
Online Measurement of Real-Time Cytotoxic Responses Induced by Multi-Component Matrices, such as Natural Products, through Electric Cell-Substrate Impedance Sensing (ECIS)


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Optimization of the Cell-Substrate Impedance Sensing (ECIS) Assay

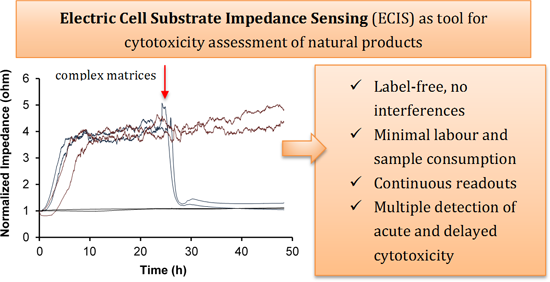{kind=link}
{kind=link}
{kind=link}
| Cell Concentration (Cells/mL) × 105 | Time (h) | 8W10E | 8W1E | ||||
|---|---|---|---|---|---|---|---|
| Z′ | S/N | S/B | Z′ | S/N | S/B | ||
| 4 | 24 | 0.53 | 6.88 | 1.88 | 0.92 | 44.13 | 4.09 |
| 48 | 0.56 | 7.36 | 2.52 | 0.89 | 38.00 | 3.92 | |
| 2 | 24 | 0.11 | 2.81 | 1.86 | 0.63 | 10.11 | 3.24 |
| 48 | 0.16 | 3.74 | 2.17 | 0.26 | 4.46 | 3.56 | |
| 1 | 24 | 0.17 | 4.05 | 1.34 | 0.42 | 2.38 | 2.23 |
| 48 | 0.23 | 4.36 | 1.53 | 0.04 | 3.48 | 2.93 | |
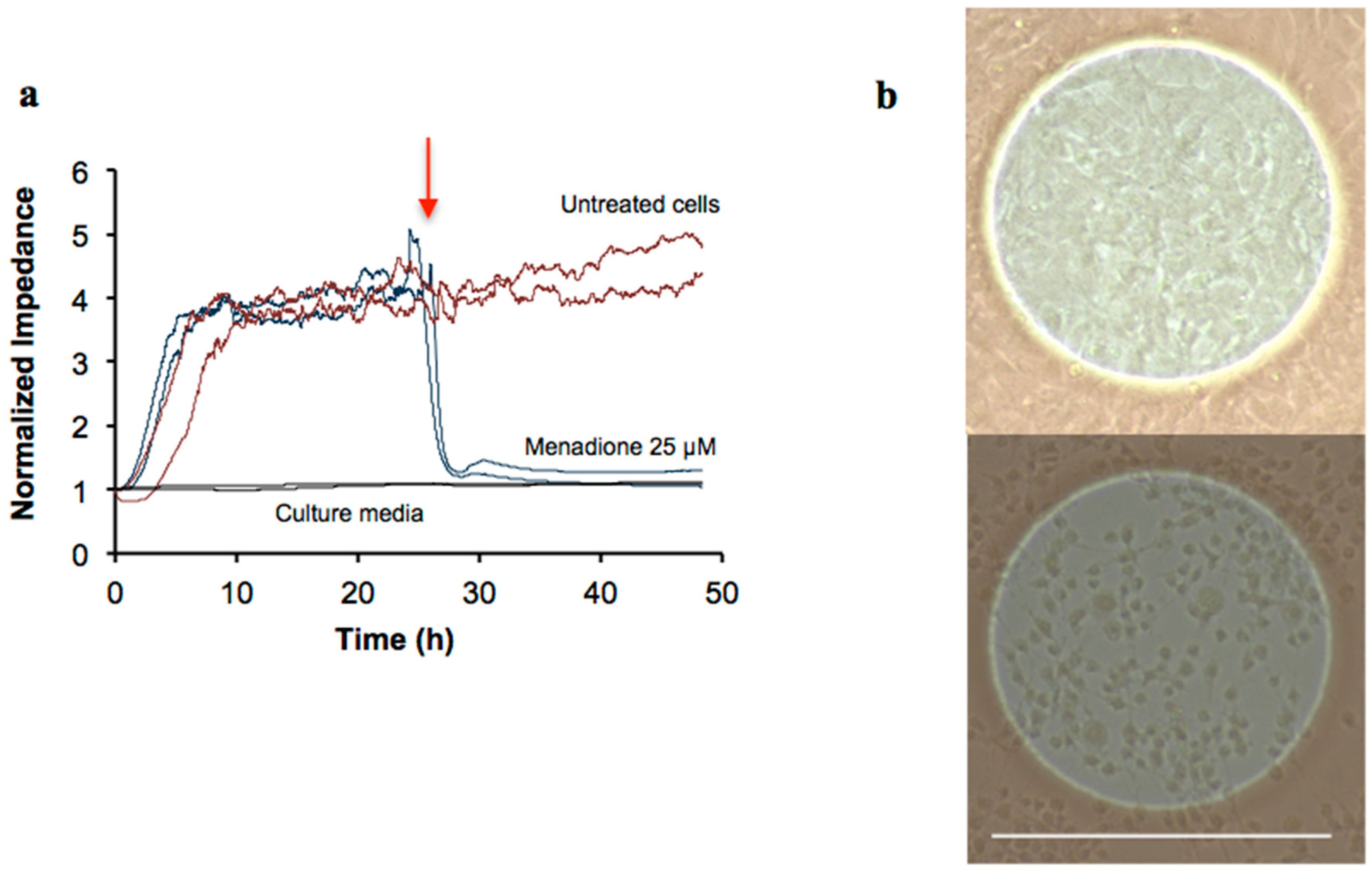
2.2. Cytotoxicity Profiling of Four Natural Products Using the ECIS Assay
| Sample (Extract) | Resazurin Reduction Assay (RFU) | ATP Luminometric Assay (RLU) | LIVE/DEAD Fluorescence Assay (RFU) | |
|---|---|---|---|---|
| Calcein | EthD-1 | |||
| Probe control | 83.8 ± 4.9 | 137.4 ± 4.6 | 6.671 ± 0.074 | 0.121 ± 0.017 |
| NP1 | 176.6 ± 6.6 *** | 1758.9 ± 65.3 *** | 10.200 ± 0.092 *** | 0.433 ± 0.013 *** |
| NP2 | 106.8 ± 2.9 | 119.4 ± 1.6 | 6.581 ± 0.112 | 0.151 ± 0.009 |
| NP3 | 102.7 ± 28.8 | 122.0 ± 4.8 | 7.131 ± 0.134 * | 0.143 ± 0.008 |
| NP4 | 93.8 ± 6.6 | 122.0 ± 4.8 | 6.754 ± 0.102 | 0.190 ± 0.013 * |
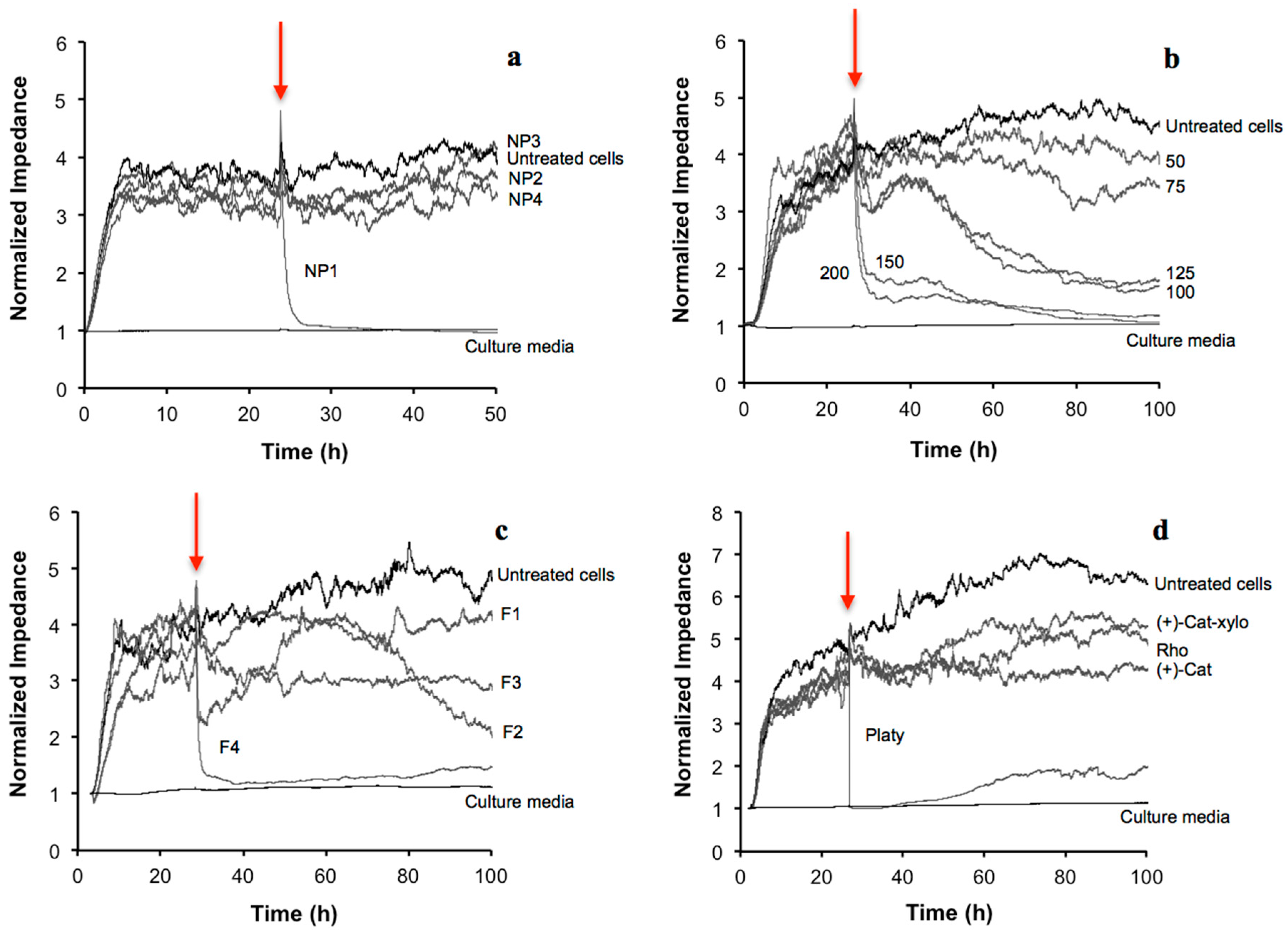
2.3. Detailed Cytotoxicity Study of B. pendula Extract (NP1) Using the ECIS Assay
| Sample (Extract or Fraction) | (+)-Catechin | Rhododendrin | Platyphylloside | Procyanidins |
|---|---|---|---|---|
| (+)-Catechin 7-O-β-d-Xylopyranoside | ||||
| Crude extract | 0.0581 ± 0.0018 | 0.0794 ± 0.0010 | 0.210 ± 0.010 | 0.28 ± 0.05 |
| F1 | 0.00446 ± 0.00010 | 0.169 ± 0.007 | 0.142 ± 0.005 | 0.17 ± 0.03 |
| F2 | 0.195 ± 0.006 | – | 0.533 ± 0.018 | 0.014 ± 0.007 |
| F3 | 0.050 ± 0.004 | – | – | 0.51 ± 0.07 |
| F4 | – | – | – | 1.00 ± 0.08 |
3. Experimental Section
3.1. Betula Pendula Extract
3.2. Commercially-Standardized Natural Extracts
3.3. Cell Culture Conditions
3.4. Impedance Measurements with ECIS
3.5. ECIS Optimization Trials and Repeatability
3.6. ECIS Signal Authentication by Imaging
3.7. ECIS Cytotoxicity Studies with Natural Products
3.8. Interferences of Optical Readouts by Natural Extracts
3.9. Data Processing and Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Potterat, O.; Hamburger, M. Natural products in drug discovery—Concepts and approaches for tracking bioactivity. Curr. Org. Chem. 2006, 10, 899–920. [Google Scholar] [CrossRef]
- Fallarero, A.; Hanski, L.; Vuorela, P. How to translate a bioassay into a screening assay for natural products: General considerations and implementation of antimicrobial screens. Planta Med. 2014, 80, 1182–1199. [Google Scholar] [CrossRef] [PubMed]
- Kraus, B.; Ziegler, M.; Wolff, H. Linear fluorescence unmixing in cell biological research. In Modern Research and Educational Topics in Microscopy; Méndez-Vilas, A., Díaz, J., Eds.; Formatex: Badajoz, Spain, 2007; Volume 2, pp. 863–872. [Google Scholar]
- Bruggisser, R.; von Daeniken, K.; Jundt, G.; Schaffner, W.; Tullberg-Reinert, H. Interference of plant extracts, phytoestrogens and antioxidants with the MTT tetrazolium assay. Planta Med. 2002, 68, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Soriano, F.X.; Galbete, J.L.; Forloni, G. Effect of β-amyloid on endothelial cells: Lack of direct toxicity, enhancement of MTT-induced cell death and intracellular accumulation. Neurochem. Int. 2003, 43, 251–261. [Google Scholar] [CrossRef]
- Shoemaker, M.; Cohen, I.; Campbell, M. Reduction of MTT by aqueous herbal extracts in the absence of cells. J. Ethnopharmacol. 2004, 93, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.L. Stem cells and drug discovery: The beginning of a new era? Cell 2008, 132, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Shiau, A.K.; Massari, M.E.; Ozbal, C.C. Back to basics: Label-free technologies for small molecule screening. Comb. Chem. High Throughput Screen. 2008, 11, 231–237. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Bhalerao, S.V.; Lidstone, E.A.; Ahmad, I.S.; Abbasi, A.; Cunningham, B.T.; Watkin, K.L. Cytotoxicity screening of Bangladeshi medicinal plant extracts on pancreatic cancer cells. BMC Complement. Altern. Med. 2010, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.L.; George, S.; Ahmad, I.; Gosangari, S.L.; Abbasi, A.; Cunningham, B.T.; Watkin, K.L. Cytotoxicity effects of Amoora rohituka and chittagonga on breast and pancreatic cancer cells. Evid.-Based Complement. Altern. Med. 2011, 2011, 860605. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.L.; Gosangari, S.L.; Watkin, K.L.; Cunningham, B.T. A label-free photonic crystal biosensor imaging method for detection of cancer cell cytotoxicity and proliferation. Apoptosis 2007, 12, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Kling, B.; Bücherl, D.; Palatzky, P.; Matysik, F.M.; Decker, M.; Wegener, J.; Heilmann, J. Flavonoids, flavonoid metabolites, and phenolic acids inhibit oxidative stress in the neuronal cell line HT-22 monitored by ECIS and MTT assay: A comparative study. J. Nat. Prod. 2014, 77, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Giaever, I.; Keese, C.R. Monitoring fibroblast behavior in tissue culture with an applied electric field. Proc. Natl. Acad. Sci. USA 1984, 81, 3761–3764. [Google Scholar] [CrossRef] [PubMed]
- Arndt, S.; Seebach, J.; Psathaki, K.; Galla, H.J.; Wegener, J. Bioelectrical impedance assay to monitor changes in cell shape during apoptosis. Biosens. Bioelectron. 2004, 19, 583–594. [Google Scholar] [CrossRef]
- Campbell, C.E.; Laane, M.M.; Haugarvoll, E.; Giaever, I. Monitoring viral-induced cell death using electric cell-substrate impedance sensing. Biosens. Bioelectron. 2007, 23, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Keese, C.R.; Wegener, J.; Walker, S.R.; Giaever, I. Electrical wound-healing assay for cells in vitro. Proc. Natl. Acad. Sci. USA 2004, 101, 1554–1559. [Google Scholar] [CrossRef] [PubMed]
- Opp, D.; Wafula, B.; Lim, J.; Huang, E.; Lo, J.C.; Lo, C.M. Use of electric cell-substrate impedance sensing to assess in vitro cytotoxicity. Biosens. Bioelectron. 2009, 24, 2625–2629. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Luong, J.H. On-line monitoring of cell growth and cytotoxicity using electric cell-substrate impedance sensing (ECIS). Biotechnol. Prog. 2003, 19, 1000–1005. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Lachance, B.; Sunahara, G.; Luong, J.H. Assessment of cytotoxicity using electric cell-substrate impedance sensing: Concentration and time response function approach. Anal. Chem. 2002, 74, 5748–5753. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Thirion, C.; Pfaffl, M.W. Electric cell-substrate impedance sensing (ECIS) based real-time measurement of titer dependent cytotoxicity induced by adenoviral vectors in an IPI-2I cell culture model. Biosens. Bioelectron. 2011, 26, 2000–2005. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Xu, Y.; Wang, L.; Mitchelson, K.; Xing, W.; Cheng, J. Use of cellular electrical impedance sensing to assess in vitro cytotoxicity of anticancer drugs in a human kidney cell nephrotoxicity model. Analyst 2012, 137, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Tam, J.C.; Sanders, A.J.; Ko, C.H.; Fung, K.P.; Leung, P.C.; Harding, K.G.; Jiang, W.G.; Lau, C.B. Molecular angiogenic events of a two-herb wound healing formula involving MAPK and Akt signaling pathways in human vascular endothelial cells. Wound Repair Regen. 2013, 21, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.L.; Chiang, Y.C.; Huang, C.C.; Fang, J.Y.; Chen, D.F.; Hung, C.F. Zeaxanthin inhibits PDGF-BB-induced migration in human dermal fibroblasts. Exp. Dermatol. 2010, 19, e173–e181. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.G.; Ye, L.; Ji, K.; Ruge, F.; Wu, Y.; Gao, Y.; Ji, J.; Mason, M.D. Antitumour effects of Yangzheng Xiaoji in human osteosarcoma: The pivotal role of focal adhesion kinase signalling. Oncol. Rep. 2013, 30, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Nagalingam, A.; Arbiser, J.L.; Bonner, M.Y.; Saxena, N.K.; Sharma, D. Honokiol activates AMP-activated protein kinase in breast cancer cells via an LKB1-dependent pathway and inhibits breast carcinogenesis. Breast Cancer Res. 2012, 14, R35. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.M.; Huang, J.H.; Chiang, H.S.; Wu, W.B.; Lin, H.H.; Hong, J.Y.; Hung, C.F. Effects of (−)-epigallocatechin gallate on RPE cell migration and adhesion. Mol. Vis. 2010, 16, 586–595. [Google Scholar] [PubMed]
- Fallarero, A.; Ainasoja, M.; Sandberg, M.; Teeri, T.H.; Vuorela, P.M. GT1-7 cell-based cytoxicity screening assay on 96-well microplates as a platform for the safety assessment of genetically modified Gerbera hybrida extracts. Drug Chem. Toxicol. 2009, 32, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Leeds, P.; Chen, R.W.; Wei, W.; Leng, Y.; Bredesen, D.E.; Chuang, D.M. Neuronal apoptosis induced by pharmacological concentrations of 3-hydroxykynurenine: Characterization and protection by dantrolene and Bcl-2 overexpression. J. Neurochem. 2000, 75, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Sathishkumar, K.; Murthy, S.N.; Uppu, R.M. Cytotoxic effects of oxysterols produced during ozonolysis of cholesterol in murine GT1-7 hypothalamic neurons. Free Radic. Res. 2007, 41, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Sortino, M.A.; Battaglia, A.; Pamparana, F.; Carfagna, N.; Post, C.; Canonico, P.L. Neuroprotective effects of nicergoline in immortalized neurons. Eur. J. Pharmacol. 1999, 368, 285–290. [Google Scholar] [CrossRef]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Giaever, I.; Keese, C.R. Micromotion of mammalian cells measured electrically. Proc. Natl. Acad. Sci. USA 1991, 88, 7896–7900. [Google Scholar] [CrossRef] [PubMed]
- Wegener, J.; Keese, C.R.; Giaever, I. Electric cell-substrate impedance sensing (ECIS) as a noninvasive means to monitor the kinetics of cell spreading to artificial surfaces. Exp. Cell Res. 2000, 259, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Hillegass, J.M.; Shukla, A.; Lathrop, S.A.; MacPherson, M.B.; Fukagawa, N.K.; Mossman, B.T. Assessing nanotoxicity in cells in vitro. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2010, 2, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Calliste, C.A.; Trouillas, P.; Allais, D.P.; Simon, A.; Duroux, J.L. Free radical scavenging activities measured by electron spin resonance spectroscopy and B16 cell antiproliferative behaviors of seven plants. J. Agric. Food Chem. 2001, 49, 3321–3327. [Google Scholar] [CrossRef] [PubMed]
- Raudonė, L.; Raudonis, R.; Janulis, V.; Viškelis, P. Quality evaluation of different preparations of dry extracts of birch (Betula pendula Roth) leaves. Nat. Prod. Res. 2014, 28, 1645–1648. [Google Scholar] [CrossRef] [PubMed]
- Svobodová, A.; Walterová, D.; Psotová, J. Influence of silymarin and its flavonolignans on H2O2-induced oxidative stress in human keratinocytes and mouse fibroblasts. Burns 2006, 32, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Soria, E.A.; Eynard, A.R.; Bongiovanni, G.A. Cytoprotective effects of silymarin on epithelial cells against arsenic-induced apoptosis in contrast with quercetin cytotoxicity. Life Sci. 2010, 87, 309–315. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.; Augustin, K.M.; Nobbe, J.; Westervelt, P. Silybum marianum (milk thistle) in the management and prevention of hepatotoxicity in a patient undergoing reinduction therapy for acute myelogenous leukemia. J. Oncol. Pharm. Pract. 2012, 18, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Perrinjaquet-Moccetti, T.; Busjahn, A.; Schmidlin, C.; Schmidt, A.; Bradl, B.; Aydogan, C. Food supplementation with an olive (Olea europaea L.) leaf extract reduces blood pressure in borderline hypertensive monozygotic twins. Phytother. Res. 2008, 22, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Seddik, L.; Bah, T.M.; Aoues, A.; Slimani, M.; Benderdour, M. Elucidation of mechanisms underlying the protective effects of olive leaf extract against lead-induced neurotoxicity in Wistar rats. J. Toxicol. Sci. 2011, 36, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Shimazawa, M.; Chikamatsu, S.; Morimoto, N.; Mishima, S.; Nagai, H.; Hara, H. Neuroprotection by Brazilian green propolis against in vitro and in vivo ischemic neuronal damage. Evid. Based Complement. Altern. Med. 2005, 2, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Lopez, B.G.; de Lourenço, C.C.; Alves, D.A.; Machado, D.; Lancellotti, M.; Sawaya, A.C. Antimicrobial and cytotoxic activity of red propolis: An alert for its safe use. J. Appl. Microbiol. 2015, 119, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Shimazawa, M.; Mishima, S.; Hara, H. Water extract of propolis and its main constituents, caffeoylquinic acid derivatives, exert neuroprotective effects via antioxidant actions. Life Sci. 2007, 80, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Touriño, S.; Selga, A.; Jiménez, A.; Juliá, L.; Lozano, C.; Lizárraga, D.; Cascante, M.; Torres, J.L. Procyanidin fractions from pine (Pinus pinaster) bark: Radical scavenging power in solution, antioxidant activity in emulsion, and antiproliferative effect in melanoma cells. J. Agric. Food Chem. 2005, 53, 4728–4735. [Google Scholar] [CrossRef] [PubMed]
- Lizarraga, D.; Lozano, C.; Briedé, J.J.; van Delft, J.H.; Touriño, S.; Centelles, J.J.; Torres, J.L.; Cascante, M. The importance of polymerization and galloylation for the antiproliferative properties of procyanidin-rich natural extracts. FEBS J. 2007, 274, 4802–4811. [Google Scholar] [CrossRef] [PubMed]
- Mshvildadze, V.; Legault, J.; Lavoie, S.; Gauthier, C.; Pichette, A. Anticancer diarylheptanoid glycosides from the inner bark of Betula papyrifera. Phytochemistry 2007, 68, 2531–2536. [Google Scholar] [CrossRef] [PubMed]
- Liimatainen, J.; Karonen, M.; Sinkkonen, J.; Helander, M.; Salminen, J.-P. Characterization of phenolic compounds from inner bark of Betula pendula. Holzforschung 2012, 66, 171–181. [Google Scholar] [CrossRef]
- Karonen, M.; Ossipov, V.; Sinkkonen, J.; Loponen, J.; Haukioja, E.; Pihlaja, K. Quantitative analysis of polymeric proanthocyanidins in birch leaves with normal-phase HPLC. Phytochem. Anal. 2006, 17, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Karonen, M.; Liimatainen, J.; Sinkkonen, J. Birch inner bark procyanidins can be resolved with enhanced sensitivity by hydrophilic interaction HPLC-MS. J. Sep. Sci. 2011, 34, 3158–3165. [Google Scholar] [CrossRef] [PubMed]
- Mellon, P.L.; Windle, J.J.; Goldsmith, P.C.; Padula, C.A.; Roberts, J.L.; Weiner, R.I. Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 1990, 5, 1–10. [Google Scholar] [CrossRef]
- Loikkanen, J.J.; Naarala, J.; Savolainen, K.M. Modification of glutamate-induced oxidative stress by lead: The role of extracellular calcium. Free Radic. Biol. Med. 1998, 24, 377–384. [Google Scholar] [CrossRef]
- Bollini, S.; Herbst, J.J.; Gaughan, G.T.; Verdoorn, T.A.; Ditta, J.; Dubowchik, G.M.; Vinitsky, A. High-throughput fluorescence polarization method for identification of FKBP12 ligands. J. Biomol. Screen. 2002, 7, 526–530. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fallarero, A.; Batista-González, A.E.; Hiltunen, A.K.; Liimatainen, J.; Karonen, M.; Vuorela, P.M. Online Measurement of Real-Time Cytotoxic Responses Induced by Multi-Component Matrices, such as Natural Products, through Electric Cell-Substrate Impedance Sensing (ECIS). Int. J. Mol. Sci. 2015, 16, 27044-27057. https://doi.org/10.3390/ijms161126014
Fallarero A, Batista-González AE, Hiltunen AK, Liimatainen J, Karonen M, Vuorela PM. Online Measurement of Real-Time Cytotoxic Responses Induced by Multi-Component Matrices, such as Natural Products, through Electric Cell-Substrate Impedance Sensing (ECIS). International Journal of Molecular Sciences. 2015; 16(11):27044-27057. https://doi.org/10.3390/ijms161126014
Chicago/Turabian StyleFallarero, Adyary, Ana E. Batista-González, Anna K. Hiltunen, Jaana Liimatainen, Maarit Karonen, and Pia M. Vuorela. 2015. "Online Measurement of Real-Time Cytotoxic Responses Induced by Multi-Component Matrices, such as Natural Products, through Electric Cell-Substrate Impedance Sensing (ECIS)" International Journal of Molecular Sciences 16, no. 11: 27044-27057. https://doi.org/10.3390/ijms161126014
APA StyleFallarero, A., Batista-González, A. E., Hiltunen, A. K., Liimatainen, J., Karonen, M., & Vuorela, P. M. (2015). Online Measurement of Real-Time Cytotoxic Responses Induced by Multi-Component Matrices, such as Natural Products, through Electric Cell-Substrate Impedance Sensing (ECIS). International Journal of Molecular Sciences, 16(11), 27044-27057. https://doi.org/10.3390/ijms161126014