The Transcriptomes of the Crucian Carp Complex (Carassius auratus) Provide Insights into the Distinction between Unisexual Triploids and Sexual Diploids

Abstract
:1. Introduction
2. Results and Discussion
2.1. Transcriptome Sequencing and Assembly
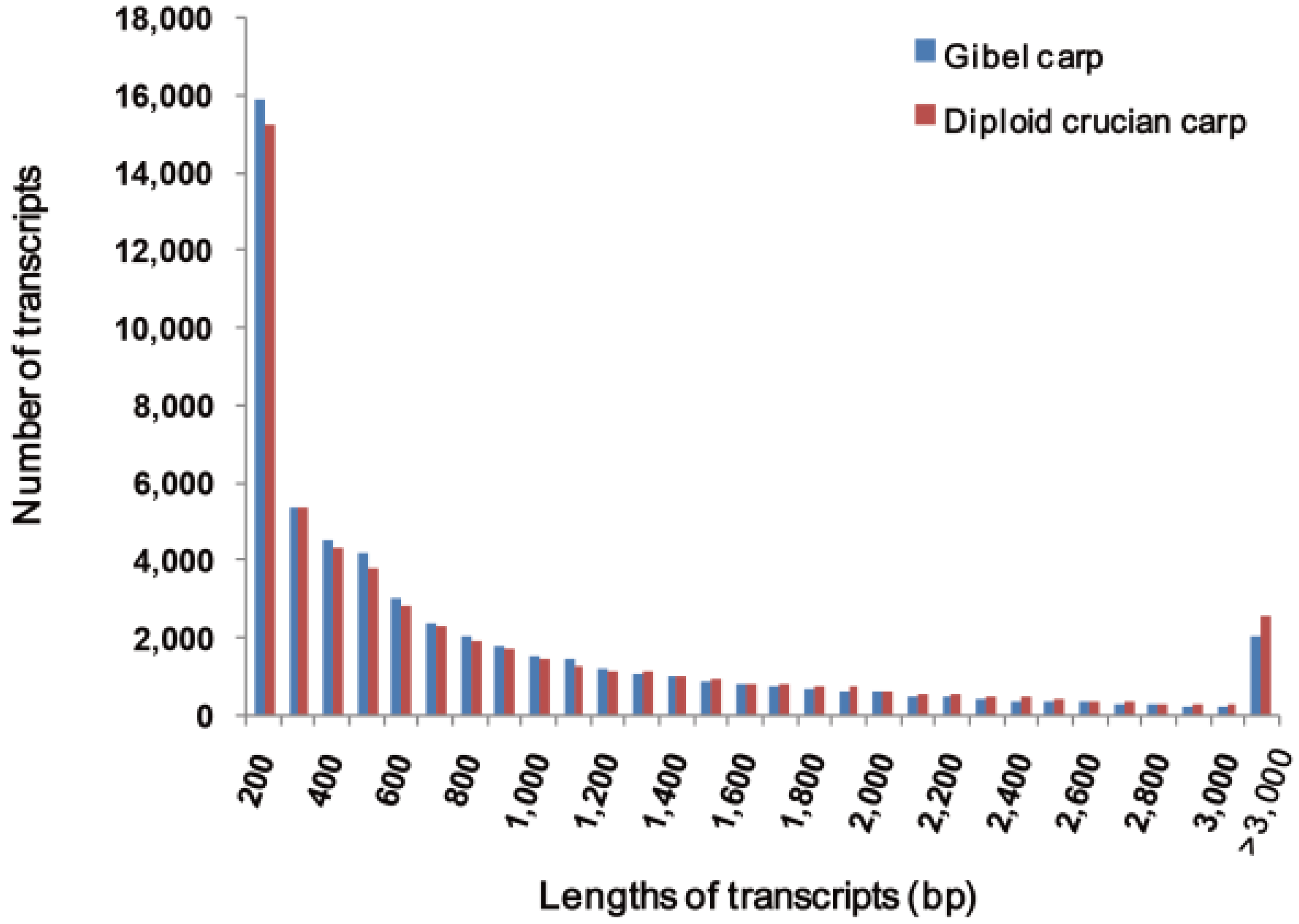
2.2. Sequence Annotation and Validation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Gibel Carp | Diploid Crucian Carp | |
|---|---|---|---|
| Homolog search | Fish Ensembl proteins | 32,030 | 31,620 |
| Fish Ensembl transcripts | 13,513 | 10,956 | |
| NCBI “nr” protein database | 235 | 56 | |
| UTRdb | 852 | 1770 | |
| NONCODE database | 336 | 491 | |
| NCBI “nt” transcript database | 4004 | 1386 | |
| ab initio prediction | BESTORF | 433 | 1260 |
| Unknown | 3056 | 6300 | |
| Total | 54,459 | 53,839 |
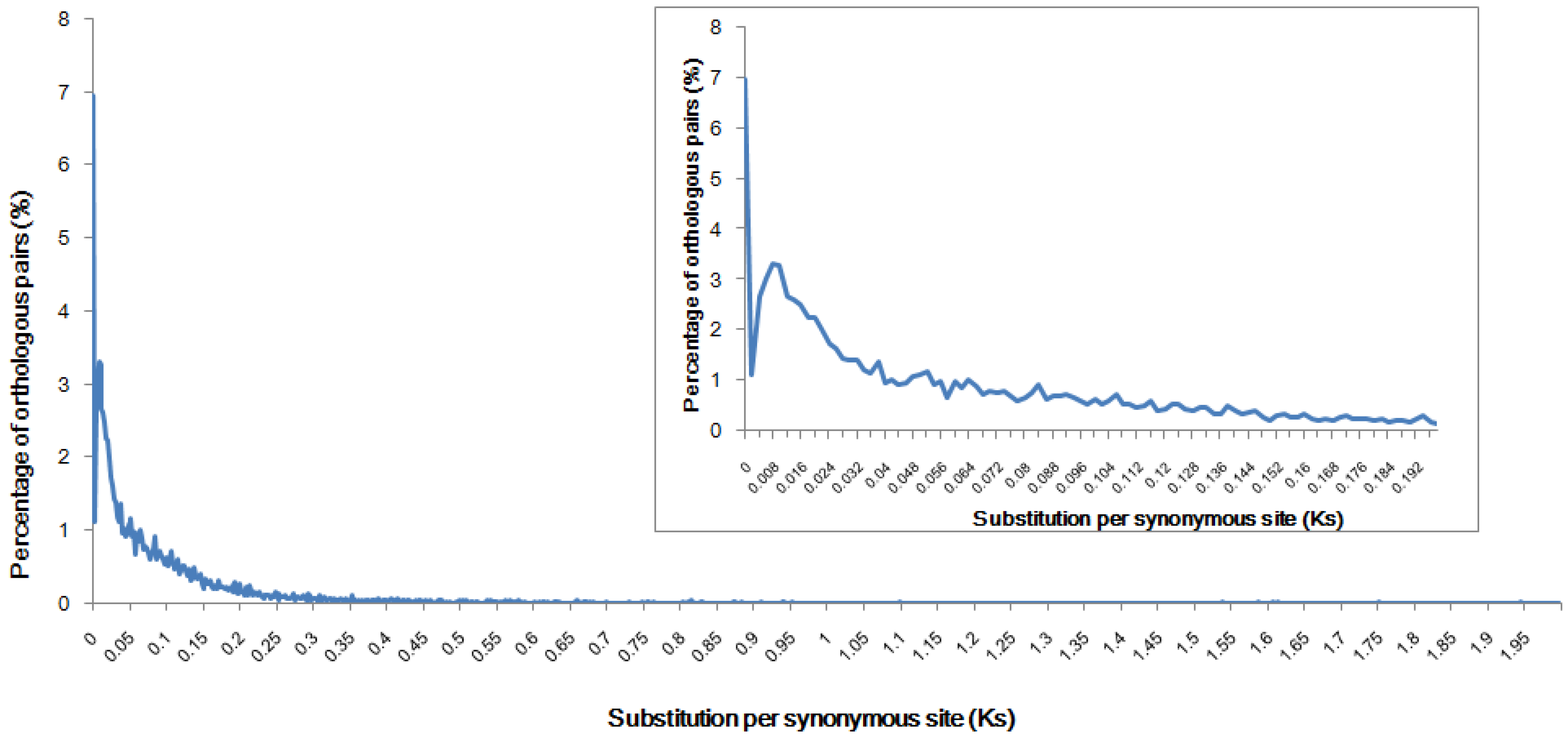
2.3. Genome Speciation Event Deduced from Orthologous Pairs

2.4. Low Tri-Allelic Polymorphism and Heterozygosity in Gibel Carp
2.5. Few Gene Expansions in Gibel Carp
| Species | GO Term | GO ID | Percentage of Transcripts (%) * | p Value |
|---|---|---|---|---|
| Gibel carp | nucleic acid binding | GO:0003676 | 13.9:13.2 | 0.009 |
| transferase activity | GO:0016740 | 10.4:9.8 | 0.012 | |
| transferase activity, transferring phosphorus-containing groups | GO:0016772 | 6.6:6.2 | 0.028 | |
| DNA binding | GO:0003677 | 7.6:7.1 | 0.033 | |
| Diploid crucian carp | transporter activity | GO:0005215 | 4.1:4.7 | 0.000 |
| transmembrane transporter activity | GO:0022857 | 3.2:3.9 | 0.000 | |
| passive transmembrane transporter activity | GO:0022803 | 1.0:1.4 | 0.000 | |
| substrate-specific transmembrane transporter activity | GO:0022891 | 2.7:3.3 | 0.000 | |
| substrate-specific transporter activity | GO:0022892 | 4.1:4.7 | 0.001 |
| Species | KEGG Pathway | Percentage of Gibel Carp Transcripts (%) | Percentage of Diploid Crucian Carp Transcripts (%) | p Value |
|---|---|---|---|---|
| Gibel carp | Ubiquitin mediated proteolysis | 1.70 | 1.38 | 0.013 |
| Progesterone-mediated oocyte maturation | 1.13 | 0.89 | 0.018 | |
| Fanconi anemia pathway | 0.61 | 0.46 | 0.038 | |
| Diploid crucian carp | Neuroactive ligand-receptor interaction | 1.07 | 1.53 | 0.000 |
| Cell adhesion molecules | 0.91 | 1.18 | 0.010 | |
| Ribosome | 0.55 | 0.74 | 0.024 | |
| Tight junction | 1.51 | 1.76 | 0.045 |
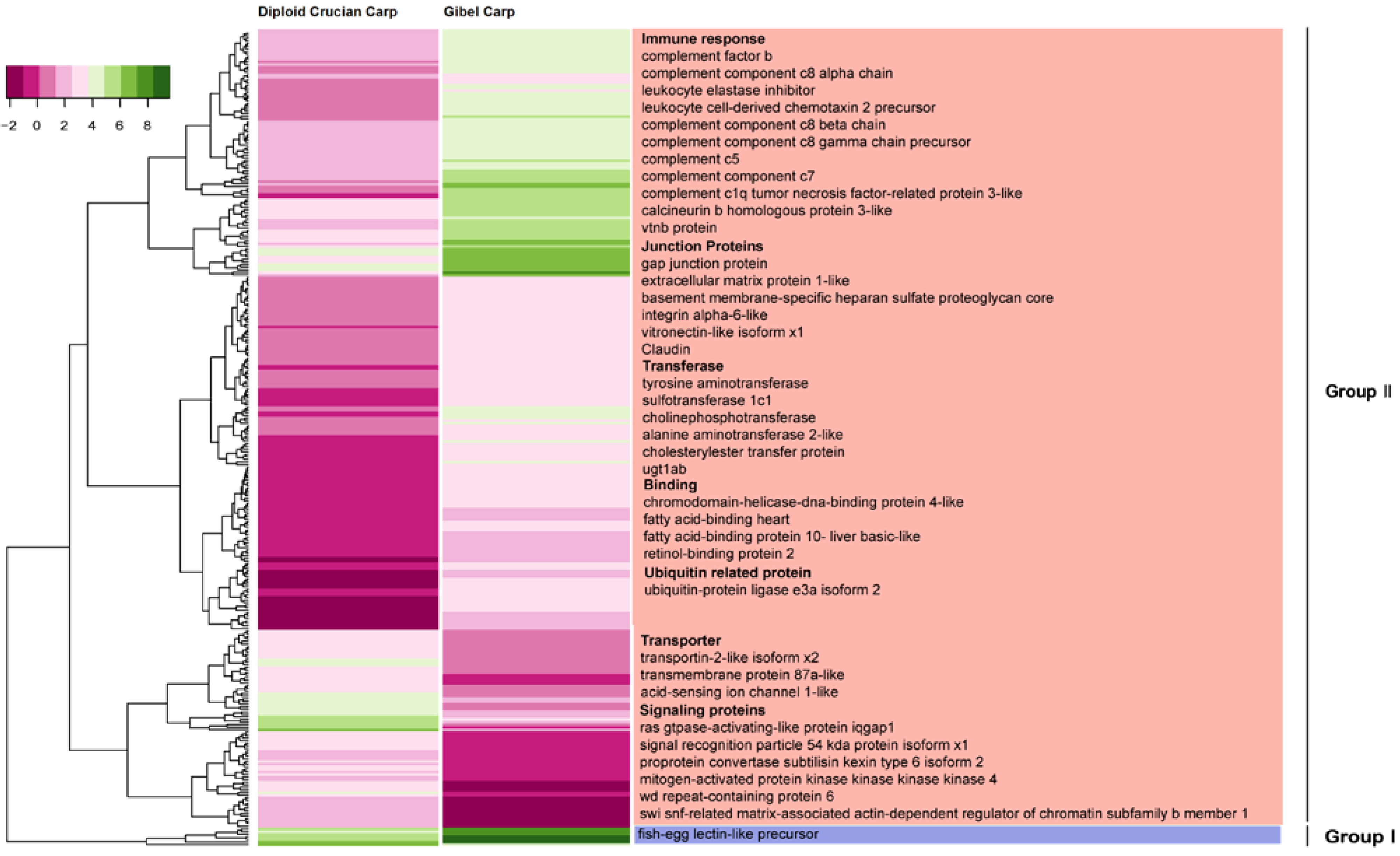
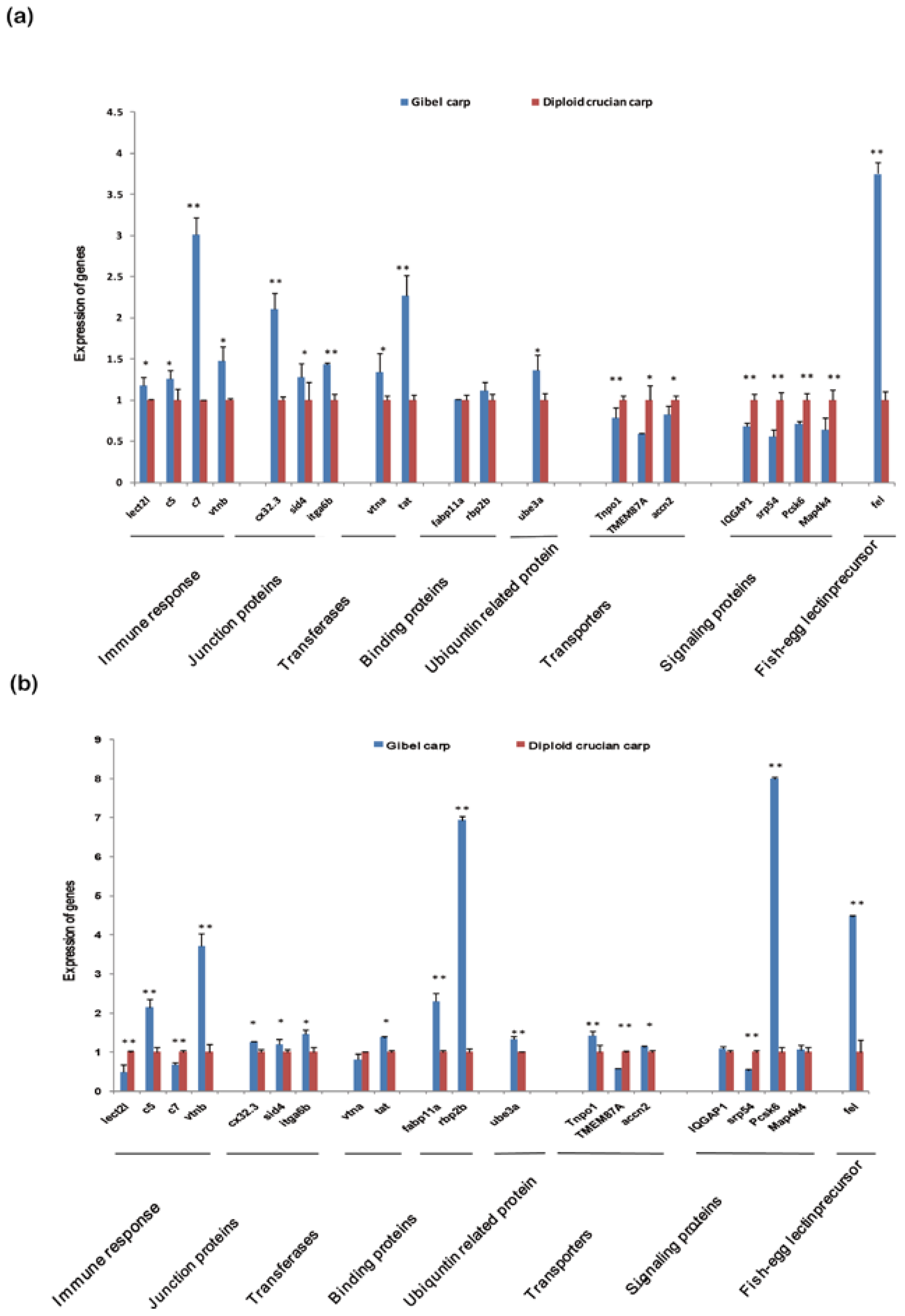
2.6. Differentially Expressed Orthologous Genes in Pooled Tissues and Gonads
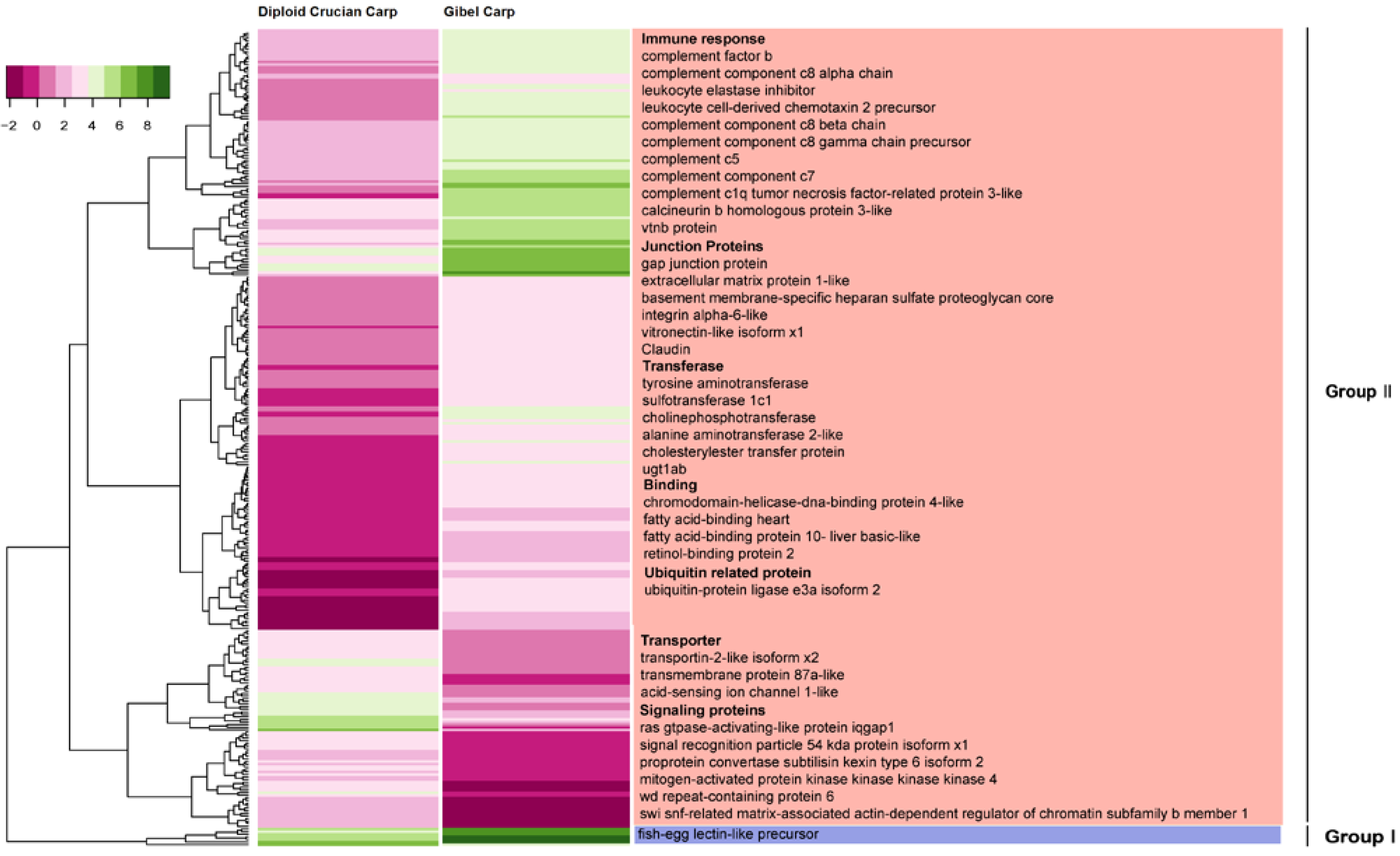
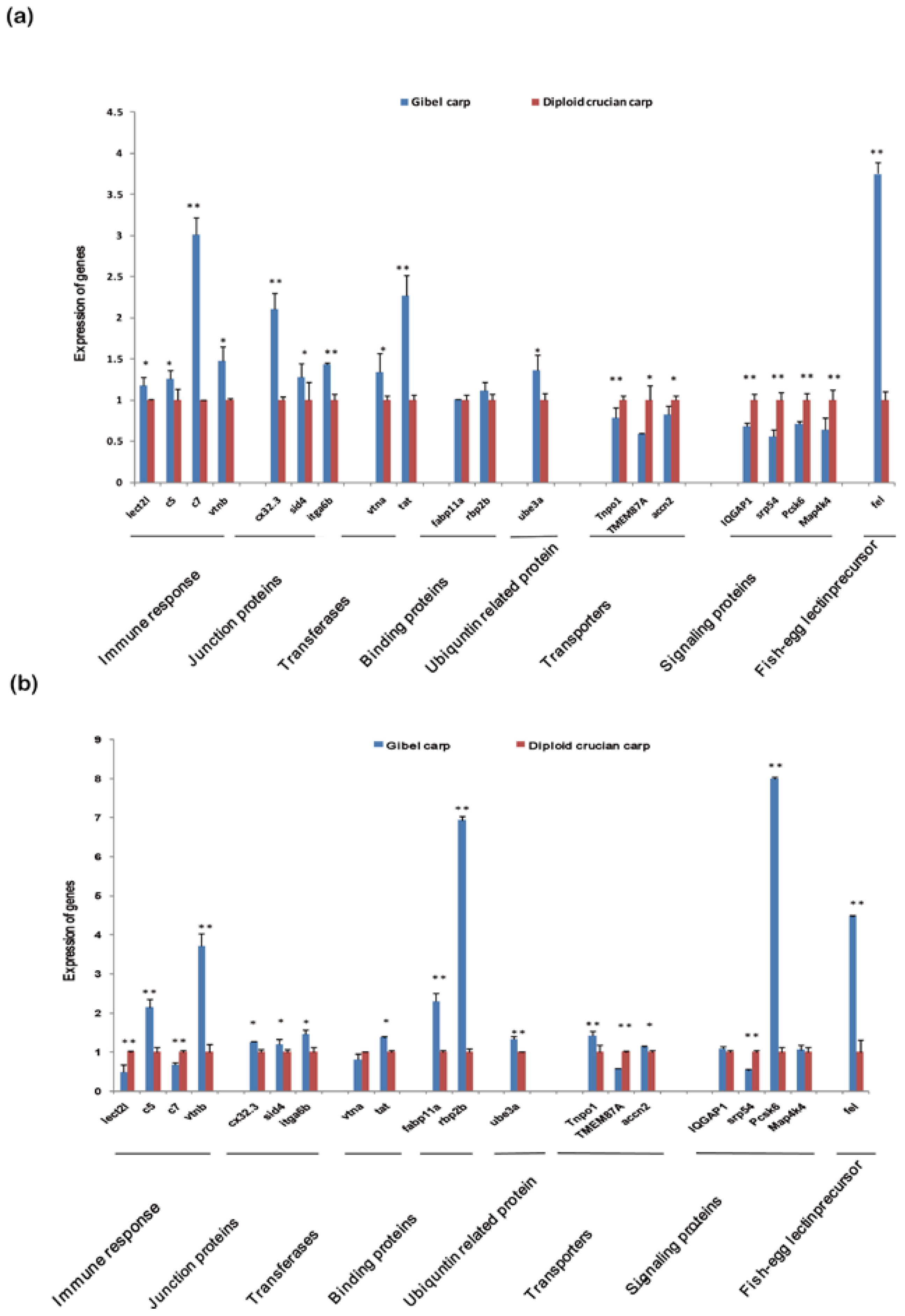
3. Experimental Section
3.1. Sample Collection and Transcriptome Sequencing
3.2. Transcriptome Assembly, Annotation, and Validation
3.3. Genome Speciation Event Deduced from the Ks Distribution of Orthologous Pairs
3.4. The Analysis of Tri-Allelic Polymorphism and Heterozygosity in Gibel Carp
3.5. Gene Expansions in Special Molecular Functions and Pathways
3.6. Analysis and Validation of Differentially Expressed Orthologous Genes
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Neaves, W.B.; Baumann, P. Unisexual reproduction among vertebrates. Trends Genet. 2011, 27, 81–88. [Google Scholar] [CrossRef]
- Lamatsch, D.K.; Stöck, M. Sperm-dependent parthenogenesis and hybridogenesis in teleost fishes. In Lost Sex; Springer: Dordrecht, The Netherlands, 2009; pp. 399–432. [Google Scholar]
- Xiao, J.; Zou, T.; Chen, Y.; Chen, L.; Liu, S.; Tao, M.; Zhang, C.; Zhao, R.; Zhou, Y.; Long, Y.; et al. Coexistence of diploid, triploid and tetraploid crucian carp (Carassius auratus) in natural waters. BMC Genet. 2011, 12, 20. [Google Scholar]
- Jiang, F.F.; Wang, Z.W.; Zhou, L.; Jiang, L.; Zhang, X.J.; Apalikova, O.V.; Brykov, V.A.; Gui, J.F. High male incidence and evolutionary implications of triploid form in northeast Asia Carassius auratus complex. Mol. Phylogenet. Evol. 2013, 66, 350–359. [Google Scholar] [CrossRef]
- Chun, F.L.; Yang, S.T.; Gui, J.F. Differential screening and characterization analysis of the egg envelope glycoprotein ZP3 cDNAs between gynogenetic and gonochoristic crucian carp. Cell Res. 2001, 11, 17–27. [Google Scholar] [CrossRef]
- Yi, M.S.; Li, Y.Q.; Liu, J.D.; Zhou, L.; Yu, Q.X.; Gui, J.F. Molecular cytogenetic detection of paternal chromosome fragments in allogynogenetic gibel carp, Carassius auratus gibelio Bloch. Chromosome Res. 2003, 11, 665–671. [Google Scholar]
- Zhou, L.; Gui, J.F. Karyotypic diversity in polyploid gibel carp, Carassius auratus gibelio Bloch. Genetica 2002, 115, 223–232. [Google Scholar] [CrossRef]
- Zhu, H.P.; Ma, D.M.; Gui, J.F. Triploid origin of the gibel carp as revealed by 5S rDNA localization and chromosome painting. Chromosome Res. 2006, 14, 767–776. [Google Scholar] [CrossRef]
- Gui, J.F.; Zhu, Z.Y. Molecular basis and genetic improvement of economically important traits in aquaculture animals. Chin. Sci. Bull. 2012, 57, 1751–1760. [Google Scholar] [CrossRef]
- LI, C.J.; GUI, J.F. Comparative studies on in vitro sperm decondensation and pronucleus formation in egg extracts between gynogenetic and bisexual fish. Cell Res. 2003, 13, 159–169. [Google Scholar] [CrossRef]
- Wang, D.; Mao, H.L.; Peng, J.X.; Li, X.Y.; Zhou, L.; Gui, J.F. Discovery of a male-biased mutant family and identification of a male-specific SCAR marker in gynogenetic gibel carp Carassius auratus gibelio. Prog. Nat. Sci. 2009, 19, 1537–1544. [Google Scholar] [CrossRef]
- Yuan, J.A.; He, Z.Z.; Yuan, X.N.; Jiang, X.Y.; Sun, X.W.; Zou, S.M. Speciation of polyploid cyprinidae fish of common carp, crucian carp, and silver crucian carp derived from duplicated hox genes. J. Exp. Zool. Part B Mol. Dev. Evol. 2010, 314B, 445–456. [Google Scholar] [CrossRef]
- Bickler, P.E.; Buck, L.T. Hypoxia tolerance in reptiles, amphibians, and fishes: Life with variable oxygen availability. Annu. Rev. Phys. 2007, 69, 145–170. [Google Scholar] [CrossRef]
- Liao, X.; Cheng, L.; Xu, P.; Lu, G.; Wachholtz, M.; Sun, X.; Chen, S. Transcriptome analysis of Crucian Carp (Carassius auratus), an important aquaculture and hypoxia-tolerant species. PLoS One 2013, 8, e62308. [Google Scholar]
- Zhou, Z.C.; Dong, Y.; Sun, H.J.; Yang, A.F.; Chen, Z.; Gao, S.; Jiang, J.W.; Guan, X.Y.; Jiang, B.; Wang, B. Transcriptome sequencing of sea cucumber (Apostichopus japonicus) and the identification of gene-associated markers. Mol. Ecol. Resour. 2014, 14, 127–138. [Google Scholar] [CrossRef]
- Robertson, L.S.; Cornman, R.S. Transcriptome resources for the frogs Lithobates clamitans and Pseudacris regilla, emphasizing antimicrobial peptides and conserved loci for phylogenetics. Mol. Ecol. Resour. 2014, 14, 178–183. [Google Scholar] [CrossRef]
- Wang, J.T.; Li, J.T.; Zhang, X.F.; Sun, X.W. Transcriptome analysis reveals the time of the fourth round of genome duplication in common carp (Cyprinus carpio). BMC Genomics 2012, 13, 96. [Google Scholar] [CrossRef]
- Blanc, G.; Wolfe, K.H. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 2004, 16, 1667–1678. [Google Scholar] [CrossRef]
- David, L.; Blum, S.; Feldman, M.W.; Lavi, U.; Hillel, J. Recent duplication of the common carp (Cyprinus carpio L.) genome as revealed by analyses of microsatellite loci. 2003, 20, 1425–1434. [Google Scholar]
- Pamilo, P.; Bianchi, N. Evolution of the Zfx and Zfy genes: Rates and interdependence between the genes. Mol. Biol. Evolut. 1993, 10, 271–281. [Google Scholar]
- Sinclair, E.A.; Pramuk, J.B.; Bezy, R.L.; Crandall, K.A.; Sites, J.W., Jr. DNA evidence for nonhybrid origins of parthenogenesis in natural populations of vertebrates. Evolution 2010, 64, 1346–1357. [Google Scholar]
- Watts, P.C.; Buley, K.R.; Sanderson, S.; Boardman, W.; Ciofi, C.; Gibson, R. Parthenogenesis in Komodo dragons. Nature 2006, 444, 1021–1022. [Google Scholar] [CrossRef]
- Bai, Z.Y.; Liu, F.; Li, J.L.; Yue, G.H. Identification of Triploid Individuals and Clonal Lines in Carassius auratus Complex Using Microsatellites. Int. J. Biol. Sci. 2011, 7, 279–285. [Google Scholar]
- Wolfe, K.H. Yesterday’s polyploids and the mystery of diploidization. Nat. Rev. Genet. 2001, 2, 333–341. [Google Scholar] [CrossRef]
- Zhu, Y.P.; Xue, W.; Wang, J.T.; Wan, Y.M.; Wang, S.L.; Xu, P.; Zhang, Y.; Li, J.T.; Sun, X.W. Identification of common carp (Cyprinus carpio) microRNAs and microRNA-related SNPs. BMC Genomics 2012, 13, 413. [Google Scholar] [CrossRef]
- Voronina, E.; Marzluff, W.F.; Wessel, G.M. Cyclin B synthesis is required for sea urchin oocyte maturation. Dev. Biol. 2003, 256, 258–275. [Google Scholar] [CrossRef]
- Yang, Z.A.; Li, Q.H.; Wang, Y.F.; Gui, J.F. Comparative investigation on spindle behavior and MPF activity changes during oocyte maturation between gynogenetic and amphimictic crucian carp. Cell Res. 1999, 9, 145–154. [Google Scholar] [CrossRef]
- Morrill, G.A.; Dowd, T.L.; Kostellow, A.B.; Gupta, R.K. Progesterone-induced changes in the phosphoryl potential during the meiotic divisions in amphibian oocytes: Role of Na/K-ATPase. BMC Dev. Biol. 2011, 11, 67. [Google Scholar] [CrossRef]
- Pace, M.C.; Thomas, P. Steroid-induced oocyte maturation in Atlantic croaker (Micropogonias undulatus) is dependent on activation of the phosphatidylinositol 3-kinase/Akt signal transduction pathway. Biol. Reprod. 2005, 73, 988–996. [Google Scholar] [CrossRef]
- Glotzer, M.; Murray, A.W.; Kirschner, M.W. Cyclin is degraded by the ubiquitin pathway. Nature 1991, 349, 132–138. [Google Scholar] [CrossRef]
- Moldovan, G.L.; D’Andrea, A.D. How the fanconi anemia pathway guards the genome. Annu. Rev. Genet. 2009, 43, 223–249. [Google Scholar] [CrossRef]
- Galliano, M.; Minchiotti, L.; Campagnoli, M.; Sala, A.; Visai, L.; Amoresano, A.; Pucci, P.; Casbarra, A.; Cauci, M.; Perduca, M.; et al. Structural and biochemical characterization of a new type of lectin isolated from carp eggs. Biochem. J. 2003, 376, 433–440. [Google Scholar] [CrossRef]
- Chen, B.; Gui, J. Identification of a novel C1q family member in color crucian carp (Carassius auratus) ovary. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2004, 138, 285–293. [Google Scholar] [CrossRef]
- Dong, C.H.; Yang, S.T.; Yang, Z.A.; Zhang, L.; Gui, J.F. A C-type lectin associated and translocated with cortical granules during oocyte maturation and egg fertilization in fish. Dev. Biol. 2004, 265, 341–354. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Sammeth, M.; Bouckenooghe, T.; Bottu, G.; Sisino, G.; Igoillo-Esteve, M.; Ortis, F.; Santin, I.; Colli, M.L.; Barthson, J. The human pancreatic islet transcriptome: Expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 2012, 8, e1002552. [Google Scholar] [CrossRef]
- Cox, M.P.; Peterson, D.A.; Biggs, P.J. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 2010, 11, 485. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef]
- Sandvik, G.K.; Tomter, A.B.; Bergan, J.; Zoppellaro, G.; Barra, A.L.; Rohr, A.K.; Kolberg, M.; Ellefsen, S.; Andersson, K.K.; Nilsson, G.E. Studies of ribonucleotide reductase in crucian carp-an oxygen dependent enzyme in an anoxia tolerant vertebrate. PLoS One 2012, 7, e42784. [Google Scholar] [CrossRef]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef]
- Enright, A.J.; van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Flicek, P.; Ahmed, I.; Amode, M.R.; Barrell, D.; Beal, K.; Brent, S.; Carvalho-Silva, D.; Clapham, P.; Coates, G.; Fairley, S.; et al. Ensembl 2013. Nucleic Acids Res. 2013, 41, D48–D55. [Google Scholar] [CrossRef]
- Meyer, L.R.; Zweig, A.S.; Hinrichs, A.S.; Karolchik, D.; Kuhn, R.M.; Wong, M.; Sloan, C.A.; Rosenbloom, K.R.; Roe, G.; Rhead, B.; et al. The UCSC Genome Browser database: Extensions and updates 2013. Nucleic Acids Res. 2013, 41, D64–D69. [Google Scholar] [CrossRef]
- Pesole, G.; Liuni, S.; Grillo, G.; Ippedico, M.; Larizza, A.; Makalowski, W.; Saccone, C. UTRdb: A specialized database of 5' and 3' untranslated regions of eukaryotic mRNAs. Nucleic Acids Res. 1999, 27, 188–191. [Google Scholar] [CrossRef]
- Bu, D.; Yu, K.; Sun, S.; Xie, C.; Skogerbo, G.; Miao, R.; Xiao, H.; Liao, Q.; Luo, H.; Zhao, G.; et al. NONCODE v3.0 : Integrative annotation of long noncoding RNAs. Nucleic Acids Res. 2012, 40, D210–D215. [Google Scholar] [CrossRef]
- Dinger, M.E.; Pang, K.C.; Mercer, T.R.; Mattick, J.S. Differentiating Protein-Coding and Noncoding RNA: Challenges and Ambiguities. PLoS Comput. Biol. 2008, 4, e1000176. [Google Scholar] [CrossRef]
- Zhang, G.F.; Fang, X.D.; Guo, X.M.; Li, L.; Luo, R.B.; Xu, F.; Yang, P.C.; Zhang, L.L.; Wang, X.T.; Qi, H.G.; et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 2012, 490, 49–54. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.Z.; Huang, J.J.; Ding, Y.; Wu, J.M.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L.P. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Östlund, G.; Schmitt, T.; Forslund, K.; Köstler, T.; Messina, D.N.; Roopra, S.; Frings, O.; Sonnhammer, E.L. InParanoid 7: New algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res. 2010, 38, D196–D203. [Google Scholar] [CrossRef]
- Chen, F.; Mackey, A.J.; Vermunt, J.K.; Roos, D.S. Assessing performance of orthology detection strategies applied to eukaryotic genomes. PLoS One 2007, 2, e383. [Google Scholar]
- Srivastava, A.; Rogers, W.L.; Breton, C.M.; Cai, L.M.; Malmberg, R.L. Transcriptome analysis of sarracenia, an insectivorous plant. DNA Res. 2011, 18, 253–261. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The european molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Zheng, X.; Kuang, Y.; Zhao, Z.; Zhao, L.; Li, C.; Jiang, L.; Cao, D.; Lu, C.; et al. A consensus linkage map provides insights on genome character and evolution in common carp (Cyprinus carpio L.). Mar. Biotechnol. 2013, 15, 275–312. [Google Scholar] [CrossRef]
- Ye, J.; Fang, L.; Zheng, H.K.; Zhang, Y.; Chen, J.; Zhang, Z.J.; Wang, J.; Li, S.T.; Li, R.Q.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M.; Ragg, T. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006, 7, 3. [Google Scholar] [CrossRef]
- Fleige, S.; Pfaffl, M.W. RNA integrity and the effect on the real-time qRT-PCR performance. Mol. Asp. Med. 2006, 27, 126–139. [Google Scholar] [CrossRef]
- Cheng, H.L.; Wang, X.; Peng, Y.X.; Meng, X.P.; Sun, S.P.; Shi, X.Y. Molecular cloning and tissue distribution of lipoprotein lipase full-length cDNA from Pengze crucian carp (Carassius auratus var. Pengze). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2009, 153, 109–115. [Google Scholar] [CrossRef]
- Somamoto, T.; Yoshiura, Y.; Sato, A.; Nakao, M.; Nakanishi, T.; Okamoto, N.; Ototake, M. Expression profiles of TCRbeta and CD8α mRNA correlate with virus-specific cell-mediated cytotoxic activity in ginbuna crucian carp. Virology 2006, 348, 370–377. [Google Scholar] [CrossRef]
- An, L.H.; Lei, K.; Zheng, B.H. Use of heat shock protein mRNA expressions as biomarkers in wild crucian carp for monitoring water quality. Environ. Toxicol. Pharmacol. 2014, 37, 248–255. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, Y.B.; Liu, T.K.; Sun, F.; Gui, J.F. Subcellular localization and functional characterization of a fish IRF9 from crucian carp Carassius auratus. Fish. Shellfish. Immunol. 2012, 33, 258–266. [Google Scholar] [CrossRef]
- Bustin, S. Transparency of reporting in molecular diagnostics. Int. J. Mol. Sci. 2013, 14, 15878–15884. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.; et al. The need for transparency and good practices in the qPCR literature. Nat. Methods 2013, 10, 1063–1067. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, C.-Y.; Li, J.-T.; Kuang, Y.-Y.; Xu, R.; Zhao, Z.-X.; Hou, G.-Y.; Liang, H.-W.; Sun, X.-W. The Transcriptomes of the Crucian Carp Complex (Carassius auratus) Provide Insights into the Distinction between Unisexual Triploids and Sexual Diploids. Int. J. Mol. Sci. 2014, 15, 9386-9406. https://doi.org/10.3390/ijms15069386
Li C-Y, Li J-T, Kuang Y-Y, Xu R, Zhao Z-X, Hou G-Y, Liang H-W, Sun X-W. The Transcriptomes of the Crucian Carp Complex (Carassius auratus) Provide Insights into the Distinction between Unisexual Triploids and Sexual Diploids. International Journal of Molecular Sciences. 2014; 15(6):9386-9406. https://doi.org/10.3390/ijms15069386
Chicago/Turabian StyleLi, Chun-Yan, Jiong-Tang Li, You-Yi Kuang, Ru Xu, Zi-Xia Zhao, Guang-Yuan Hou, Hong-Wei Liang, and Xiao-Wen Sun. 2014. "The Transcriptomes of the Crucian Carp Complex (Carassius auratus) Provide Insights into the Distinction between Unisexual Triploids and Sexual Diploids" International Journal of Molecular Sciences 15, no. 6: 9386-9406. https://doi.org/10.3390/ijms15069386