The Peroxisome Proliferator-Activated Receptor (PPAR) α Agonist Fenofibrate Suppresses Chemically Induced Lung Alveolar Proliferative Lesions in Male Obese Hyperlipidemic Mice

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Results
2.1.1. General Observation
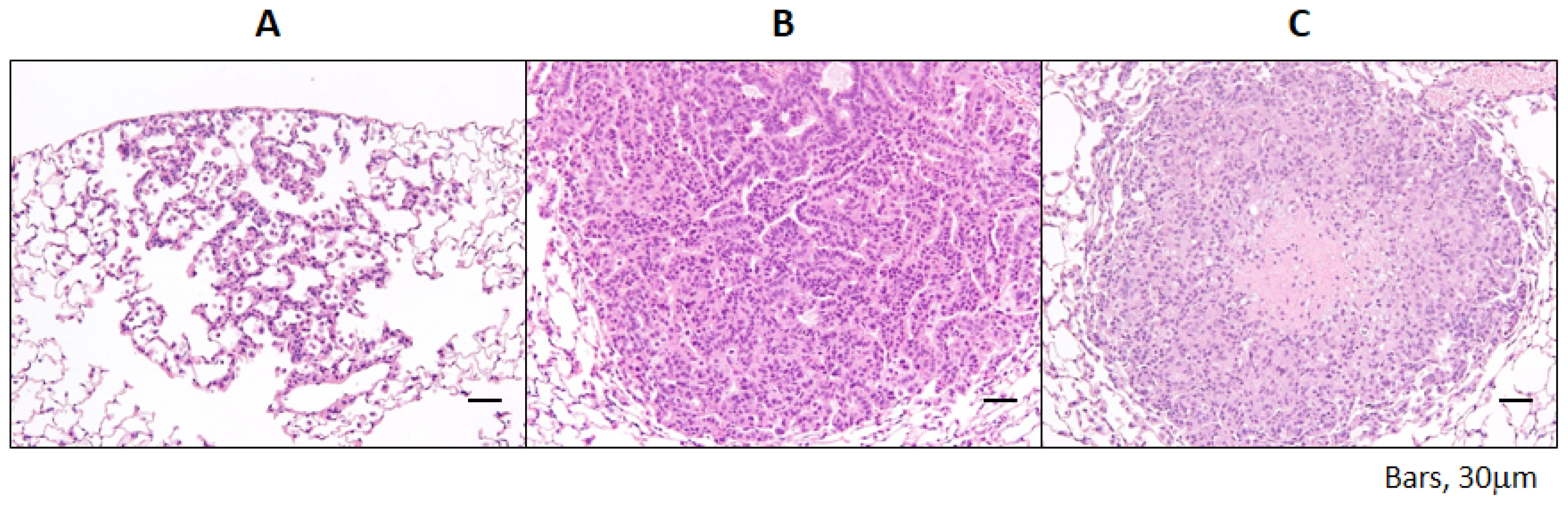
2.1.2. Incidence and Multiplicity of Pulmonary Proliferative Lesions
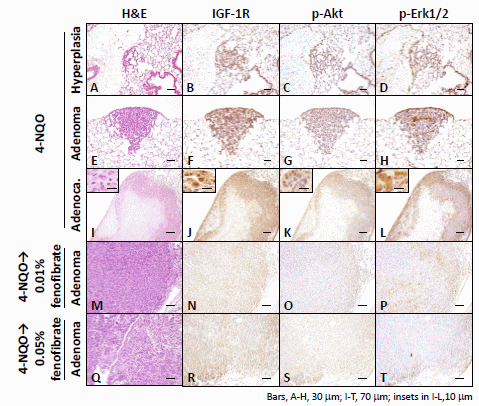
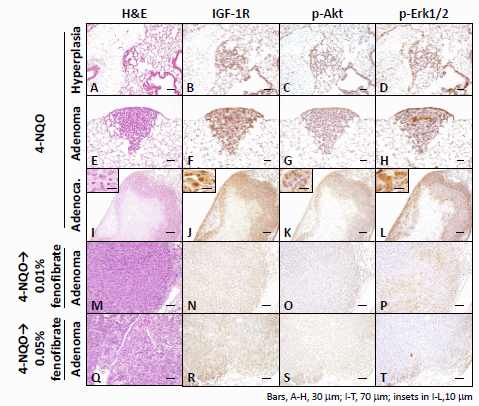
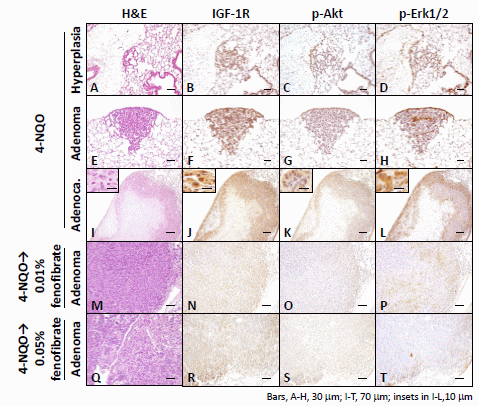
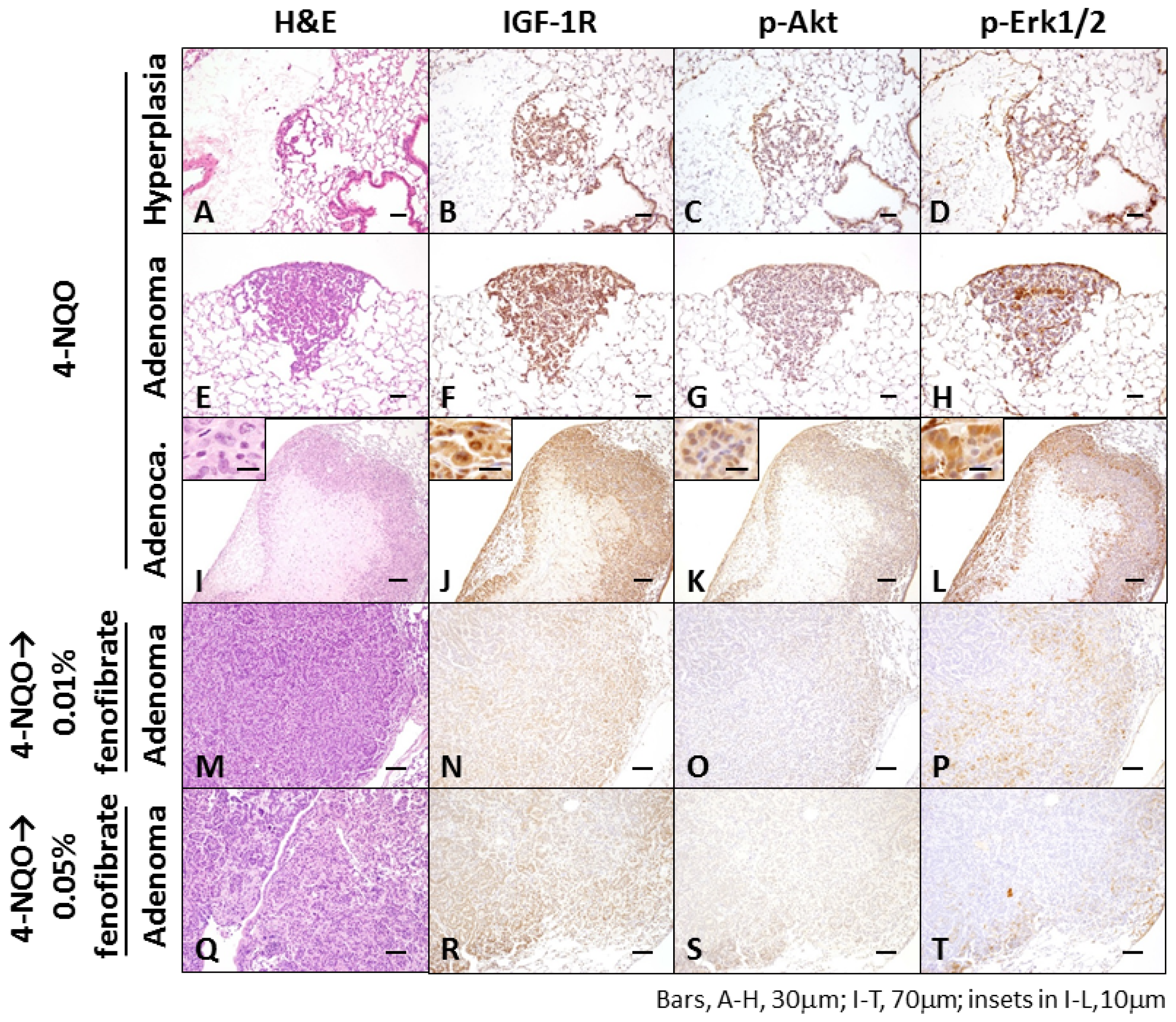
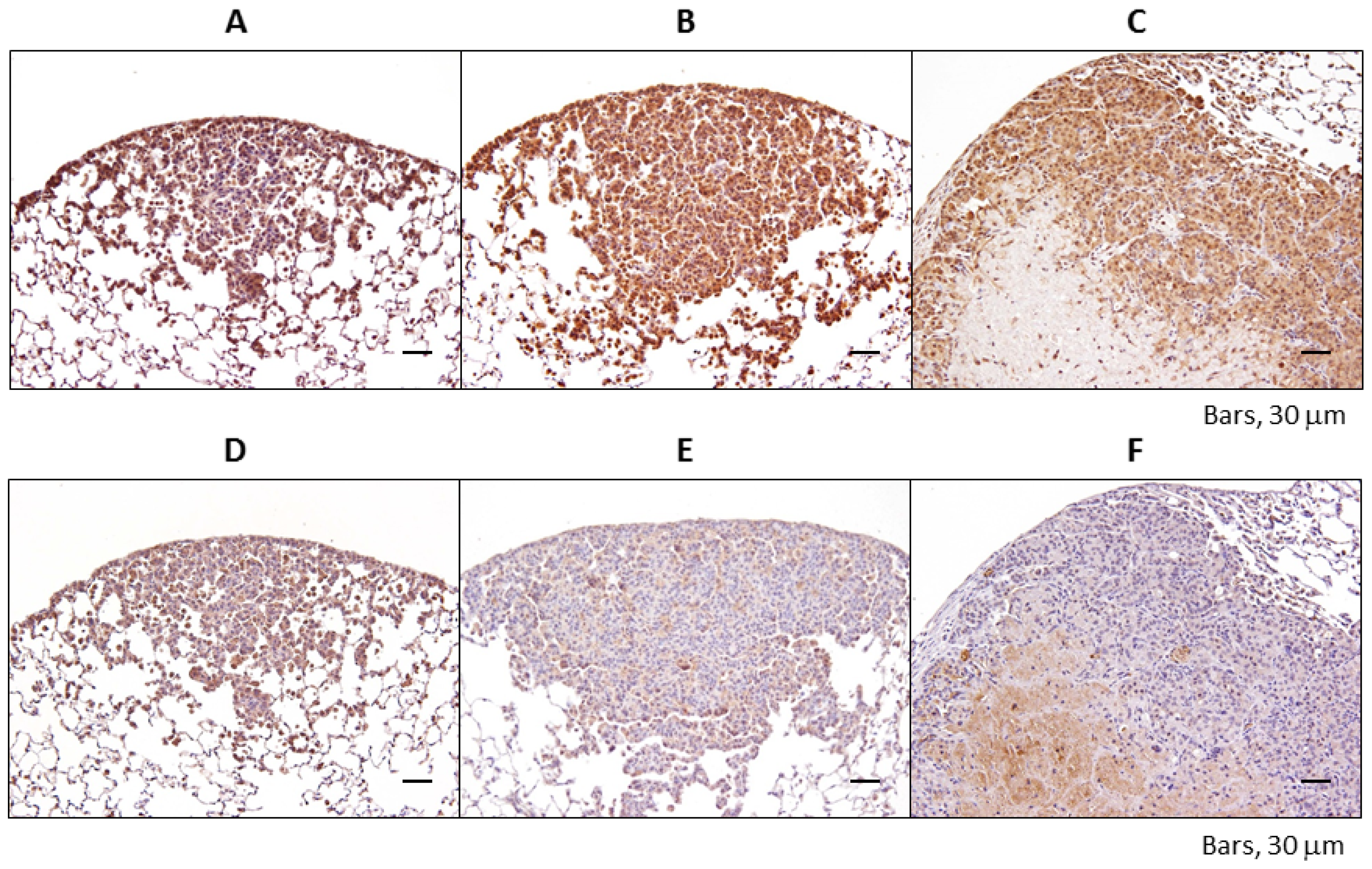
2.1.3. Immunohistochemical Expression of IGF-1R, p-Akt, p-Erk1/2, PPARα, and PPARγ in Pulmonary Proliferative Lesions Induced by 4-NQO
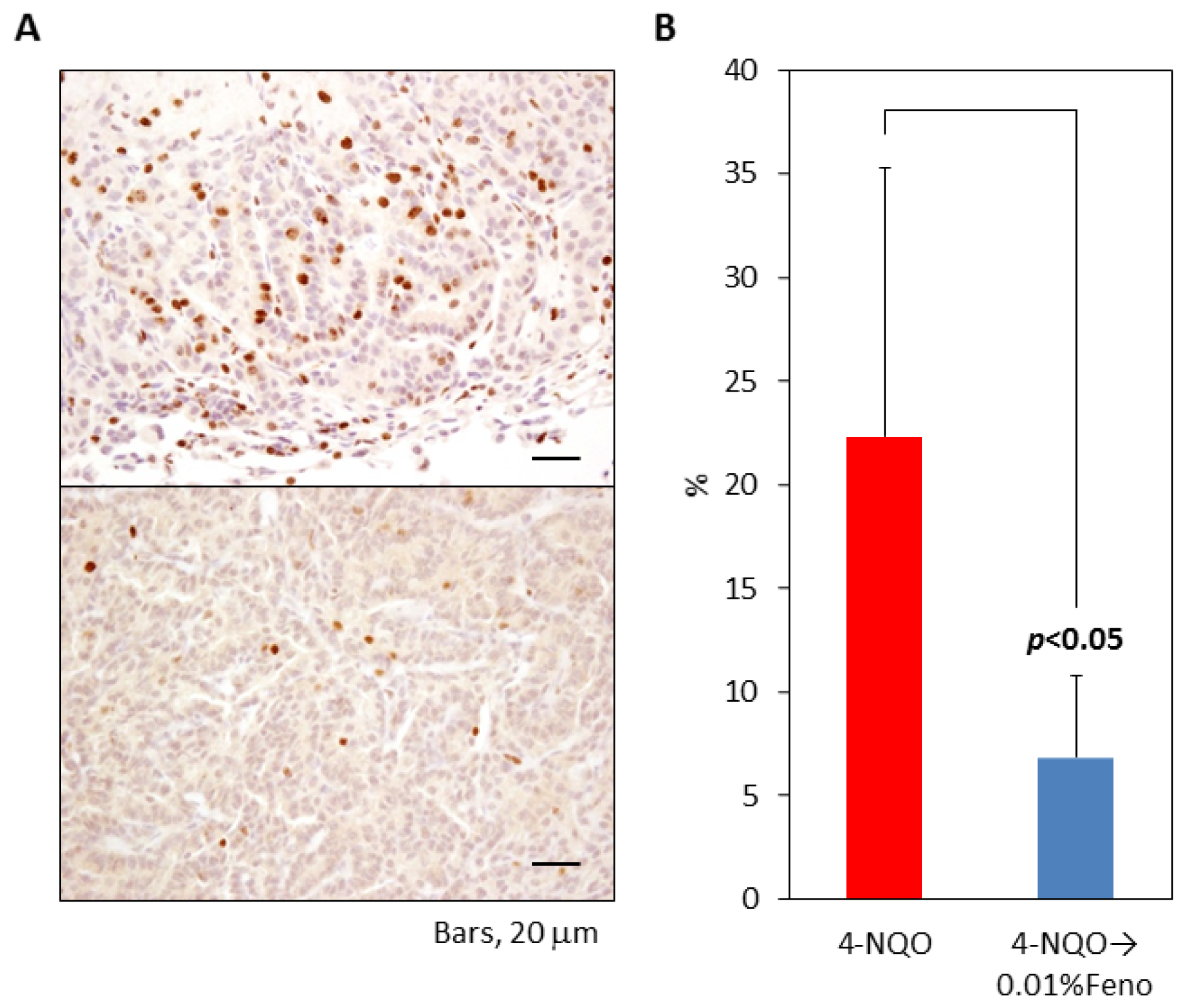
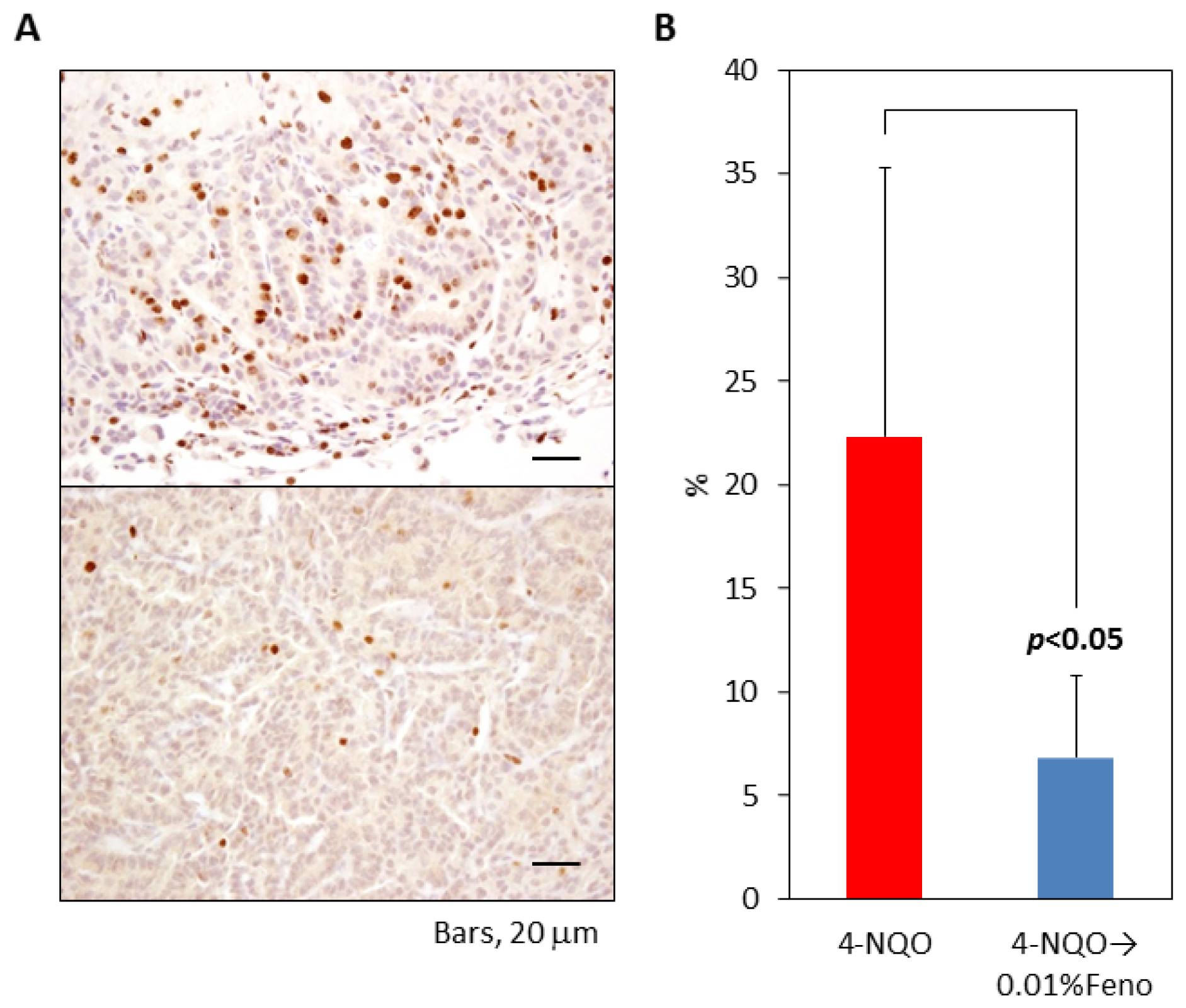
2.1.4. Ki-67 Labeling Index of Pulmonary Proliferative Lesions
2.1.5. Serum Triglyceride, Free Fatty Acid, Total Cholesterol, Glucose, Insulin, and Insulin-Like Growth Factor-1 Levels
2.2. Discussion
3. Experimental Section
3.1. Animals, Diet, and Chemicals
3.2. Experimental Design
3.3. Immunohistochemistry
3.4. Plasma IGF-1 and Insulin Levels
3.5. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsToshiya Kuno, Kazuya Hata, Yoshinobu Hirose, and Takuji Tanaka designed the study, performed the experiments and drafted the manuscript. Toshiya Kuno and Takuji Tanaka carried out data interpretation. Manabu Takamatsu, Akira Hara, Satoru Takahashi, and Katsumi Imaida provide technical support on experimental design and important comments in improving the manuscript. All authors read and approved the final manuscript.
References
- Siegel, R.; Ward, E.; Brawley, O.; Jemal, A. Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer Clin 2011, 61, 212–236. [Google Scholar]
- Katanoda, K.; Matsuda, T.; Matsuda, A.; Shibata, A.; Nishino, Y.; Fujita, M.; Soda, M.; Ioka, A.; Sobue, T.; Nishimoto, H. An updated report of the trends in cancer incidence and mortality in Japan. Jpn. J. Clin. Oncol 2013, 43, 492–507. [Google Scholar]
- Gainor, J.F.; Shaw, A.T. Emerging paradigms in the development of resistance to tyrosine kinase inhibitors in lung cancer. J. Clin. Oncol 2013, 31, 3987–3996. [Google Scholar]
- Keith, R.L.; Miller, Y.E. Lung cancer chemoprevention: current status and future prospects. Nat. Rev. Clin. Oncol 2013, 10, 334–343. [Google Scholar]
- Luo, J.; Chlebowski, R.; Wactawski-Wende, J.; Schlecht, N.F.; Tinker, L.; Margolis, K.L. Diabetes and lung cancer among postmenopausal women. Diabetes Care 2012, 35, 1485–1491. [Google Scholar]
- Ulmer, H.; Borena, W.; Rapp, K.; Klenk, J.; Strasak, A.; Diem, G.; Concin, H.; Nagel, G. Serum triglyceride concentrations and cancer risk in a large cohort study in Austria. Br. J. Cancer 2009, 101, 1202–1206. [Google Scholar]
- Petridou, E.T.; Sergentanis, T.N.; Antonopoulos, C.N.; Dessypris, N.; Matsoukis, I.L.; Aronis, K.; Efremidis, A.; Syrigos, C.; Mantzoros, C.S. Insulin resistance: An independent risk factor for lung cancer? Metabolism 2011, 60, 1100–1106. [Google Scholar]
- Becker, S.; Dossus, L.; Kaaks, R. Obesity related hyperinsulinaemia and hyperglycaemia and cancer development. Arch. Physiol. Biochem 2009, 115, 86–96. [Google Scholar]
- Memmott, R.M.; Mercado, J.R.; Maier, C.R.; Kawabata, S.; Fox, S.D.; Dennis, P.A. Metformin prevents tobacco carcinogen-induced lung tumorigenesis. Cancer Prev. Res 2010, 3, 1066–1076. [Google Scholar]
- Noto, H.; Goto, A.; Tsujimoto, T.; Noda, M. Cancer risk in diabetic patients treated with metformin: A systematic review and meta-analysis. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Fidler, M.J.; Shersher, D.D.; Borgia, J.A.; Bonomi, P. Targeting the insulin-like growth factor receptor pathway in lung cancer: Problems and pitfalls. Ther. Adv. Med. Oncol 2012, 4, 51–60. [Google Scholar]
- Siwicky, M.D.; Petrik, J.J.; Moorehead, R.A. The function of IGF-IR in NNK-mediated lung tumorigenesis. Lung Cancer 2011, 71, 11–18. [Google Scholar]
- Bailyes, E.M.; Nave, B.T.; Soos, M.A.; Orr, S.R.; Hayward, A.C.; Siddle, K. Insulin receptor/IGF-I receptor hybrids are widely distributed in mammalian tissues: Quantification of individual receptor species by selective immunoprecipitation and immunoblotting. Biochem. J 1997, 327, 209–215. [Google Scholar]
- Ding, J.; Tang, J.; Chen, X.; Men, H.T.; Luo, W.X.; Du, Y.; Ge, J.; Li, C.; Chen, Y.; Cheng, K.; et al. Expression characteristics of proteins of the insulin-like growth factor axis in non-small cell lung cancer patients with preexisting type 2 diabetes mellitus. Asian Pac. J. Cancer Prev 2013, 14, 5675–5680. [Google Scholar]
- McKeage, K.; Keating, G.M. Fenofibrate: A review of its use in dyslipidaemia. Drugs 2011, 71, 1917–1946. [Google Scholar]
- Keating, G.M. Fenofibrate: A review of its lipid-modifying effects in dyslipidemia and its vascular effects in type 2 diabetes mellitus. Am. J. Cardiovasc. Drugs 2011, 11, 227–247. [Google Scholar]
- Urbanska, K.; Pannizzo, P.; Grabacka, M.; Croul, S.; del Valle, L.; Khalili, K.; Reiss, K. Activation of PPARalpha inhibits IGF-I-mediated growth and survival responses in medulloblastoma cell lines. Int. J. Cancer 2008, 123, 1015–1024. [Google Scholar]
- Panigrahy, D.; Kaipainen, A.; Huang, S.; Butterfield, C.E.; Barnes, C.M.; Fannon, M.; Laforme, A.M.; Chaponis, D.M.; Folkman, J.; Kieran, M.W. PPARα agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 985–990. [Google Scholar]
- Yamasaki, D.; Kawabe, N.; Nakamura, H.; Tachibana, K.; Ishimoto, K.; Tanaka, T.; Aburatani, H.; Sakai, J.; Hamakubo, T.; Kodama, T.; et al. Fenofibrate suppresses growth of the human hepatocellular carcinoma cell via PPARα-independent mechanisms. Eur. J. Cell Biol 2011, 90, 657–664. [Google Scholar]
- Tanaka, T. Preclinical cancer chemoprevention studies using animal model of inflammation-associated colorectal carcinogenesis. Cancers 2012, 4, 673–700. [Google Scholar]
- Roberts, W.C. Safety of fenofibrate—US and worldwide experience. Cardiology 1989, 76, 169–179. [Google Scholar]
- El Azzouzi, H.; Leptidis, S.; Bourajjaj, M.; Armand, A.S.; van der Nagel, R.; van Bilsen, M.; da Costa Martins, P.A.; de Windt, L.J. Peroxisome proliferator-activated receptor (PPAR) gene profiling uncovers insulin-like growth factor-1 as a PPARalpha target gene in cardioprotection. J. Biol. Chem 2011, 286, 14598–14607. [Google Scholar]
- Suzuki, W.; Iizuka, S.; Tabuchi, M.; Funo, S.; Yanagisawa, T.; Kimura, M.; Sato, T.; Endo, T.; Kawamura, H. A new mouse model of spontaneous diabetes derived from ddY strain. Exp. Anim 1999, 48, 181–189. [Google Scholar]
- Tanaka, T.; Ishigamori, R. Understanding carcinogenesis for fighting oral cancer. J. Oncol 2011, 2011. [Google Scholar] [CrossRef]
- Yano, T.; Obata, Y.; Ishikawa, G.; Ichikawa, T. Enhancing effect of high dietary iron on lung tumorigenesis in mice. Cancer Lett 1994, 76, 57–62. [Google Scholar]
- Malkinson, A.M. The genetic basis of susceptibility to lung tumors in mice. Toxicology 1989, 54, 241–271. [Google Scholar]
- Li, M.Y.; Yuan, H.; Ma, L.T.; Kong, A.W.; Hsin, M.K.; Yip, J.H.; Underwood, M.J.; Chen, G.G. Roles of peroxisome proliferator-activated receptor-α and -γ in the development of non-small cell lung cancer. Am. J. Respir. Cell Mol. Biol 2010, 43, 674–683. [Google Scholar]
- Chang, N.W.; Tsai, M.H.; Lin, C.; Hsu, H.T.; Chu, P.Y.; Yeh, C.M.; Chiu, C.F.; Yeh, K.T. Fenofibrate exhibits a high potential to suppress the formation of squamous cell carcinoma in an oral-specific 4-nitroquinoline 1-oxide/arecoline mouse model. Biochim. Biophys. Acta 2011, 1812, 558–564. [Google Scholar]
- Gunter, M.J.; Hoover, D.R.; Yu, H.; Wassertheil-Smoller, S.; Rohan, T.E.; Manson, J.E.; Li, J.; Ho, G.Y.; Xue, X.; Anderson, G.L.; et al. Insulin, insulin-like growth factor-I, and risk of breast cancer in postmenopausal women. J. Nat. Cancer Inst 2009, 101, 48–60. [Google Scholar]
- Irwin, M.L.; Duggan, C.; Wang, C.Y.; Smith, A.W.; McTiernan, A.; Baumgartner, R.N.; Baumgartner, K.B.; Bernstein, L.; Ballard-Barbash, R. Fasting C-peptide levels and death resulting from all causes and breast cancer: The health, eating, activity, and lifestyle study. J. Clin. Oncol 2011, 29, 47–53. [Google Scholar]
- Currie, C.J.; Poole, C.D.; Gale, E.A. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia 2009, 52, 1766–1777. [Google Scholar]
- Chang, C.H.; Lin, J.W.; Wu, L.C.; Lai, M.S.; Chuang, L.M. Oral insulin secretagogues, insulin, and cancer risk in type 2 diabetes mellitus. J. Clin. Endocrinol. Metab 2012, 97, E1170–E1175. [Google Scholar]
- Camidge, D.R.; Dziadziuszko, R.; Hirsch, F.R. The rationale and development of therapeutic insulin-like growth factor axis inhibition for lung and other cancers. Clin. Lung Cancer 2009, 10, 262–272. [Google Scholar]
- LeRoith, D.; Roberts, C.T., Jr. The insulin-like growth factor system and cancer. Cancer Lett 2003, 195, 127–137. [Google Scholar]
- Gallagher, E.J.; LeRoith, D. The proliferating role of insulin and insulin-like growth factors in cancer. Trends Endocrinol. Metab 2010, 21, 610–618. [Google Scholar]
- Terai, K.; Sakamoto, K.; Goto, M.; Matsuda, M.; Kasamaki, S.; Shinmura, K.; Takita, N.; Kamano, T. Greater development of 1,2-dimethylhydrazine-induced colon cancer in a rat model of type 2 diabetes mellitus. J. Int. Med. Res 2006, 34, 385–389. [Google Scholar]
- Koh, E.H.; Kim, M.S.; Park, J.Y.; Kim, H.S.; Youn, J.Y.; Park, H.S.; Youn, J.H.; Lee, K.U. Peroxisome proliferator-activated receptor (PPAR)-α activation prevents diabetes in OLETF rats: Comparison with PPAR-γ activation. Diabetes 2003, 52, 2331–2337. [Google Scholar]
- Mohr, U. International Classification of Rodent Tumors. Part II. The Mouse; Springer: New York, NY, USA, 2001. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Treatment (number of mice examined) | Number of mice with proliferative lung lesions (%) | Number of proliferative lung lesion/mouse | |||
|---|---|---|---|---|---|---|
| Hyperplasia | Adenoma | Adenocarcinoma | Total | |||
| 1 | 4-NQO (24) | 8/24 (33) | 0.21 ± 0.41 a | 0.17 ± 0.48 | 0.08 ± 0.28 | 0.46 ± 0.78 |
| 2 | 4-NQO—0.01% fenofibrate (24) | 4/24 (17) | 0.13 ± 0.45 | 0.08 ± 0.28 | 0 | 0.21 ± 0.51 |
| 3 | 4-NQO—0.05% fenofibrate (25) | 1/25 (4) b | 0 | 0.04 ± 0.20 | 0 | 0.04 ± 0.20 c |
| 4 | 0.05% fenofibrate (13) | 1/13 (8) | 0 | 0.08 ± 0.28 | 0 | 0.08 ± 0.28 |
| 5 | Non-treatment (13) | 1/13 (8) | 0 | 0.08 ± 0.28 | 0 | 0.08 ± 0.28 |
| Group | Treatment (number of mice examined) | Triglyceride (mg/dL) | Free fatty acid (mEq/L) | Total cholesterol (mg/dL) | Glucose (mg/dL) | Insulin (ng/dL) | IGF-1 (ng/dL) |
|---|---|---|---|---|---|---|---|
| 1 | 4-NQO (5) | 153.6 ± 43.2 a | 2235.6 ± 542.5 | 266.0 ± 48.2 | 128.4 ± 9.1 | 7.89 ± 2.71 | 547.4 ± 58.4 |
| 2 | 4-NQO—0.01% fenofibrate (5) | 161.2 ± 17.0 | 1540.8 ± 365.5 b | 250.0 ± 25.0 | 130.2 ± 24.4 | 1.31 ± 0.83 c | 456.3 ± 15.7 c |
| 3 | 4-NQO—0.05% fenofibrate (5) | 60.8 ± 19.0 c,d | 1154.4 ± 80.0 c | 201.8 ± 27.4 | 131.0 ± 11.7 | 0.16 ± 0.05 c | 367.1 ± 16.0 c |
| 4 | 0.05% fenofibrate (5) | 119.8 ± 28.0 e | 1218.4 ± 148.1 f | 217.4 ± 48.3 | 109.0 ± 14.7 | 0.05 ± 0.04 e | 317.7 ± 47.8 e |
| 5 | Non-treatment (5) | 208.2 ± 44.7 | 1876.8 ± 267.7 | 238.0 ± 60.4 | 113.6 ± 16.2 | 10.09 ± 1.07 | 573.8 ± 23.6 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kuno, T.; Hata, K.; Takamatsu, M.; Hara, A.; Hirose, Y.; Takahashi, S.; Imaida, K.; Tanaka, T. The Peroxisome Proliferator-Activated Receptor (PPAR) α Agonist Fenofibrate Suppresses Chemically Induced Lung Alveolar Proliferative Lesions in Male Obese Hyperlipidemic Mice. Int. J. Mol. Sci. 2014, 15, 9160-9172. https://doi.org/10.3390/ijms15059160
Kuno T, Hata K, Takamatsu M, Hara A, Hirose Y, Takahashi S, Imaida K, Tanaka T. The Peroxisome Proliferator-Activated Receptor (PPAR) α Agonist Fenofibrate Suppresses Chemically Induced Lung Alveolar Proliferative Lesions in Male Obese Hyperlipidemic Mice. International Journal of Molecular Sciences. 2014; 15(5):9160-9172. https://doi.org/10.3390/ijms15059160
Chicago/Turabian StyleKuno, Toshiya, Kazuya Hata, Manabu Takamatsu, Akira Hara, Yoshinobu Hirose, Satoru Takahashi, Katsumi Imaida, and Takuji Tanaka. 2014. "The Peroxisome Proliferator-Activated Receptor (PPAR) α Agonist Fenofibrate Suppresses Chemically Induced Lung Alveolar Proliferative Lesions in Male Obese Hyperlipidemic Mice" International Journal of Molecular Sciences 15, no. 5: 9160-9172. https://doi.org/10.3390/ijms15059160