
Intra-Genomic Internal Transcribed Spacer Region Sequence Heterogeneity and Molecular Diagnosis in Clinical Microbiology

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Clinical Characteristics
2.2. Direct PCR Product Sequencing
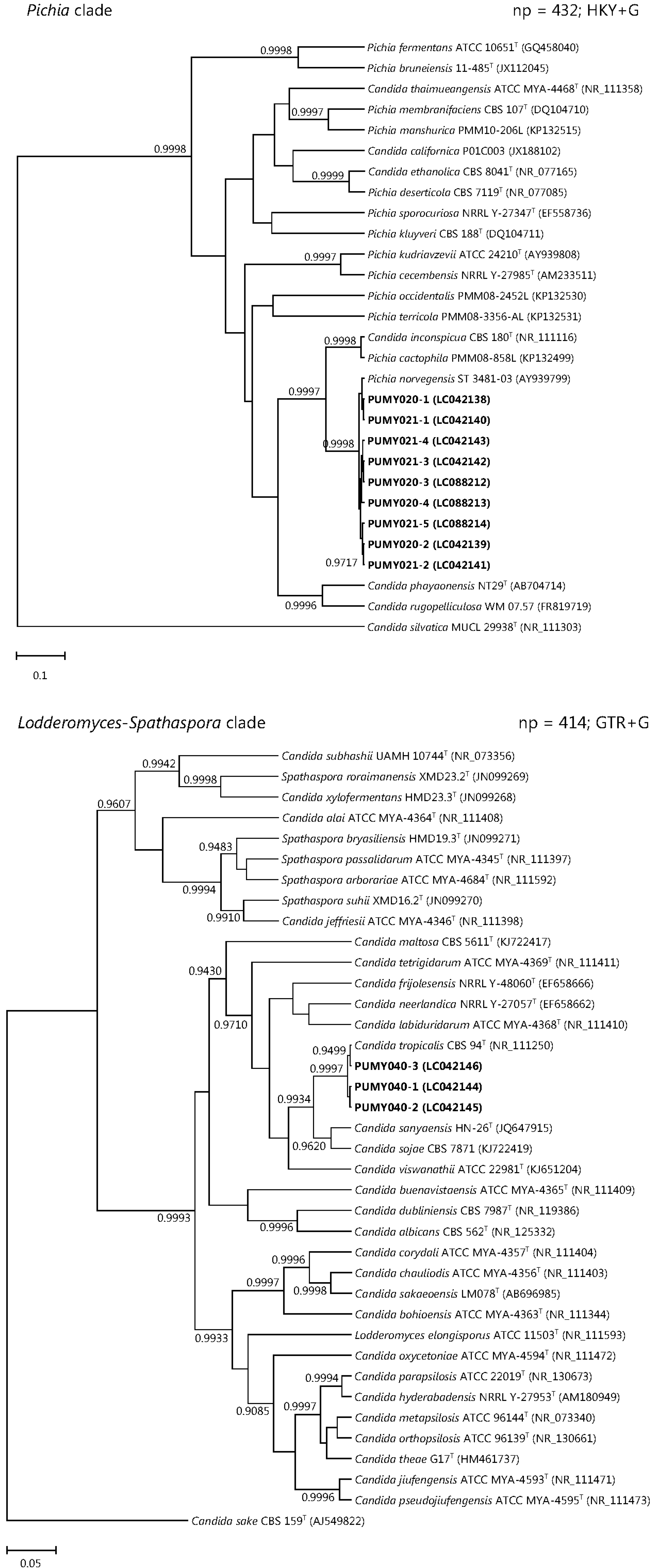
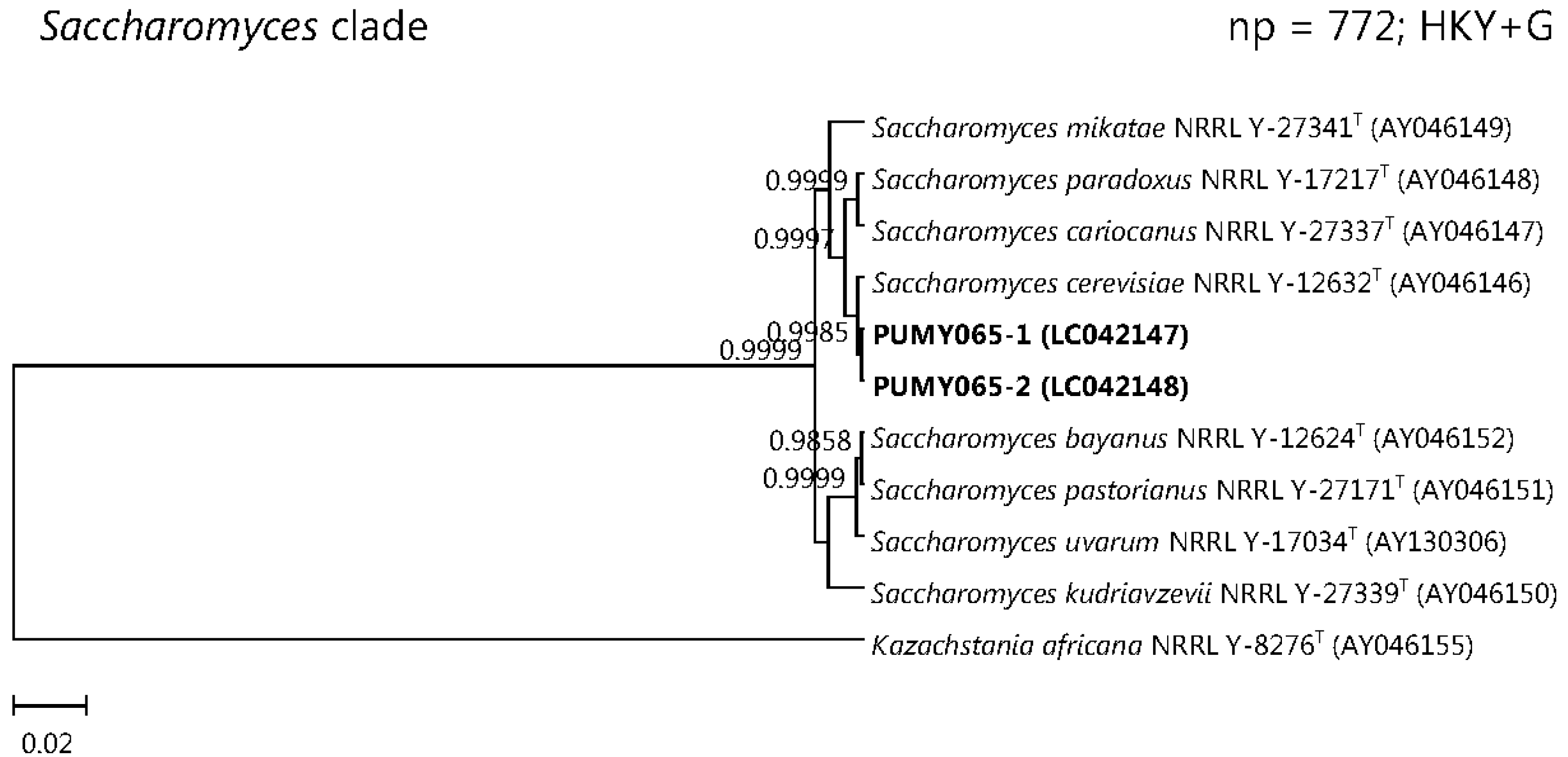
2.3. Sequencing of Cloned PCR Products and Phylogenetic Analyses



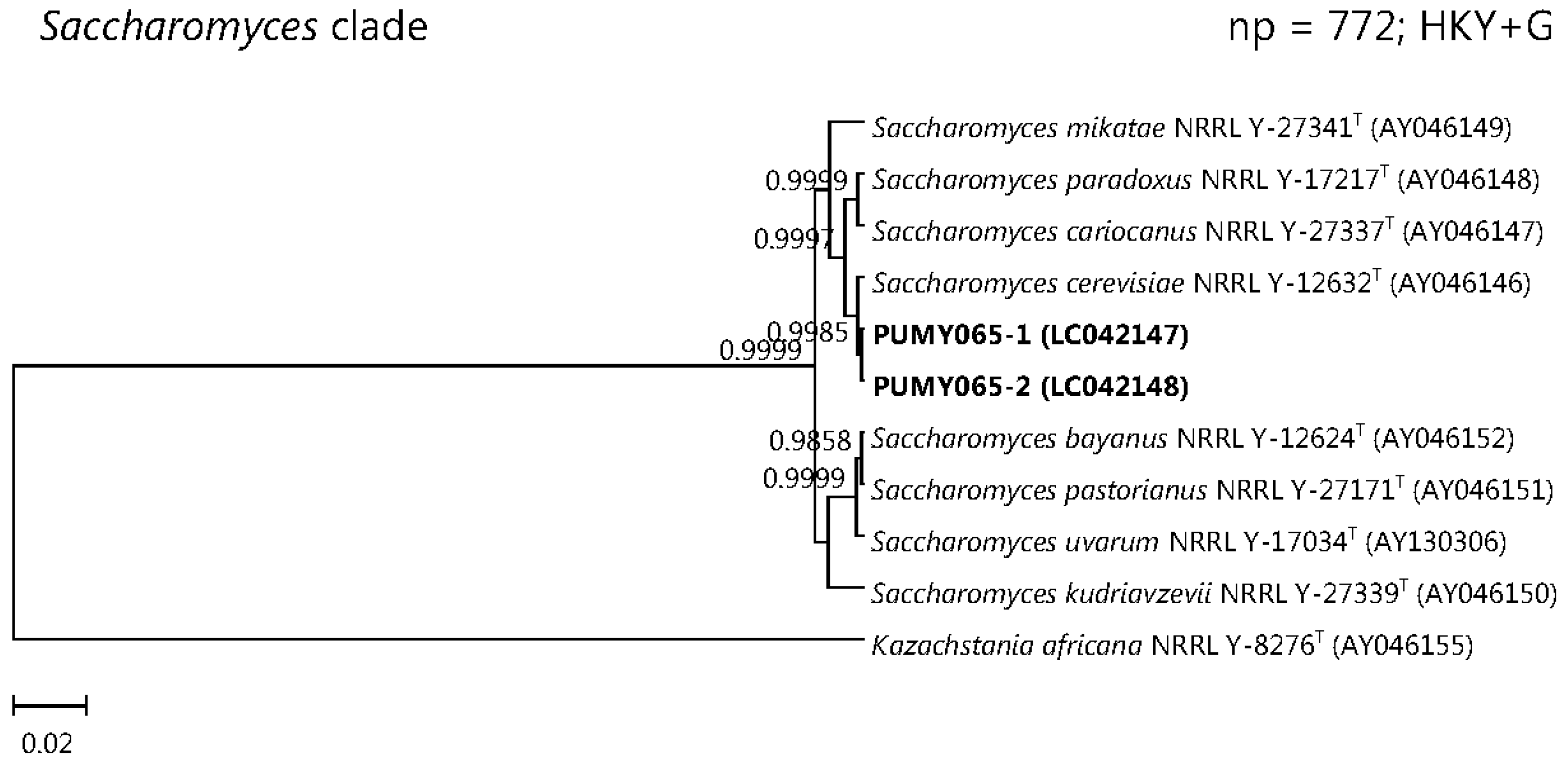
3. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Sex/Age a | Underlying Diseases b | Clinical Diagnosis | Clinical Specimens | Strains c | Number of Intra-Genomic ITS Copies d | Number of Intra-Genomic ITS Polymorphic Sites d | Location of Intra-Genomic ITS Polymorphic Sites d | Final Identification |
|---|---|---|---|---|---|---|---|---|---|
| 1 | F/30 | Carcinoma of cervix, post-total abdominal hysterectomy and bilateral salpingo-oophorectomy and radiotherapy | Suppurative peritonitis | Peritoneal fluid | PUMY010 | 7 | 4 | ITS1 and ITS2 | Candida glabrata |
| 2 | F/86 | N/A | Intestinal obstruction | Intraperitoneal drainage fluid | PUMY011 | 3 | 3 | ITS1 | Candida glabrata |
| 3 | M/66 | Type 2 diabetes mellitus | Esophageal perforation and mediastinitis | Pleural fluid | PUMY020 | 4 | 3 | ITS1 and ITS2 | Pichia (Candida) norvegensis |
| Blood | PUMY021 | 5 | 3 | ITS1 and ITS2 | Pichia (Candida) norvegensis | ||||
| 4 | M/60 | Chronic renal failure | Peritonitis | Peritoneal fluid | PUMY040 | 3 | 2 | ITS2 | Candida tropicalis |
| 5 | F/58 | Type 2 diabetes mellitus | Lung abscess and empyema | Empyema pus | PUMY065 | 2 | 1 | ITS1 | Saccharomyces cerevisiae |
4. Materials and Methods
4.1. Patients and Strains
4.2. DNA Extraction, PCR and Direct PCR Product Sequencing
4.3. Cloning and Sequencing
4.4. Comparative Sequence Identity Analyses and Phylogenetic Analyses
4.5. Nucleotide Sequence Accession Number
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interests
References
- Kwiatkowski, N.P.; Babiker, W.M.; Merz, W.G.; Carroll, K.C.; Zhang, S.X. Evaluation of nucleic acid sequencing of the D1/D2 region of the large subunit of the 28S rDNA and the internal transcribed spacer region using SmartGene IDNS software for identification of filamentous fungi in a clinical laboratory. J. Mol. Diagn. 2012, 14, 393–401. [Google Scholar] [CrossRef]
- Irinyi, L.; Serena, C.; Garcia-Hermoso, D.; Arabatzis, M.; Desnos-Ollivier, M.; Vu, D.; Cardinali, G.; Arthur, I.; Normand, A.C.; Giraldo, A.; et al. International Society of Human and Animal Mycology (ISHAM)-ITS reference DNA barcoding database—The quality controlled standard tool for routine identification of human and animal pathogenic fungi. Med. Mycol. 2015, 53, 313–337. [Google Scholar] [CrossRef] [PubMed]
- Black, J.; Dean, T.; Byfield, G.; Foarde, K.; Menetrez, M. Determining fungi rRNA copy number by PCR. J. Biomol. Tech. 2013, 24, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Carlson, E.; Pappagianis, D. Determination of ribosomal DNA copy number and comparison among strains of Coccidioides. Mycopathologia 2015, 179, 45–51. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.; Cigelnik, E. Two divergent intragenomic rDNA ITS2 types within a monophyletic lineage of the fungus Fusarium are nonorthologous. Mol. Phylogenet. Evol. 1997, 7, 103–116. [Google Scholar] [CrossRef]
- Ganley, A.R.D.; Kobayashi, T. Highly efficient concerted evolution in the ribosomal DNA repeats: Total rDNA repeat variation revealed by whole-genome shotgun sequence data. Genom. Res. 2007, 17, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Simon, U.K.; Weiß, M. Intragenomic variation of fungal ribosomal genes is higher than previously thought. Mol. Biol. Evol. 2008, 25, 2251–2254. [Google Scholar] [CrossRef] [PubMed]
- Alper, I.; Frenette, M.; Labrie, S. Ribosomal DNA polymorphisms in the yeast Geotrichum candidum. Fungal Biol. 2011, 115, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, K.; Steenkamp, E.T.; Coetzee, M.P.A.; Wingfield, M.J.; Wingfield, B.D. Concerted evolution in the ribosomal RNA cistron. PLoS ONE 2013, 8, e59355. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.M.; Yao, Y.J. Intrastrain internal transcribed spacer heterogeneity in Ganoderma species. Can. J. Microbiol. 2005, 51, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Pannecoucque, J.; Höfte, M. Detection of rDNA ITS polymorphism in Rhizoctonia solani AG 2-1 isolates. Mycologia 2009, 101, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Lindner, D.L.; Banik, M.T. Intragenomic variation in the ITS rDNA region obscures phylogenetic relationships and inflates estimates of operational taxonomic units in genus Laetiporus. Mycologia 2011, 103, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Vydryakova, G.A.; Van, D.T.; Shoukouhi, P.; Psurtseva, N.V.; Bissett, J. Intergenomic and intragenomic ITS sequence heterogeneity in Neonothopanus nambi (Agaricales) from Vietnam. Mycology 2011, 3, 89–99. [Google Scholar]
- Sanders, I.R.; Alt, M.; Groppe, K.; Boller, T.; Wiemken, A. Identification of ribosomal DNA polymorphisms among and within spores of the Glomales: Application to studies on the genetic diversity of arbuscular mycorrhizal fungal communities. New Phytol. 1995, 130, 419–427. [Google Scholar] [CrossRef]
- Lloyd-Macgilp, S.A.; Chambers, S.M.; Dodd, J.C.; Fitter, A.H.; Walker, C.; Young, J.P.W. Diversity of the ribosomal internal transcribed spacers within and among isolates of Glomus mosseae and related mycorrhizal fungi. New Phytol. 1996, 133, 103–111. [Google Scholar] [CrossRef]
- Redecker, D.; Thierfelder, H.; Walker, C.; Werner, D. Restriction analysis of PCR-amplified internal transcribed spacers of ribosomal DNA as a tool for species identification in different genera of the order Glomales. Appl. Environ. Microbiol. 1997, 63, 1756–1761. [Google Scholar] [PubMed]
- Lanfranco, L.; Delpero, M.; Bonfante, P. Intrasporal variability of ribosomal sequences in the endomycorrhizal fungus Gigaspora margarita. Mol. Ecol. 1999, 8, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Redecker, D.; Hijri, M.; Dulieu, H.; Sanders, I.R. Phylogenetic analysis of a dataset of fungal 5.8S rDNA sequences shows that highly divergent copies of internal transcribed spacers reported from Scutellospora castanea are of ascomycete origin. Fungal Genet. Biol. 1999, 28, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Hijri, M.; Hosny, M.; van Tuinen, D.; Dulieu, H. Intraspecific ITS polymorphism in Scutellospora castanea (Glomales, Zygomycota) is structured within multinucleate spores. Fungal Genet. Biol. 1999, 26, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Jansa, J.; Mozafar, A.; Banke, S.; McDonald, B.A.; Frossard, E. Intra- and intersporal diversity of ITS rDNA sequences in Glomus intraradices assessed by cloning and sequencing, and by SSCP analysis. Mycol. Res. 2002, 106, 670–681. [Google Scholar]
- O’Mahony, E.M.; Tay, W.T.; Paxton, R.J. Multiple rRNA variants in a single spore of the Microsporidian Nosema bombi. J. Eukaryot. Microbiol. 2007, 54, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Tay, W.T.; O’Mahony, E.M.; Paxton, R.J. Complete rRNA gene sequences reveal that the microsporidium Nosema bombi infects diverse bumblebee (Bombus spp.) hosts and contains multiple polymorphic sites. J. Eukaryot. Microbiol. 2005, 52, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Leung, S.Y.; To, K.K.W.; Chan, J.F.W.; Ngan, A.H.Y.; Cheng, V.C.C.; Lau, S.K.P.; Yuen, K.Y. Internal transcribed spacer region sequence heterogeneity in Rhizopus microsporus: implications for molecular diagnosis in clinical microbiology laboratories. J. Clin. Microbiol. 2010, 48, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Ngan, A.H.Y.; Tsang, C.C.C.; Ling, I.W.H.; Chan, J.F.W.; Leung, S.Y.; Yuen, K.Y.; Lau, S.K.P. Clinical spectrum of Exophiala infections and a novel Exophiala species, Exophiala hongkongensis. J. Clin. Microbiol. 2013, 51, 260–267. [Google Scholar] [CrossRef] [PubMed]
- White, T.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M., Gelfand, D., Shinsky, J., White, T., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Tsang, C.-C.; Xiao, M.; Cheng, J.; Xu, Y.; Lau, S.K.P.; Woo, P.C.Y. Intra-Genomic Internal Transcribed Spacer Region Sequence Heterogeneity and Molecular Diagnosis in Clinical Microbiology. Int. J. Mol. Sci. 2015, 16, 25067-25079. https://doi.org/10.3390/ijms161025067
Zhao Y, Tsang C-C, Xiao M, Cheng J, Xu Y, Lau SKP, Woo PCY. Intra-Genomic Internal Transcribed Spacer Region Sequence Heterogeneity and Molecular Diagnosis in Clinical Microbiology. International Journal of Molecular Sciences. 2015; 16(10):25067-25079. https://doi.org/10.3390/ijms161025067
Chicago/Turabian StyleZhao, Ying, Chi-Ching Tsang, Meng Xiao, Jingwei Cheng, Yingchun Xu, Susanna K. P. Lau, and Patrick C. Y. Woo. 2015. "Intra-Genomic Internal Transcribed Spacer Region Sequence Heterogeneity and Molecular Diagnosis in Clinical Microbiology" International Journal of Molecular Sciences 16, no. 10: 25067-25079. https://doi.org/10.3390/ijms161025067