The Role of 8-Oxoguanine DNA Glycosylase-1 in Inflammation


 and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Defense against the Accumulation of 8-oxo-7,8-Dihydroguanine (8-oxoG) in the Genome
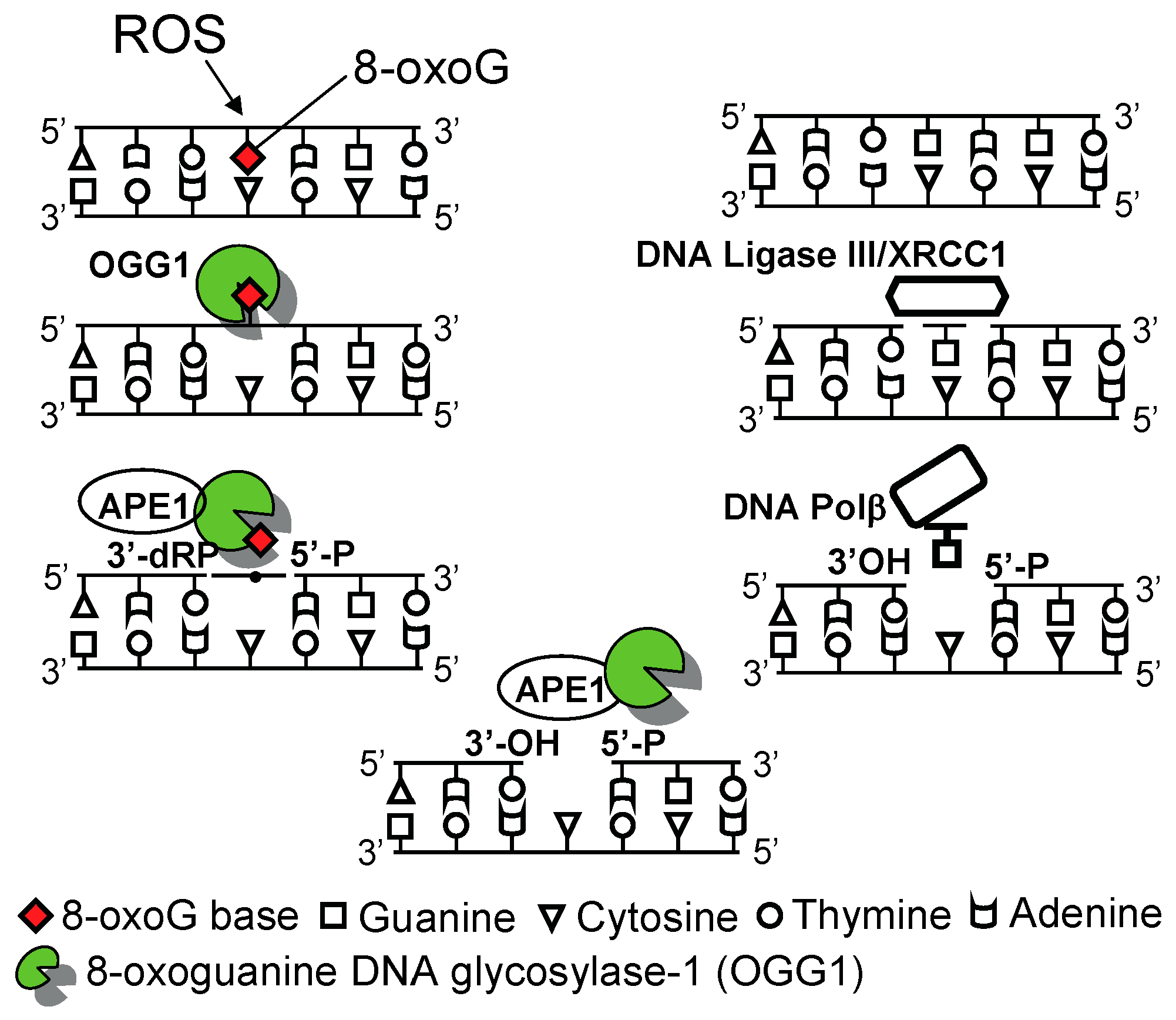
2.2. 8-oxoG Is a Biomarker of Oxidative Stress
2.2.1. 8-oxoG Is a Biomarker of Environmental Lung Exposures
2.2.2. Oxidatively Modified Guanine Lesions Are Signatures of Chronic Lung Inflammation
2.3. 8-Oxoguanine DNA Glycosylase-1 (OGG1): Role in Inflammatory Processes
2.3.1. Lack of 8-oxoG Repair by OGG1 Confers Resistance to Inflammation in Mouse Model
2.3.2. Decreased Allergic Immune Responses in the Lungs of Ogg1 Knockout (Ogg1−/−) Mice
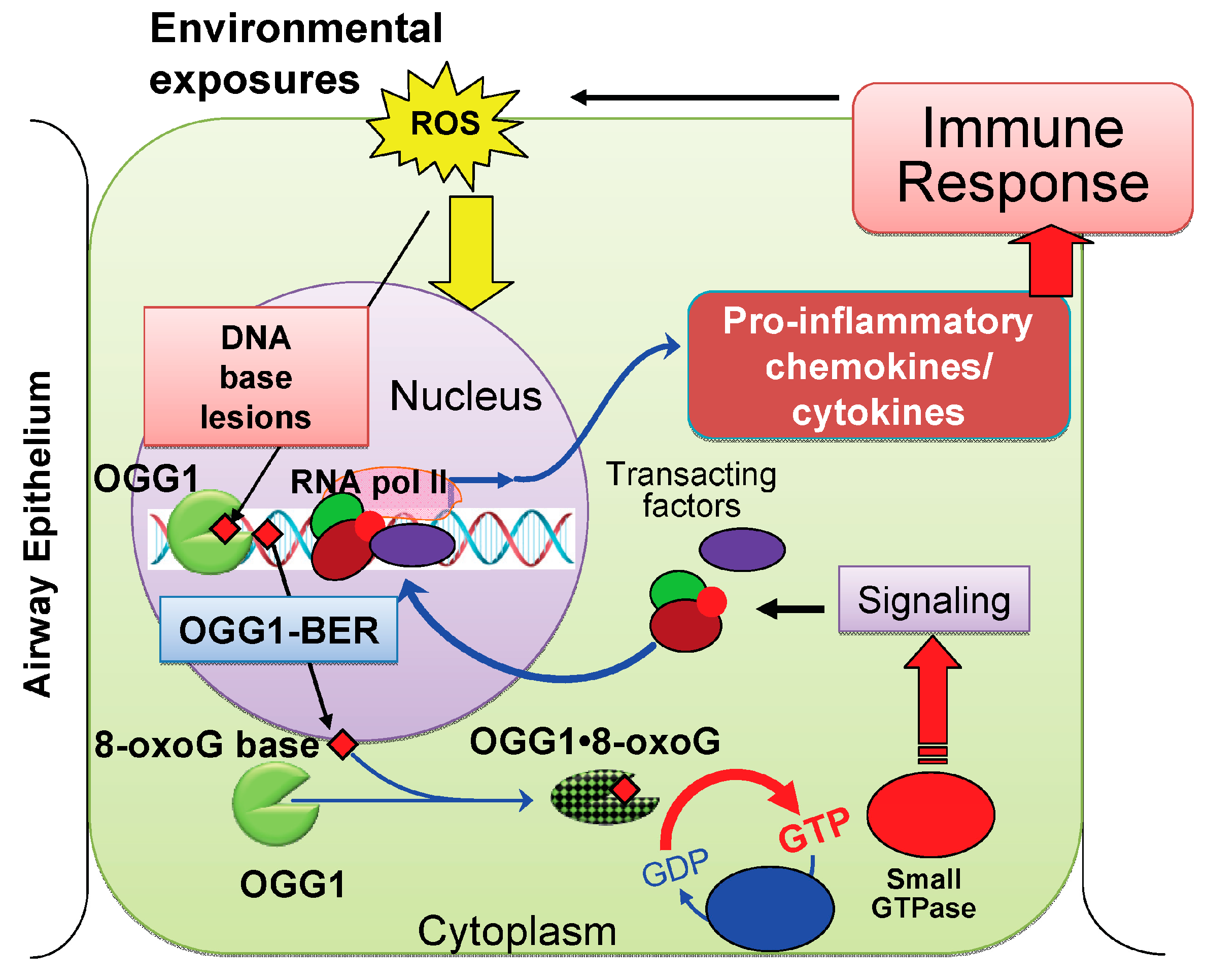
2.4. OGG1-Initiated DNA Base Excision Repair Pathway (OGG1–BER): A Link to Pro-Inflammatory Signaling
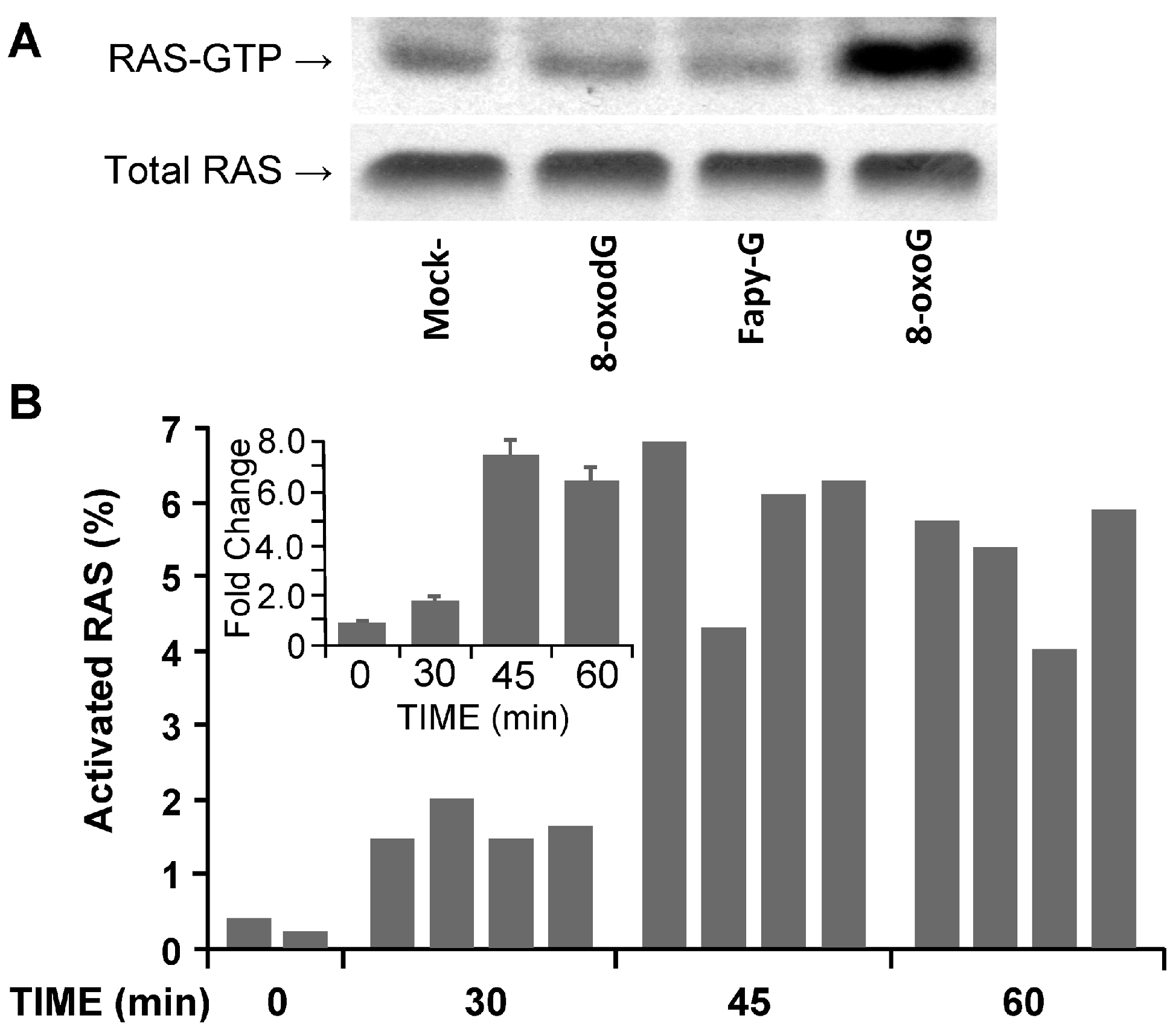
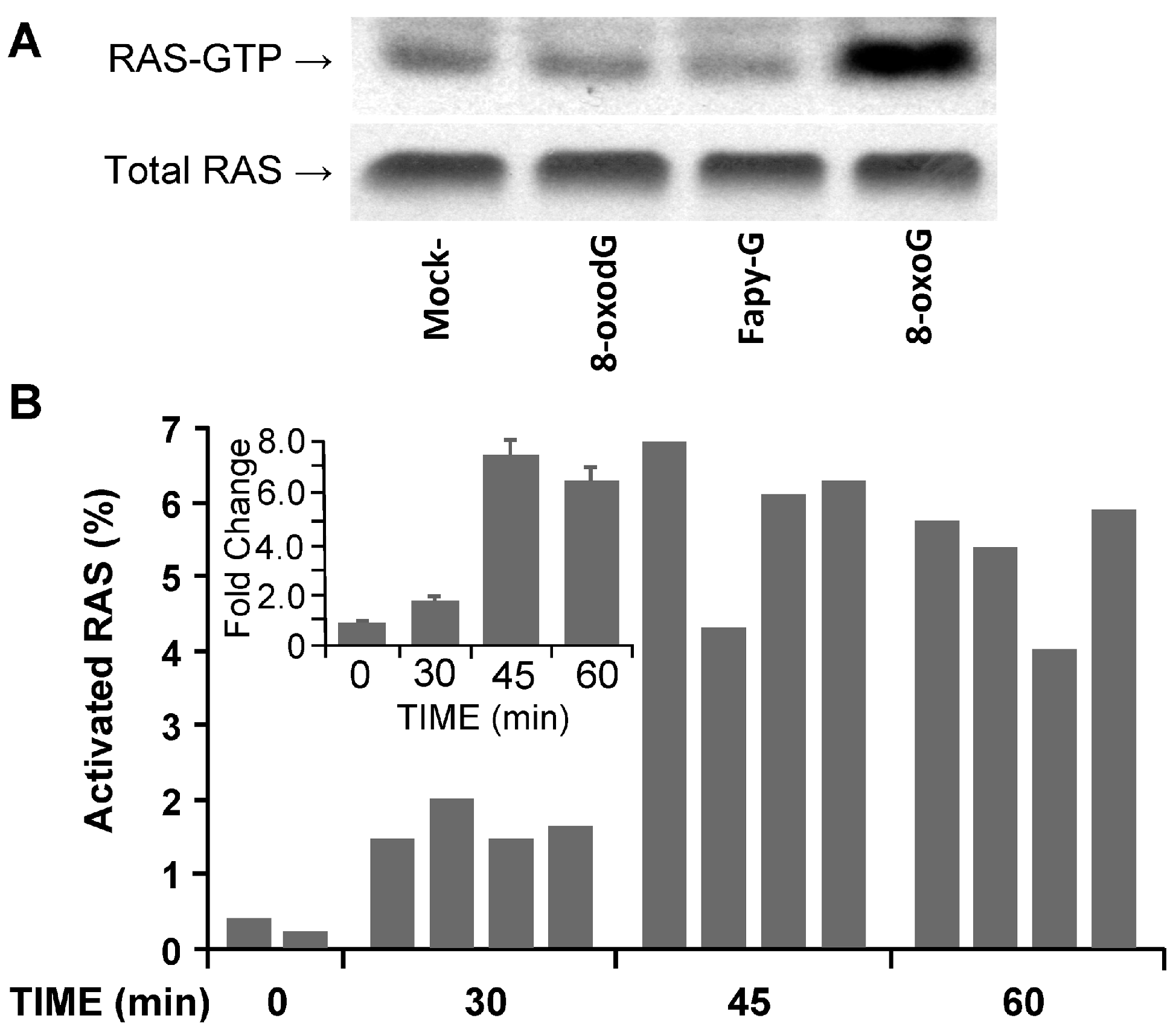
2.4.1. OGG1–BER Results in Activation of Small Guanosine Triphosphatases (GTPases)
2.4.2. Activation of Rat Sarcoma (RAS) Family GTPases by OGG1–BER Product 8-oxoG in Lungs
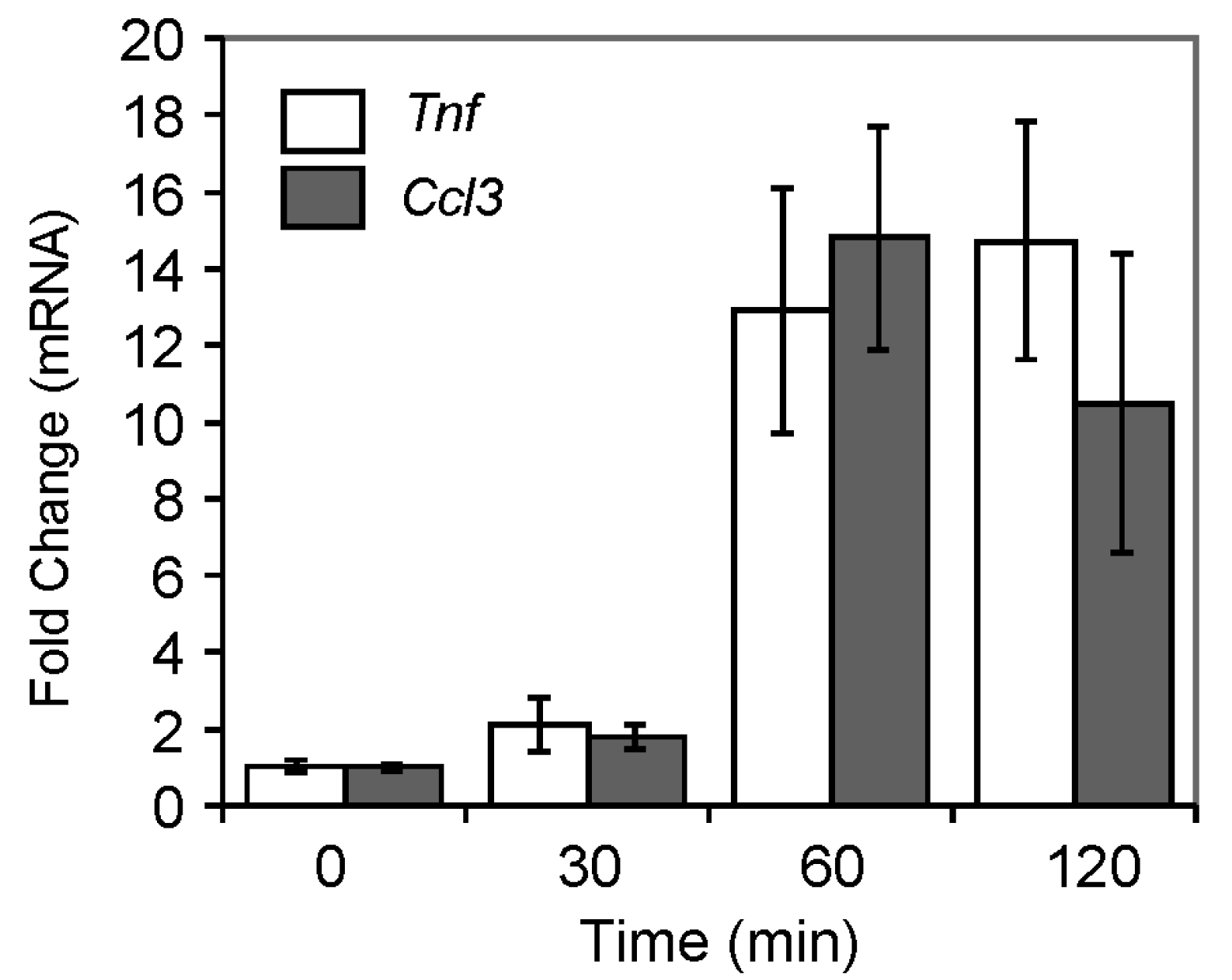
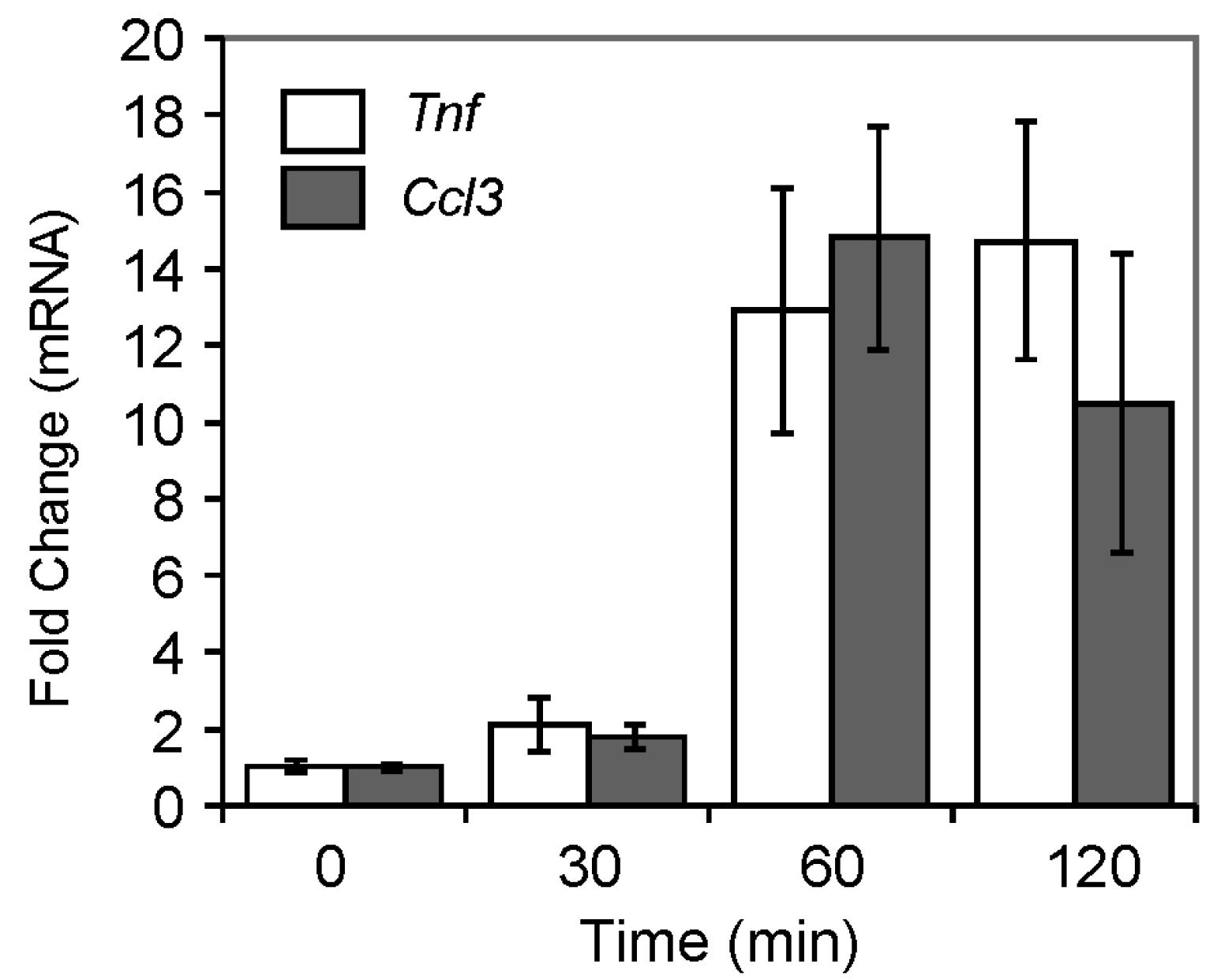
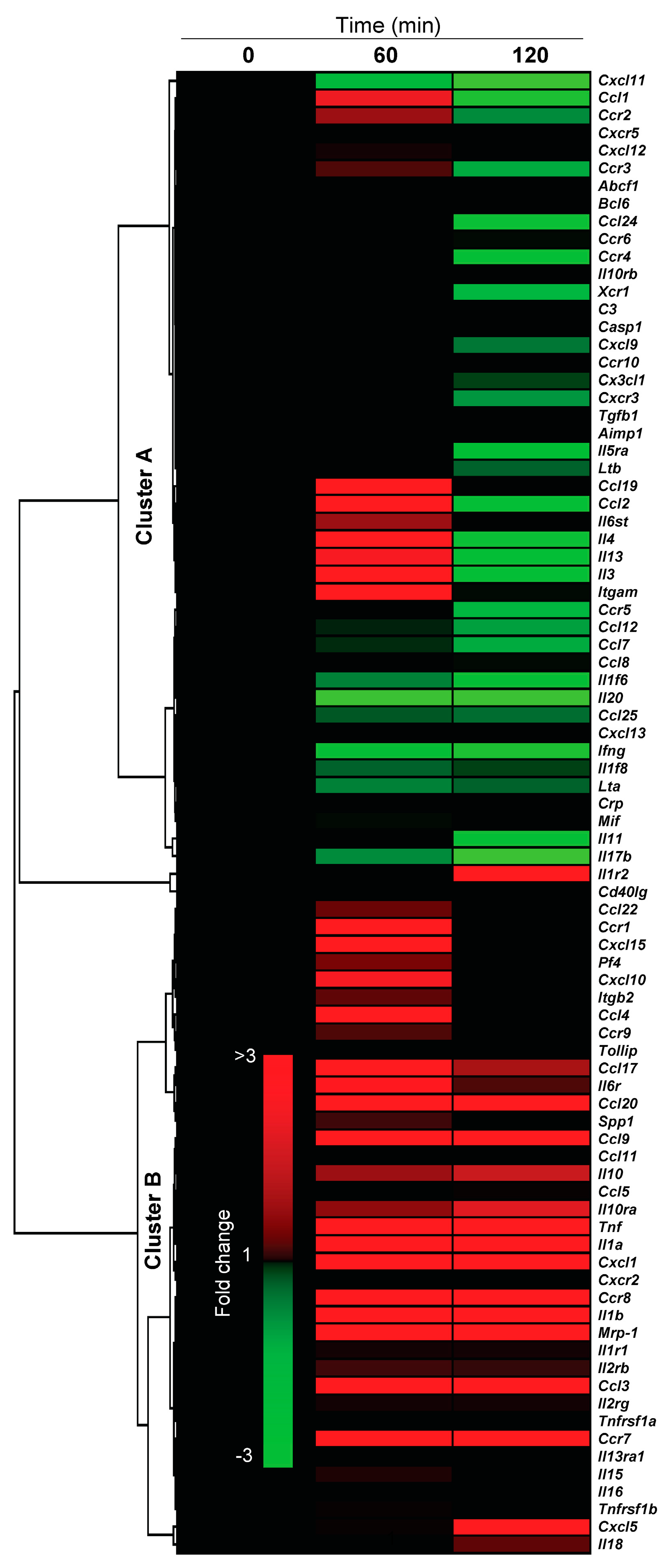
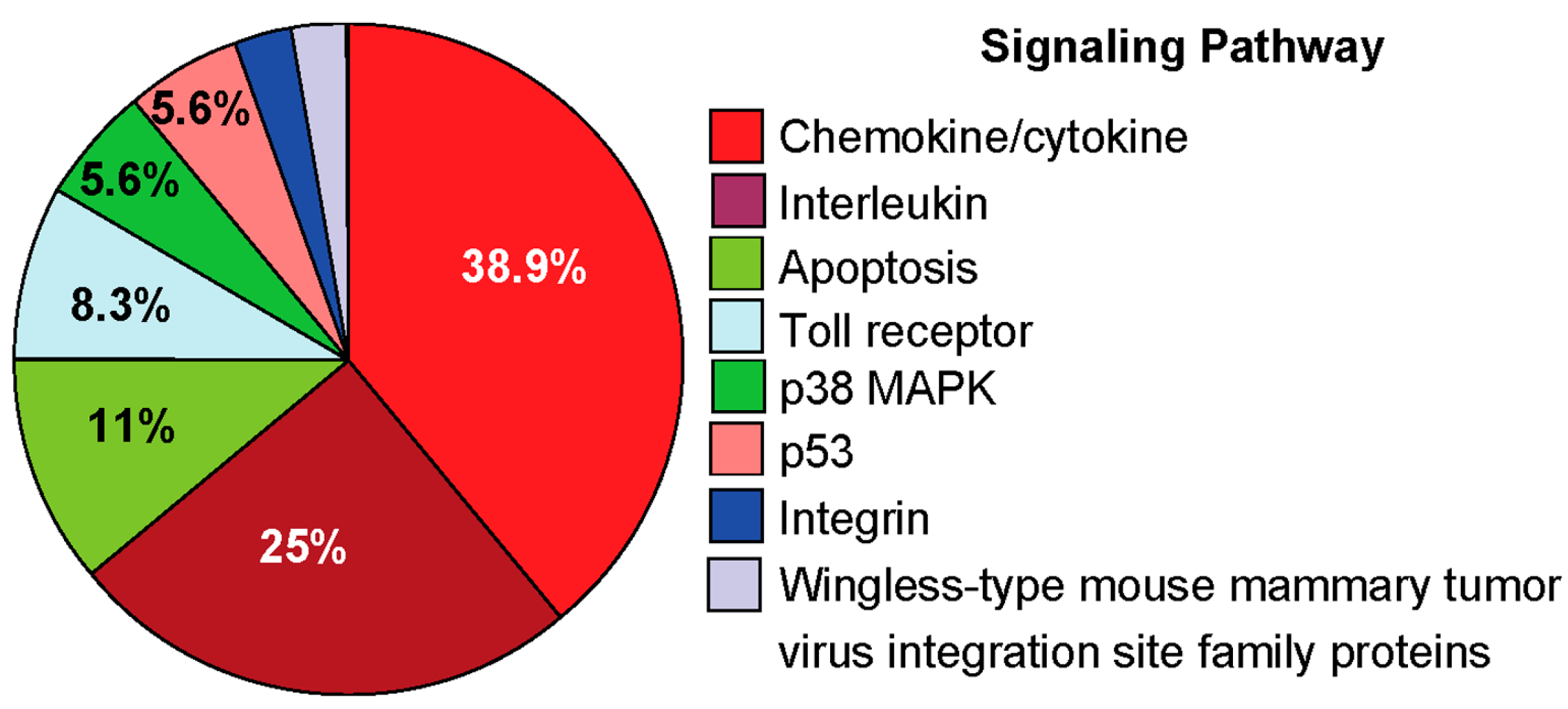
2.4.3. Pro-Inflammatory Gene Expression in the Lungs upon Challenge with 8-oxoG Base

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | RefSeq ID | Name | 60 min | 120 min |
|---|---|---|---|---|
| Fold Change | ||||
| Ccl17 | NM_011332 | Chemokine (C–C motif) ligand 17 | 2.25 | 1.62 |
| Ccl19 | NM_011888 | Chemokine (C–C motif) ligand 19 | 2.28 | −1.04 |
| Ccl20 | NM_016960 | Chemokine (C–C motif) ligand 20 | 7.30 | 5.02 |
| Ccl3 | NM_011337 | Chemokine (C–C motif) ligand 3 | 10.53 | 10.53 |
| Ccl4 | NM_013652 | Chemokine (C–C motif) ligand 4 | 2.16 | 1.02 |
| Ccl6 | NM_009139 | Chemokine (C–C motif) ligand 6 | 2.00 | 1.97 |
| Ccl9 | NM_011338 | Chemokine (C–C motif) ligand 9 | 2.09 | 2.22 |
| Ccr1 | NM_009912 | Chemokine (C–C motif) receptor 1 | 2.03 | 1.29 |
| Ccr7 | NM_007719 | Chemokine (C–C motif) receptor 7 | 2.50 | 2.28 |
| Ccr8 | NM_007720 | Chemokine (C–C motif) receptor 8 | 2.76 | 2.84 |
| Cxcl1 | NM_008176 | Chemokine (C–X–C motif) ligand 1 | 41.27 | 65.21 |
| Cxcl5 | NM_009141 | Chemokine (C–X–C motif) ligand 5 | 1.32 | 2.47 |
| IL1α | NM_010554 | Interleukin 1 α | 4.09 | 5.29 |
| IL1β | NM_008361 | Interleukin 1 β | 4.31 | 4.40 |
| IL1r2 | NM_010555 | Interleukin 1 receptor, type II | −1.07 | 2.01 |
| IL3 | NM_010556 | Interleukin 3 | 2.28 | −2.12 |
| IL4 | NM_021283 | Interleukin 4 | 2.10 | −2.47 |
| Itgam | NM_008401 | Integrin alpha M | 2.01 | −1.26 |
| Tnf | NM_013693 | Tumor necrosis factor | 7.90 | 9.93 |
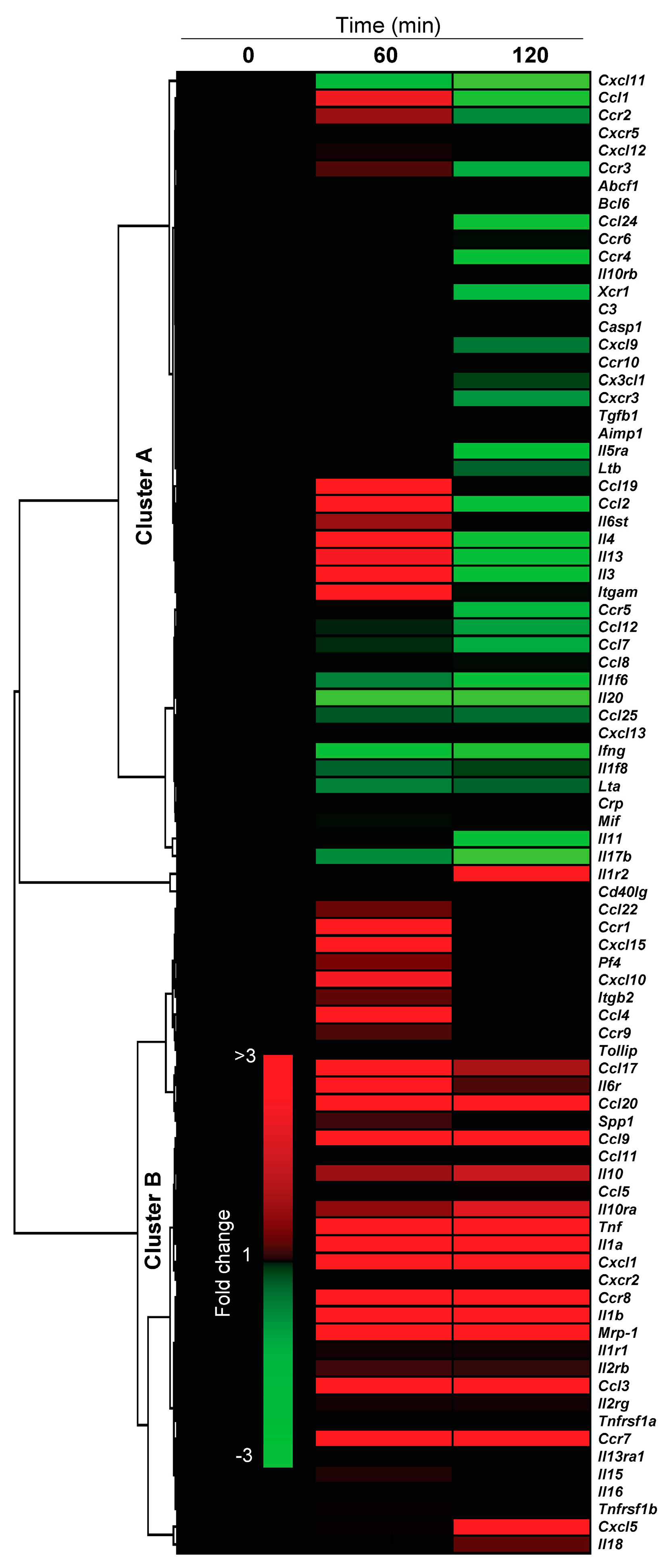
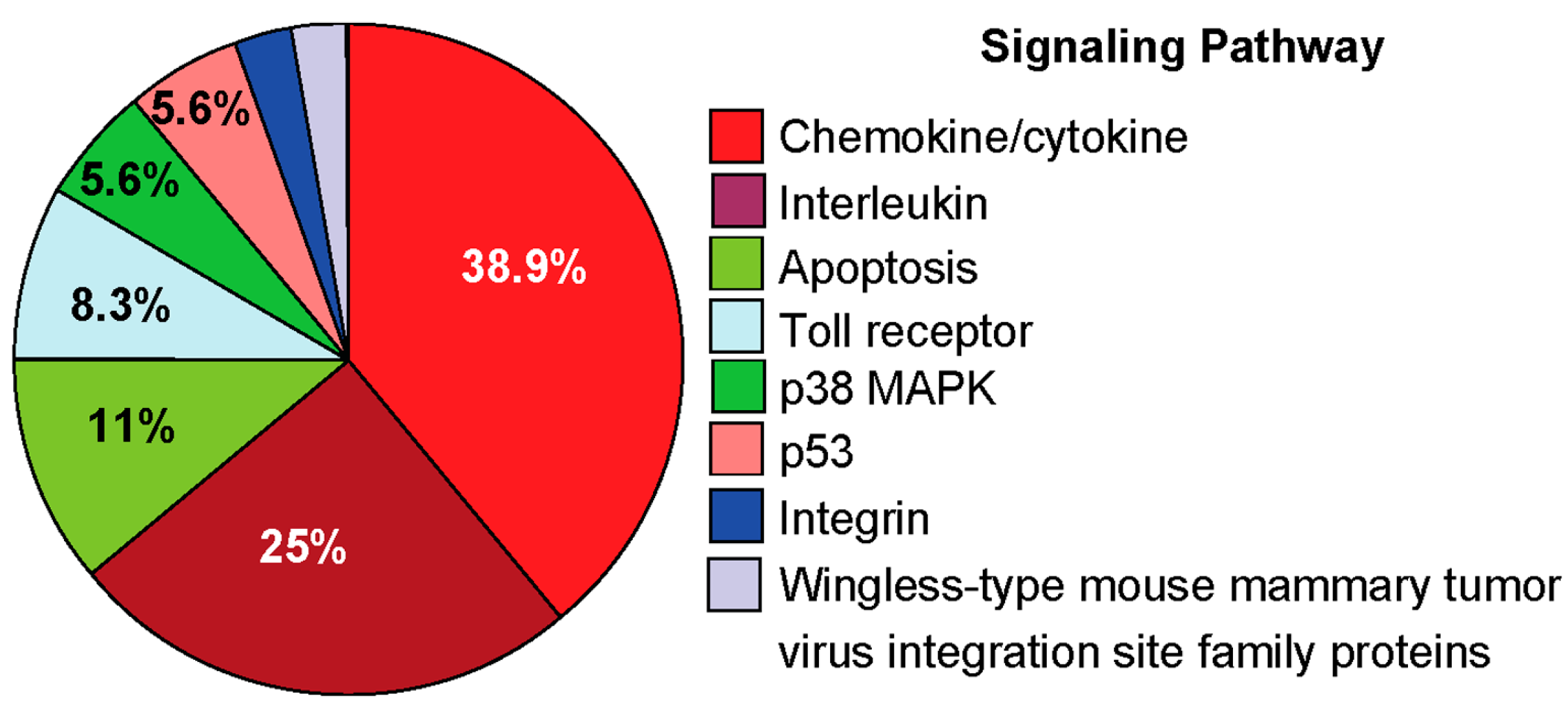
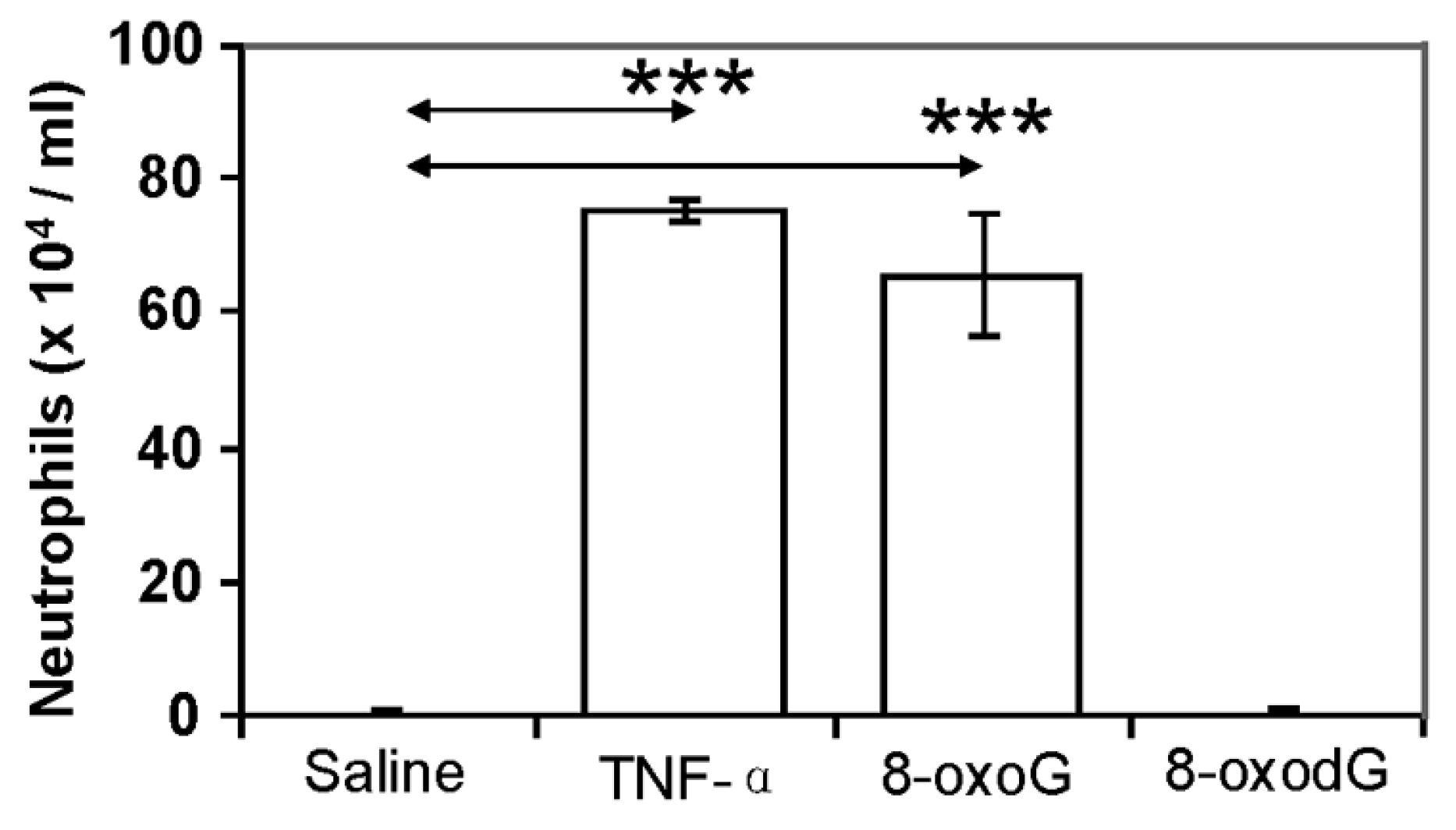
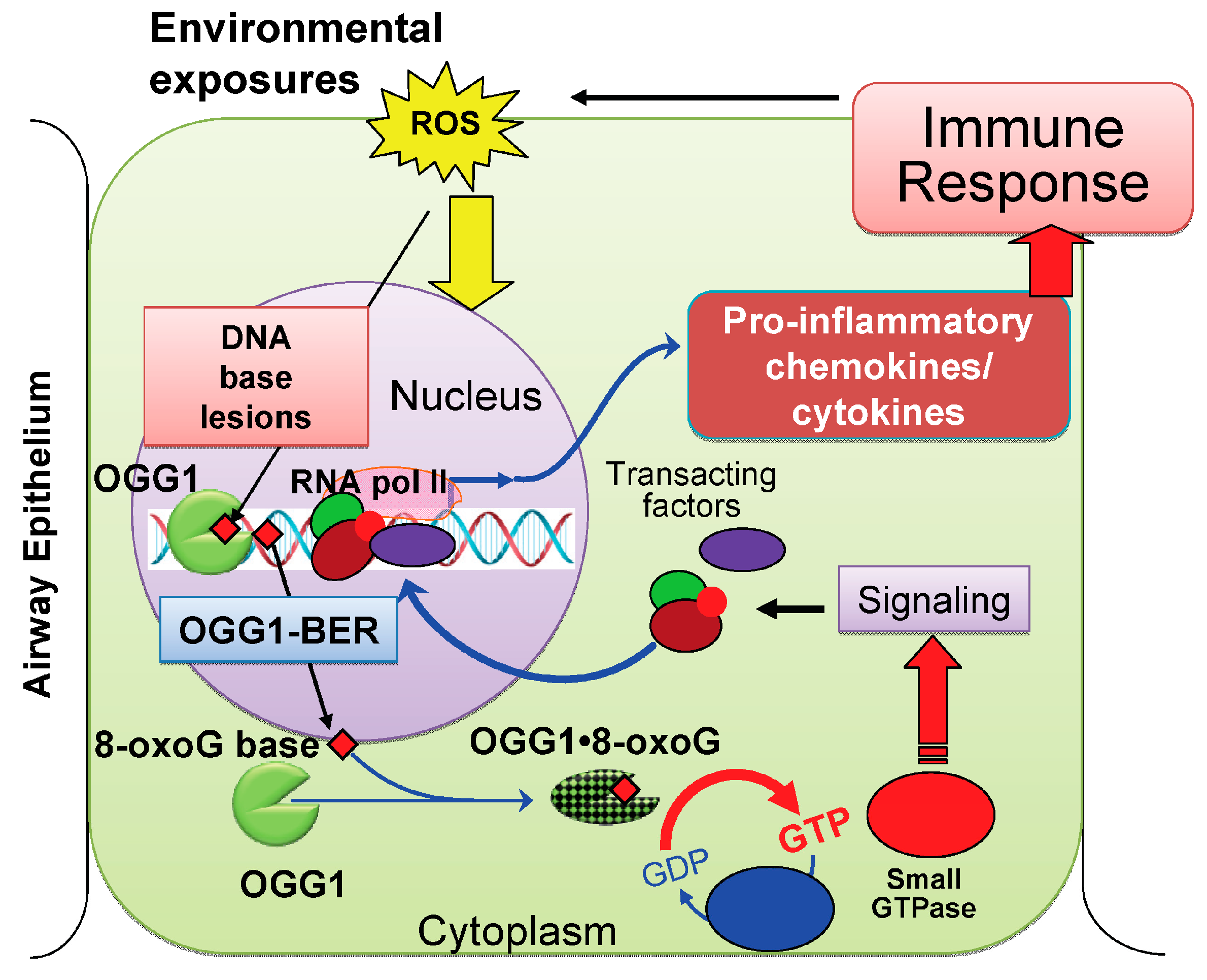
3. Conclusions
Abbreviations
| 8-oxoG | 8-oxo-7,8-dihydroguanine |
| 8-oxodG | 7,8-dihydro-8-oxo-2'-deoxyguanosine |
| AE | airway epithelia |
| AHR | airway hyperresponsiveness |
| APE1 | apurinic/apyrimidinic endonuclease 1 |
| AP sites | apurinic/apyrimidinic site; BER, base excision repair |
| BALF | bronchoalveolar lavage fluid |
| CXCL | C–X–C-motif containing chemokine ligands Cxcls, gene or mRNA encoding CXCLs |
| COPD | chronic obstructive pulmonary disease |
| DC | dendritic cells |
| FapyG | 2,6-diamino-4-hydroxy-5-formamidopyrimidine |
| GEF | Guanine nucleotide exchange factor |
| ERK1/2 | extracellular signal-regulated kinase 1/2 |
| H-Ras and K-Ras | mammalian homolog of Harvey and Kirsten sarcoma virus oncogene |
| IIR | innate immune response |
| IL | interleukin |
| LPS | lipopolysaccharides |
| N-Ras | Neuroblastoma RAS viral oncogene homolog |
| MAPK | mitogen activated kinase(s) |
| MEK1/2 | mitogen-activated kinase, kinase 1/2 |
| OGG1 | 8-oxoguanine DNA glycosylase-1 |
| OGG1D | deficient in OGG1 expression |
| OGG1P | Proficient in OGG1 expression |
| OS | oxidative stress |
| RHO | Ras homology (Rho) family of small GTPases |
| RAC1 | Ras-related C3 botulinum toxin substrate 1 GTPase |
| ROS | reactive oxygen species |
| PI3K | phosphatidyl inositol 3 kinase |
| RWPE | ragweed pollen grain extract |
| SSBs | DNA single-strand breaks |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Holgate, S.T.; Roberts, G.; Arshad, H.S.; Howarth, P.H.; Davies, D.E. The role of the airway epithelium and its interaction with environmental factors in asthma pathogenesis. Proc. Am. Thorac. Soc. 2009, 6, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Immunology of asthma and chronic obstructive pulmonary disease. Nat. Rev. Immunol. 2008, 8, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Holgate, S.T. The sentinel role of the airway epithelium in asthma pathogenesis. Immunol. Rev. 2011, 242, 205–219. [Google Scholar] [CrossRef] [PubMed]
- D’Autreaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell. Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S. DNA glycosylases: Specificity and mechanisms. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 189–192. [Google Scholar] [PubMed]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Boldogh, I. 8-oxo-7,8-Dihydroguanine: Links to gene expression, aging, and defense against oxidative stress. Free Radic. Biol. Med. 2010, 49, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Margolin, Y.; Cloutier, J.F.; Shafirovich, V.; Geacintov, N.E.; Dedon, P.C. Paradoxical hotspots for guanine oxidation by a chemical mediator of inflammation. Nat. Chem. Biol. 2006, 2, 365–366. [Google Scholar] [CrossRef] [PubMed]
- Steenken, S.; Jovanovic, S.V. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J. Am. Chem. Soc. 1997, 119, 617–618. [Google Scholar] [CrossRef]
- Hickerson, R.P.; Prat, F.; Muller, J.G.; Foote, C.S.; Burrows, C.J. Sequence and stacking dependence of 8-oxoguanine oxidation: Comparison of one-electron vs. singlet oxygen mechanisms. singlet oxygen mechanisms. Am. Chem. Soc. 1999, 121, 9423–9428. [Google Scholar] [CrossRef]
- Burrows, C.J.; Muller, J.G. Oxidative nucleobase modifications leading to strand scission. Chem. Rev. 1998, 98, 1109–1152. [Google Scholar] [CrossRef] [PubMed]
- Kino, K.; Sugiyama, H. GC->CG transversion mutation might be caused by 8-oxoguanine oxidation product. Nucleic Acids Symp. Ser. 2000, 44, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M. Formation of an 8-hydroxyguanine moiety in deoxyribonucleic acid on gamma-irradiation in aqueous solution. Biochemistry 1985, 24, 4476–4481. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.B.; Holmlin, R.E.; Barton, J.K. Oxidative DNA damage through long-range electron transfer. Nature 1996, 382, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Izumi, T.; Wiederhold, L.R.; Roy, G.; Roy, R.; Jaiswal, A.; Bhakat, K.K.; Mitra, S.; Hazra, T.K. Mammalian DNA base excision repair proteins: Their interactions and role in repair of oxidative DNA damage. Toxicology 2003, 193, 43–65. [Google Scholar] [CrossRef]
- Dizdaroglu, M.; Kirkali, G.; Jaruga, P. Formamidopyrimidines in DNA: Mechanisms of formation, repair, and biological effects. Free Radic. Biol. Med. 2008, 45, 1610–1621. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, K.; Ohtsubo, T.; Oda, H.; Fujiwara, T.; Kang, D.; Sugimachi, K.; Nakabeppu, Y. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell 1999, 10, 1637–1652. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Hazra, T.K.; Roy, R.; Ikeda, S.; Biswas, T.; Lock, J.; Boldogh, I.; Izumi, T. Complexities of DNA base excision repair in mammalian cells. Mol. Cells 1997, 7, 305–312. [Google Scholar] [PubMed]
- Bruner, S.D.; Norman, D.P.; Verdine, G.L. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature 2000, 403, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Yang, W.; Karplus, M.; Verdine, G.L. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature 2005, 434, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Nehls, P.; Seiler, F.; Rehn, B.; Greferath, R.; Bruch, J. Formation and persistence of 8-oxoguanine in rat lung cells as an important determinant for tumor formation following particle exposure. Environ. Health Perspect. 1997, 105, S1291–S1296. [Google Scholar] [CrossRef]
- Lunec, J. ESCODD: European standards committee on oxidative DNA damage. Free Radic. Res. 1998, 29, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Seiler, J.P. Chirality—From molecules to organisms. Arch. Toxicol. 1995, 17, 491–498. [Google Scholar]
- Loft, S.; Poulsen, H.E.; Vistisen, K.; Knudsen, L.E. Increased urinary excretion of 8-oxo-2'-deoxyguanosine, a biomarker of oxidative DNA damage, in urban bus drivers. Mutat. Res. 1999, 441, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Pang, B.; McFaline, J.L.; Taghizadeh, K.; Dedon, P.C. Surveying the damage: The challenges of developing nucleic acid biomarkers of inflammation. Mol. Biosyst. 2008, 4, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Barbato, D.L.; Tomei, G.; Tomei, F.; Sancini, A. Traffic air pollution and oxidatively generated DNA damage: Can urinary 8-oxo-7,8-dihydro-2-deoxiguanosine be considered a good biomarker? A meta-analysis. Biomarkers 2010, 15, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Inoue, Y.; Suzuki, S. Changes in the urinary excretion level of 8-hydroxyguanine by exposure to reactive oxygen-generating substances. Free Radic. Biol. Med. 1995, 18, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.W.; Chen, M.L.; Lung, S.C.; Tsai, C.J.; Yin, X.J.; Mao, I.F. Exposure assessment of PM2.5 and urinary 8-OHdG for diesel exhaust emission inspector. Sci. Total Environ. 2010, 408, 505–510. [Google Scholar]
- Wei, Y.; Han, I.K.; Hu, M.; Shao, M.; Zhang, J.J.; Tang, X. Personal exposure to particulate PAHs and anthraquinone and oxidative DNA damages in humans. Chemosphere 2010, 81, 1280–1285. [Google Scholar] [CrossRef] [PubMed]
- Malayappan, B.; Garrett, T.J.; Segal, M.; Leeuwenburgh, C. Urinary analysis of 8-oxoguanine, 8-oxoguanosine, fapy-guanine and 8-oxo-2'-deoxyguanosine by high-performance liquid chromatography–electrospray tandem mass spectrometry as a measure of oxidative stress. J. Chromatogr. A 2007, 1167, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Andreoli, R.; Protano, C.; Manini, P.; de Palma, G.; Goldoni, M.; Petyx, M.; Rondinone, B.M.; Vitali, M.; Mutti, A. Association between environmental exposure to benzene and oxidative damage to nucleic acids in children. Med. Lav. 2012, 103, 324–337. [Google Scholar]
- Tsurudome, Y.; Hirano, T.; Yamato, H.; Tanaka, I.; Sagai, M.; Hirano, H.; Naqata, N.; Itoh, H.; Kasai, H. Changes in levels of 8-hydroxyguanine in DNA, its repair and OGG1 mRNA in rat lungs after intratracheal administration of diesel exhaust particles. Carcinogenesis 1999, 20, 1573–1576. [Google Scholar] [CrossRef] [PubMed]
- Moller, P.; Danielsen, P.H.; Jantzen, K.; Roursgaard, M.; Loft, S. Oxidatively damaged DNA in animals exposed to particles. Crit. Rev. Toxicol. 2013, 43, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Gernez, Y.; Tirouvanziam, R.; Chanez, P. Neutrophils in chronic inflammatory airway diseases: Can we target them and how? Respir. J. 2010, 35, 467–469. [Google Scholar] [CrossRef]
- Yu, S.; Kim, H.Y.; Chang, Y.J.; DeKruyff, R.H.; Umetsu, D.T. Innate lymphoid cells and asthma. J. Allergy Clin. Immunol. 2014, 133, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Battaile, J.T.; Patel, A.C.; You, Y.; Agapov, E.; Grayson, M.H.; Benoit, L.A.; Byers, D.E.; Alevy, Y.; Tucker, J.; et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat. Med. 2008, 14, 633–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.Y.; DeKruyff, R.H.; Umetsu, D.T. The many paths to asthma: Phenotype shaped by innate and adaptive immunity. Nat. Immunol. 2010, 11, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Balmes, J.R.; Aris, R.M.; Chen, L.L.; Scannell, C.; Tager, I.B.; Finkbeiner, W.; Christian, D.; Kelly, T.; Hearne, P.Q. Effects of ozone on normal and potentially sensitive human subjects. Part I: Airway inflammation and responsiveness to ozone in normal and asthmatic subjects. Res. Rep. Health Eff. Inst. 1997, 78, 1–37. [Google Scholar]
- Ilumets, H.; Rytila, P.H.; Sovijarvi, A.R.; Tervahartiala, T.; Myllarniemi, M.; Sorsa, A.R.; Kinnula, V.L. Transient elevation of neutrophil proteinases in induced sputum during COPD exacerbation. Scand. J. Clin. Lab. Investig. 2008, 68, 618–623. [Google Scholar] [CrossRef]
- Kirkham, P.; Rahman, I. Oxidative stress in asthma and COPD: Antioxidants as a therapeutic strategy. Pharmacol. Ther. 2006, 111, 476–494. [Google Scholar] [CrossRef] [PubMed]
- Zeyrek, D.; Cakmak, A.; Atas, A.; Kocyigit, A.; Erel, O. DNA damage in children with asthma bronchiale and its association with oxidative and antioxidative measurements. Pediatr. Allergy Immunol. 2009, 20, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Hasbal, C.; Aksu, B.Y.; Himmetoglu, S.; Dincer, Y.; Koc, E.E.; Hatipoqlu, S.; Akcay, T. DNA damage and glutathione level in children with asthma bronchiale: Effect of antiasthmatic therapy. Pediatr. Allergy Immunol. 2010, 21, e674–e678. [Google Scholar] [CrossRef] [PubMed]
- Al-Afaleg, N.O.; Al-Senaidy, A.; El-Ansary, A. Oxidative stress and antioxidant status in Saudi asthmatic patients. Clin. Biochem. 2011, 44, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Proklou, A.; Soulitzis, N.; Neofytou, E.; Rovina, N.; Zervas, E.; Gaga, M.; Siafakas, N.M.; Tzortzaki, E.G. Granule cytotoxic activity and oxidative DNA damage in smoking and nonsmoking patients with asthma. Chest 2013, 144, 1230–1237. [Google Scholar] [PubMed]
- Deslee, G.; Woods, J.C.; Moore, C.; Conradi, S.H.; Gierada, D.S.; Atkinson, J.J.; Battaile, J.T.; Liu, L.; Patterson, G.A.; Adair-Kirk, T.L.; et al. Oxidative damage to nucleic acids in severe emphysema. Chest 2009, 135, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Deslee, G.; Adair-Kirk, T.L.; Betsuyaku, T.; Woods, J.C.; Moore, C.H.; Gierada, D.S.; Conradi, S.H.; Atkinson, J.J.; Toennies, H.M.; Battaile, J.T.; et al. Cigarette smoke induces nucleic acid oxidation in lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 2010, 43, 576–584. [Google Scholar] [CrossRef]
- Igishi, T.; Hitsuda, Y.; Kato, K.; Sako, T.; Burioka, N.; Yasuda, K.; Sano, H.; Shigeoka, Y.; Nakanishi, H.; Shimizu, E. Elevated urinary 8-hydroxydeoxyguanosine, a biomarker of oxidative stress, and lack of association with antioxidant vitamins in chronic obstructive pulmonary disease. Respirology 2003, 8, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Caramori, G.; Adcock, I.M.; Casolari, P.; Ito, K.; Jazrawi, E.; Tsaprouni, L.; Villetti, G.; Civelli, M.; Carnini, C.; Chung, K.F.; et al. Unbalanced oxidant-induced DNA damage and repair in COPD: A link towards lung cancer. Thorax 2011, 66, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Mabalirajan, U.; Rehman, R.; Singh, B.K.; Parmar, V.S.; Prasad, A.K.; Biswal, S.; Ghosh, B. A novel cinnamate derivative attenuates asthma features and reduces bronchial epithelial injury in mouse model. Int. Immunopharmacol. 2013, 15, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Klungland, A.; Rosewell, I.; Hollenbach, S.; Larsen, E.; Daly, G.; Epe, B.; Seeberg, E.; Lindahl, T.; Barnes, D.E. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc. Natl. Acad. Sci. USA 1999, 96, 13300–13305. [Google Scholar] [CrossRef] [PubMed]
- Minowa, O.; Arai, T.; Hirano, M.; Monden, Y.; Nakai, S.; Fukuda, M.; Itoh, M.; Takano, H.; Hippou, Y.; Aburatani, H.; et al. Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 4156–4161. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Kelly, V.P.; Komoro, K.; Minowa, O.; Noda, T.; Nishimura, S. Cell proliferation in liver of Mmh/Ogg1-deficient mice enhances mutation frequency because of the presence of 8-hydroxyguanine in DNA. Cancer Res. 2003, 63, 4287–4292. [Google Scholar] [PubMed]
- Arai, T.; Kelly, V.P.; Minowa, O.; Noda, T.; Nishimura, S. High accumulation of oxidative DNA damage, 8-hydroxyguanine, in Mmh/Ogg1 deficient mice by chronic oxidative stress. Carcinogenesis 2002, 23, 2005–2010. [Google Scholar] [CrossRef] [PubMed]
- De Souza-Pinto, N.C.; Eide, L.; Hogue, B.A.; Thybo, T.; Stevnsner, T.; Seeberg, E.; Klungland, A.; Bohr, V.A. Repair of 8-oxodeoxyguanosine lesions in mitochondrial dna depends on the oxoguanine dna glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial dna of OGG1-defective mice. Cancer Res. 2001, 61, 5378–5381. [Google Scholar]
- Touati, E.; Michel, V.; Thiberge, J.M.; Ave, P.; Huerre, M.; Bourgade, F.; Klungland, A.; Labigne, A. Deficiency in OGG1 protects against inflammation and mutagenic effects associated with H. pylori infection in mouse. Helicobacter 2006, 11, 494–505. [Google Scholar] [CrossRef]
- Mabley, J.G.; Pacher, P.; Deb, A.; Wallace, R.; Elder, R.H.; Szabo, C. Potential role for 8-oxoguanine DNA glycosylase in regulating inflammation. FASEB J. 2005, 19, 290–292. [Google Scholar] [PubMed]
- Innocenti, F.; Fabbri, A.; Anichini, R.; Tuci, S.; Pettina, G.; Vannucci, F.; de Giorgio, L.A.; Seghieri, G. Indications of reduced pulmonary function in type 1 (insulin-dependent) diabetes mellitus. Diabetes Res. Clin. Pract. 1994, 25, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Lange, P.; Groth, S.; Kastrup, J.; Mortensen, J.; Appleyard, M.; Nyboe, J.; Jensen, G.; Schnohr, P. Diabetes mellitus, plasma glucose and lung function in a cross-sectional population study. Eur. Respir. J. 1989, 2, 14–19. [Google Scholar] [PubMed]
- Litonjua, A.A.; Lazarus, R.; Sparrow, D.; Demolles, D.; Weiss, S.T. Lung function in type 2 diabetes: The normative aging study. Respir. Med. 2005, 99, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yuan, K.; Yan, C.; Fox, J., 3rd; Gaid, M.; Breitwieser, W.; Bansal, A.K.; Zeng, H.; Gao, H.; Wu, M. 8-Oxoguanine-DNA glycosylase 1 deficiency modifies allergic airway inflammation by regulating STAT6 and IL-4 in cells and in mice. Free Radic. Biol. Med. 2012, 52, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Swamy, M.; Jamora, C.; Havran, W.; Hayday, A. Epithelial decision makers: In search of the “epimmunome”. Nat. Immunol. 2012, 11, 656–665. [Google Scholar] [CrossRef]
- Holgate, S.T. Epithelium dysfunction in asthma. J. Allergy Clin. Immunol. 2007, 120, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Bacsi, A.; Aguilera-Aguirre, L.; Szczesny, B.; Radak, Z.; Hazra, T.K.; Sur, S.; Ba, X.; Boldogh, I. Down-regulation of 8-oxoguanine DNA glycosylase 1 expression in the airway epithelium ameliorates allergic lung inflammation. DNA Repair 2013, 12, 18–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacsi, A.; Choudhury, B.K.; Dharajiya, N.; Sur, S.; Boldogh, I. Subpollen particles: Carriers of allergenic proteins and oxidases. J. Allergy Clin. Immunol. 2006, 118, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Bacsi, A.; Dharajiya, N.; Choudhury, B.K.; Sur, S.; Boldogh, I. Effect of pollen-mediated oxidative stress on immediate hypersensitivity reactions and late-phase inflammation in allergic conjunctivitis. J. Allergy Clin. Immunol. 2005, 116, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Boldogh, I.; Bacsi, A.; Choudhury, B.K.; Dharajiya, N.; Alam, R.; Hazra, T.K.; Mitra, S.; Goldblum, R.M.; Sur, S. ROS generated by pollen NADPH oxidase provide a signal that augments antigen-induced allergic airway inflammation. J. Clin. Investig. 2005, 115, 2169–2179. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.W.; Hazra, T.K.; Izumi, T.; Mitra, S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: Potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001, 29, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Izumi, T.; Schein, C.H.; Oezguen, N.; Feng, Y.; Braun, W. Effects of backbone contacts 3' to the abasic site on the cleavage and the product binding by human apurinic/apyrimidinic endonuclease (APE1). Biochemistry 2004, 43, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Izumi, T.; Boldogh, I.; Bhakat, K.K.; Hill, J.W.; Hazra, T.K. Choreography of oxidative damage repair in mammalian genomes. Free Radic. Biol. Med. 2002, 33, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Hegde, P.M.; Rao, K.S.; Mitra, S. Oxidative genome damage and its repair in neurodegenerative diseases: Function of transition metals as a double-edged sword. J. Alzheimerʼs Dis. 2011, 24, 183–198. [Google Scholar]
- Sidorenko, V.S.; Nevinsky, G.A.; Zharkov, D.O. Mechanism of interaction between human 8-oxoguanine-DNA glycosylase and AP endonuclease. DNA Repair 2007, 6, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef] [PubMed]
- Jamaluddin, M.; Wang, S.; Boldogh, I.; Tian, B.; Brasier, A.R. TNF-α-induced NF-κB/RelA Ser (276) phosphorylation and enhanceosome formation is mediated by an ROS-dependent PKAc pathway. Cell. Signal. 2007, 19, 1419–1433. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xia, F.; Hermance, N.; Mabb, A.; Simonson, S.; Morrissey, S.; Gandhi, P.; Munson, M.; Miyamoto, S.; Kelliher, M.A. A cytosolic ATM/NEMO/RIP1 complex recruits TAK1 to mediate the NF-kappaB and p38 mitogen-activated protein kinase (MAPK)/MAPK-activated protein 2 responses to DNA damage. Mol. Cell. Biol. 2011, 31, 2774–2786. [Google Scholar] [CrossRef] [PubMed]
- Biton, S.; Ashkenazi, A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-α feedforward signaling. Cell 2011, 145, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Hadian, K.; Krappmann, D. Signals from the nucleus: Activation of NF-κB by cytosolic ATM in the DNA damage response. Sci. Signal. 2011, 4, pe2. [Google Scholar] [PubMed]
- Oumouna, M.; Datta, R.; Oumouna-Benachour, K.; Suzuki, Y.; Hans, C.; Mattews, K.; Fallon, K.; Boulares, H. Poly(ADP-ribose) polymerase-1 inhibition prevents eosinophil recruitment by modulating Th2 cytokines in a murine model of allergic airway inflammation: A potential specific effect on IL-5. J. Immunol. 2006, 177, 6489–6496. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [PubMed]
- Boldogh, I.; Hajas, G.; Aguilera-Aguirre, L.; Hegde, M.L.; Radak, Z.; Bacsi, A.; Sur, S.; Hazra, T.K.; Mitra, S. Activation of ras signaling pathway by 8-oxoguanine DNA glycosylase bound to its excision product, 8-oxoguanine. J. Biol. Chem. 2012, 287, 20769–20773. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; de Souza-Pinto, N.C.; Haraguchi, K.; Hogue, B.A.; Jaruga, P.; Greenberg, M.M.; Dizdaroglu, M.; Bohr, V.A. Repair of formamidopyrimidines in DNA involves different glycosylases: Role of the OGG1, NTH1, and NEIL1 enzymes. J. Biol. Chem. 2005, 280, 40544–40551. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.C.; Boguski, M.; Broek, D.; Powers, S. Influence of guanine nucleotides on complex formation between Ras and CDC25 proteins. Mol. Cell. Biol. 1993, 13, 1345–1352. [Google Scholar] [PubMed]
- Mosteller, R.D.; Han, J.; Broek, D. Identification of residues of the H-ras protein critical for functional interaction with guanine nucleotide exchange factors. Mol. Cell. Biol. 1994, 14, 1104–1112. [Google Scholar] [PubMed]
- German, P.; Szaniszlo, P.; Hajas, G.; Radak, Z.; Bacsi, A.; Hazra, T.K.; Hegde, M.L.; Ba, X.; Boldogh, I. Activation of cellular signaling by 8-oxoguanine DNA glycosylase-1-initiated DNA base excision repair. DNA Repair 2013, 12, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Heo, J. Redox control of GTPases: From molecular mechanisms to functional significance in health and disease. Antioxid. Redox Signal. 2011, 14, 689–724. [Google Scholar] [CrossRef] [PubMed]
- Hajas, G.; Bacsi, A.; Aguilera-Aguirre, L.; Hegde, M.L.; Tapas, K.H.; Sur, S.; Radak, Z.; Ba, X.; Boldogh, I. 8-Oxoguanine DNA glycosylase-1 links DNA repair to cellular signaling via the activation of the small GTPase Rac1. Free Radic. Biol. Med. 2013, 61, 384–394. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Hosoki, K.; Bacsi, A.; Radak, Z.; Hegde, M.L.; Sur, S.; Hazra, T.K.; Brasier, A.R.; Ba, X.; Boldogh, I. 8-Oxoguanine DNA glycosylase-1-mediated DNA repair is associated with Rho GTPase activation and alpha-smooth muscle actin polymerization. Free Radic. Biol. Med. 2014, 73, 430–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandita, T.K. Unraveling the novel function of the DNA repair enzyme 8-oxoguanine-DNA glycosylase in activating key signaling pathways. Free Radic. Biol. Med. 2014, 73, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M. Base-excision repair of oxidative DNA damage by DNA glycosylases. Mutat. Res. 2005, 591, 45–59. [Google Scholar] [CrossRef]
- Kim, H.J.; Yoon, S.H.; Ryu, H.O.; Yoon, B.H.; Choi, S.; Ye, S.K.; Chung, M.H. 8-oxo-7,8-dihydroguanosine triphosphate (8-oxoGTP) down-regulates respiratory burst of neutrophils by antagonizing GTP toward Rac, a small GTP binding protein. Free Radic. Res. 2007, 41, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kim, D.Y.; Lee, J.K.; Ro, J.Y.; Chung, M.H. 8-oxo-2'-Deoxyguanosine suppresses allergy-induced lung tissue remodeling in mice. Eur. J. Pharmacol. 2011, 651, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Hajas, G.; Bacsi, A.; Aguilerra-Aguirre, L.; German, P.; Radak, Z.; Sur, S.; Hazra, T.K.; Boldogh, I. Biochemical identification of a hydroperoxide derivative of the free 8-oxo-7,8-dihydroguanine base. Free Radic. Biol. Med. 2012, 52, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; App, H.; Zhang, X.F.; Banerjee, P.; Brautigan, D.L.; Rapp, U.R.; Avruch, J. Raf-1 activates MAP kinase-kinase. Nature 1992, 358, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Yu, J.; Field, J. Signals from the Ras, Rac, and Rho GTPases converge on the Pak protein kinase in Rat-1 fibroblasts. Mol. Cell. Biol. 1999, 19, 1881–1891. [Google Scholar] [PubMed]
- Birukova, A.A.; Fu, P.; Xing, J.; Cokic, I.; Birukov, K.G. Lung endothelial barrier protection by iloprost in the 2-hit models of ventilator-induced lung injury (VILI) involves inhibition of Rho signaling. Transl. Res. 2010, 155, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.W.; Shin, M.G.; Lee, S.; Kim, J.R.; Park, W.S.; Cho, K.H.; Meyer, T.; Heo, W.D. Cooperative activation of PI3K by Ras and Rho family small GTPases. Mol. Cell 2012, 47, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.R.; Kasteler, S.D.; Schmitt, R.E.; Hoidal, J.R. Receptor for advanced glycation end-products signals through Ras during tobacco smoke-induced pulmonary inflammation. Am. J. Respir. Cell Mol. Biol. 2011, 45, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.S.; Chen, Y.H. Ras family of small GTPases in immunity and inflammation. Curr. Opin. Pharmacol. 2012, 12, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Mor, A.; Aizman, E.; Chapman, J.; Kloog, Y. Immunomodulatory properties of farnesoids: The new steroids? Curr. Med. Chem. 2013, 20, 1218–1224. [Google Scholar]
- Duan, W.; Chan, J.H.; Wong, C.H.; Leung, B.P.; Wong, W.S. Anti-inflammatory effects of mitogen-activated protein kinase kinase inhibitor U0126 in an asthma mouse model. J. Immunol. 2004, 172, 7053–7059. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Cao, Y.X.; Xu, C.B.; Zhang, Y. The Raf-1 inhibitor GW5074 and dexamethasone suppress sidestream smoke-induced airway hyperresponsiveness in mice. Respir. Res. 2008, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Tudhope, S.J.; Finney-Hayward, T.K.; Nicholson, A.G.; Mayer, R.J.; Barnette, M.S.; Barnes, P.J.; Donnnelly, L.E. Different mitogen-activated protein kinase-dependent cytokine responses in cells of the monocyte lineage. J. Pharmacol. Exp. Ther. 2008, 324, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.F.; Deem, T.L.; Bruce, A.C.; Reutershan, J.; Wu, D.; Ley, K. Leukocyte phosphoinositide-3 kinase γ is required for chemokine-induced, sustained adhesion under flow in vivo. J. Leukoc. Biol. 2006, 80, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.J.; Resnick, R.J.; Shalloway, D. Nonradioactive determination of RAS–GTP levels using activated ras interaction assay. Methods Enzymol. 2001, 333, 333–342. [Google Scholar] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (−ΔΔCt) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Aguilera-Aguirre, L.; Bacsi, A.; Saavedra-Molina, A.; Kurosky, A.; Sur, S.; Boldogh, I. Mitochondrial dysfunction increases allergic airway inflammation. J. Immunol. 2009, 183, 5379–5387. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Thomas, P.D. PANTHER in 2013: Modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013, 41, D377–D386. [Google Scholar] [CrossRef] [PubMed]
- Ba, X.; Bacsi, A.; Luo, J.; Aguilera-Aguirre, L.; Zeng, X.; Radak, Z.; Brasier, A.R.; Boldogh, I. 8-Oxoguanine DNA glycosylase-1 augments proinflammatory gene expression by facilitating the recruitment of site-specific transcription factors. J. Immunol. 2014, 192, 2384–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ba, X.; Aguilera-Aguirre, L.; Rashid, Q.T.A.N.; Bacsi, A.; Radak, Z.; Sur, S.; Hosoki, K.; Hegde, M.L.; Boldogh, I. The Role of 8-Oxoguanine DNA Glycosylase-1 in Inflammation. Int. J. Mol. Sci. 2014, 15, 16975-16997. https://doi.org/10.3390/ijms150916975
Ba X, Aguilera-Aguirre L, Rashid QTAN, Bacsi A, Radak Z, Sur S, Hosoki K, Hegde ML, Boldogh I. The Role of 8-Oxoguanine DNA Glycosylase-1 in Inflammation. International Journal of Molecular Sciences. 2014; 15(9):16975-16997. https://doi.org/10.3390/ijms150916975
Chicago/Turabian StyleBa, Xueqing, Leopoldo Aguilera-Aguirre, Qura Tul Ain Nmi Rashid, Attila Bacsi, Zsolt Radak, Sanjiv Sur, Koa Hosoki, Muralidhar L. Hegde, and Istvan Boldogh. 2014. "The Role of 8-Oxoguanine DNA Glycosylase-1 in Inflammation" International Journal of Molecular Sciences 15, no. 9: 16975-16997. https://doi.org/10.3390/ijms150916975