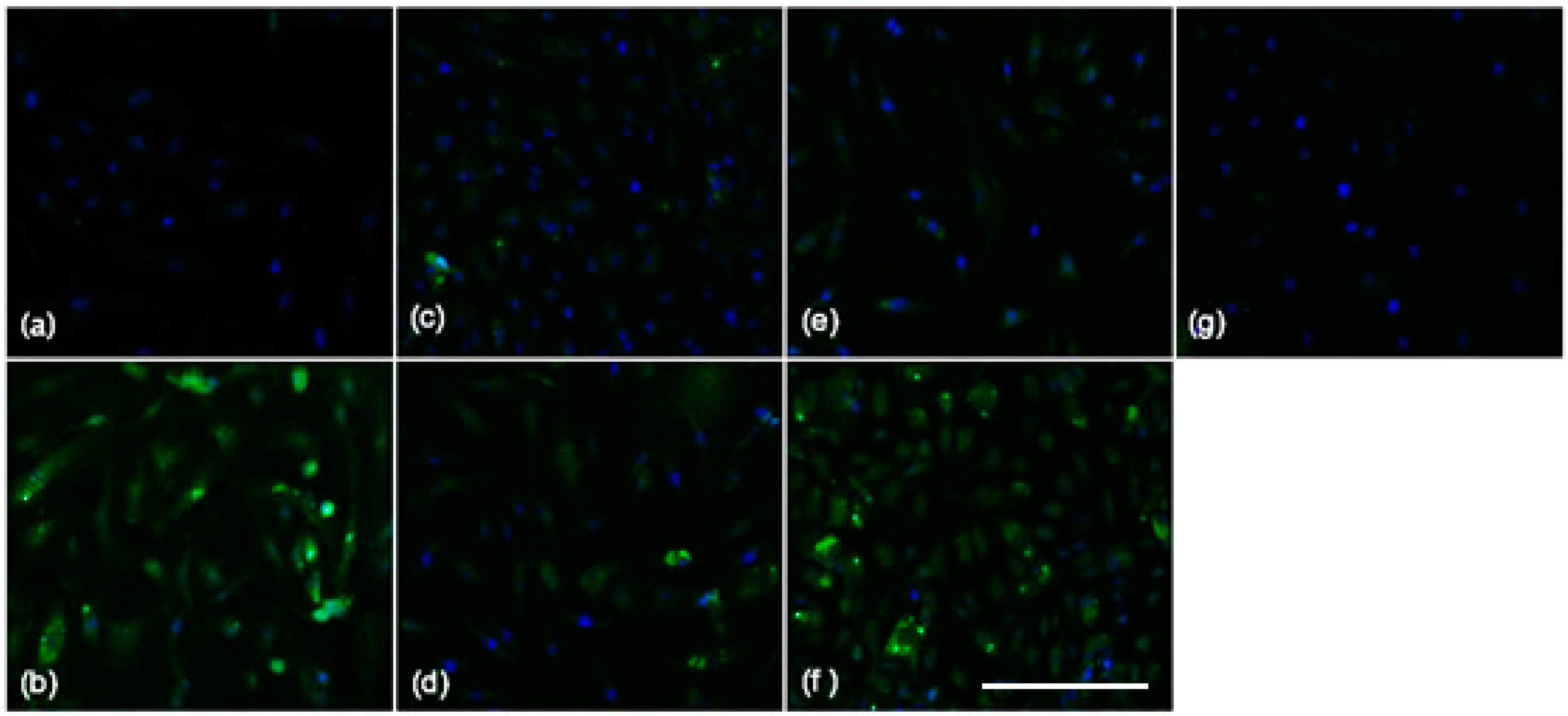
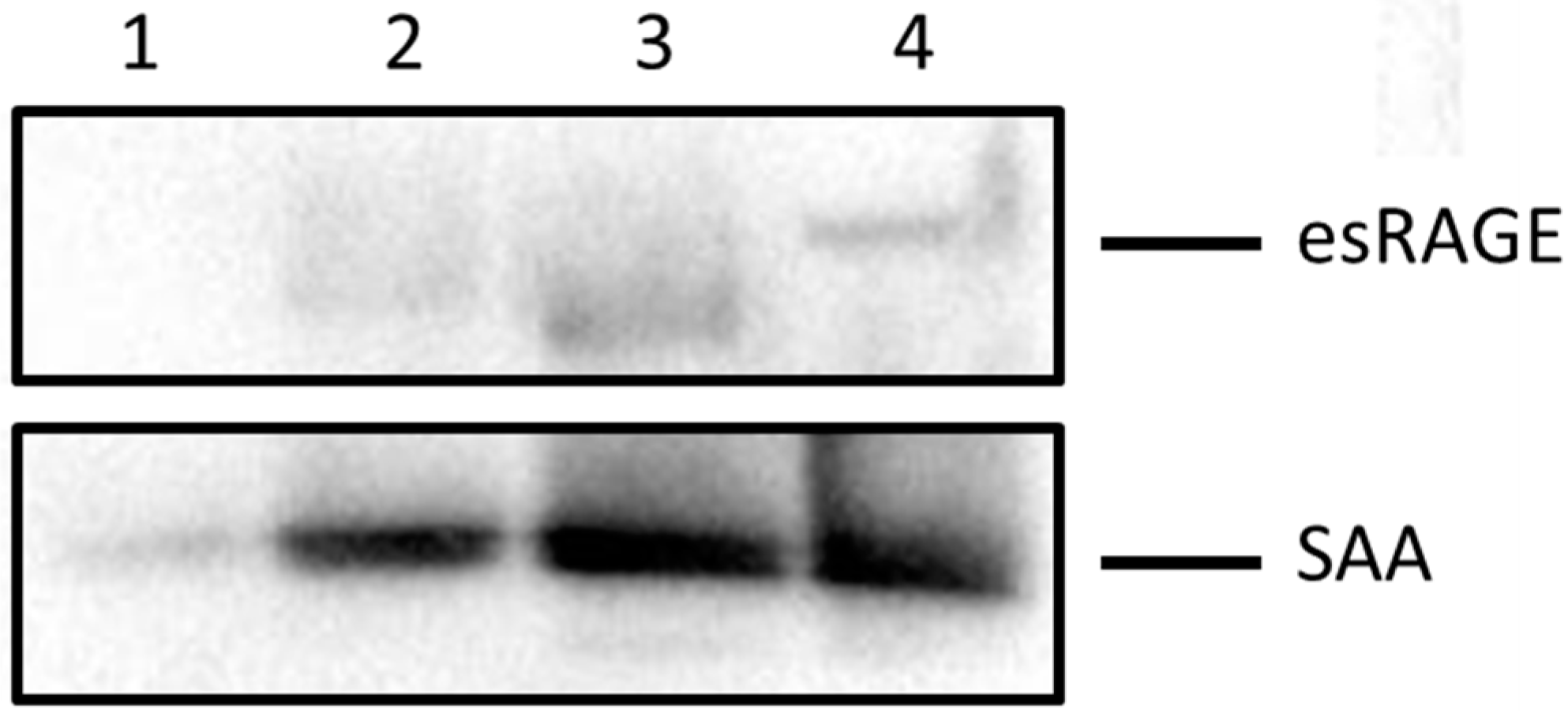
2.3. Blocking SAA Activation of RAGE Largely Fails to Inhibit SAA Activities on HCtAEC
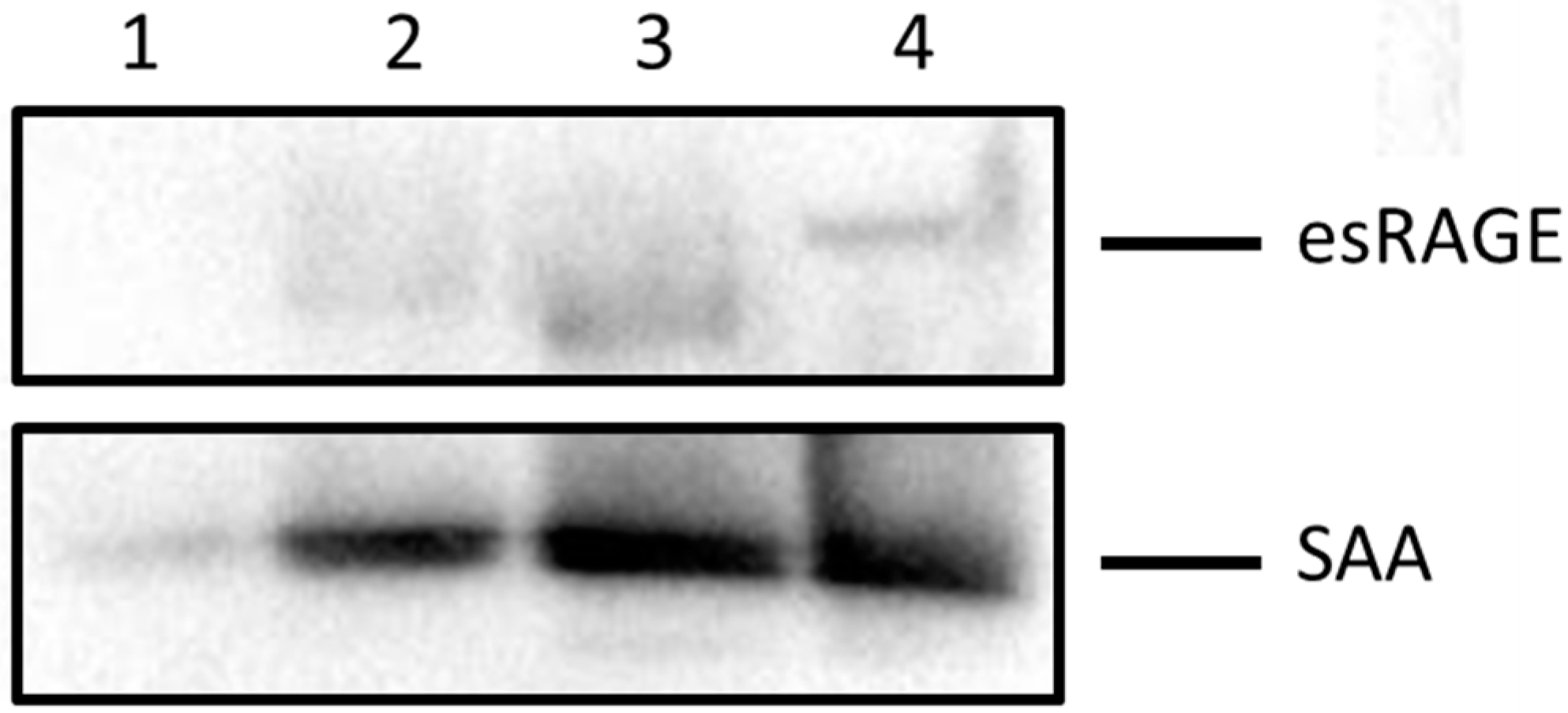
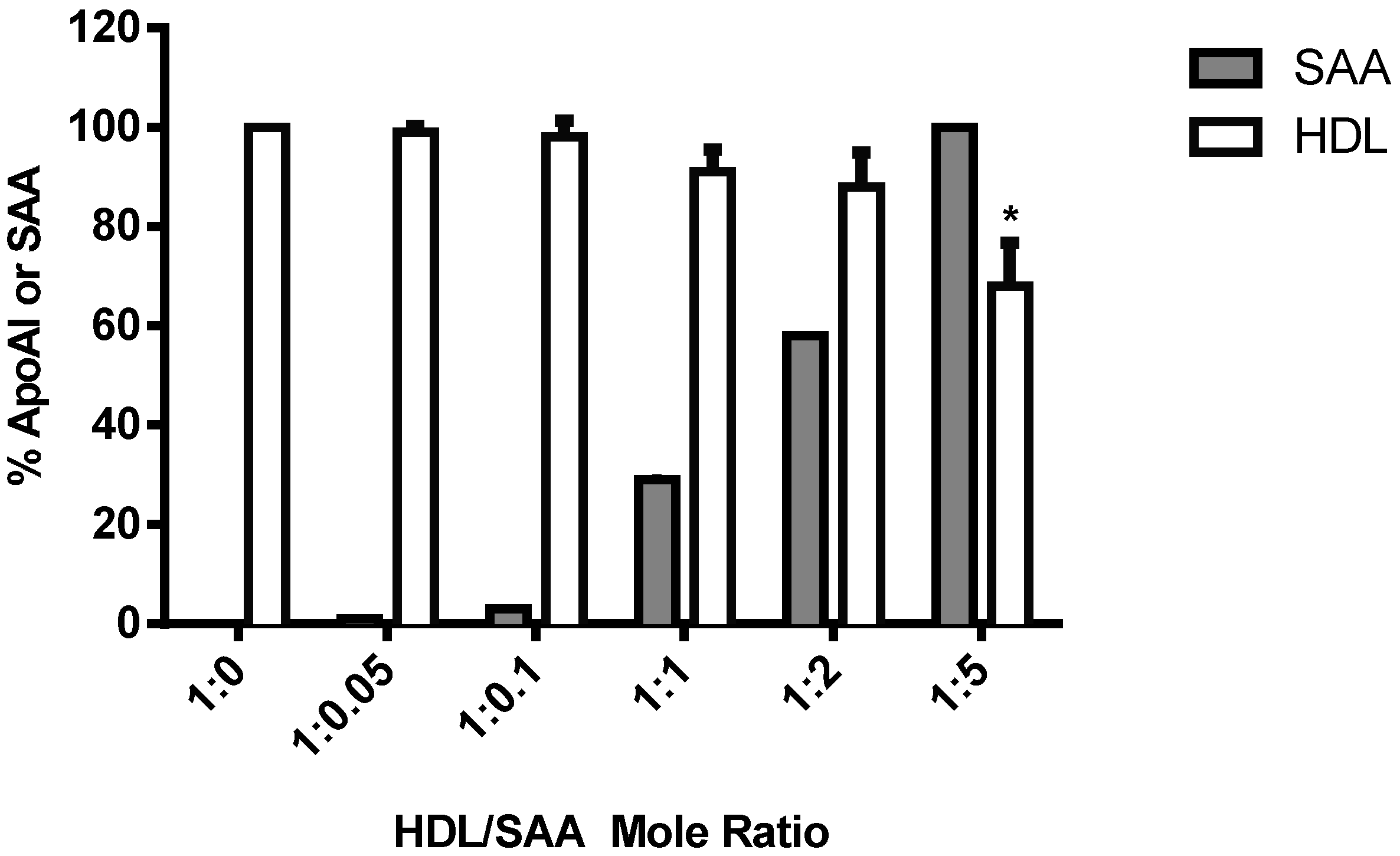
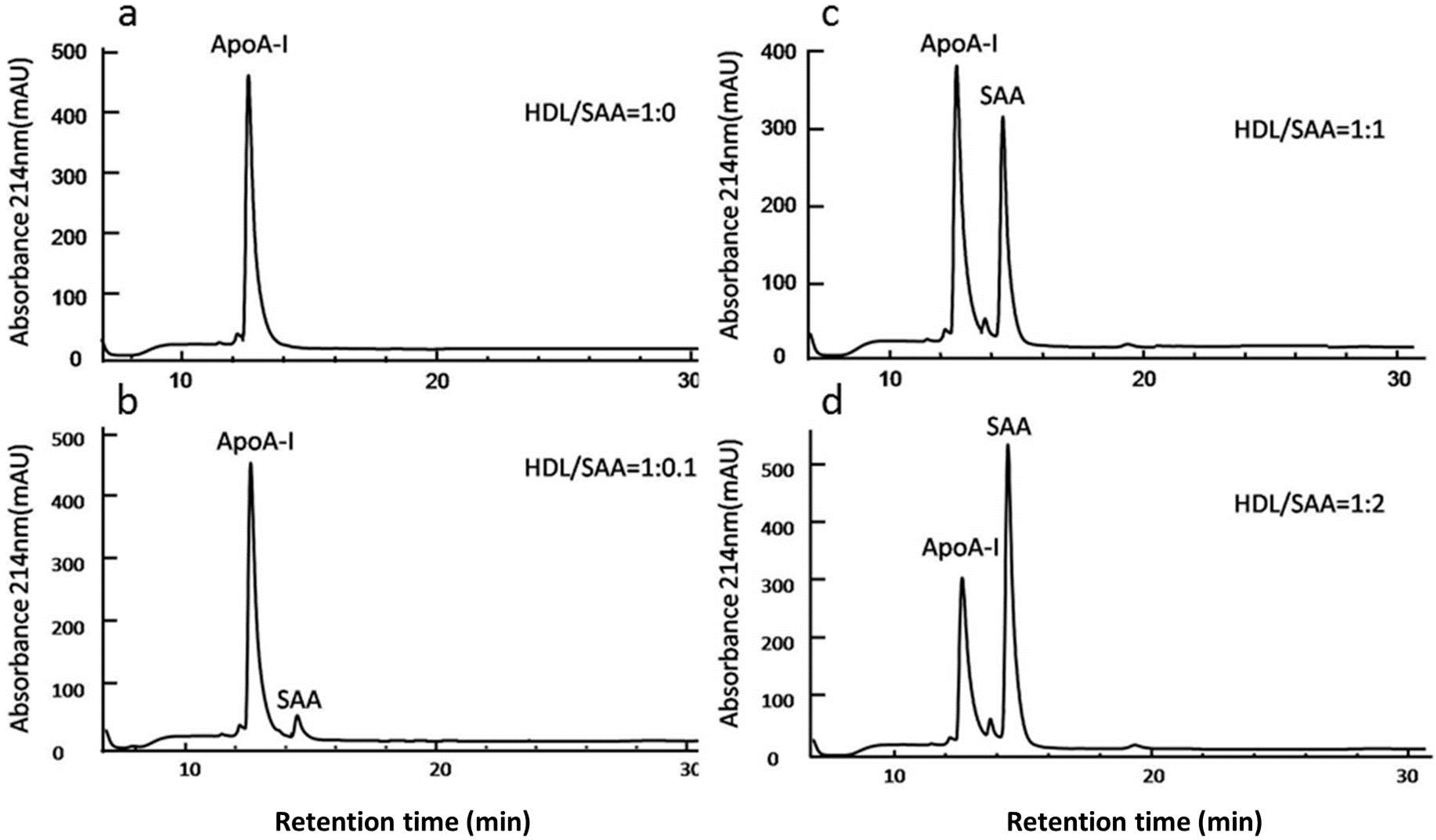
2.4. SAA Displaces Apolipoprotein A-I (ApoA-I) in HDL in a Dose-Dependent Manner

3. Discussion
4. Methods
4.1. Materials
4.2. Preparation of the Soluble Antagonist Targeting RAGE (esRAGE)
4.3. Cell Culture
4.4. Isolation of Native HDL
4.5. Measurement of Cellular Protein Content
4.6. Treatment of HCtAEC with SAA, HDL and Specific Receptor Inhibitors
4.7. Synthesis of Cloned DNA
4.8. Gene Analysis
| cDNA | Forward (5' → 3') | Reverse (5' → 3') |
|---|---|---|
| β-Actin | GGACTTCGAGCAAGA | AGCACTGTGTTGGCG |
| TF | GTGACCTCACCGACGAGATT | CCGAGGTTTGTCTCCAGGTA |
| TNF | CAGAGGGCCTGTACCTCATC | GGAAGACCCCTCCCAGATAG |
| NFκB | CTGGAAGCACGAATGACAGA | TGAGGTCCATCTCCTTGGTC |
4.9. Direct ELISA Quantification of Tissue Factor and Vascular Endothelial Growth Factor
4.10. Immunocytochemistry
4.11. Protein Complex Immunoprecipitation (Co-IP)
4.12. SDS-PAGE and Western Blotting
4.13. Analysis of HDL Apolipoproteins ApoAI and SAA with Liquid Chromatography
4.14. Statistics
5. Conclusions
Acknowledgments
Author Contributions
Abbreviations
| SAA | Serum Amyloid A |
| EC | Endothelial cells |
| CVD | Cardiovascular disease |
| esRAGE | Endogenous secretory RAGE |
| TF | Tissue factor |
| HCtAEC | Human carotid artery endothelial cells |
| FPRL-1 | Formyl peptide receptor-like-1 |
| TLR2/4 | Toll-like receptors-2/4 |
| HDL | High-density lipoprotein |
| NO | Nitric oxide |
| TNF | Tumour necrosis factor |
| TLRs | Toll-like receptors |
| RAGE | Receptor for advanced glycation end products |
| ApoA-I | Apolipoprotein A-I |
| NFκB | Nuclear factor κ B |
| HUVEC | Human umbilical vein endothelial |
| Egr-1 | Epidermal growth response-1 |
| AP-1 | Activator protein-1 |
| VEGF | Vascular endothelial growth factor |
| HBSS | Hanks’ balanced salt solution |
| DAPI | 4',6-diamidino-2-phenylindole |
| Co-IP | Co-Immunoprecipitation |
| PVDF | Polyvinylidene difluoride nitrocellulose |
| FITC | Fluorescein isothiocyanate |
Conflicts of Interest
References
- Mashima, R.; Witting, P.K.; Stocker, R. Oxidants and antioxidants in atherosclerosis. Curr. Opin. Lipidol. 2001, 12, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Ludmer, P.L.; Selwyn, A.P.; Shook, T.L.; Wayne, R.R.; Mudge, G.H.; Alexander, R.W.; Ganz, P. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N. Engl. J. Med. 1986, 315, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.R.; Witting, P.K.; Drummond, G.R. Redox control of endothelial function and dysfunction: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2008, 10, 1713–1765. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Raines, E.W.; Plump, A.S.; Breslow, J.L.; Ross, R. Upregulation of VCAM-1 and ICAM-1 at atherosclerosis-prone sites on the endothelium in the ApoE-deficient mouse. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; de Nigris, F.; Williams-Ignarro, S.; Pignalosa, O.; Sica, V.; Ignarro, L.J. Nitric oxide and atherosclerosis: An update. Nitric Oxide 2006, 15, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Favero, G.; Paganelli, C.; Buffoli, B.; Rodella, L.F.; Rezzani, R. Endothelium and its alterations in cardiovascular diseases: Life style intervention. Biomed. Res. Int. 2014, 2014, 801896. [Google Scholar] [CrossRef] [PubMed]
- Khan, B.V.; Harrison, D.G.; Olbrych, M.T.; Alexander, R.W.; Medford, R.M. Nitric oxide regulates vascular cell adhesion molecule 1 gene expression and redox-sensitive transcriptional events in human vascular endothelial cells. Proc. Natl. Acad. Sci. USA 1996, 93, 9114–9119. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Keaney, J.F., Jr. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef] [PubMed]
- Mackay, F.; Loetscher, H.; Stueber, D.; Gehr, G.; Lesslauer, W. Tumor necrosis factor alpha (TNF-alpha)-induced cell adhesion to human endothelial cells is under dominant control of one TNF receptor type, TNF-R55. J. Exp. Med. 1993, 177, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Nawroth, P.P.; Stern, D.M. Modulation of endothelial cell hemostatic properties by tumor necrosis factor. J. Exp. Med. 1986, 163, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Mechtcheriakova, D.; Schabbauer, G.; Lucerna, M.; Clauss, M.; de Martin, R.; Binder, B.R.; Hofer, E. Specificity, diversity, and convergence in VEGF and TNF-alpha signaling events leading to tissue factor up-regulation via EGR-1 in endothelial cells. FASEB J. 2001, 15, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Liuzzo, G.; Biasucci, L.M.; Gallimore, J.R.; Grillo, R.L.; Rebuzzi, A.G.; Pepys, M.B.; Maseri, A. The prognostic value of C-reactive protein and serum amyloid a protein in severe unstable angina. N. Engl. J. Med. 1994, 331, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Hennekens, C.H.; Buring, J.E.; Rifai, N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N. Engl. J. Med. 2000, 342, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Edfeldt, K. Toll to be paid at the gateway to the vessel wall. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1085–1087. [Google Scholar] [CrossRef] [PubMed]
- Willerson, J.T.; Ridker, P.M. Inflammation as a cardiovascular risk factor. Circulation 2004, 109, II2–II10. [Google Scholar] [CrossRef] [PubMed]
- Maier, W.; Altwegg, L.A.; Corti, R.; Gay, S.; Hersberger, M.; Maly, F.E.; Sutsch, G.; Roffi, M.; Neidhart, M.; Eberli, F.R.; et al. Inflammatory markers at the site of ruptured plaque in acute myocardial infarction: Locally increased interleukin-6 and serum amyloid A but decreased C-reactive protein. Circulation 2005, 111, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Badolato, R.; Wang, J.M.; Murphy, W.J.; Lloyd, A.R.; Michiel, D.F.; Bausserman, L.L.; Kelvin, D.J.; Oppenheim, J.J. Serum amyloid A is a chemoattractant: Induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J. Exp. Med. 1994, 180, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Mullan, R.H.; Bresnihan, B.; Golden-Mason, L.; Markham, T.; O’Hara, R.; FitzGerald, O.; Veale, D.J.; Fearon, U. Acute-phase serum amyloid A stimulation of angiogenesis, leukocyte recruitment, and matrix degradation in rheumatoid arthritis through an NF-kappaB-dependent signal transduction pathway. Arthritis Rheum. 2006, 54, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Song, C.; Endoh, I.; Goyette, J.; Jessup, W.; Freedman, S.B.; McNeil, H.P.; Geczy, C.L. Serum amyloid A induces monocyte tissue factor. J. Immunol. 2007, 178, 1852–1860. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Wu, T.; Qin, W.; An, C.; Wang, Z.; Zhang, M.; Zhang, Y.; Zhang, C.; An, F. Serum amyloid A directly accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Mol. Med. 2011, 17, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dong, Z.; Liu, H.; Xia, Y.F.; Liu, X.M.; Luo, B.B.; Wang, W.K.; Li, B.; Gao, F.; Zhang, C.; et al. Serum amyloid A stimulates lipoprotein-associated phospholipase A2 expression in vitro and in vivo. Atherosclerosis 2013, 228, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Shen, Y.; Yamen, E.; Hsu, K.; Yan, W.; Witting, P.K.; Geczy, C.L.; Freedman, S.B. Serum amyloid A may potentiate prothrombotic and proinflammatory events in acute coronary syndromes. Atherosclerosis 2009, 202, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Hsu, K.; Yamen, E.; Yan, W.; Fock, J.; Witting, P.K.; Geczy, C.L.; Freedman, S.B. Serum amyloid A induction of cytokines in monocytes/macrophages and lymphocytes. Atherosclerosis 2009, 207, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Witting, P.K.; Song, C.; Hsu, K.; Hua, S.; Parry, S.N.; Aran, R.; Geczy, C.; Freedman, S.B. The acute-phase protein serum amyloid A induces endothelial dysfunction that is inhibited by high-density lipoprotein. Free Radic. Biol. Med. 2011, 51, 1390–1398. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chai, H.; Wang, Z.; Lin, P.H.; Yao, Q.; Chen, C. Serum amyloid A induces endothelial dysfunction in porcine coronary arteries and human coronary artery endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2399–H2408. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Kim, M.K.; Park, K.S.; Bae, Y.H.; Yun, J.; Park, J.I.; Kwak, J.Y.; Bae, Y.S. Serum amyloid A stimulates matrix-metalloproteinase-9 upregulation via formyl peptide receptor like-1-mediated signaling in human monocytic cells. Biochem. Biophys. Res. Commun. 2005, 330, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; He, R.; Tian, J.; Ye, P.P.; Ye, R.D. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J. Immunol. 2008, 181, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Sandri, S.; Rodriguez, D.; Gomes, E.; Monteiro, H.P.; Russo, M.; Campa, A. Is serum amyloid A an endogenous TLR4 agonist? J. Leukoc. Biol. 2008, 83, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Zhu, H.; Zhu, A.; Golabek, A.; Du, H.; Roher, A.; Yu, J.; Soto, C.; Schmidt, A.M.; Stern, D.; et al. Receptor-dependent cell stress and amyloid accumulation in systemic amyloidosis. Nat. Med. 2000, 6, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Nicholls, S.; Rye, K.A.; Anantharamaiah, G.M.; Navab, M.; Fogelman, A.M. Antiinflammatory properties of HDL. Circ. Res. 2004, 95, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.M.; Goyette, J.; Tedla, N.; Hsu, K.; Geczy, C.L. S100A12 suppresses pro-inflammatory, but not pro-thrombotic functions of serum amyloid A. PLoS ONE 2013, 8, e62372. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; Song, C.; Geczy, C.L.; Freedman, S.B.; Witting, P.K. A role for acute-phase serum amyloid A and high-density lipoprotein in oxidative stress, endothelial dysfunction and atherosclerosis. Redox. Rep. 2009, 14, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Ashby, D.; Gamble, J.; Vadas, M.; Fidge, N.; Siggins, S.; Rye, K.; Barter, P.J. Lack of effect of serum amyloid A (SAA) on the ability of high-density lipoproteins to inhibit endothelial cell adhesion molecule expression. Atherosclerosis 2001, 154, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Chang, M.Y.; Wang, S.; Wight, T.N.; McMillen, T.S.; Oram, J.F.; Vaisar, T.; Heinecke, J.W.; de Beer, F.C.; de Beer, M.C.; et al. Serum amyloid A facilitates the binding of high-density lipoprotein from mice injected with lipopolysaccharide to vascular proteoglycans. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1326–1332. [Google Scholar] [CrossRef] [PubMed]
- Tolle, M.; Huang, T.; Schuchardt, M.; Jankowski, V.; Prufer, N.; Jankowski, J.; Tietge, U.J.; Zidek, W.; van der Giet, M. High-density lipoprotein loses its anti-inflammatory capacity by accumulation of pro-inflammatory-serum amyloid A. Cardiovasc. Res. 2012, 94, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Puranik, R.; Bao, S.; Nobecourt, E.; Nicholls, S.J.; Dusting, G.J.; Barter, P.J.; Celermajer, D.S.; Rye, K.A. Low dose apolipoprotein A-I rescues carotid arteries from inflammation in vivo. Atherosclerosis 2008, 196, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Ohishi, M.; Takeya, Y.; Onishi, M.; Ito, N.; Oguro, R.; Yamamoto, K.; Kamide, K.; Rakugi, H. Carotid plaque score and intima media thickness as predictors of stroke and mortality in hypertensive patients. Hypertens. Res. 2013, 36, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Plichart, M.; Celermajer, D.S.; Zureik, M.; Helmer, C.; Jouven, X.; Ritchie, K.; Tzourio, C.; Ducimetiere, P.; Empana, J.P. Carotid intima-media thickness in plaque-free site, carotid plaques and coronary heart disease risk prediction in older adults. The Three-City Study. Atherosclerosis 2011, 219, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Freedman, S.B.; Witting, P.K. Serum amyloid A stimulates cultured endothelial cells to migrate and proliferate: Inhibition by the multikinase inhibitor BIBF1120. Clin. Exp. Pharmacol. Physiol. 2013, 40, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.; Gotto, A.M.; LaRosa, J.C.; Maroni, J.; Szarek, M.; Grundy, S.M.; Kastelein, J.J.; Bittner, V.; Fruchart, J.C. Treating to New Targets I. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N. Engl. J. Med. 2007, 357, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Sang, H.; Ye, R.D. Serum amyloid A induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R. Blood 2003, 101, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhou, S.; Heng, C.K. Impact of serum amyloid A on tissue factor and tissue factor pathway inhibitor expression and activity in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1645–1650. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Prince, A. Activation of Ca2+-dependent signaling by TLR2. J. Immunol. 2006, 177, 1330–1337. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.S.; Wang, J.M.; Murphy, P.M.; Gao, J.L. Serum amyloid A is a chemotactic agonist at FPR2, a low-affinity N-formylpeptide receptor on mouse neutrophils. Biochem. Biophys. Res. Commun. 2000, 270, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Riddell, J.R.; Maier, P.; Sass, S.N.; Moser, M.T.; Foster, B.A.; Gollnick, S.O. Peroxiredoxin 1 stimulates endothelial cell expression of VEGF via TLR4 dependent activation of HIF-1alpha. PLoS ONE 2012, 7, e50394. [Google Scholar] [CrossRef] [PubMed]
- Foncea, R.; Carvajal, C.; Almarza, C.; Leighton, F. Endothelial cell oxidative stress and signal transduction. Biol. Res. 2000, 33, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Fiuza, C.; Bustin, M.; Talwar, S.; Tropea, M.; Gerstenberger, E.; Shelhamer, J.H.; Suffredini, A.F. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood 2003, 101, 2652–2660. [Google Scholar] [CrossRef] [PubMed]
- Lakota, K.; Mrak-Poljsak, K.; Bozic, B.; Tomsic, M.; Sodin-Semrl, S. Serum amyloid A activation of human coronary artery endothelial cells exhibits a neutrophil promoting molecular profile. Microvasc. Res. 2013, 90, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.G.; Sandri, S.; Campa, A. High-density lipoprotein prevents SAA-induced production of TNF-alpha in THP-1 monocytic cells and peripheral blood mononuclear cells. Mem. Inst. Oswaldo Cruz 2011, 106, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.G.; Fidge, N.H.; Gallon-Beaumier, G.; Kemp, B.E.; Kingwell, B.A. High-density lipoprotein and apolipoprotein AI increase endothelial NO synthase activity by protein association and multisite phosphorylation. Proc. Natl. Acad. Sci. USA 2004, 101, 6999–7004. [Google Scholar] [CrossRef] [PubMed]
- Crauwels, H.M.; van Hove, C.E.; Holvoet, P.; Herman, A.G.; Bult, H. Plaque-associated endothelial dysfunction in apolipoprotein E-deficient mice on a regular diet. Effect of human apolipoprotein AI. Cardiovasc. Res. 2003, 59, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; di Bartolo, B.A.; Nakhla, S.; Heather, A.K.; Mitchell, T.W.; Jessup, W.; Celermajer, D.S.; Barter, P.J.; Rye, K.A. Anti-inflammatory effects of apolipoprotein A-I in the rabbit. Atherosclerosis 2010, 212, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Reimers, G.J.; Jackson, C.L.; Rickards, J.; Chan, P.Y.; Cohn, J.S.; Rye, K.A.; Barter, P.J.; Rodgers, K.J. Inhibition of rupture of established atherosclerotic plaques by treatment with apolipoprotein A-I. Cardiovasc. Res. 2011, 91, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Wang, N.; Tall, A.R. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Welch, C.; Pagler, T.A.; Ranalletta, M.; Lamkanfi, M.; Han, S.; Ishibashi, M.; Li, R.; Wang, N.; Tall, A.R. Increased inflammatory gene expression in ABC transporter-deficient macrophages: Free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation 2008, 118, 1837–1847. [Google Scholar] [CrossRef] [PubMed]
- Sano, O.; Kobayashi, A.; Nagao, K.; Kumagai, K.; Kioka, N.; Hanada, K.; Ueda, K.; Matsuo, M. Sphingomyelin-dependence of cholesterol efflux mediated by ABCG1. J. Lipid Res. 2007, 48, 2377–2384. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ishibashi, M.; Seimon, T.; Lee, M.; Sharma, S.M.; Fitzgerald, K.A.; Samokhin, A.O.; Wang, Y.; Sayers, S.; Aikawa, M.; et al. Free cholesterol accumulation in macrophage membranes activates Toll-like receptors and p38 mitogen-activated protein kinase and induces cathepsin K. Circ. Res. 2009, 104, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, D.; Binnington, B.; Rog, T.; Vattulainen, I.; Grzybek, M.; Coskun, U.; Lingwood, C.A.; Simons, K. Cholesterol modulates glycolipid conformation and receptor activity. Nat. Chem. Biol. 2011, 7, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Tam, S.P.; Flexman, A.; Hulme, J.; Kisilevsky, R. Promoting export of macrophage cholesterol: The physiological role of a major acute-phase protein, serum amyloid A 2.1. J. Lipid Res. 2002, 43, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Hayat, S.; Raynes, J.G. Acute phase serum amyloid A protein increases high density lipoprotein binding to human peripheral blood mononuclear cells and an endothelial cell line. Scand. J. Immunol. 2000, 51, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Artl, A.; Marsche, G.; Lestavel, S.; Sattler, W.; Malle, E. Role of serum amyloid A during metabolism of acute-phase HDL by macrophages. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Ahlin, S.; Olsson, M.; Wilhelmson, A.S.; Skalen, K.; Boren, J.; Carlsson, L.M.; Svensson, P.A.; Sjoholm, K. Adipose tissue-derived human serum amyloid A does not affect atherosclerotic lesion area in hSAA1+/-/ApoE-/- mice. PLoS ONE 2014, 9, e95468. [Google Scholar] [CrossRef] [PubMed]
- De Beer, M.C.; Wroblewski, J.M.; Noffsinger, V.P.; Rateri, D.L.; Howatt, D.A.; Balakrishnan, A.; Ji, A.; Shridas, P.; Thompson, J.C.; van der Westhuyzen, D.R.; et al. Deficiency of endogenous acute phase serum amyloid A does not affect atherosclerotic lesions in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Garner, B.; Witting, P.K.; Waldeck, A.R.; Christison, J.K.; Raftery, M.; Stocker, R. Oxidation of high density lipoproteins. I. Formation of methionine sulfoxide in apolipoproteins AI and AII is an early event that accompanies lipid peroxidation and can be enhanced by alpha-tocopherol. J. Biol. Chem. 1998, 273, 6080–6087. [Google Scholar] [CrossRef] [PubMed]
- Rayner, B.S.; Duong, T.T.; Myers, S.J.; Witting, P.K. Protective effect of a synthetic anti-oxidant on neuronal cell apoptosis resulting from experimental hypoxia re-oxygenation injury. J. Neurochem. 2006, 97, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chami, B.; Barrie, N.; Cai, X.; Wang, X.; Paul, M.; Morton-Chandra, R.; Sharland, A.; Dennis, J.M.; Freedman, S.B.; Witting, P.K. Serum Amyloid A Receptor Blockade and Incorporation into High-Density Lipoprotein Modulates Its Pro-Inflammatory and Pro-Thrombotic Activities on Vascular Endothelial Cells. Int. J. Mol. Sci. 2015, 16, 11101-11124. https://doi.org/10.3390/ijms160511101
Chami B, Barrie N, Cai X, Wang X, Paul M, Morton-Chandra R, Sharland A, Dennis JM, Freedman SB, Witting PK. Serum Amyloid A Receptor Blockade and Incorporation into High-Density Lipoprotein Modulates Its Pro-Inflammatory and Pro-Thrombotic Activities on Vascular Endothelial Cells. International Journal of Molecular Sciences. 2015; 16(5):11101-11124. https://doi.org/10.3390/ijms160511101
Chicago/Turabian StyleChami, Belal, Nicola Barrie, Xiaoping Cai, Xiaosuo Wang, Moumita Paul, Rebecca Morton-Chandra, Alexandra Sharland, Joanne M. Dennis, Saul B. Freedman, and Paul K. Witting. 2015. "Serum Amyloid A Receptor Blockade and Incorporation into High-Density Lipoprotein Modulates Its Pro-Inflammatory and Pro-Thrombotic Activities on Vascular Endothelial Cells" International Journal of Molecular Sciences 16, no. 5: 11101-11124. https://doi.org/10.3390/ijms160511101
APA StyleChami, B., Barrie, N., Cai, X., Wang, X., Paul, M., Morton-Chandra, R., Sharland, A., Dennis, J. M., Freedman, S. B., & Witting, P. K. (2015). Serum Amyloid A Receptor Blockade and Incorporation into High-Density Lipoprotein Modulates Its Pro-Inflammatory and Pro-Thrombotic Activities on Vascular Endothelial Cells. International Journal of Molecular Sciences, 16(5), 11101-11124. https://doi.org/10.3390/ijms160511101