Proteome Analysis of Subsarcolemmal Cardiomyocyte Mitochondria: A Comparison of Different Analytical Platforms

Abstract
:1. Introduction
2. Results
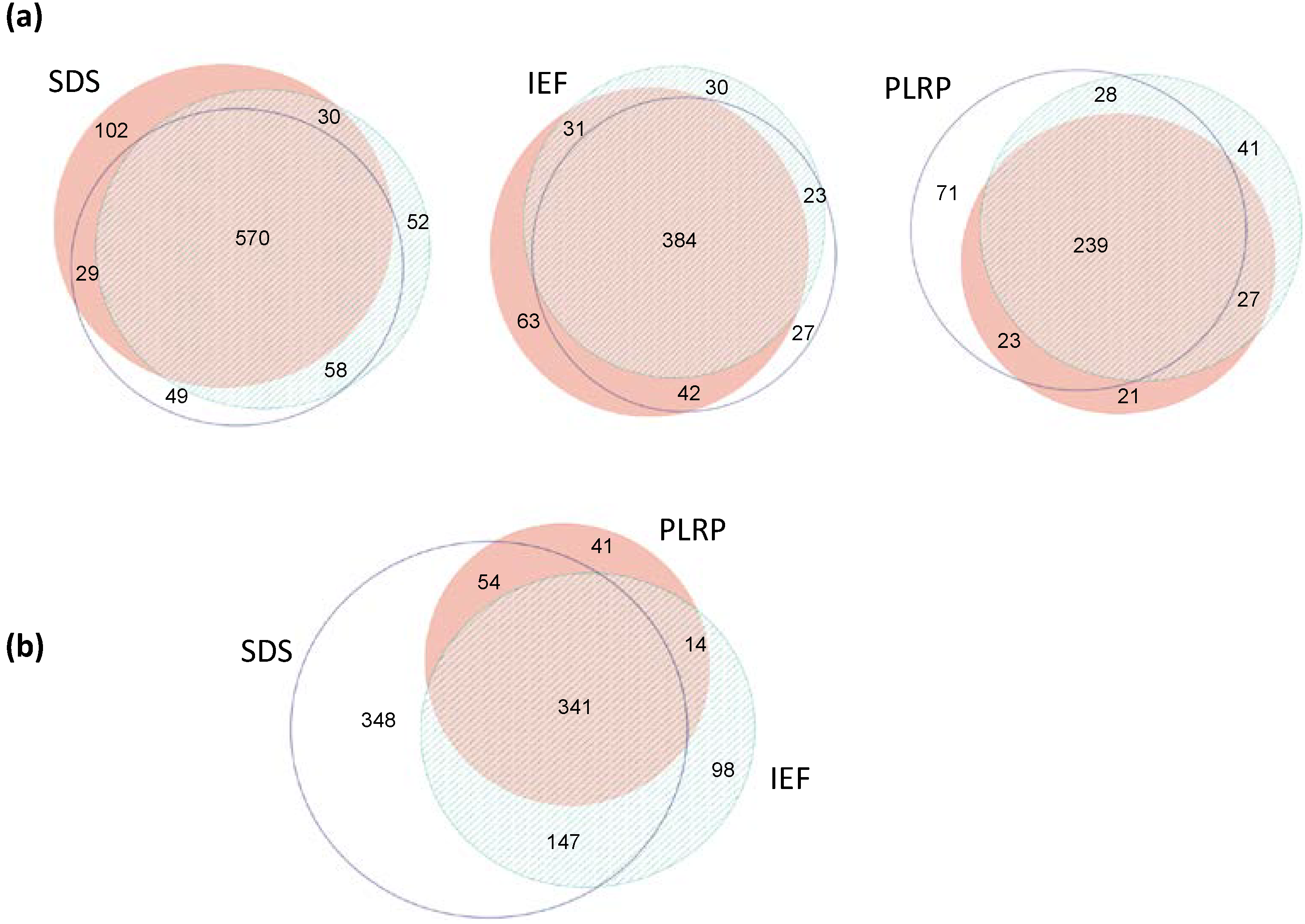
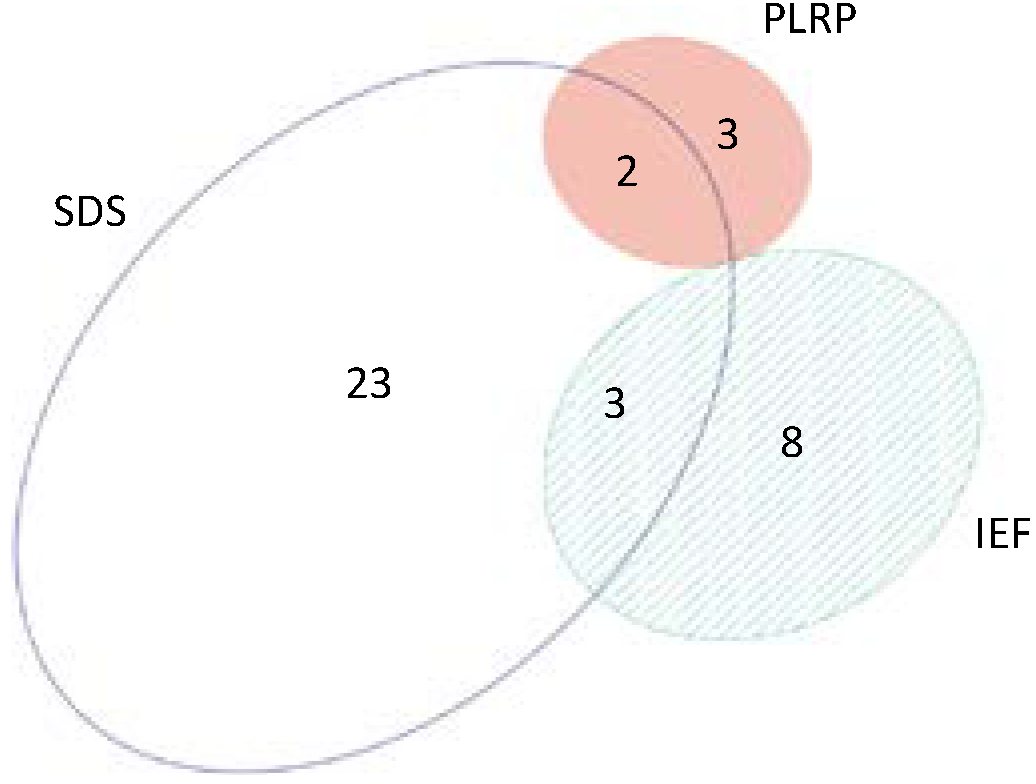
2.1. Summary of Protein Identification Results
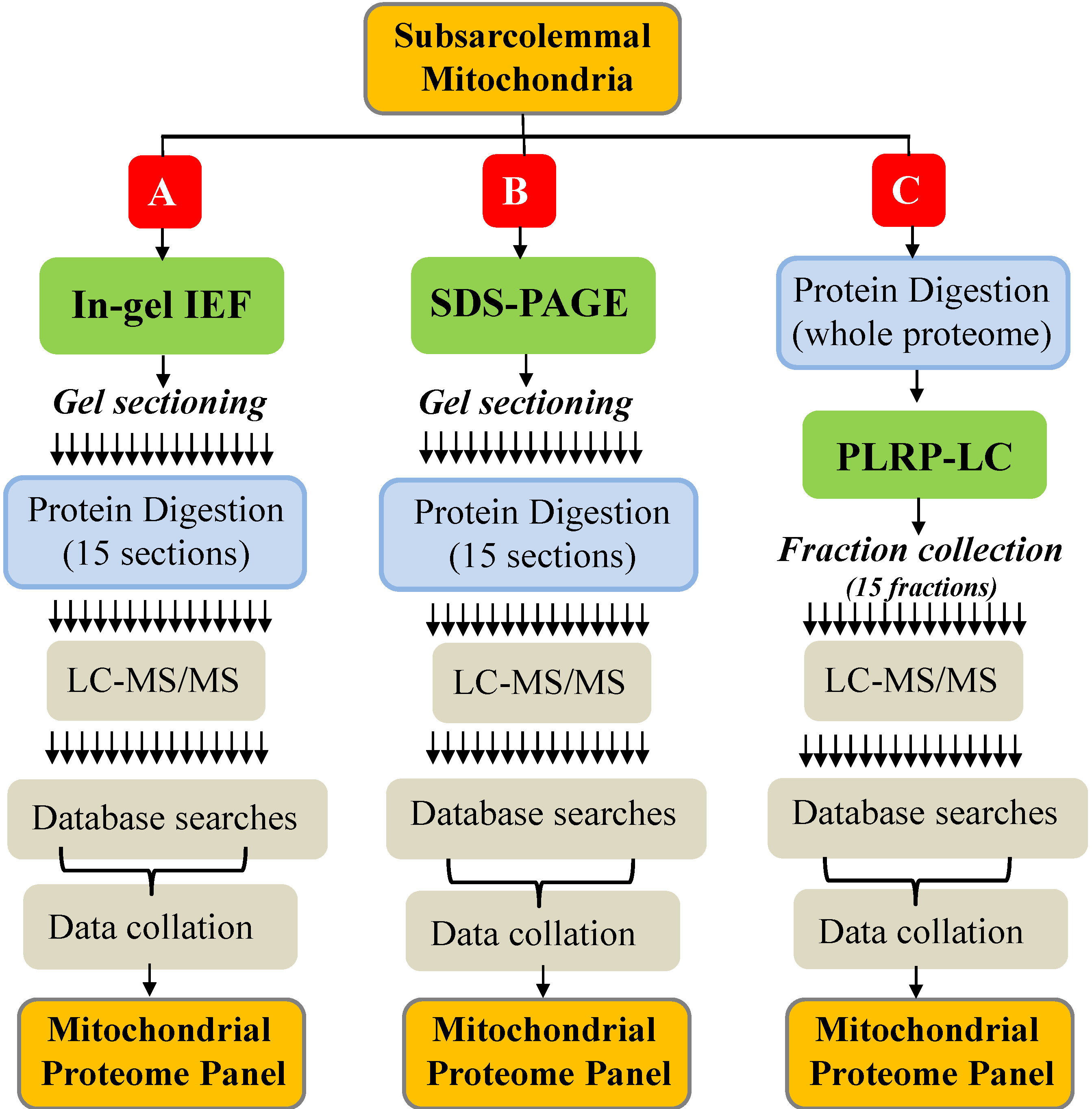

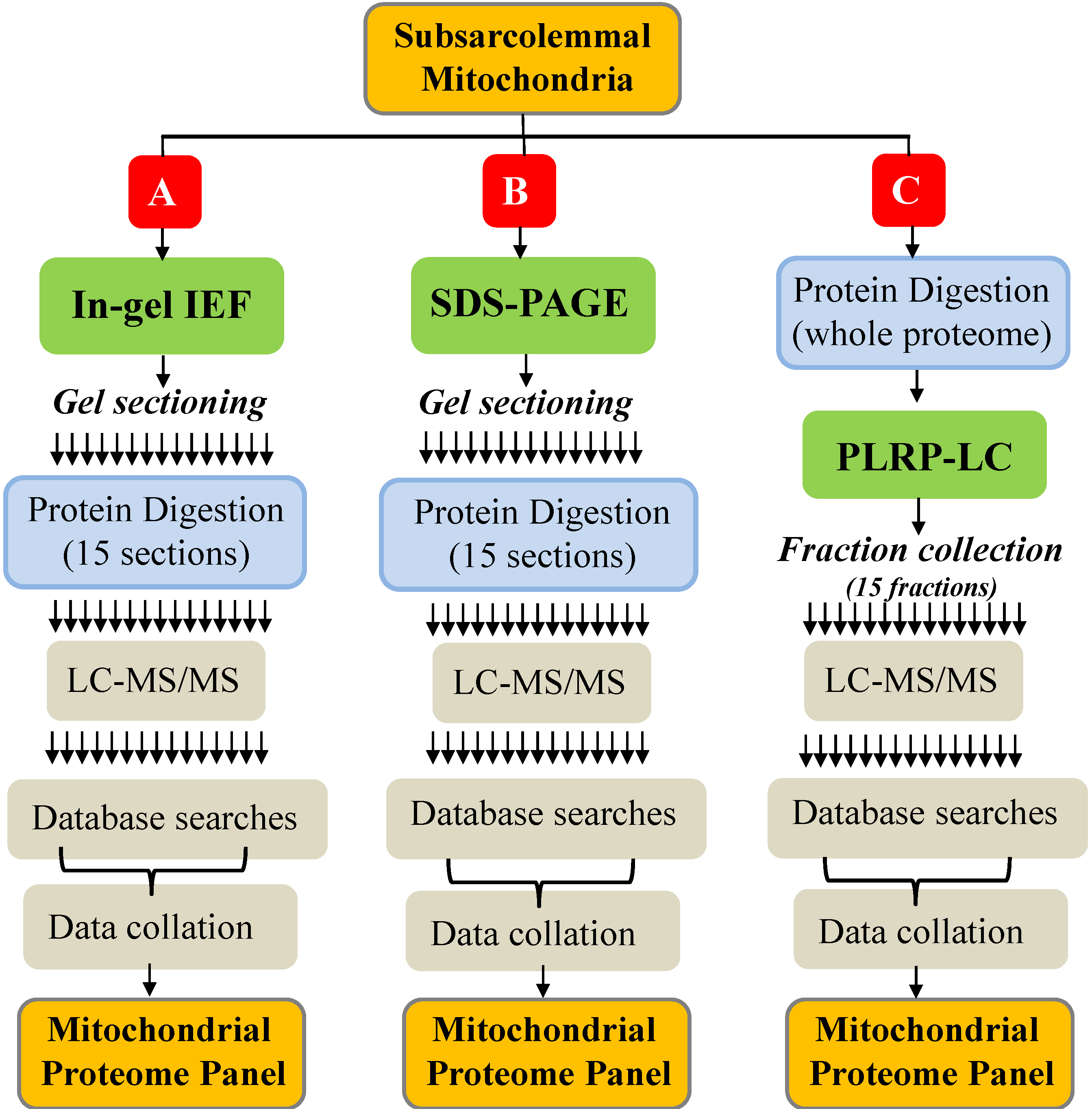{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Performance Characteristic | SDS-PAGE | IEF | PLRP |
|---|---|---|---|
| Number of proteins—replicate 1 | 708 | 520 | 315 |
| Number of proteins—replicate 2 | 726 | 468 | 330 |
| Number of proteins—replicate 3 | 696 | 476 | 361 |
| Average ± STD | 710 ± 15 | 488 ± 28 | 335 ± 23 |
| C.V. | 2.1% | 5.7% | 7.0% |
| Total number of proteins (all runs) | 890 | 600 | 450 |
| Total number of peptides (all runs) | 6023 | 3110 | 1671 |
| Proteome coverage overlap | 64% | 63% | 53% |
| Total number of proteins (all platforms) | 1043 | – | – |
| Proteome coverage overlap (all platforms) | 32.7% | – | – |

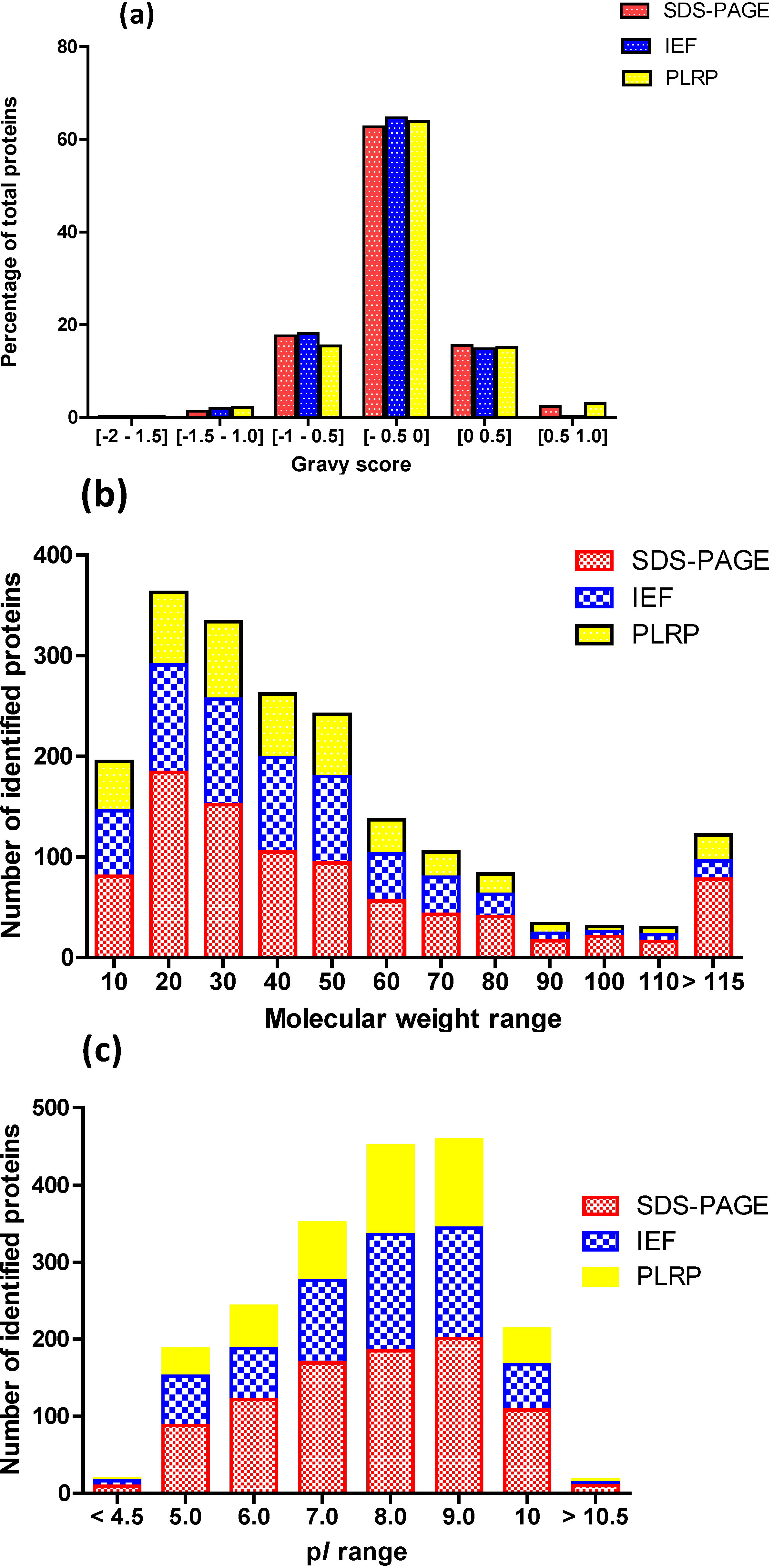
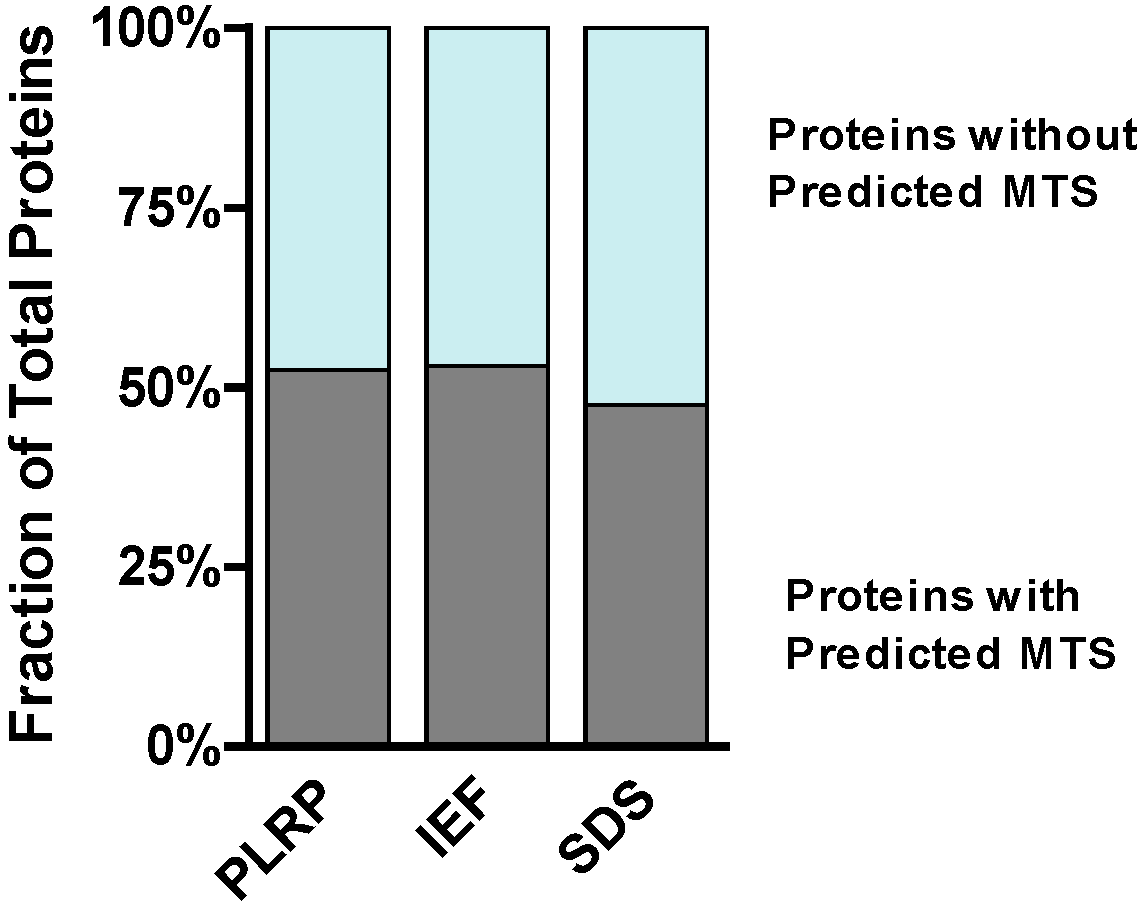
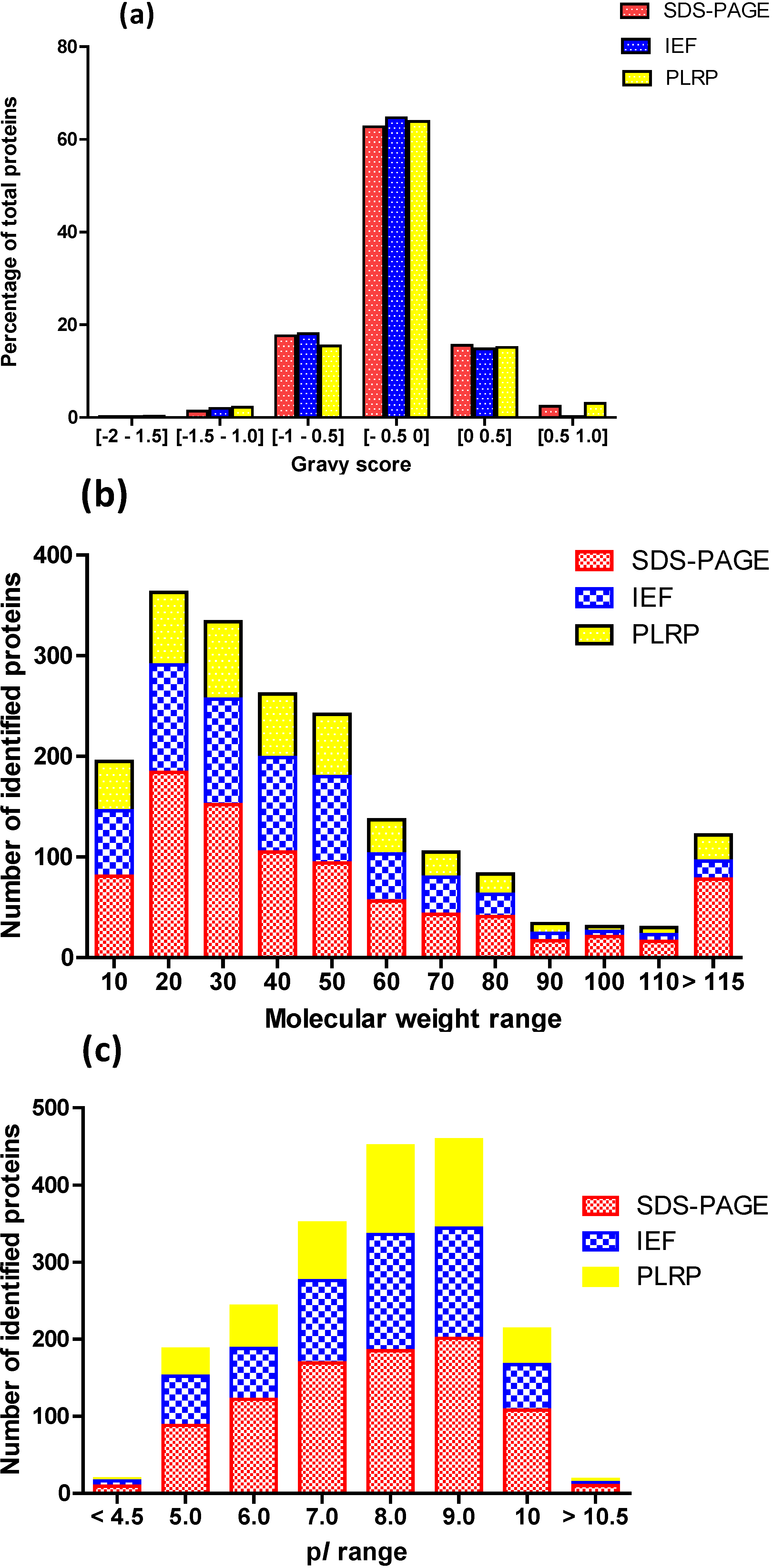
2.2. Intrinsic Chemical Properties of the Identified Proteins
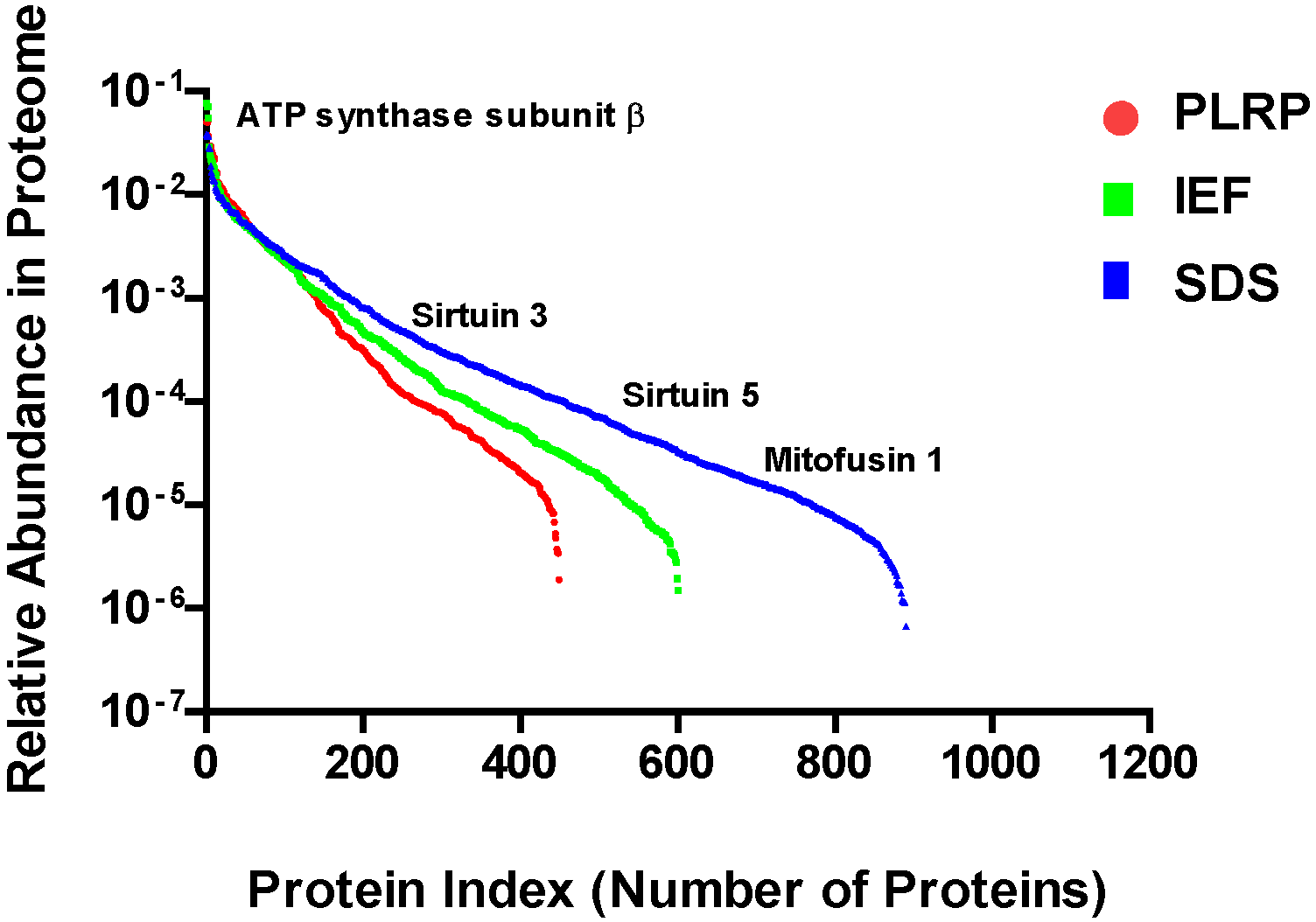
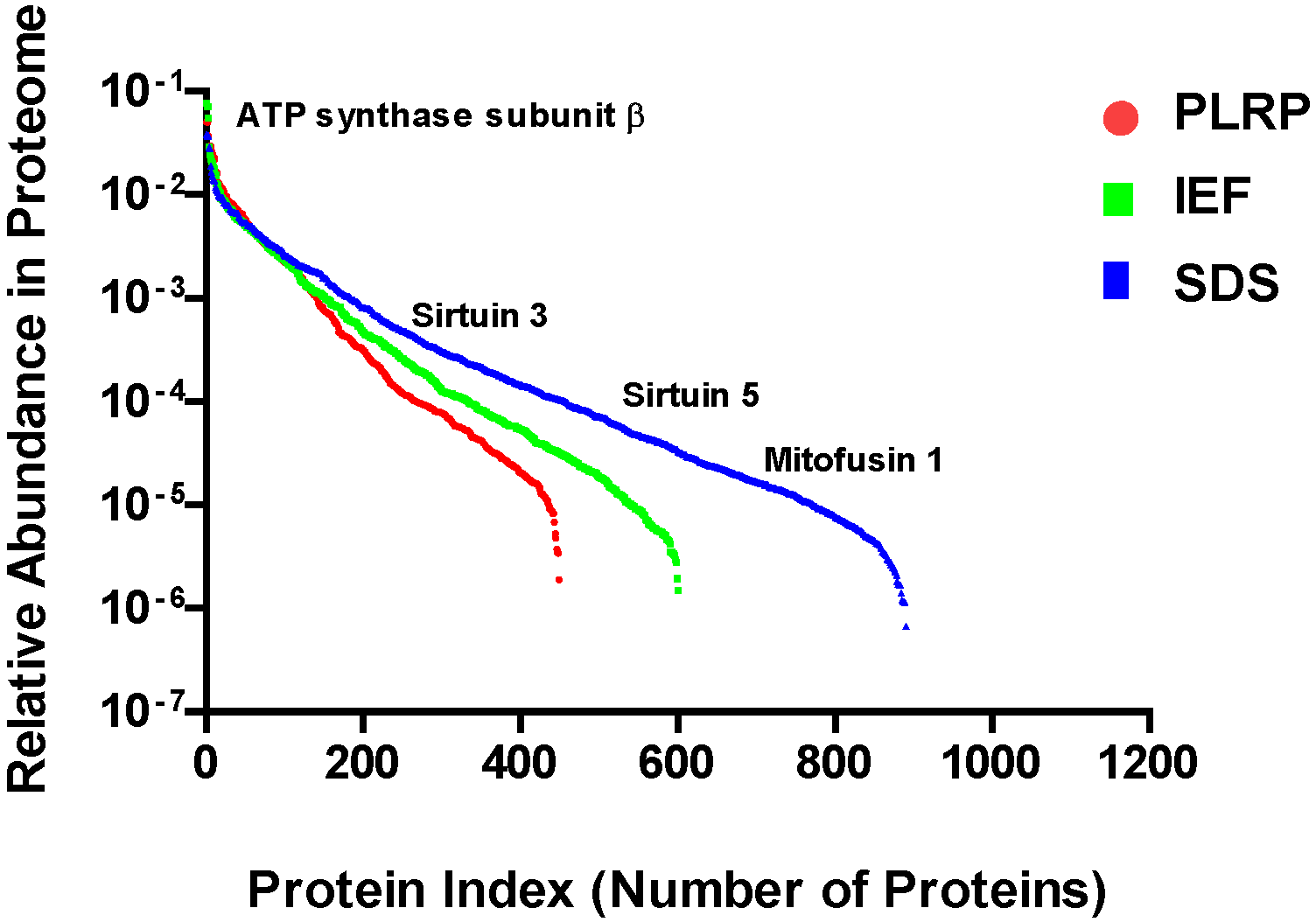
2.3. Mitochondrial Proteome Abundance
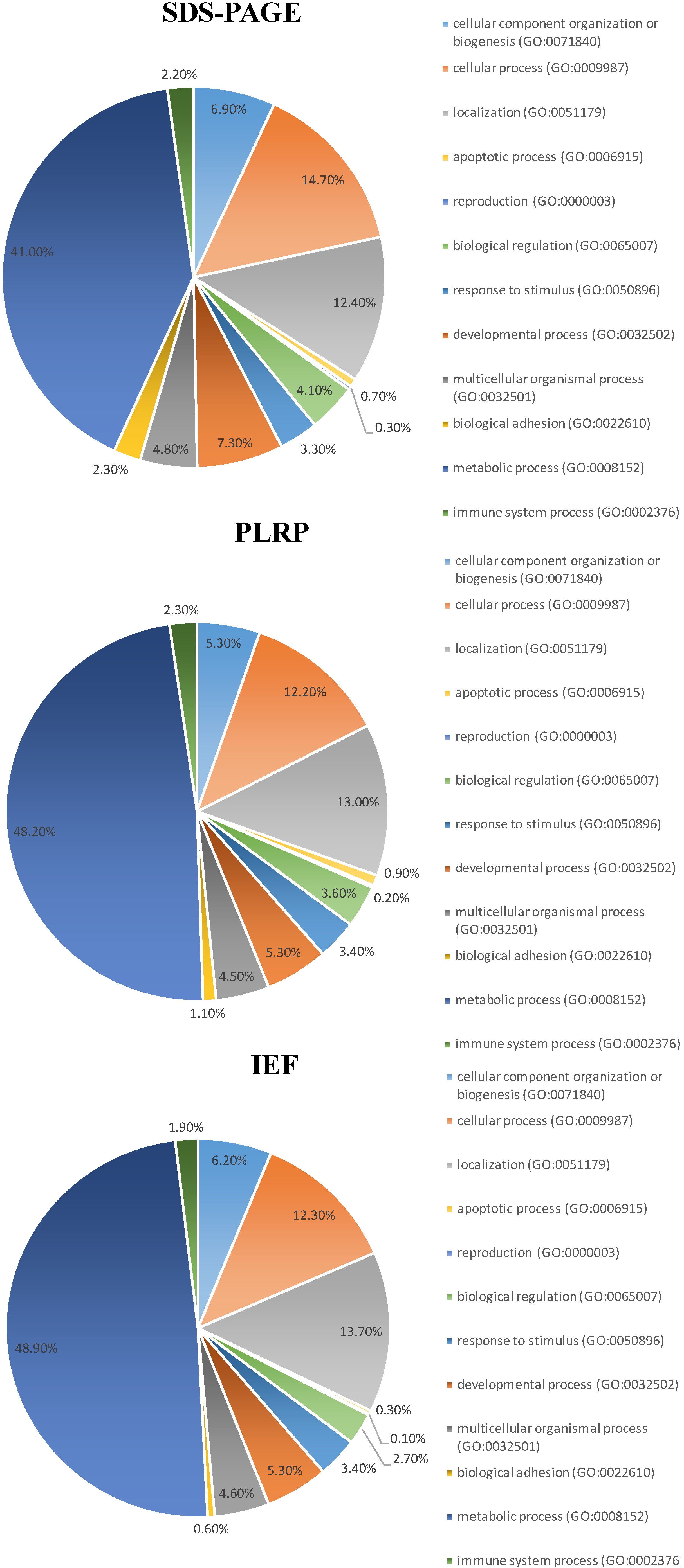
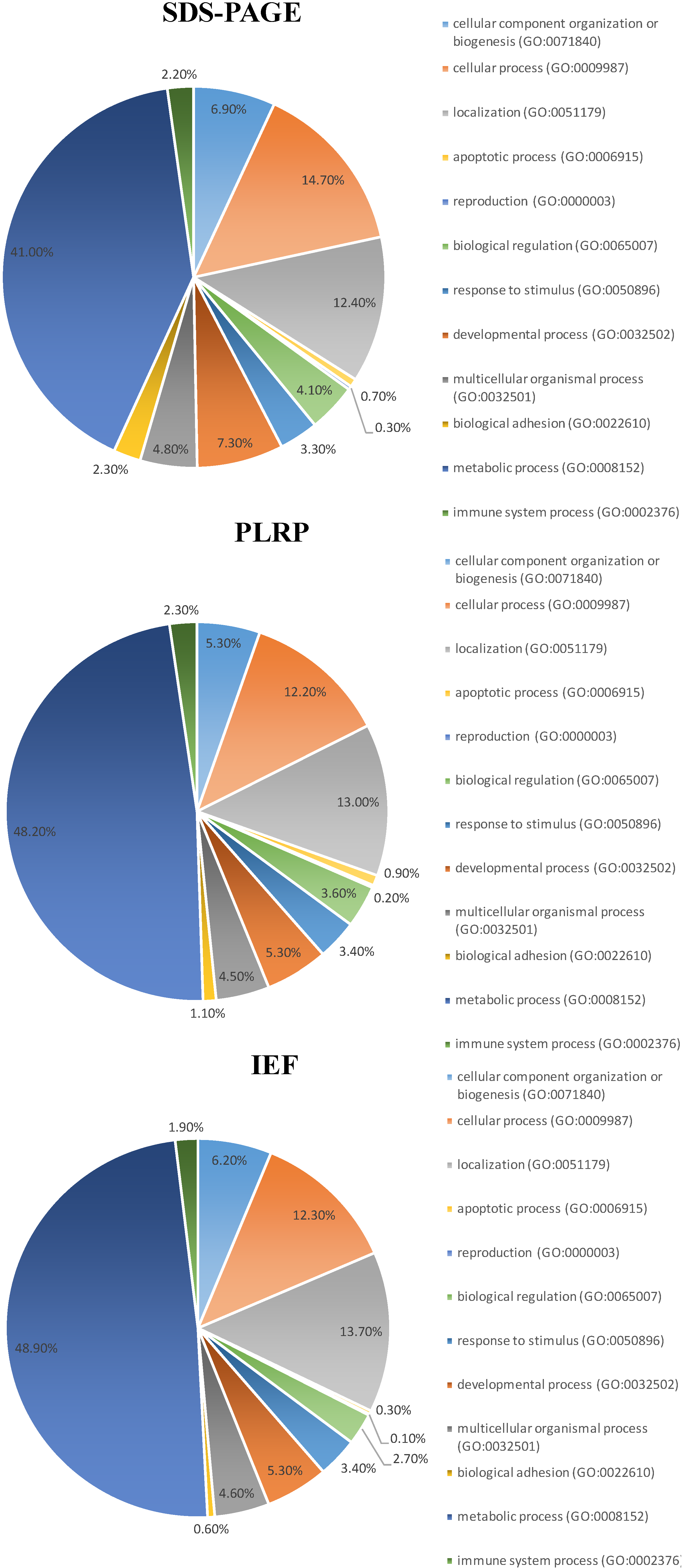
2.4. Categorization of the Identified Proteins
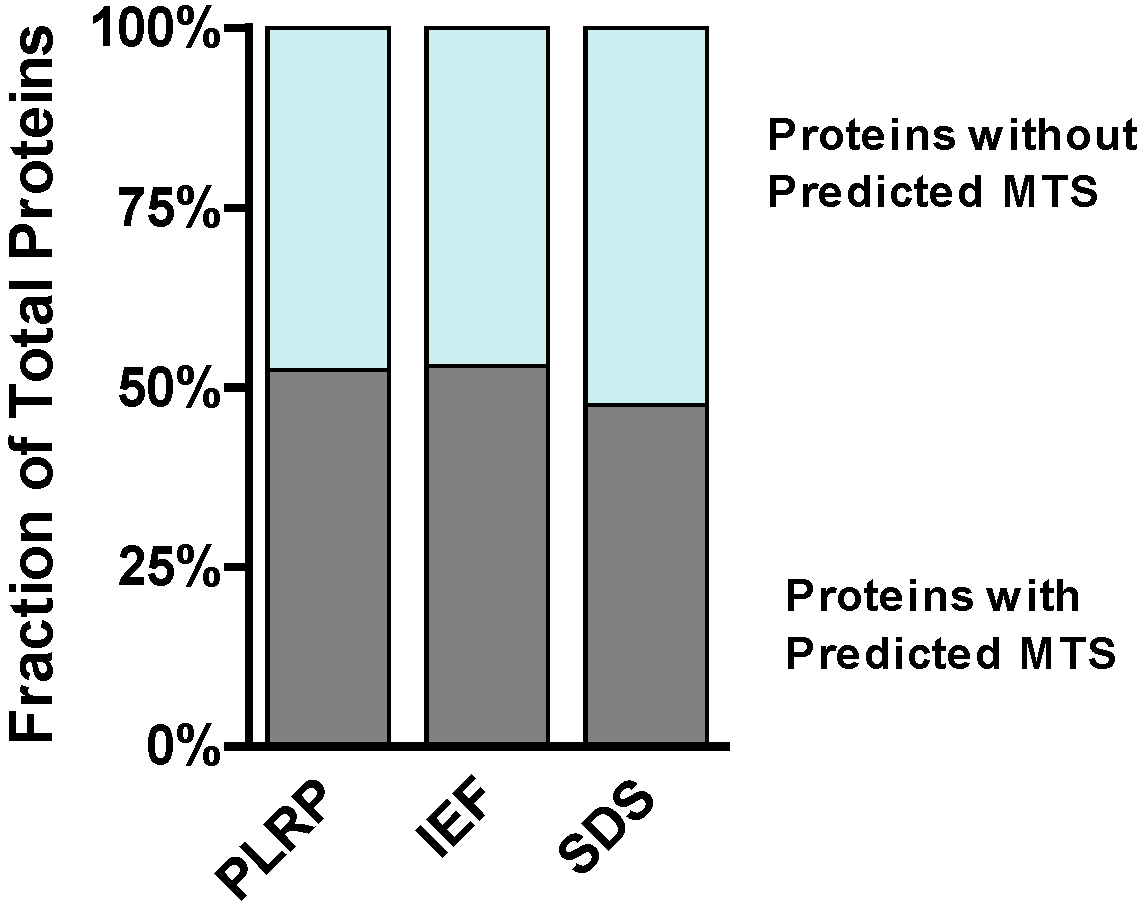
2.5. Selected Functional Characteristics of the Mitochondrial Proteome Panel
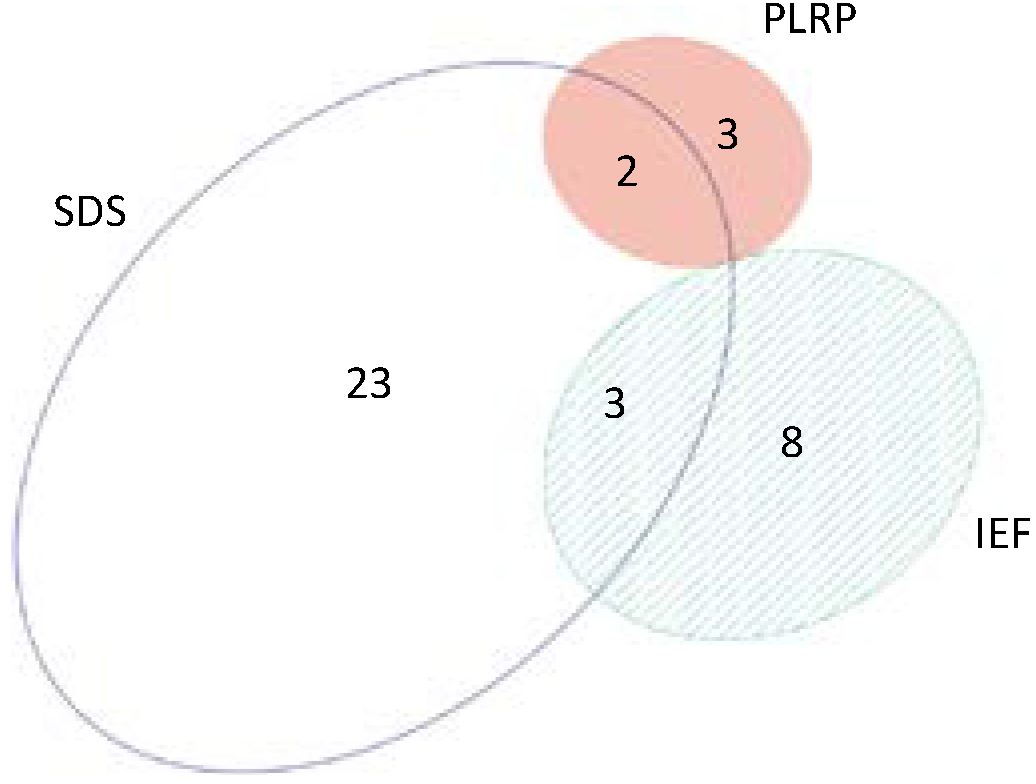
2.6. Beyond Protein Identity—Protein Acetylation
3. Discussion
4. Experimental Section
4.1. Mitochondria Isolation and Protein Extraction
4.2. Isoelectric Focusing (IEF)
4.3. Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE)
4.4. In-Solution Protein Digestion
4.5. In-Gel Protein Digestion
4.6. High-pH Reversed-Phase Chromatography (PLRP)
4.7. Liquid Chromatography-Mass Spectrometry (LC-MS/MS)
4.8. Bioinformatics
5. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Duchen, M.R.; Szabadkai, G. Roles of mitochondria in human disease. Essays Biochem. 2010, 47, 115–137. [Google Scholar] [CrossRef]
- Khan, M.U.; Cheema, Y.; Shahbaz, A.U.; Ahokas, R.A.; Sun, Y.; Gerling, I.C.; Bhattacharya, S.K.; Weber, K.T. Mitochondria play a central role in nonischemic cardiomyocyte necrosis: Common to acute and chronic stressor states. Pflugers Arch. 2012, 464, 123–131. [Google Scholar] [CrossRef]
- Gandhi, M.S.; Kamalov, G.; Shahbaz, A.U.; Bhattacharya, S.K.; Ahokas, R.A.; Sun, Y.; Gerling, I.C.; Weber, K.T. Cellular and molecular pathways to myocardial necrosis and replacement fibrosis. Heart Fail. Rev. 2011, 16, 23–34. [Google Scholar] [CrossRef]
- Marin-Garcia, J. Mitochondria and the Heart; Springer: New York, NY, USA, 2005; Volume 256. [Google Scholar]
- Rosca, M.G.; Hoppel, C.L. Mitochondria in heart failure. Cardiovasc. Res. 2010, 88, 40–50. [Google Scholar] [CrossRef]
- Neubauer, S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef]
- Dugo, P.; Cacciola, F.; Kumm, T.; Dugo, G.; Mondello, L. Comprehensive multidimensional liquid chromatography: Theory and applications. J. Chromatogr. 2008, 1184, 353–368. [Google Scholar] [CrossRef]
- Tran, J.C.; Zamdborg, L.; Ahlf, D.R.; Lee, J.E.; Catherman, A.D.; Durbin, K.R.; Tipton, J.D.; Vellaichamy, A.; Kellie, J.F.; Li, M.; et al. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature 2011, 480, 254–258. [Google Scholar] [CrossRef]
- Chen, E.I.; Hewel, J.; Felding-Habermann, B.; Yates, J.R. Large scale protein profiling by combination of protein fractionation and multidimensional protein identification technology (MudPIT). Mol. Cell. Proteomics 2006, 5, 53–56. [Google Scholar]
- Gilar, M.; Olivova, P.; Daly, A.E.; Gebler, J.C. Two-dimensional separation of peptides using RP–RP–HPLC system with different pH in first and second separation dimensions. J. Sep. Sci. 2005, 28, 1694–1703. [Google Scholar] [CrossRef]
- Gilar, M.; Olivova, P.; Chakraborty, A.B.; Jaworski, A.; Geromanos, S.J.; Gebler, J.C. Comparison of 1-D and 2-D LC MS/MS methods for proteomic analysis of human serum. Electrophoresis 2009, 30, 1157–1167. [Google Scholar] [CrossRef]
- Nakamura, T.; Kuromitsu, J.; Oda, Y. Evaluation of comprehensive multidimensional separations using reversed-phase, reversed-phase liquid chromatography/mass spectrometry for shotgun proteomics. J. Proteome Res. 2008, 7, 1007–1011. [Google Scholar] [CrossRef]
- Beranova-Giorgianni, S.; Zhao, Y.; Desiderio, D.M.; Giorgianni, F. Phosphoproteomic analysis of the human pituitary. Pituitary 2006, 9, 109–120. [Google Scholar] [CrossRef]
- Zhao, Y.; Giorgianni, F.; Desiderio, D.M.; Fang, B.; Beranova-Giorgianni, S. Toward a global analysis of the human pituitary proteome by multiple gel-based technology. Anal. Chem. 2005, 77, 5324–5331. [Google Scholar] [CrossRef]
- Giorgianni, F.; Desiderio, D.M.; Beranova-Giorgianni, S. Proteome analysis using isoelectric focusing in immobilized pH gradient gels followed by mass spectrometry. Electrophoresis 2003, 24, 253–259. [Google Scholar] [CrossRef]
- Rezaul, K.; Wu, L.; Mayya, V.; Hwang, S.-I.; Han, D. A systematic characterization of mitochondrial proteome from human T leukemia cells. Mol. Cell. Proteomics 2005, 4, 169–181. [Google Scholar]
- Kohrs, F.; Heyer, R.; Magnussen, A.; Benndorf, D.; Muth, T.; Behne, A.; Rapp, E.; Kausmann, R.; Heiermann, M.; Klocke, M.; et al. Sample prefractionation with liquid isoelectric focusing enables in depth microbial metaproteome analysis of mesophilic and thermophilic biogas plants. Anaerobe 2014. [Google Scholar] [CrossRef]
- Gandhi, M.S.; Deshmukh, P.A.; Kamalov, G.; Zhao, T.; Zhao, W.; Whaley, J.T.; Tichy, J.R.; Bhattacharya, S.K.; Ahokas, R.A.; Sun, Y.; et al. Causes and consequences of zinc dyshomeostasis in rats with chronic aldosteronism. J. Cardiovasc. Pharmacol. 2008, 52, 245–252. [Google Scholar] [CrossRef]
- Brosch, M.; Yu, L.; Hubbard, T.; Choudhary, J. Accurate and sensitive peptide identification with mascot percolator. J. Proteome Res. 2009, 8, 3176–3181. [Google Scholar] [CrossRef]
- Spivak, M.; Weston, J.; Bottou, L.; Kall, L.; Noble, W.S. Improvements to the percolator algorithmfor peptide identification from shotgun proteomics data sets. J. Proteome Res. 2009, 8, 3737–3745. [Google Scholar] [CrossRef]
- Forner, F.; Foster, L.J.; Campanaro, S.; Valle, G.; Mann, M. Quantitative proteomic comparison of rat mitochondria from muscle, heart, and liver. Mol. Cell. Proteomics 2006, 5, 608–619. [Google Scholar]
- Paoletti, A.C.; Parmely, T.J.; Tomomori-Sato, C.; Sato, S.; Zhu, D.; Conaway, R.C.; Conaway, J.W.; Florens, L.; Washburn, M.P. Quantitative proteomic analysis of distinct mammalian mediator complexes using normalized spectral abundance factors. Proc. Natl. Acad. Sci. USA 2006, 103, 18928–18933. [Google Scholar] [CrossRef]
- Emanuelsson, O.; Brunak, S.; von Heijne, G.; Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar] [CrossRef]
- Neupert, W.; Herrmann, J.M. Translocation of proteins into mitochondria. Annu. Rev. Biochem. 2007, 76, 723–749. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Thomas, P.D.; Kejariwal, A.; Campbell, M.J.; Mi, H.; Diemer, K.; Guo, N.; Ladunga, I.; Ulitsky-Lazareva, B.; Muruganujan, A.; Rabkin, S. PANTHER: A browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 2003, 31, 334–341. [Google Scholar] [CrossRef]
- Ozcan, U. Mitofusins: Mighty regulators of metabolism. Cell 2013, 155, 17–18. [Google Scholar] [CrossRef]
- Huang, J.-Y.; Hirschey, M.D.; Shimazu, T.; Ho, L.; Verdin, E. Mitochondrial sirtuins. Biochim. Biophys. Acta 2010, 1804, 1645–1651. [Google Scholar] [CrossRef]
- Giorgianni, F.; Usman Khan, M.; Weber, K.T.; Gerling, I.C.; Beranova-Giorgianni, S. Phosphoproteome mapping of cardiomyocyte mitochondria in a rat model of heart failure. Mol. Cell. Biochem. 2014, 389, 159–167. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, J.; Lu, L.; Chen, S.S.; Quinn, M.T.; Weber, K.T. Aldosterone-induced inflammation in the rat heart: Role of oxidative stress. Am. J. Pathol. 2002, 161, 1773–1781. [Google Scholar] [CrossRef]
- Ferreira, R.; Rocha, H.; Almeida, V.; Padrão, A.I.; Santa, C.; Vilarinho, L.; Amado, F.; Vitorino, R. Mitochondria proteome profiling: A comparative analysis between gel-and gel-free approaches. Talanta 2013, 115, 277–283. [Google Scholar] [CrossRef]
- Reinders, J.; Zahedi, R.P.; Pfanner, N.; Meisinger, C.; Sickmann, A. Toward the complete yeast mitochondrial proteome: Multidimensional separation techniques for mitochondrial proteomics. J. Proteome Res. 2006, 5, 1543–1554. [Google Scholar] [CrossRef]
- McQueen, P.; Krokhin, O. Optimal selection of 2D reversed-phase-reversed-phase HPLC separation techniques in bottom-up proteomics. Expert Rev. Proteomics 2012, 9, 125–128. [Google Scholar] [CrossRef]
- Beranova-Giorgianni, S.; Desiderio, D.M.; Giorgianni, F. Phosphoproteome analysis by in-gel isoelectric focusing and tandem mass spectrometry. Methods Mol. Biol. 2009, 519, 383–396. [Google Scholar] [CrossRef]
- Gautier, V.; Mouton-Barbosa, E.; Bouyssié, D.; Delcourt, N.; Beau, M.; Girard, J.-P.; Cayrol, C.; Burlet-Schiltz, O.; Monsarrat, B.; de Peredo, A.G. Label-free quantification and shotgun analysis of complex proteomes by one-dimensional SDS-PAGE/nanoLC-MS evaluation for the large scale analysis of inflammatory human endothelial cells. Mol. Cell. Proteomics 2012, 11, 527–539. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- PANTHER Classification System. Available online: http://www.pantherdb.org (accessed on 19 March 2014).
- TargetP 1.1 Server. Available online: http://www.cbs.dtu.dk/services/TargetP (accesed on 21 March 2014).
- Cytoscape. Available online: http://www.cytoscape.org (accessed on 20 March 2014).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Giorgianni, F.; Koirala, D.; Weber, K.T.; Beranova-Giorgianni, S. Proteome Analysis of Subsarcolemmal Cardiomyocyte Mitochondria: A Comparison of Different Analytical Platforms. Int. J. Mol. Sci. 2014, 15, 9285-9301. https://doi.org/10.3390/ijms15069285
Giorgianni F, Koirala D, Weber KT, Beranova-Giorgianni S. Proteome Analysis of Subsarcolemmal Cardiomyocyte Mitochondria: A Comparison of Different Analytical Platforms. International Journal of Molecular Sciences. 2014; 15(6):9285-9301. https://doi.org/10.3390/ijms15069285
Chicago/Turabian StyleGiorgianni, Francesco, Diwa Koirala, Karl T. Weber, and Sarka Beranova-Giorgianni. 2014. "Proteome Analysis of Subsarcolemmal Cardiomyocyte Mitochondria: A Comparison of Different Analytical Platforms" International Journal of Molecular Sciences 15, no. 6: 9285-9301. https://doi.org/10.3390/ijms15069285