
Oxidative Stress, Bone Marrow Failure, and Genome Instability in Hematopoietic Stem Cells

{kind=link}
{kind=link}
Abstract
:1. Introduction
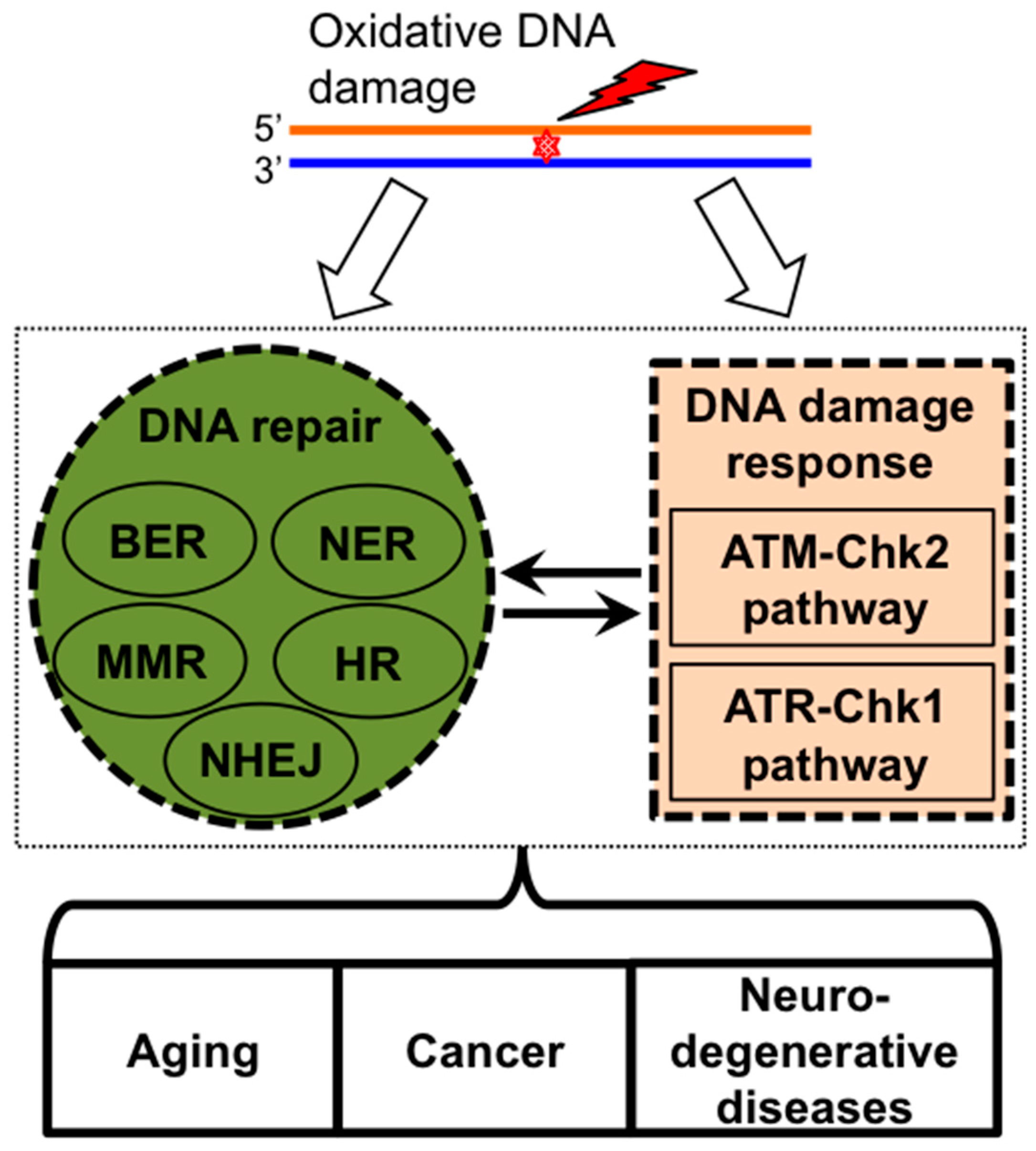
2. ROS Induces DNA Damage Signaling and Repair
2.1. Oxidative Stress and Oxidative DNA Damage
2.2. DNA Damage Response Pathways and DNA Repair
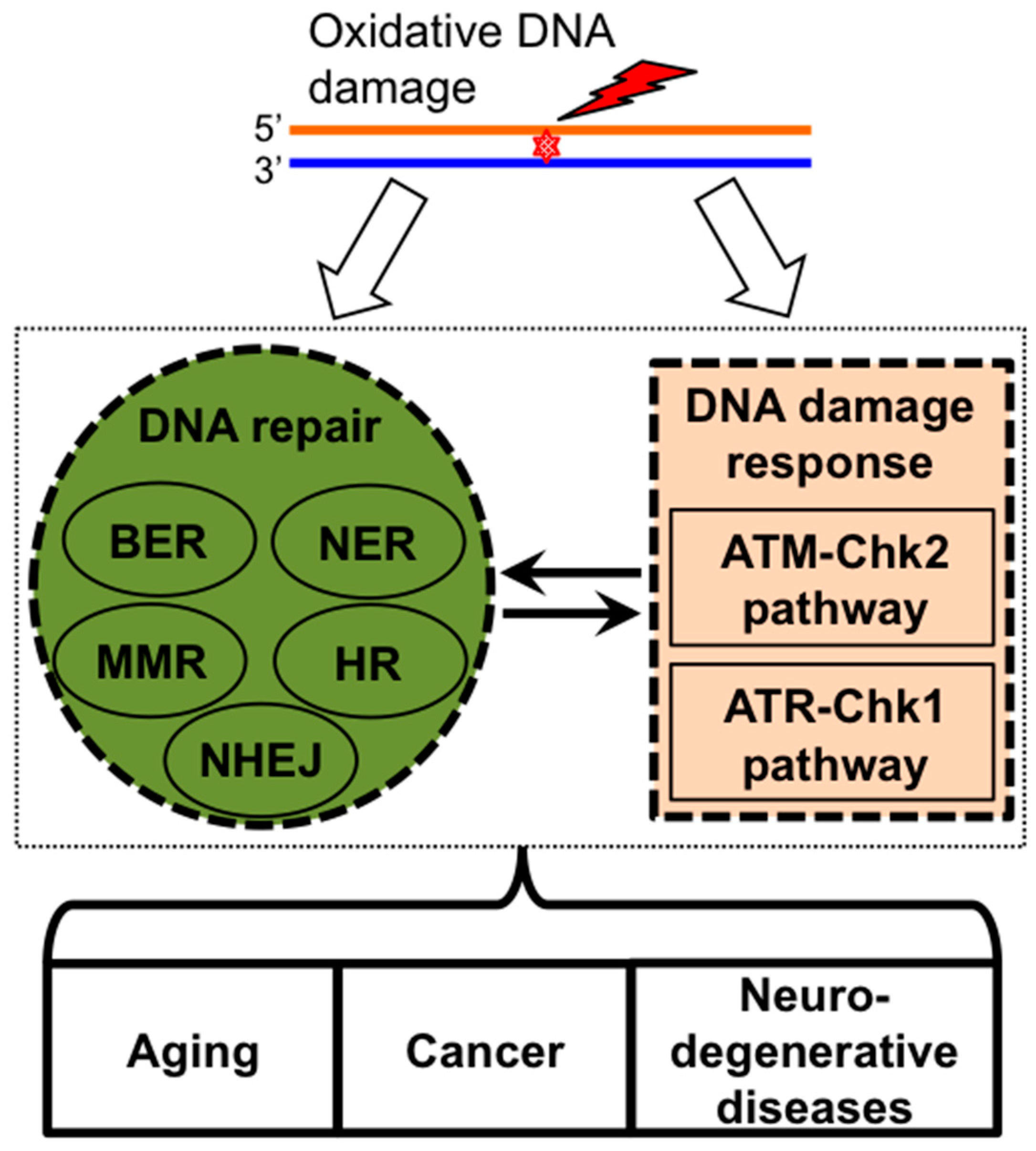
3. Bone Marrow Failure Syndromes
3.1. Fanconi Anemia
3.2. Additional Degenerative Excision Repair Defects
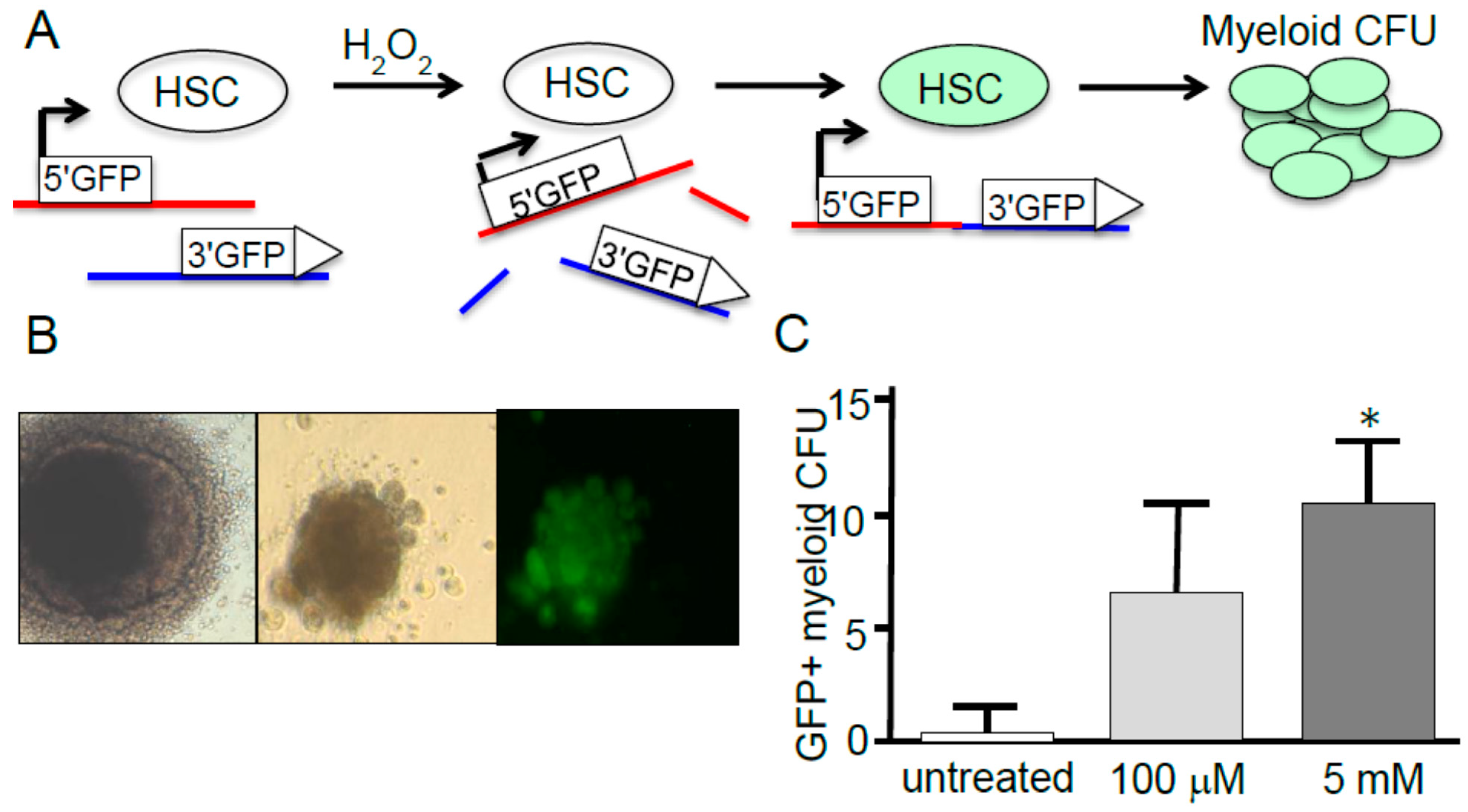
4. Hematopoietic Stem Cell Response to ROS
4.1. HSC Response to ROS
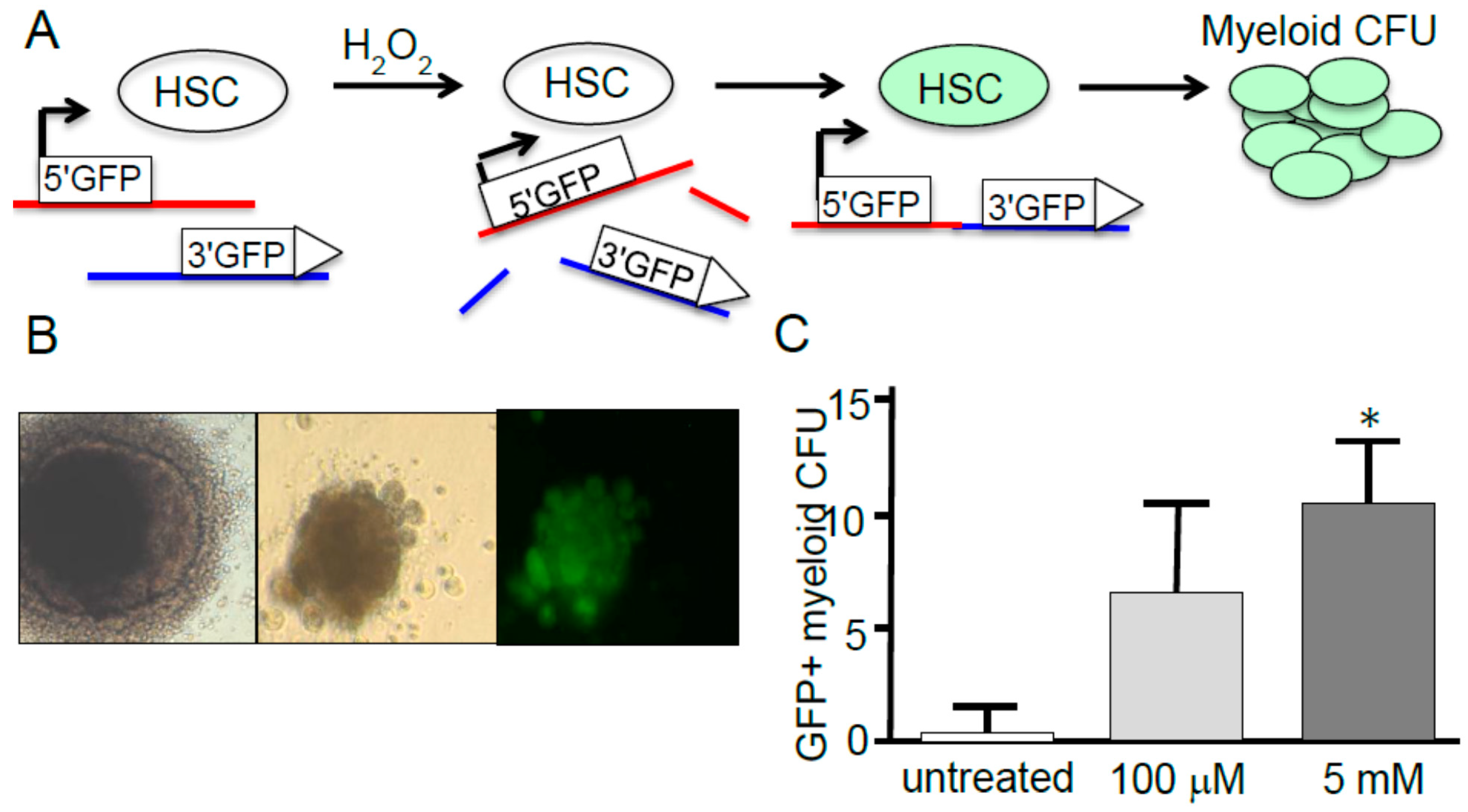
4.2. HSC Response to ROS and Aging
4.3. Leukemic Cells and ROS
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jackson, A.L.; Loeb, L.A. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat. Res. 2001, 477, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J. Oxidative stress: The paradox of aerobic life. Biochem. Soc. Symp. 1995, 61, 1–31. [Google Scholar] [PubMed]
- Beckman, K.B.; Ames, B.N. Oxidative decay of DNA. J. Biol. Chem. 1997, 272, 19633–19636. [Google Scholar] [CrossRef] [PubMed]
- Olinski, R.; Gackowski, D.; Foksinski, M.; Rozalski, R.; Roszkowski, K.; Jaruga, P. Oxidative DNA damage: Assessment of the role in carcinogenesis, atherosclerosis, and acquired immunodeficiency syndrome. Free Radic. Biol. Med. 2002, 33, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.C.; Bohmann, D.; Jasper, H. Jnk signaling confers tolerance to oxidative stress and extends lifespan in drosophila. Dev. Cell 2003, 5, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Jamieson, C.H.; Weissman, I.L. Stems cells and the pathways to aging and cancer. Cell 2008, 132, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; McMurray, C.T. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 2007, 145, 1233–1248. [Google Scholar] [CrossRef] [PubMed]
- Er, T.K.; Tsai, S.M.; Wu, S.H.; Chiang, W.; Lin, H.C.; Lin, S.F.; Wu, S.H.; Tsai, L.Y.; Liu, T.Z. Antioxidant status and superoxide anion radical generation in acute myeloid leukemia. Clin. Biochem. 2007, 40, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, M.; Sun, C.; Francisco, L.; Chakraborty, S.; Sabado, M.; McDonald, T.; Gyorffy, J.; Chang, K.; Wang, S.; et al. Altered hematopoietic cell gene expression precedes development of therapy-related myelodysplasia/acute myeloid leukemia and identifies patients at risk. Cancer Cell 2011, 20, 591–605. [Google Scholar] [CrossRef]
- Zhou, F.L.; Zhang, W.G.; Wei, Y.C.; Meng, S.; Bai, G.G.; Wang, B.Y.; Yang, H.Y.; Tian, W.; Meng, X.; Zhang, H.; et al. Involvement of oxidative stress in the relapse of acute myeloid leukemia. J. Biol. Chem. 2010, 285, 15010–15015. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol. 1997, 82, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Betteridge, D.J. What is oxidative stress? Metabolism 2000, 49, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef] [PubMed]
- De Bandeira, M.S.; da Fonseca, L.J.; da Guedes, S.G.; Rabelo, L.A.; Goulart, M.O.; Vasconcelos, S.M. Oxidative stress as an underlying contributor in the development of chronic complications in diabetes mellitus. Int. J. Mol. Sci. 2013, 14, 3265–3284. [Google Scholar] [CrossRef] [PubMed]
- Agnez-Lima, L.F.; Melo, J.T.; Silva, A.E.; Oliveira, A.H.; Timoteo, A.R.; Lima-Bessa, K.M.; Martinez, G.R.; Medeiros, M.H.; di Mascio, P.; Galhardo, R.S.; et al. DNA damage by singlet oxygen and cellular protective mechanisms. Mutat. Res. 2012, 751, 15–28. [Google Scholar] [CrossRef]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar] [CrossRef] [PubMed]
- Berquist, B.R.; Wilson, D.M., 3rd. Pathways for repairing and tolerating the spectrum of oxidative DNA lesions. Cancer Lett. 2012, 327, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Riley, P.A. Free radicals in biology: Oxidative stress and the effects of ionizing radiation. Int. J. Radiat. Biol. 1994, 65, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M. Oxidatively induced DNA damage: Mechanisms, repair and disease. Cancer Lett. 2012, 327, 26–47. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.A.; Gius, D.; Wink, D.A.; Krishna, M.C.; Russo, A.; Mitchell, J.B. Oxidative stress, redox, and the tumor microenvironment. Semin. Radiat. Oncol. 2004, 14, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C. DNA damage and repair. Nature 2003, 421, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Ravanat, J.L.; TavernaPorro, M.; Menoni, H.; Angelov, D. Oxidatively generated complex DNA damage: Tandem and clustered lesions. Cancer Lett. 2012, 327, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Gutowski, M.; Kowalczyk, S. A study of free radical chemistry: Their role and pathophysiological significance. Acta Biochim. Pol. 2013, 60, 1–16. [Google Scholar] [PubMed]
- Rubattu, S.; Mennuni, S.; Testa, M.; Mennuni, M.; Pierelli, G.; Pagliaro, B.; Gabriele, E.; Coluccia, R.; Autore, C.; Volpe, M.; et al. Pathogenesis of chronic cardiorenal syndrome: Is there a role for oxidative stress? Int. J. Mol. Sci. 2013, 14, 23011–23032. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Sorrell, M.; Berman, Z. Functional interplay between ATM/ATR-mediated DNA damage response and DNA repair pathways in oxidative stress. Cell. Mol. Life Sci. CMLS 2014, 71, 3951–3967. [Google Scholar] [CrossRef]
- Barzilai, A.; Yamamoto, K. DNA damage responses to oxidative stress. DNA Repair (Amst) 2004, 3, 1109–1115. [Google Scholar] [CrossRef]
- Chen, B.P.; Li, M.; Asaithamby, A. New insights into the roles of ATM and DNA-PKCs in the cellular response to oxidative stress. Cancer Lett. 2012, 327, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013. [Google Scholar] [CrossRef]
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Kastan, M.B. Initiating cellular stress responses. Cell 2004, 118, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates atm through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, M.; Gatti, R.A. Pathogenesis of ataxia-telangiectasia: The next generation of ATM functions. Blood 2013, 121, 4036–4045. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, S.; Kozlov, S.; Farooqi, A.A.; Naqi, A.; Lavin, M.; Khanna, K.K. ATM protein kinase: The linchpin of cellular defenses to stress. Cell. Mol. Life Sci. CMLS 2011, 68, 2977–3006. [Google Scholar] [CrossRef]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Willis, J.; Patel, Y.; Lentz, B.L.; Yan, S. APE2 is required for ATR-chk1 checkpoint activation in response to oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 10592–10597. [Google Scholar] [CrossRef] [PubMed]
- Clerkin, J.S.; Naughton, R.; Quiney, C.; Cotter, T.G. Mechanisms of ros modulated cell survival during carcinogenesis. Cancer Lett. 2008, 266, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Panayiotidis, M. Reactive oxygen species (ROS) in multistage carcinogenesis. Cancer Lett. 2008, 266, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Fan, J.; Rassool, F.V. Genomic instability in myeloid malignancies: Increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008, 270, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Vila, M.; Jackson-Lewis, V. Neurodegeneration: What is it and where are we? J. Clin. Investig. 2003, 111, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Zhao, Z.; Goto, S.; Koltai, E. Age-associated neurodegeneration and oxidative damage to lipids, proteins, and DNA. Mol. Asp. Med. 2011, 32, 305–315. [Google Scholar] [CrossRef]
- Rulten, S.L.; Caldecott, K.W. DNA strand break repair and neurodegeneration. DNA Repair 2013, 12, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, P.S.; Tamary, H.; Alter, B.P. How high are carrier frequencies of rare recessive syndromes? Contemporary estimates for Fanconi anemia in the United States and Israel. Am. J. Med. Genet. A 2011, 155A, 1877–1883. [Google Scholar] [CrossRef]
- Alter, B.P.; Greene, M.H.; Velazquez, I.; Rosenberg, P.S. Cancer in Fanconi anemia. Blood 2003, 101, 2072. [Google Scholar] [CrossRef] [PubMed]
- Bakker, J.L.; van Mil, S.E.; Crossan, G.; Sabbaghian, N.; de Leeneer, K.; Poppe, B.; Adank, M.; Gille, H.; Verheul, H.; Meijers-Heijboer, H.; et al. Analysis of the novel Fanconi anemia gene SLX4/FancP in familial breast cancer cases. Hum. Mutat. 2013, 34, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.C.; van de Vrugt, H.J.; van der Valk, M.A.; Oostra, A.B.; Krimpenfort, P.; de Vries, Y.; Joenje, H.; Berns, A.; Arwert, F. Mice with a targeted disruption of the Fanconi Anemia homolog FancA. Hum. Mol. Genet. 2000, 9, 1805–1811. [Google Scholar] [CrossRef] [PubMed]
- Hanna, L.A.; Foreman, R.K.; Tarasenko, I.A.; Kessler, D.S.; Labosky, P.A. Requirement for FoxD3 in maintaining pluripotent cells of the early mouse embryo. Genes Dev. 2002, 16, 2650–2661. [Google Scholar] [CrossRef] [PubMed]
- Sii-Felice, K.; Barroca, V.; Etienne, O.; Riou, L.; Hoffschir, F.; Fouchet, P.; Boussin, F.D.; Mouthon, M.A. Role of fanconi DNA repair pathway in neural stem cell homeostasis. Cell Cycle 2008, 7, 1911–1915. [Google Scholar] [CrossRef] [PubMed]
- Carreau, M. Not-so-novel phenotypes in the Fanconi anemia group D2 mouse model. Blood 2004, 103, 2430. [Google Scholar] [CrossRef] [PubMed]
- Aube, M.; Lafrance, M.; Charbonneau, C.; Goulet, I.; Carreau, M. Hematopoietic stem cells from FancC−/− mice have lower growth and differentiation potential in response to growth factors. Stem Cells 2002, 20, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Whitney, M.A.; Royle, G.; Low, M.J.; Kelly, M.A.; Axthelm, M.K.; Reifsteck, C.; Olson, S.; Braun, R.E.; Heinrich, M.C.; Rathbun, R.K.; et al. Germ cell defects and hematopoietic hypersensitivity to γ-interferon in mice with a targeted disruption of the Fanconi anemia c gene. Blood 1996, 88, 49–58. [Google Scholar] [PubMed]
- Atanassov, B.S.; Barrett, J.C.; Davis, B.J. Homozygous germ line mutation in exon 27 of murine Brca2 disrupts the FancD2-Brca2 pathway in the homologous recombination-mediated DNA interstrand cross-links’ repair but does not affect meiosis. Genes Chromosomes Cancer 2005, 44, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Navarro, S.; Meza, N.W.; Quintana-Bustamante, O.; Casado, J.A.; Jacome, A.; McAllister, K.; Puerto, S.; Surralles, J.; Segovia, J.C.; Bueren, J.A.; et al. Hematopoietic dysfunction in a mouse model for Fanconi anemia group D1. Mol. Ther. 2006, 14, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Houghtaling, S.; Timmers, C.; Noll, M.; Finegold, M.J.; Jones, S.N.; Meyn, M.S.; Grompe, M. Epithelial cancer in Fanconi anemia complementation group D2 (FancD2) knockout mice. Genes Dev. 2003, 17, 2021–2035. [Google Scholar] [CrossRef] [PubMed]
- Godthelp, B.C.; Wiegant, W.W.; Waisfisz, Q.; Medhurst, A.L.; Arwert, F.; Joenje, H.; Zdzienicka, M.Z. Inducibility of nuclear Rad51 foci after DNA damage distinguishes all Fanconi anemia complementation groups from D1/Brca2. Mutat. Res. 2006, 594, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Kumari, U.; Ya Jun, W.; Huat Bay, B.; Lyakhovich, A. Evidence of mitochondrial dysfunction and impaired ROS detoxifying machinery in fanconi anemia cells. Oncogene 2014, 33, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, J.; Sejas, D.P.; Pang, Q. Hypoxia-reoxygenation induces premature senescence in FA bone marrow hematopoietic cells. Blood 2005, 106, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Hadjur, S.; Ung, K.; Wadsworth, L.; Dimmick, J.; Rajcan-Separovic, E.; Scott, R.W.; Buchwald, M.; Jirik, F.R. Defective hematopoiesis and hepatic steatosis in mice with combined deficiencies of the genes encoding FancC and Cu/Zn superoxide dismutase. Blood 2001, 98, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Langevin, F.; Crossan, G.P.; Rosado, I.V.; Arends, M.J.; Patel, K.J. FancD2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 2011, 475, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, K.H.; Patronas, N.J.; Schiffmann, R.; Brooks, B.P.; Tamura, D.; DiGiovanna, J.J. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: A complex genotype-phenotype relationship. Neuroscience 2007, 145, 1388–1396. [Google Scholar] [CrossRef] [PubMed]
- Van der Horst, G.T.; Meira, L.; Gorgels, T.G.; de Wit, J.; Velasco-Miguel, S.; Richardson, J.A.; Kamp, Y.; Vreeswijk, M.P.; Smit, B.; Bootsma, D.; et al. UVb radiation-induced cancer predisposition in Cockayne syndrome group A (Csa) mutant mice. DNA Repair (Amst) 2002, 1, 143–157. [Google Scholar] [CrossRef]
- De Waard, H.; de Wit, J.; Gorgels, T.G.; van den Aardweg, G.; Andressoo, J.O.; Vermeij, M.; van Steeg, H.; Hoeijmakers, J.H.; van der Horst, G.T. Cell type-specific hypersensitivity to oxidative damage in Csb and XPA mice. DNA Repair (Amst) 2003, 2, 13–25. [Google Scholar] [CrossRef]
- De Waard, H.; de Wit, J.; Andressoo, J.O.; van Oostrom, C.T.; Riis, B.; Weimann, A.; Poulsen, H.E.; van Steeg, H.; Hoeijmakers, J.H.; van der Horst, G.T.; et al. Different effects of Csa and Csb deficiency on sensitivity to oxidative DNA damage. Mol. Cell. Biol. 2004, 24, 7941–7948. [Google Scholar] [CrossRef] [PubMed]
- Spivak, G.; Hanawalt, P.C. Host cell reactivation of plasmids containing oxidative DNA lesions is defective in Cockayne syndrome but normal in UV-sensitive syndrome fibroblasts. DNA Repair (Amst) 2006, 5, 13–22. [Google Scholar] [CrossRef]
- D’Errico, M.; Parlanti, E.; Teson, M.; Degan, P.; Lemma, T.; Calcagnile, A.; Iavarone, I.; Jaruga, P.; Ropolo, M.; Pedrini, A.M.; et al. The role of Csa in the response to oxidative DNA damage in human cells. Oncogene 2007, 26, 4336–4343. [Google Scholar] [CrossRef] [PubMed]
- Melis, J.P.; van Steeg, H.; Luijten, M. Oxidative DNA damage and nucleotide excision repair. Antioxid. Redox Signal. 2013, 18, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Hollander, M.C.; Philburn, R.T.; Patterson, A.D.; Velasco-Miguel, S.; Friedberg, E.C.; Linnoila, R.I.; Fornace, A.J., Jr. Deletion of XPC leads to lung tumors in mice and is associated with early events in human lung carcinogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 13200–13205. [Google Scholar] [CrossRef] [PubMed]
- Melis, J.P.; Wijnhoven, S.W.; Beems, R.B.; Roodbergen, M.; van den Berg, J.; Moon, H.; Friedberg, E.; van der Horst, G.T.; Hoeijmakers, J.H.; Vijg, J.; et al. Mouse models for xeroderma pigmentosum group a and group C show divergent cancer phenotypes. Cancer Res. 2008, 68, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.L.; Kumar, M.A.; Day, T.W.; Hardy, T.M.; Hamilton, S.; Besch-Williford, C.; Safa, A.R.; Pollok, K.E.; Smith, M.L. The Xpc gene markedly affects cell survival in mouse bone marrow. Mutagenesis 2009, 24, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M. Roles of oxidative stress in xeroderma pigmentosum. Adv. Exp. Med. Biol. 2008, 637, 120–127. [Google Scholar] [PubMed]
- Walne, A.J.; Dokal, I. Advances in the understanding of dyskeratosis congenita. Br. J. Haematol. 2009, 145, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Tummala, H.; Kirwan, M.; Walne, A.J.; Hossain, U.; Jackson, N.; Pondarre, C.; Plagnol, V.; Vulliamy, T.; Dokal, I. ERCC6L2 mutations link a distinct bone-marrow-failure syndrome to DNA repair and mitochondrial function. Am. J. Hum. Genet. 2014, 94, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Shao, L.; Spitz, D.R. Reactive oxygen species in normal and tumor cells. Adv. Cancer Res. 2014, 122, 1–67. [Google Scholar] [PubMed]
- Yahata, T.; Muguruma, Y.; Yumino, S.; Sheng, Y.; Uno, T.; Matsuzawa, H.; Ito, M.; Kato, S.; Hotta, T.; Ando, K.; et al. Quiescent human hematopoietic stem cells in the bone marrow niches organize the hierarchical structure of hematopoiesis. Stem Cells 2008, 26, 3228–3236. [Google Scholar] [CrossRef] [PubMed]
- Yahata, T.; Takanashi, T.; Muguruma, Y.; Ibrahim, A.A.; Matsuzawa, H.; Uno, T.; Sheng, Y.; Onizuka, M.; Ito, M.; Kato, S.; et al. Accumulation of oxidative DNA damage restricts the self-renewal capacity of human hematopoietic stem cells. Blood 2011, 118, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Owusu-Ansah, E.; Banerjee, U. Reactive oxygen species prime drosophila haematopoietic progenitors for differentiation. Nature 2009, 461, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.G.; Tresini, M. Oxidative stress and gene regulation. Free Radic. Biol. Med. 2000, 28, 463–499. [Google Scholar] [CrossRef] [PubMed]
- Amundson, S.A.; Do, K.T.; Vinikoor, L.; Koch-Paiz, C.A.; Bittner, M.L.; Trent, J.M.; Meltzer, P.; Fornace, A.J., Jr. Stress-specific signatures: Expression profiling of p53 wild-type and -null human cells. Oncogene 2005, 24, 4572–4579. [Google Scholar] [CrossRef] [PubMed]
- Anantharam, V.; Lehrmann, E.; Kanthasamy, A.; Yang, Y.; Banerjee, P.; Becker, K.G.; Freed, W.J.; Kanthasamy, A.G. Microarray analysis of oxidative stress regulated genes in mesencephalic dopaminergic neuronal cells: Relevance to oxidative damage in Parkinson’s disease. Neurochem. Int. 2007, 50, 834–847. [Google Scholar] [CrossRef] [PubMed]
- Birrell, G.W.; Brown, J.A.; Wu, H.I.; Giaever, G.; Chu, A.M.; Davis, R.W.; Brown, J.M. Transcriptional response of saccharomyces cerevisiae to DNA-damaging agents does not identify the genes that protect against these agents. Proc. Natl. Acad. Sci. USA 2002, 99, 8778–8783. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.Y.; Chen, Y.; Gadisetti; Chandramouli, V.R.; Cook, J.A.; Coffin, D.; Tsai, M.H.; DeGraff, W.; Yan, H.; Zhao, S.; et al. Gene expression after treatment with hydrogen peroxide, menadione, or t-butyl hydroperoxide in breast cancer cells. Cancer Res. 2002, 62, 6246–6254. [Google Scholar] [PubMed]
- Islaih, M.; Halstead, B.W.; Kadura, I.A.; Li, B.; Reid-Hubbard, J.L.; Flick, L.; Altizer, J.L.; Thom Deahl, J.; Monteith, D.K.; Newton, R.K.; et al. Relationships between genomic, cell cycle, and mutagenic responses of TK6 cells exposed to DNA damaging chemicals. Mutat. Res. 2005, 578, 100–116. [Google Scholar] [CrossRef] [PubMed]
- Purdom-Dickinson, S.E.; Lin, Y.; Dedek, M.; Morrissy, S.; Johnson, J.; Chen, Q.M. Induction of antioxidant and detoxification response by oxidants in cardiomyocytes: Evidence from gene expression profiling and activation of Nrf2 transcription factor. J. Mol. Cell. Cardiol. 2007, 42, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Weigel, A.L.; Handa, J.T.; Hjelmeland, L.M. Microarray analysis of H2O2-, HNE-, or tBH-treated ARPE-19 cells. Free Radic. Biol. Med. 2002, 33, 1419–1432. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; He, M.; Lee, N.H.; Liu, E.T. Identification of Myc-mediated death response pathways by microarray analysis. J. Biol. Chem. 2002, 277, 13059–13066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fong, C.C.; Wong, M.S.; Tzang, C.H.; Lai, W.P.; Fong, W.F.; Sui, S.F.; Yang, M. Molecular mechanisms of survival and apoptosis in Raw 264.7 macrophages under oxidative stress. Apoptosis 2005, 10, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Abbas, H.A.; Maccio, D.R.; Coskun, S.; Jackson, J.G.; Hazen, A.L.; Sills, T.M.; You, M.J.; Hirschi, K.K.; Lozano, G. Mdm2 is required for survival of hematopoietic stem cells/progenitors via dampening of ROS-induced p53 activity. Cell Stem Cell 2010, 7, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, Y.; Graves, D.T. FoxO transcription factors: Their clinical significance and regulation. BioMed Res. Int. 2014. [Google Scholar] [CrossRef]
- Miyamoto, K.; Araki, K.Y.; Naka, K.; Arai, F.; Takubo, K.; Yamazaki, S.; Matsuoka, S.; Miyamoto, T.; Ito, K.; Ohmura, M.; et al. FoxO3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 2007, 1, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Greer, C.; Secombe, J. KDM5 interacts with foxO to modulate cellular levels of oxidative stress. PLoS Genet. 2014, 10, e1004676. [Google Scholar] [CrossRef] [PubMed]
- Kocabas, F.; Zheng, J.; Thet, S.; Copeland, N.G.; Jenkins, N.A.; DeBerardinis, R.J.; Zhang, C.; Sadek, H.A. Meis1 regulates the metabolic phenotype and oxidant defense of hematopoietic stem cells. Blood 2012, 120, 4963–4972. [Google Scholar] [CrossRef] [PubMed]
- Mouzannar, R.; McCafferty, J.; Benedetto, G.; Richardson, C. Transcriptional and phospho-proteomic screens reveal stem cell activation of insulin-resistance and transformation pathways following a single minimally toxic episode of ROS. Int. J. Genomics Proteomics 2011, 2, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Burdon, R.H. Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic. Biol. Med. 1995, 18, 775–794. [Google Scholar] [CrossRef] [PubMed]
- Burdon, R.H.; Alliangana, D.; Gill, V. Hydrogen peroxide and the proliferation of BHK-21 cells. Free Radic. Res. 1995, 23, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.; Richardson, C. Multipotent hematopoietic cells susceptible to alternative double-strand break repair pathways that promote genome rearrangements. Genes Dev. 2007, 21, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Luo, Y.; Zhou, D. Hematopoietic stem cell injury induced by ionizing radiation. Antioxid. Redox Signal. 2014, 20, 1447–1462. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, L.; Pazhanisamy, S.K.; Li, H.; Meng, A.; Zhou, D. Total body iradiation causes residual bone marrow injury by induction of persistent oxidative stress in murine hematopoietic stem cells. Free Radic. Biol. Med. 2010, 48, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Floratou, K.; Giannopoulou, E.; Antonacopoulou, A.; Karakantza, M.; Adonakis, G.; Kardamakis, D.; Matsouka, P. Oxidative stress due to radiation in CD34+ hematopoietic progenitor cells: Protection by IGF-1. J. Radiat. Res. 2012, 53, 672–685. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Kashiwakura, I. Role of reactive oxygen species in the radiation response of human hematopoietic stem/progenitor cells. PLoS One 2013, 8, e70503. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, W.; Liu, H.; Yang, L.; Liao, Q.; Cui, S.; Wang, H.; Zhao, L. miR-29b suppresses tumor growth and metastasis in colorectal cancer via downregulating Tiam1 expression and inhibiting epithelial-mesenchymal transition. Cell Death Dis. 2014, 5, e1335. [Google Scholar] [CrossRef] [PubMed]
- Saretzki, G.; Armstrong, L.; Leake, A.; Lako, M.; von Zglinicki, T. Stress defense in murine embryonic stem cells is superior to that of various differentiated murine cells. Stem Cells 2004, 22, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Ergene, U.; Cagirgan, S.; Pehlivan, M.; Yilmaz, M.; Tombuloglu, M. Factors influencing engraftment in autologous peripheral hematopoetic stem cell transplantation (PBSCT). Transfus. Apher. Sci. 2007, 36, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Bryder, D.; Seita, J.; Nussenzweig, A.; Hoeijmakers, J.; Weissman, I.L. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007, 447, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Seita, J.; Czechowicz, A.; Bhattacharya, D.; Bryder, D.; Weissman, I.L. Hematopoietic stem cell quiescence attenuates DNA damage response and permits DNA damage accumulation during aging. Cell Cycle 2007, 6, 2371–2376. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.Y.; Sharkis, S.J. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 2007, 110, 3056–3063. [Google Scholar] [CrossRef] [PubMed]
- Beerman, I.; Seita, J.; Inlay, M.A.; Weissman, I.L.; Rossi, D.J. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 2014, 15, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Lee, Y.D.; Wagers, A.J. Stem cell aging: Mechanisms, regulators and therapeutic opportunities. Nat. Med. 2014, 20, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Urao, N.; Ushio-Fukai, M. Redox regulation of stem/progenitor cells and bone marrow niche. Free Radic. Biol. Med. 2013, 54, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Rehman, J. Redox and metabolic regulation of stem/progenitor cells and their niche. Antioxid. Redox Signal. 2014, 21, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Picolli, C.; D’Aprile, A.; Ripoli, M.; Scrima, R.; Boffoli, D.; Tabilo, A.; Capitanio, N. The hypoxia-inducible factor is stabilized in circulating hematopoietic stem cells under normoxic conditions. FEBS Lett. 2007, 581, 3111–3119. [Google Scholar] [CrossRef] [PubMed]
- Picolli, C.; D’Aprile, A.; Ripoli, M.; Scrima, R.; Lecce, L.; Boffoli, D.; Tabilo, A; Capitanio, N. Bone-marrow derived hematopoietic stem/progenitor cells express multiple isoforms of NADPH oxidase and produce constituitively reactive oxygen species. Biochem. Biophys. Res. Commun. 2007, 353, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Picolli, C.; Ria, R.; Ripoli, M.; Scrima, R.; Cela, O.; D’Aprile, A.; Boffoli, D.; Falzetti, F.; Tabilo, A; Capitanio, N.; et al. Characterization of mitochondrial and extra-mitochondrial oxygen consuming reactions in human hematopoietic stem cells: Novel evidence of the occurrence of NAD(P)H oxidase activity. J. Biol. Chem. 2005, 280, 26467–26476. [Google Scholar] [CrossRef] [PubMed]
- Rouault-Pierre, K.; Lopez-Onieva, L.; Foster, K.; Anjos-Afonso, F.; Lamrissi-Garcia, I.; Serrano-Sanchez, M.; Mitter, R.; Ivanovic, Z.; de Verneuil, H.; Gribben, J.; et al. HIF-2α protects human hematopoietic stem/progenitors and acute myeloid leukemic cells from apoptosis induced by endoplasmic reticulum stress. Cell Stem Cell 2013, 13, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Shen, Q.; Claret, F.X. Novel roles of reactive oxygen species in the pathogenesis of acute myeloid leukemia. J. Leukoc. Biol. 2013, 94, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Hole, P.S.; Zabkiewicz, J.; Munje, C.; Newton, Z.; Pearn, L.; White, P.; Marquez, N.; Hills, R.K.; Burnett, A.K.; Tonks, A.; et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 2013, 122, 3322–3330. [Google Scholar] [CrossRef] [PubMed]
- Juntilla, M.M.; Patil, V.D.; Calamito, M.; Joshi, R.P.; Birnbaum, M.J.; Koretzky, G.A. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood 2010, 115, 4030–4038. [Google Scholar] [CrossRef] [PubMed]
- Woolley, J.F.; Naughton, R.; Stanicka, J.; Gough, D.R.; Bhatt, L.; Dickinson, B.C.; Chang, C.J.; Cotter, T.G. H2O2 production downstream of FLT3 is mediated by p22phox in the endoplasmic reticulum and is required for STAT5 signalling. PLoS One 2012, 7, e34050. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Fan, J.; Datta, K.; Kim, K.T.; Grosu, D.; Shapiro, P.; Small, D.; Rassool, F. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ros production, DNA damage, and misrepair: Implications for poor prognosis in AML. Blood 2008, 111, 3173–3182. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Luo, L.; Proctor, S.J.; Middleton, P.G.; Blakely, E.L.; Taylor, R.W.; Turnbull, D.M. Somatic mitochondrial DNA mutations in adult-onset leukaemia. Leukemia 2003, 17, 2487–2491. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C.; Thomassen, M.; Hasselbalch Riley, C.; Kjaer, L.; Stauffer Larsen, T.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Skov, V. Whole blood transcriptional profiling reveals deregulation of oxidative and antioxidative defence genes in myelofibrosis and related neoplasms: Potential implications of downregulation of Nrf2 for genomic instability and disease progression. PLoS One 2014, 9, e112786. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, S.A.; MacEwan, D.J. HO-1 underlies resistance of aml cells to TNF-induced apoptosis. Blood 2008, 111, 3793–3801. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.J.; Robert, C.; Gough, S.M.; Rassool, F.V.; Aplan, P.D. Oxidative stress leads to increased mutation frequency in a murine model of myelodysplastic syndrome. Leuk. Res. 2014, 38, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Vadukoot, A.K.; AbdulSalam, S.F.; Wunderlich, M.; Pullen, E.D.; Landero-Figueroa, J.; Mulloy, J.C.; Merino, E.J. Design of a hydrogen peroxide-activatable agent that specifically targets cancer cells. Bioorg. Med. Chem. 2014, 22, 6885–6892. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richardson, C.; Yan, S.; Vestal, C.G. Oxidative Stress, Bone Marrow Failure, and Genome Instability in Hematopoietic Stem Cells. Int. J. Mol. Sci. 2015, 16, 2366-2385. https://doi.org/10.3390/ijms16022366
Richardson C, Yan S, Vestal CG. Oxidative Stress, Bone Marrow Failure, and Genome Instability in Hematopoietic Stem Cells. International Journal of Molecular Sciences. 2015; 16(2):2366-2385. https://doi.org/10.3390/ijms16022366
Chicago/Turabian StyleRichardson, Christine, Shan Yan, and C. Greer Vestal. 2015. "Oxidative Stress, Bone Marrow Failure, and Genome Instability in Hematopoietic Stem Cells" International Journal of Molecular Sciences 16, no. 2: 2366-2385. https://doi.org/10.3390/ijms16022366