Importance of Kupffer Cells in the Development of Acute Liver Injuries in Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
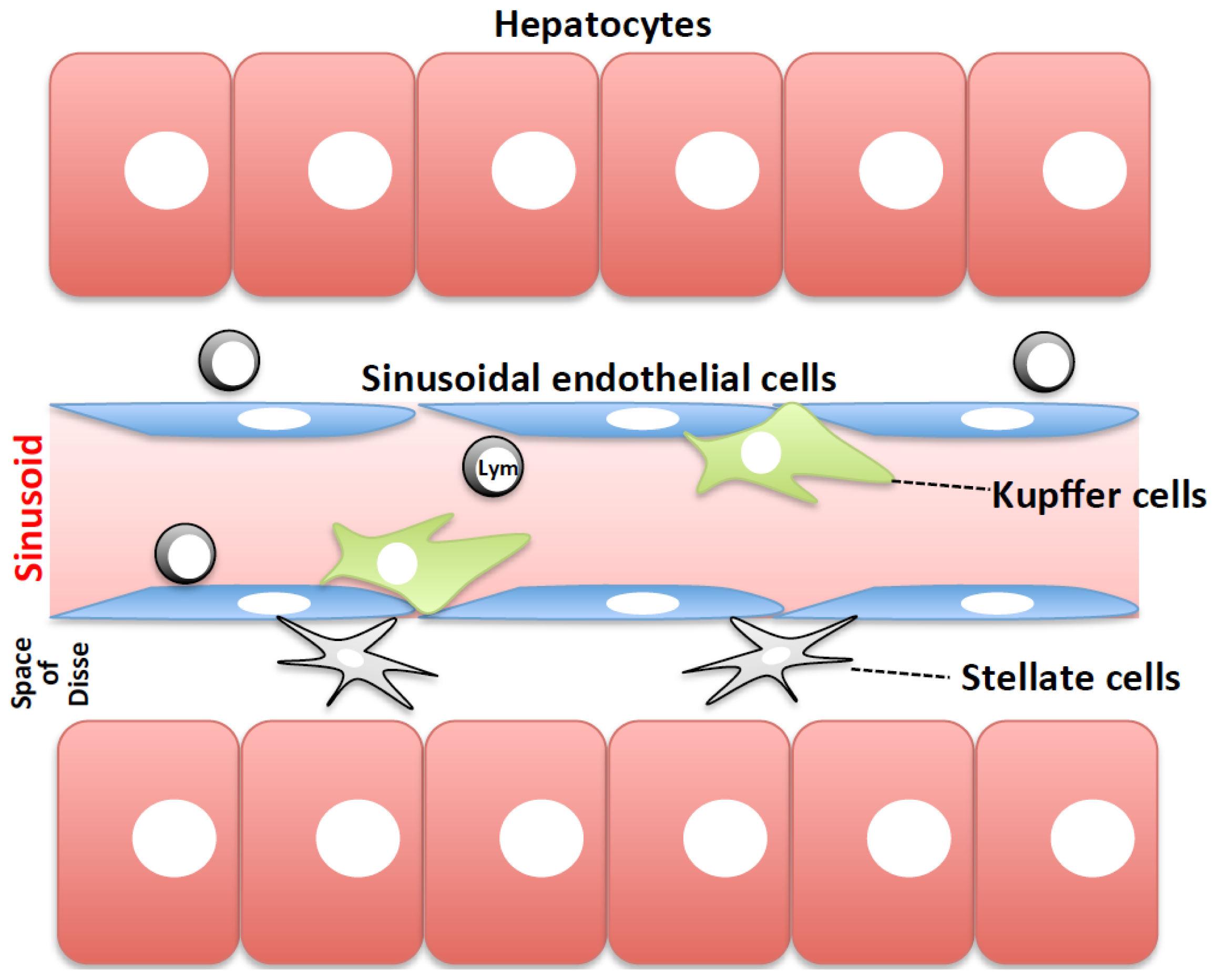
:1. Introduction
2. Endotoxin-Induced Liver Injury
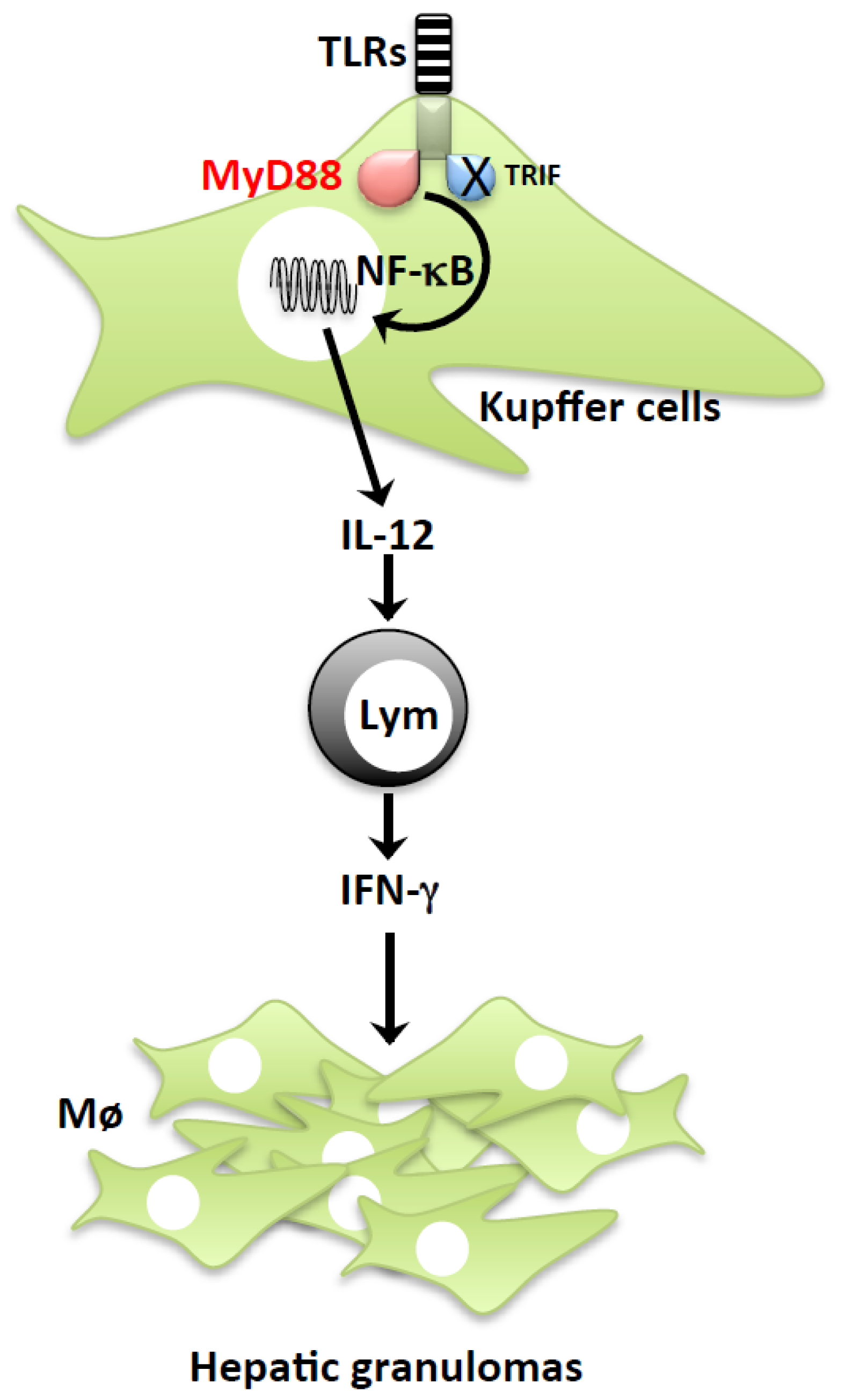
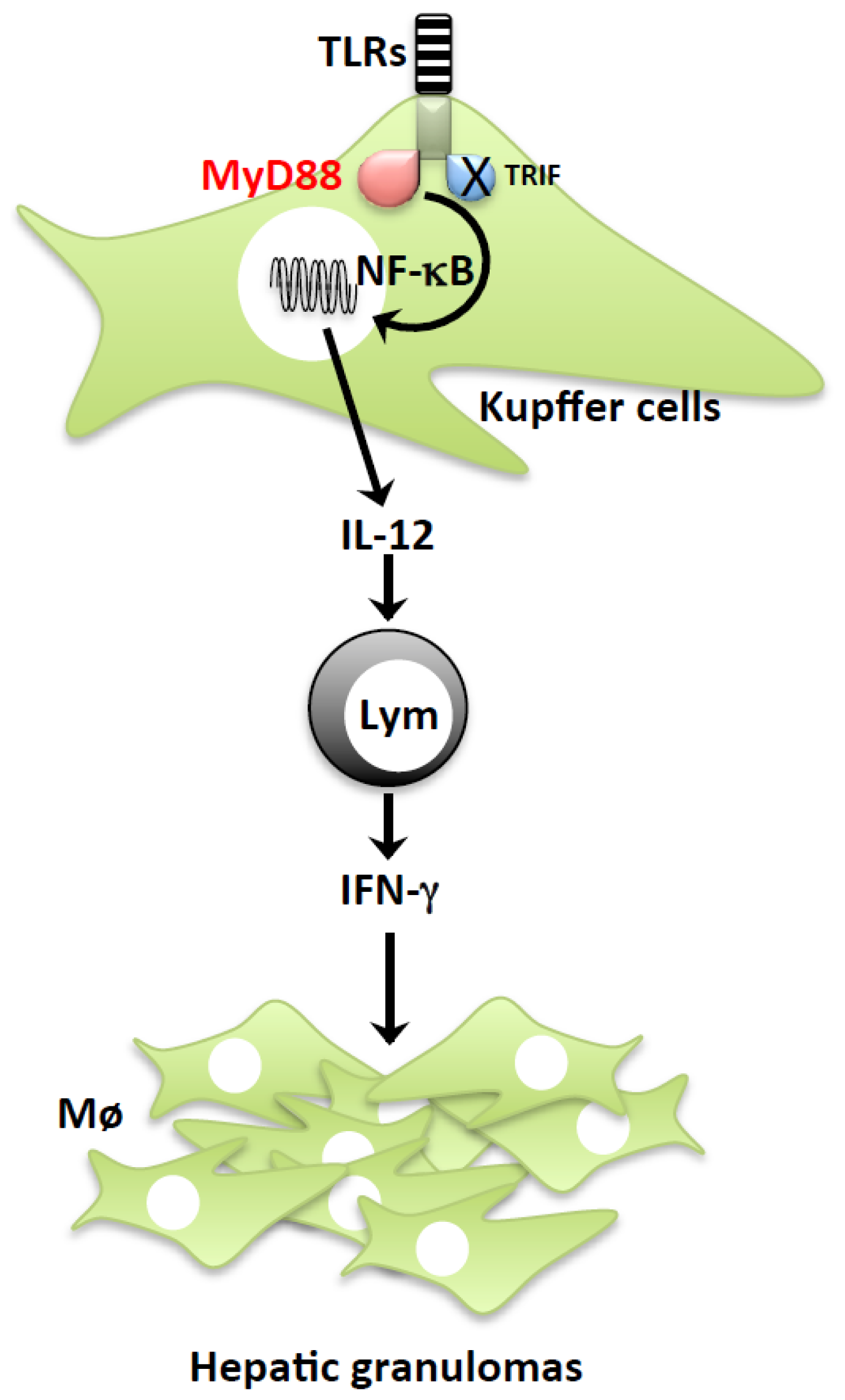
2.1. P. acnes Induction of LPS Sensitization
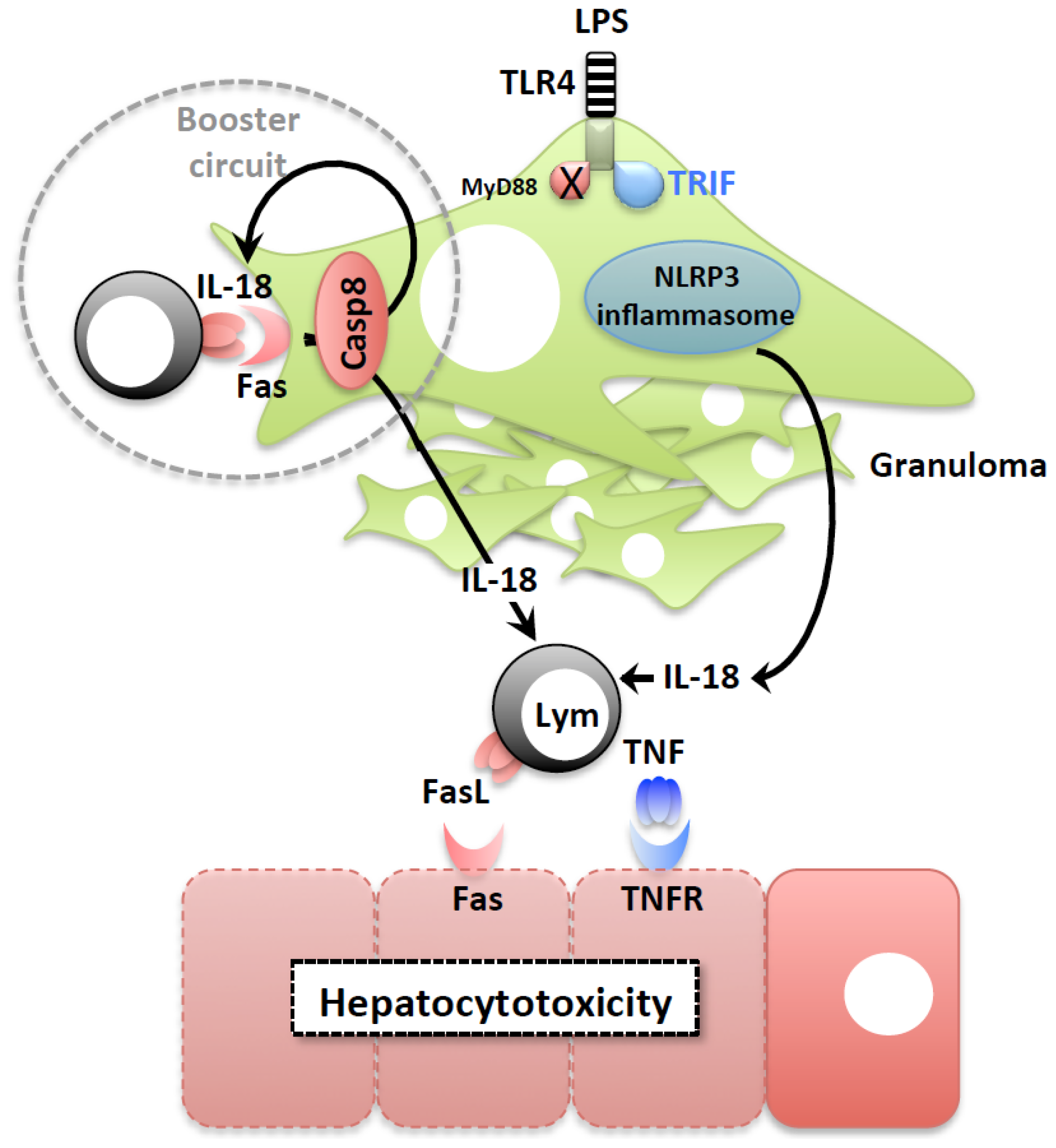
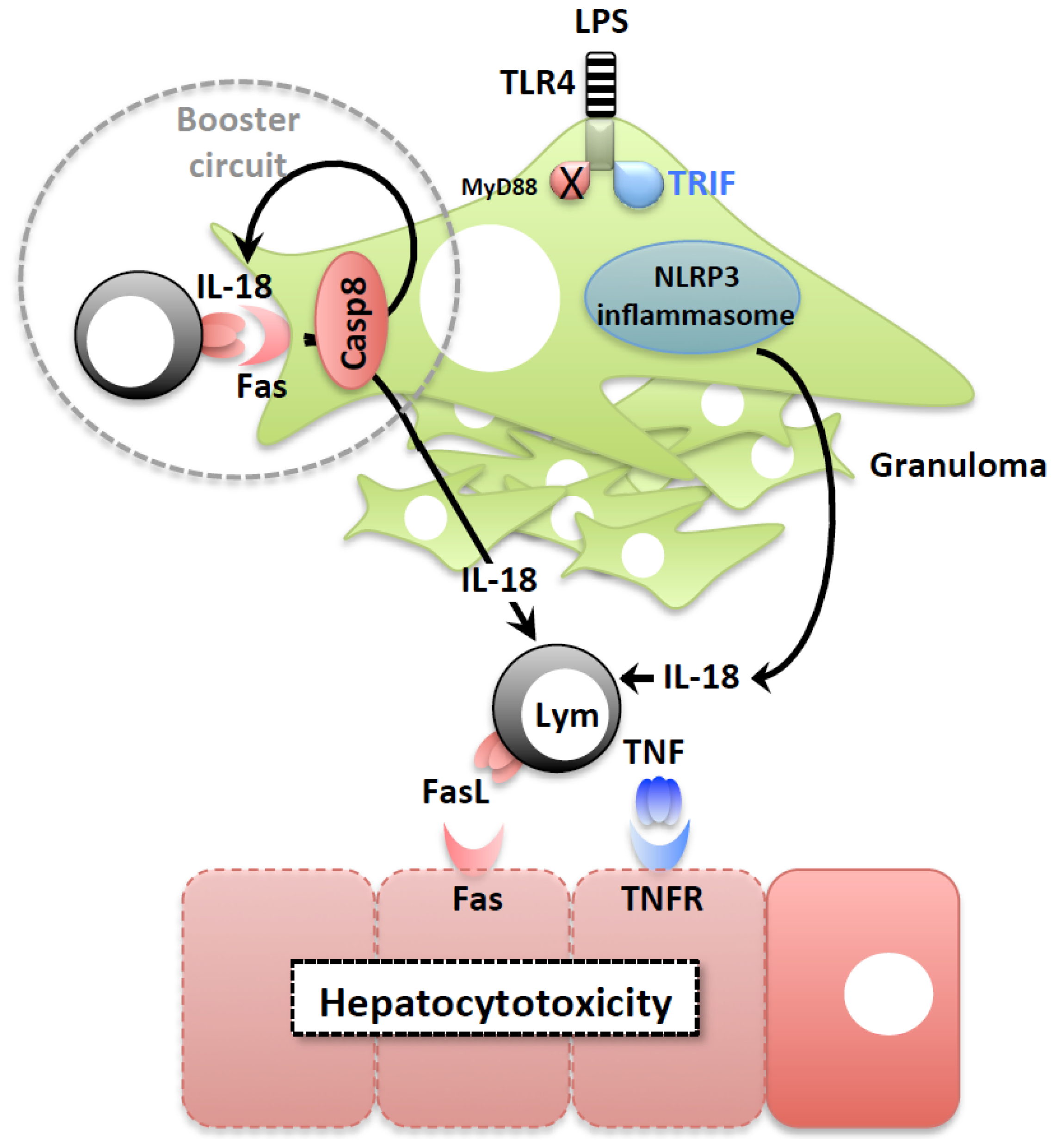
2.2. LPS-Induced Liver Injury
2.3. IL-18 Induction of FasL Accelerates the Development of Liver Injury
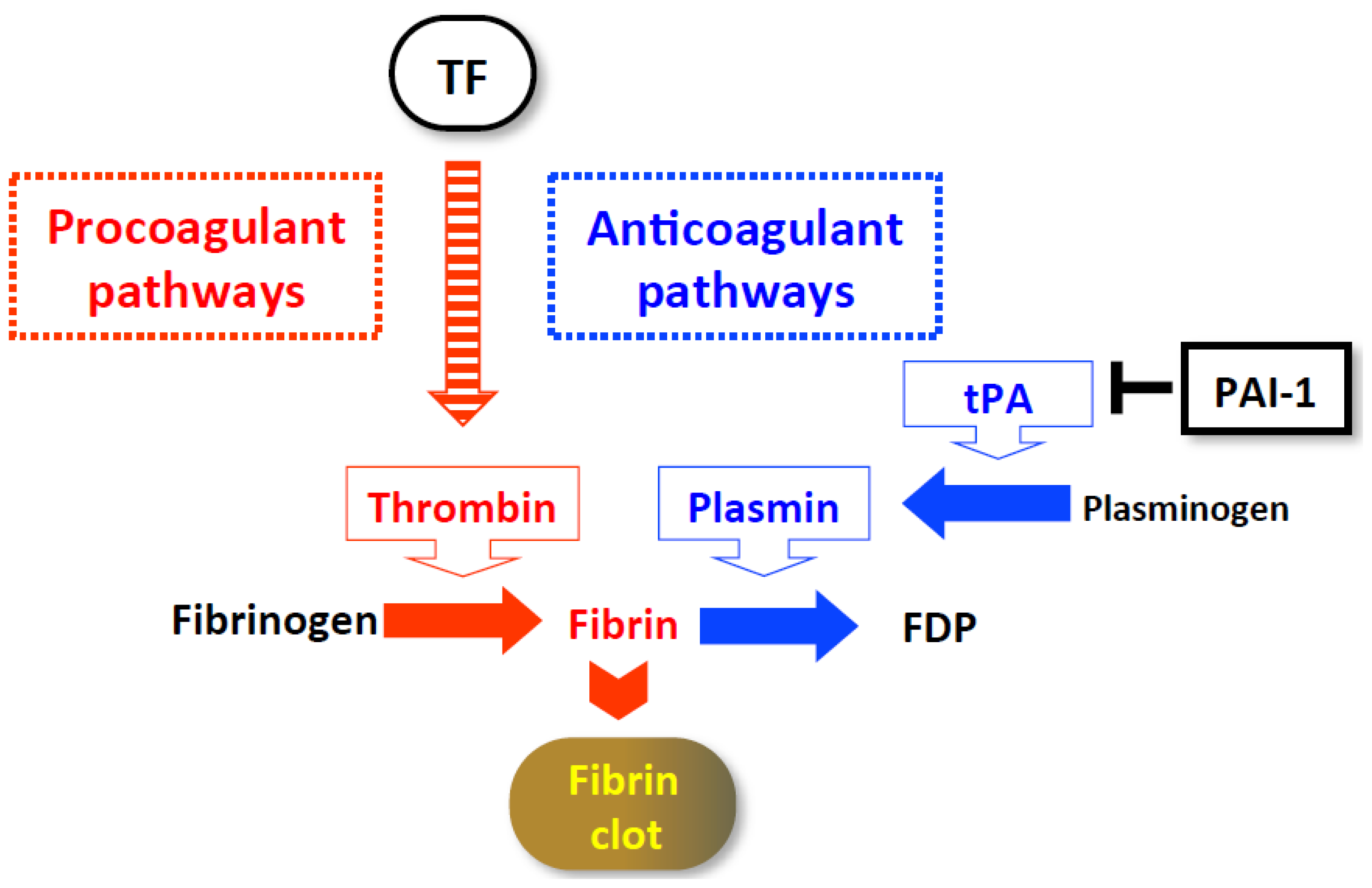
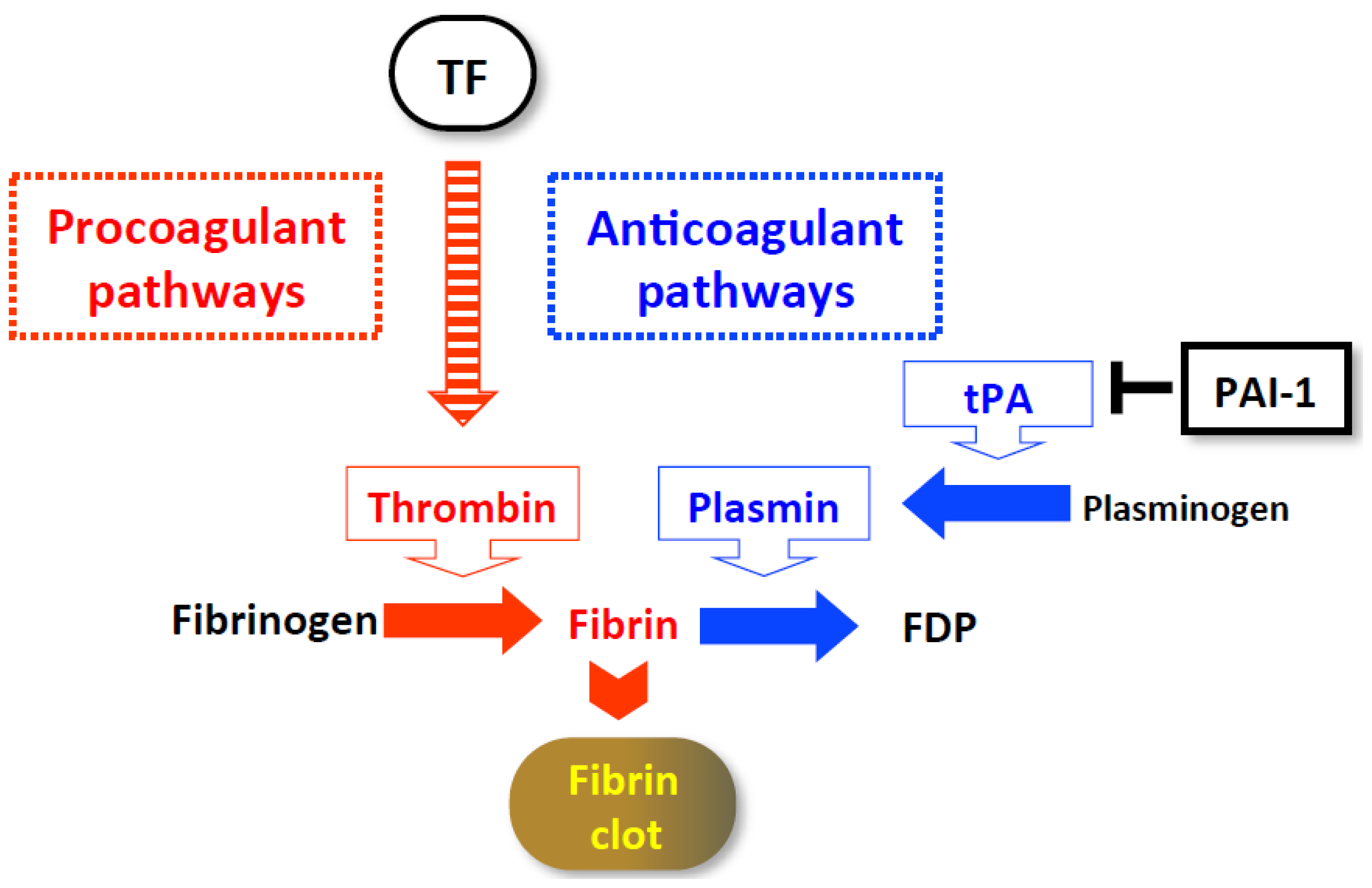
3. Hypercoagulation-Associated Acute Severe Hepatitis
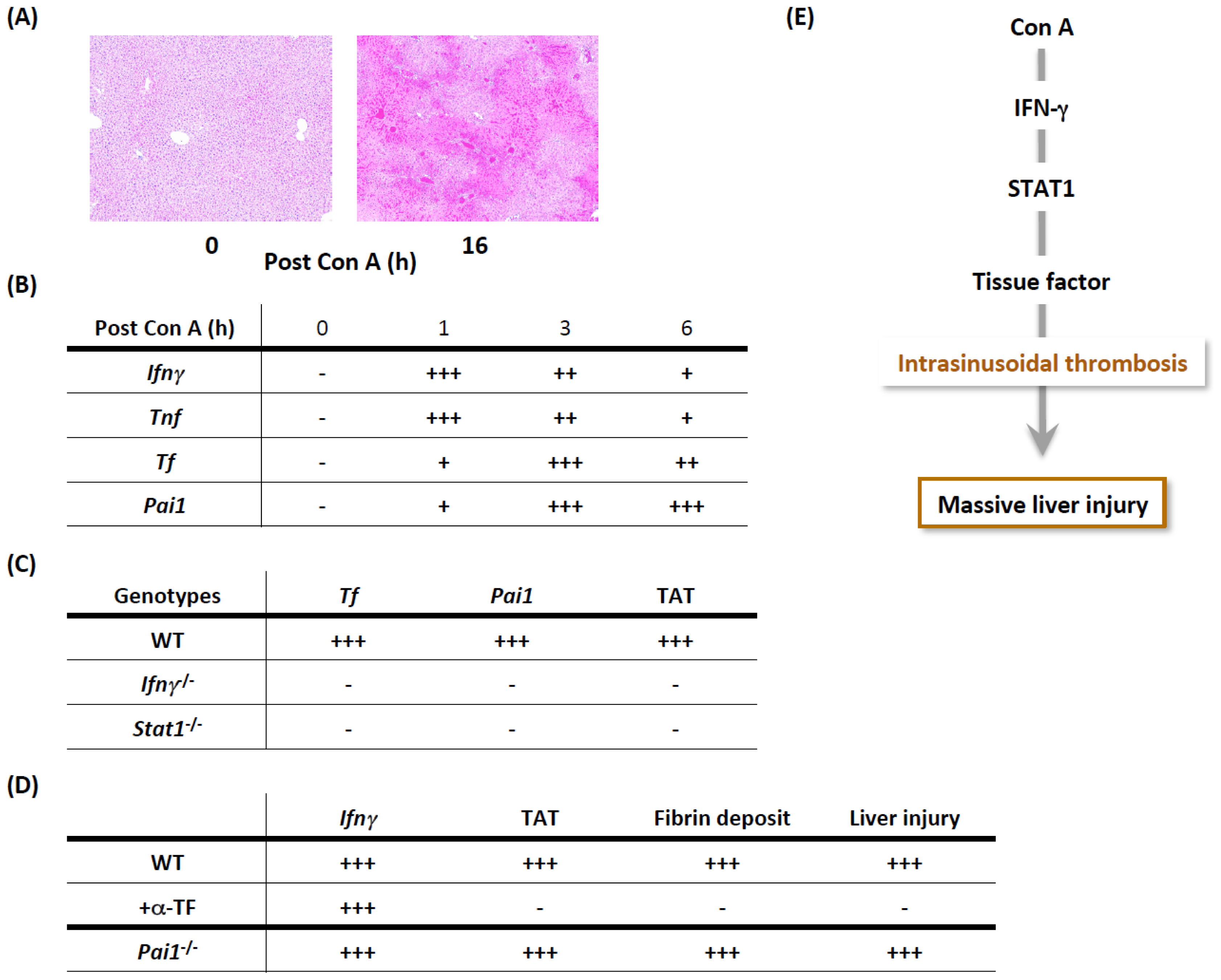
3.1. Con A-Induced Acute Liver Injury
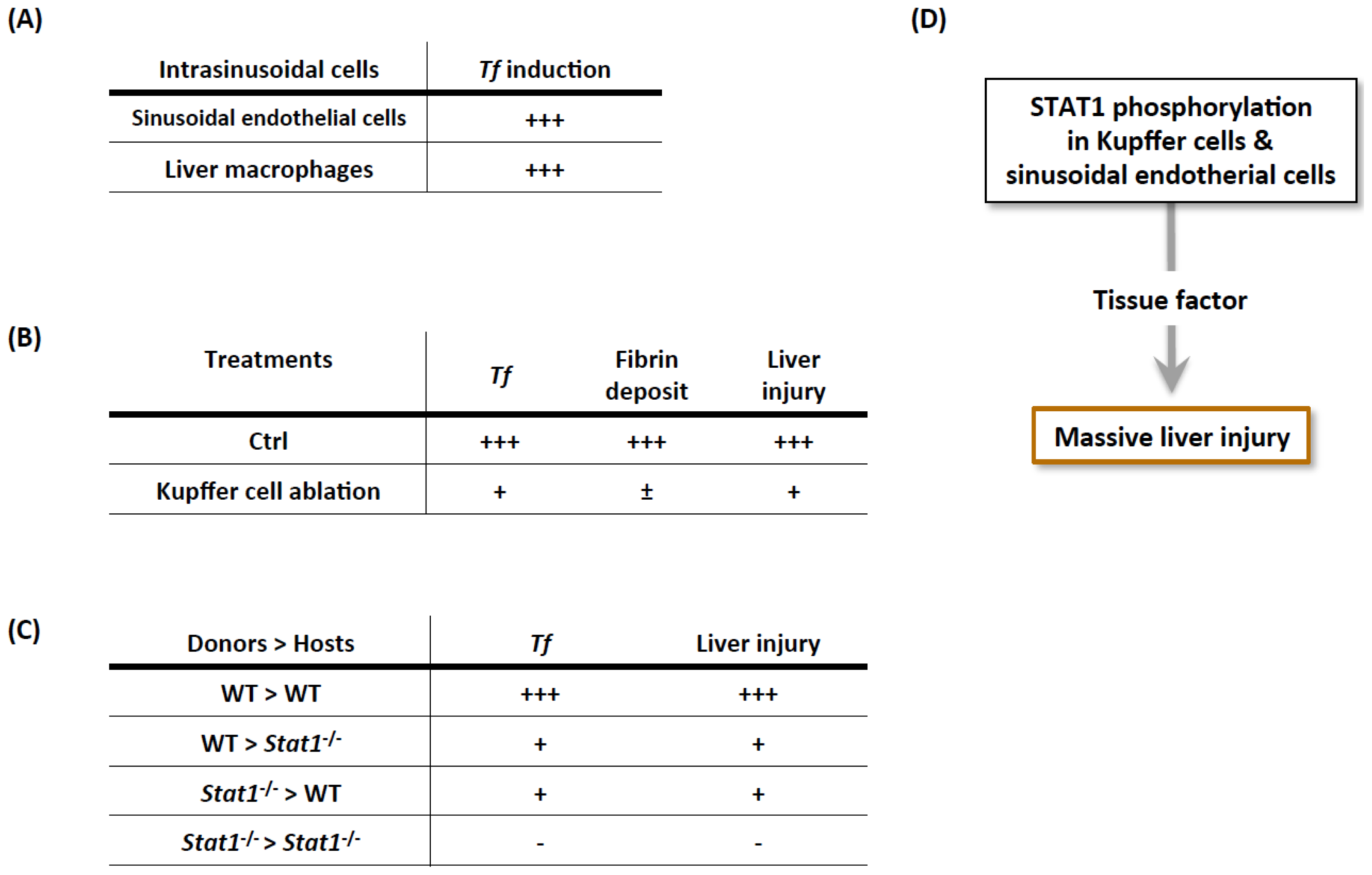
3.1.1. Importance of IFN-γ/STAT1 in the Development of Procoagulant Hepatitis
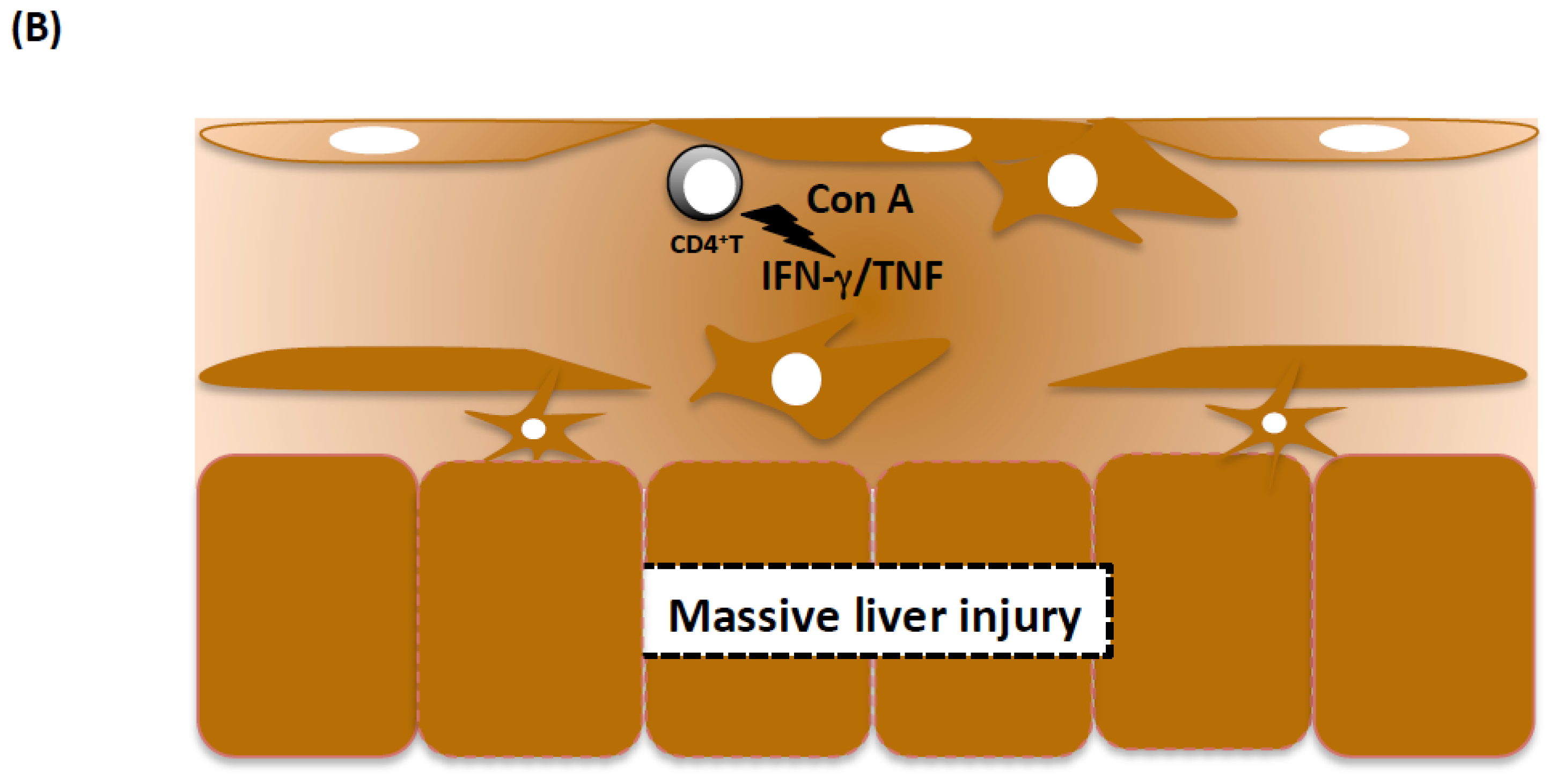
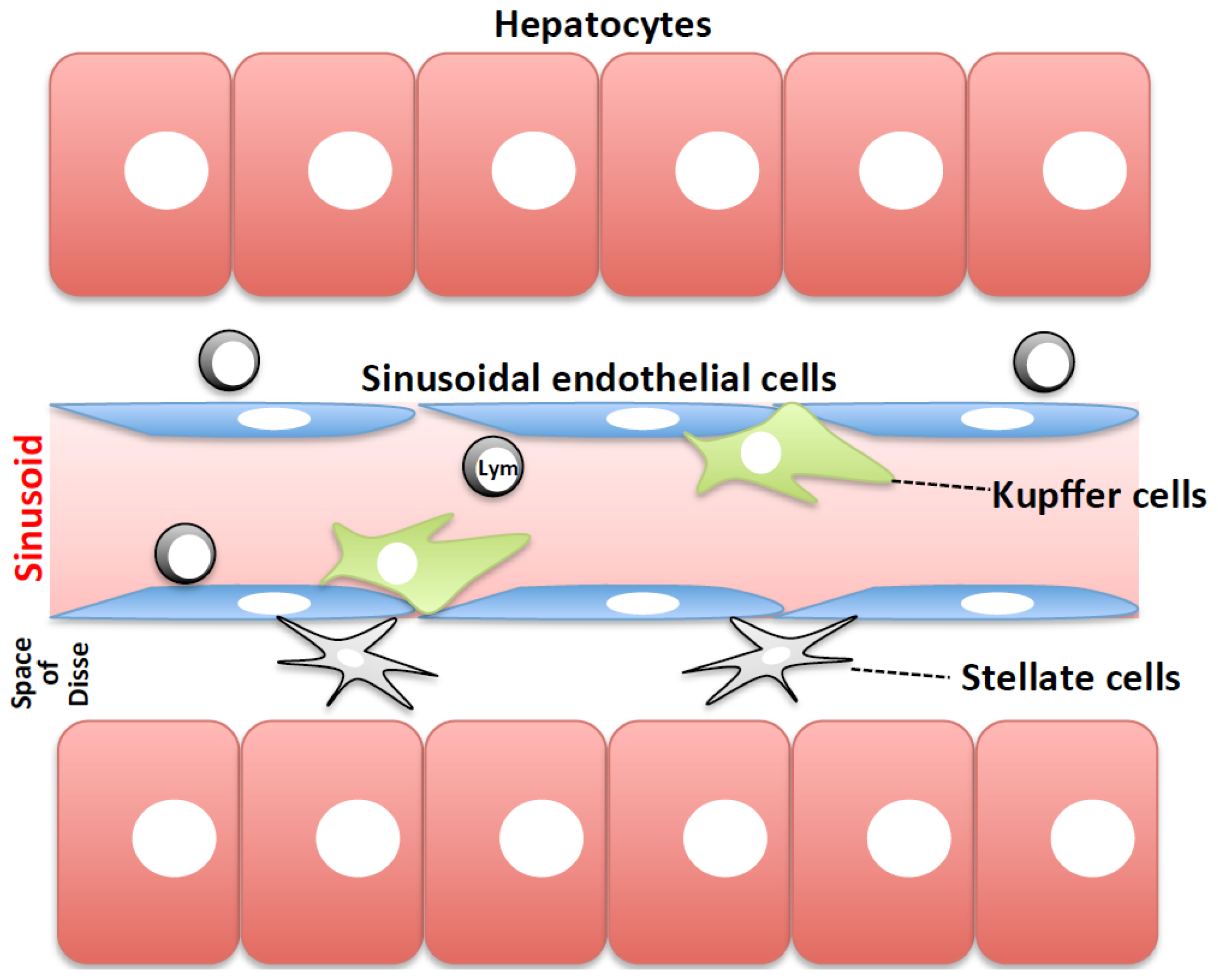
3.1.2. Cellular Mechanisms of Prothrombotic Hepatitis
3.1.3. Con A, IFN-γ, and TNF Are Necessary and Sufficient for Massive Liver Injury
3.2. APAP-Induced Acute Liver Injury
3.2.1. Hypercoagulability in APAP-Induced Hepatotoxicity
3.2.2. Kupffer Cells Play a Beneficial Role
4. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsT.H. designed and wrote the manuscript. S.N. wrote the manuscript partly.
References
- Schnabl, B.; Brenner, D.A. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 2014, 146, 1513–1524. [Google Scholar]
- Meijer, C.; Wiezer, M.J.; Diehl, A.M.; Young, S.-Q.; Schouten, H.J.; Meijer, S.; van Rooijen, N.; van Lambalgen, A.A.; Dijkstra, C.D.; van Leeuwen, P.A.M. Kupffer cell depletion by Cl2 MDP-liposomes alters hepatic cytokine expression and delays liver regeneration after partial hepatectomy. Liver 2000, 20, 66–77. [Google Scholar]
- Seki, E.; Tsutsui, H.; Iimuro, Y.; Naka, T.; Son, G.; Akira, S.; Kishimoto, T.; Nakanishi, K.; Fujimoto, J. Contribution of Toll-like receptor/Myeloid differentiation factor 88 signaling to murine liver regeneration. Hepatology 2005, 41, 443–450. [Google Scholar]
- Tanuma, Y.; Ito, T. Electron microscope study on the hepatic sinusoidal wall and fat-storeing cells in the bat. Arch. Histol. Jpn 1978, 41, 1–39. [Google Scholar]
- Kaneda, K.; Wake, K. Distribution and morphological characteristics of the pit cells in the liver of the rat. Cell Tissue Res 1983, 233, 485–498. [Google Scholar]
- Kaneda, K.; Kurioka, N.; Seki, S.; Wake, K.; Yamamoto, S. Pit cell-hepatocyte contact in autoimmune hepatitis. Hepatology 1984, 4, 955–958. [Google Scholar]
- Matsui, K.; Yoshimoto, T.; Tsutsui, H.; Hyodo, Y.; Hayashi, N.; Hiroishi, K.; Kawada, N.; Okamura, H.; Nakanishi, K.; Higashino, K. Propionibacterium acnes treatment diminishes CD4+ NK1.1+ T cells but induces type I T cells in the liver by induction of IL-12 and IL-18 production from Kupffer cells. J. Immunol 1997, 159, 97–106. [Google Scholar]
- Yoshimoto, T.; Paul, W.E. CD4pos, NK1.1pos T cells promptly produce interleukin 4 in response to in vivo challenge with anti-CD3. J. Exp. Med 1994, 179, 1285–1295. [Google Scholar]
- Santodomingo-Garzon, T.; Swaine, M.G. Role of NKT cells in atuoimmune liver disease. Autoimmun. Rev 2011, 10, 793–800. [Google Scholar]
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An evolving paradigm for stem cell biology. Cell 2008, 132, 631–644. [Google Scholar]
- Cumano, A.; Godin, I. Ontogeny of the hematopietic system. Annu. Rev. Immunol 2007, 25, 745–785. [Google Scholar]
- Hoeffel, G.; Wang, Y.; Greter, M.; See, P.; Teo, P.; Malleret, B.; Leboeuf, M.; Low, D.; Oller, G.; Almeida, F. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J. Exp. Med 2012, 209, 1167–1181. [Google Scholar]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar]
- Seki, E.; de Minicis, S.; Österreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat. Med 2007, 13, 1324–1332. [Google Scholar]
- Van Rooijen, N.; Sanders, A. Kupffer cell depletionh by liposome-delivered drugs: Comparative activity of intracellular clodronate, propamidine and ethylenediaminetetraacetic acid (EDTA). Hepatology 1996, 23, 1239–1243. [Google Scholar]
- Feruga, J.; Allison, A.C. Role of mononuclear infiltrating cells in pathogenesis of hepatitis. Lancet 1978, 312, 610–611. [Google Scholar]
- Mizoguchi, Y.; Tsutsui, H.; Sakagami, Y.; Kuboi, H.; Seki, S.; Kobayashi, K.; Yamamoto, S.; Morisawa, S. The protective effects of prostaglandin E1 in an experimental massive hepatic necrosis model. Hepatology 1987, 7, 1184–1188. [Google Scholar]
- Kawa, K.; Tsutsui, H.; Uchiyama, R.; Kato, J.; Matsui, K.; Iwakura, Y.; Matsumoto, T.; Nakanishi, K. IFN-γ is a master regulator of endotoxin shock syndrome in mice primed with heat-killed Propionibacterium acnes. Int. Immunol 2010, 22, 157–166. [Google Scholar]
- Levi, M. Disseminated intravascular coagulation: What’s new? Crit. Care Clin 2005, 21, 449–467. [Google Scholar]
- Levi, M.; Ten Cate, H. Disseminated intravascular coagulation. N. Engl. J. Med 1999, 341, 586–592. [Google Scholar]
- Tsutsui, H.; Matsui, K.; Okamura, H.; Nakanishi, K. Pathophysiological roles of interleukin-18 for inflammatory liver diseases. Immunol. Rev 2000, 174, 192–209. [Google Scholar]
- Tsutsui, H.; Imamura, M.; Fujimoto, J.; Nakanishi, K. The TLR4/TRIF-mediated activation of NLRP3 inflammasome underlies endotoxin-induced liver injury in mice. Gastroenterol. Res. Pract 2010, 2010, 641865. [Google Scholar]
- Imamura, M.; Tsutsui, H.; Yasuda, K.; Uchiyama, R.; Yumikura-Futatsugi, S.; Mitani, K.; Hayashi, S.; Akira, S.; Taniguchi, S.-I.; van Rooijen, N. Contribution of TIR domain-containing adapter inducing IFN-β-mediated IL-18 release to LPS-induced liver injury in mice. J. Hepatol 2009, 51, 333–341. [Google Scholar]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar]
- Velayudham, A.; Hritz, I.; Dolganiuc, A.; Mandrekar, P.; Kurt-Jones, E.; Szabo, G. Critical role of Toll-like receptors and the common TLR adaptor, MyD88, in induction of granulomas and liver injury. J. Hepatol 2006, 45, 813–824. [Google Scholar]
- Kalis, C.; Gumenscheimer, M.; Freudenberg, N.; Tchaptchet, S.; Fejer, G.; Heit, A.; Akira, S.; Galanos, C.; Freudenberg, M.A. Requirement for TLR9 in the immunomodulatory activity of Propionibacterium acnes. J. Immunol 2005, 174, 4295–4300. [Google Scholar]
- Ogushi, I.; Iimuro, Y.; Seki, E.; Son, G.; Hirano, T.; Hada, T.; Tsutsui, H.; Nakanishi, K.; Morishita, R.; Kaneda, Y.; et al. Nuclear factor κB decoy oligodeoxynucleotides prevent endotoxin-induced fatal liver failure in a murine model. Hepatology 2003, 38, 335–344. [Google Scholar]
- Sakao, Y.; Takeda, K.; Tsutsui, H.; Kaisho, T.; Nomura, F.; Okamura, H.; Nakanishi, K.; Akira, S. IL-18-deficient mice are resistant to endotoxin-induced liver injury but highly susceptible to endotoxin shock. Int. Immunol 1999, 11, 471–480. [Google Scholar]
- Tsuji, H.; Mukaida, N.; Harada, A.; Kaneko, S.; Matsushita, E.; Nakanuma, Y.; Tsutsui, H.; Okamura, H.; Nakanishi, K.; Tagawa, Y.-I.; et al. Alleviation of lipopolysaccharide-induced acute liver injury in Propionibacterium acnes-primed IFN-γ-deficient mice by a concomitant reduction of TNF-α, IL-12, and IL-18 production. J. Immunol 1999, 162, 1049–1055. [Google Scholar]
- Okamura, H.; Tsutsui, H.; Komatsu, T.; Yutsudo, M.; Hakura, A.; Tanimoto, T.; Torigoe, K.; Okura, T.; Nukada, Y.; Hattori, K.; et al. Cloning of a new cytokine that induces IFN-γ production by T cells. Nature 1995, 378, 88–91. [Google Scholar]
- Seki, E.; Tsutsui, H.; Nakano, H.; Tsuji, N.M.; Hoshino, K.; Adachi, O.; Adachi, K.; Futatsugi, S.; Kuida, K.; Takeuchi, O.; et al. LPS-induced IL-18 secretion from murine Kupffer cells independently of MyD88 that is critically involved in induction of production of IL-12 and IL-1β. J. Immunol 2001, 166, 2651–2657. [Google Scholar]
- Gu, Y.; Kuida, K.; Tsutsui, H.; Ku, G.; Hsiao, K.; Fleming, M.A.; Hayashi, N.; Higashino, K.; Okamura, H.; Nakanishi, K.; et al. Activation of interferon-γ inducing factor mediated by interleukin-1β converting enzyme. Science 1997, 275, 206–209. [Google Scholar]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Cande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar]
- Tschopp, J.; Martinon, F.; Burns, K. NALPs: A novel protein family involved in inflammation. Nat. Rev. Mol. Cell Biol 2003, 4, 95–104. [Google Scholar]
- Henao-Mejia, J.; Elinav, E.; Thaiss, C.A.; Flavell, R.A. Inflammasomes and metabolic disease. Annu. Rev. Physiol 2014, 76, 57–78. [Google Scholar]
- Yamamoto, M.; Yaginuma, K.; Tsutsui, H.; Sagara, J.; Guan, X.; Seki, E.; Yasuda, K.; Yamamoto, M.; Akira, S.; Nakanishi, K. ASC is essential for LPS-induced activation of procaspase-1 independently of TLR-associated signal adapter molecules. Gene. Cells 2004, 9, 1055–1067. [Google Scholar]
- Tsutsui, H.; Nakanishi, K.; Matsui, K.; Higashino, K.; Okamura, H.; Miyazawa, Y.; Kaneda, K. Interferon-γ-inducing factor up-regulates Fas ligand-mediated cytotoxic activity of murine natural killer cell clones. J. Immunol 1996, 157, 3967–3973. [Google Scholar]
- Ogasawara, J.; Watanabe-Fukunaga, R.; Adachi, M.; Matsuzawa, A.; Kasuga, T.; Kitamura, Y.; Itoh, N.; Suda, T.; Nagata, S. Lethal effect of the anti-Fas antibody in mice. Nature 1993, 364, 806–809. [Google Scholar]
- Malhi, H.; Gores, G.J.; Lemasters, J.J. Apoptosis and necrosis in the liver: A tale of two deaths? Hepatology 2006, 43, S31–S44. [Google Scholar]
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 regulates both Th1 and Th2 responses. Annu. Rev. Immunol 2001, 19, 423–474. [Google Scholar]
- Tsutsui, H.; Matsui, K.; Kawada, N.; Hyodo, Y.; Hayashi, N.; Okamura, H.; Higashino, K.; Nakanishi, K. IL-18 accounts for both TNF-α- and Fas ligand-mediated hepatotoxic pathways in endotoxin-induced liver injury in mice. J. Immunol 1997, 159, 3961–3967. [Google Scholar]
- Tsutsui, H.; Kayagaki, N.; Kuida, K.; Nakano, H.; Hayashi, N.; Takeda, K.; Matsui, K.; Kashiwamura, S.; Hada, T.; Akira, S.; et al. Caspase-1-independent, Fas/Fas ligand-mediated IL-18 secretion from macrophages causes acute liver injury in mice. Immunity 1999, 11, 359–367. [Google Scholar]
- Miwa, K.; Asano, M.; Horai, R.; Iwakura, Y.; Nagata, S.; Suda, T. Caspase-1-independent IL-1b release and inflammation induced by the apoptosis inducer Fas ligand. Nat. Med 1998, 4, 1287–1292. [Google Scholar]
- Bossaller, L.; Chiang, P.-I.; Schmidt-Lauber, C.; Ganesan, S.; Kaiser, W.J.; Rathinam, V.A.; Mocarski, E.S.; Subramanian, D.; Green, D.R.; Silverman, N.; et al. Fas (CD95) mediates noncanonical IL-1b and IL-18 maturation via caspase-8 in an RIP3-independent manner. J. Immunol 2012, 189, 5508–5512. [Google Scholar]
- Uchiyama, R.; Yonehara, S.; Tsutsui, H. Fas-mediated inflammatory response in Listeria monocytogenes infection. J. Immunol 2013, 190, 4245–4254. [Google Scholar]
- Lisman, T.; Leebeek, F.W.G.; de Groot, P.G. Haemostatic abnormalities in patients with liver disease. J. Hepatol 2002, 37, 280–287. [Google Scholar]
- Tripodi, A.; Salerno, F.; Chantarangkul, V.; Clerici, M.; Cazzaniga, M.; Primignani, M.; Mannuccio, M.P. Evidence of normal thrombin generation in cirrhosis despite abnormal conventional coagulation tests. Hepatology 2005, 41, 553–558. [Google Scholar]
- Gatt, A.; Riddell, A.; Calvaruso, V.; Tuddenham, E.G.; Makris, M.; Burroughs, A.K. Enhanced thrombin generation in patients with cirrhosis-induced coagulopathy. J. Thromb. Haemost 2010, 8, 1994–2000. [Google Scholar]
- Lisman, T.; Porte, R.J. Rebalanced hemostasis in patients with liver disease: Evidence and clinical consequences. Blood 2010, 116, 878–885. [Google Scholar]
- Stravitz, R.T.; Lisman, T.; Luketic, V.A.; Sterling, R.K.; Puri, P.; Fuchs, M.; Ibrahim, A.; Lee, W.M.; Sanyal, A.J. Minimal effects of acute liver injury/acute liver failure on hemostasis as assessed by thromboelastrography. J. Hepatol 2012, 56, 129–136. [Google Scholar]
- Lisman, T.; Bakhtiari, K.; Adelmeijer, J.; Meijers, J.C.M.; Porte, R.J.; Stravitz, T. Intact thrombin generation and decreased fibrinolytic capacity in patients with acute liver injury or acute liver failure. J. Thromb. Haemost 2012, 10, 1312–1319. [Google Scholar]
- Hugenholtz, G.G.G.; Adelmeijer, J.; Meijers, J.C.M.; Porte, R.J.; Stravitz, R.T.; Lisman, T. An unbalance between von Willebrand factor and ADAMS13 in acute liver failure: Implications for hemostasis and clinical outcome. Hepatology 2012, 58, 752–761. [Google Scholar]
- Tiegs, G.; Hentschel, J.; Wendel, A. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J. Clin. Investig 1992, 90, 196–203. [Google Scholar]
- Miyazawa, Y.; Tsutsui, H.; Mizuhara, H.; Fujiwara, H.; Kaneda, K. Involvement of intrasinusoidal hemostasis in the development of Concanavalin A-induced hepatic injury in mice. Hepatology 1997, 27, 497–506. [Google Scholar]
- Kato, J.; Okamoto, T.; Motoyama, H.; Uchiyama, R.; Kirchhofer, D.; van Rooijen, N.; Enomoto, H.; Nishiguchi, S.; Kawada, N.; Fujimoto, J. Interferon-γ-mediated tissue factor expression contributes to T-cell-mediated hepatitis through induction of hypercoagulation in mice. Hepatology 2013, 57, 362–372. [Google Scholar]
- Mackman, N. The many faces of tissue factor. J. Thromb. Haemost 2009, 7 Suppl 1, 136–139. [Google Scholar]
- Belting, M.; Ahamed, J.; Ruf, W. Signaling of the tissue factor coagulation pathway in angiogenesis and cancer. Arterioscler. Thromb. Vasc. Biol 2005, 25, 1545–1550. [Google Scholar]
- Mackman, N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler. Thromb. Vasc. Biol 2004, 24, 1015–1022. [Google Scholar]
- Van der Poll, T.; Büller, H.R.; ten Cate, H.; Wortel, C.H.; Bauer, K.A.; van Deventer, S.J.H.; Hack, C.E.; Sauerwein, H.P.; Rosenberg, R.D.; ten Cate, J.W. Activation of coagulation after administration of tumor necrosis factor to normal subjects. N. Engl. J. Med 1990, 322, 1622–1627. [Google Scholar]
- Sawdey, M.S.; Loskutoff, D.J. Regulation of murine type 1 plasminogen activator inhibitor gene expression in vivo. J. Clin. Investig. 1991, 88, 1346–1353. [Google Scholar]
- Weerasinghe, S.V.W.; Moons, D.S.; Altshuler, P.J.; Shah, Y.M.; Bishr Omary, M. Fibrinogen-γ proteolysis and solubility dinamics during apoptotic mouse liver injury: Heparin prevents and treats liver damage. Hepatology 2011, 53, 1323–1332. [Google Scholar]
- Larson, A.M.; Polson, J.; Fontana, R.J.; Davern, T.J.; Lalani, E.; Hynan, L.S.; Reisch, J.S.; Schiødt, F.V.; Ostapowicz, G.; Shakil, A.O. Acetaminophen-induced acute liver failure: Results of a united states multicenter, prospective study. Hepatology 2005, 42, 1364–1372. [Google Scholar]
- Gibson, J.D.; Pumford, N.R.; Samokyszyn, V.M.; Hinson, J.A. Mechanism of acetaminophen-induced hepatotoxicity: Covalent binding versus oxidative stress. Chem. Res. Toxicol 1996, 9, 580–585. [Google Scholar]
- Imaeda, A.B.; Watanabe, A.; Sohail, M.A.; Mahmood, S.; Mohamadnejad, M.; Sutterwala, F.S.; Flavell, R.A.; Mehal, W.Z. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J. Clin. Investig 2009, 119, 305–314. [Google Scholar]
- Ganey, P.E.; Luyendyk, J.P.; Newport, S.W.; Eagle, T.M.; Maddox, J.F.; Mackman, N.; Roth, R.A. Role of the coagulation system in acetaminophen-induced hepatotoxicity in mice. Hepatology 2007, 46, 1177–1186. [Google Scholar]
- Sullivan, B.P.; Kopec, A.K.; Joshi, N.; Brown, J.A.; Bishop, S.C.; Kassel, K.M.; Rockwell, C.; Mackman, N.; Luyendyk, J.P. Hepatocyte tissue factor activates the coagulation cascade in mice. Blood 2013, 121, 1868–1874. [Google Scholar]
- Ju, C.; Reilly, T.P.; Bourdi, M.; Radonovich, M.F.; Brady, J.N.; George, J.W.; Pohl, L.R. Protective role of Kupffer cells in acetaminophen-induced hepatic injury in mice. Chem. Res. Toxicol 2002, 15, 1504–1513. [Google Scholar]
- Holt, M.P.; Yin, H.; Ju, C. Exacerbation of acetaminophen-induced disturbances of liver sinusoidal endothelial cells in the absence of Kupffer cells in mice. Toxicol. Lett 2010, 194, 34–41. [Google Scholar]
- You, Q.; Holt, M.; Yin, H.; Li, G.; Hu, C.-J.; Ju, C. Role of hepatic resident and infiltrating macrophages in liver repair after acute injury. Biochem. Pharmacol 2013, 86, 836–843. [Google Scholar]
- Bourdi, M.; Masubuchi, Y.; Relly, T.P.; Amouzadeh, H.R.; Martin, J.L.; George, J.W.; Shah, A.G.; Pohl, L.R. Protection against acetaminophen-induced liver injury and lethality by interleukin-10: Role of inducible nitric oxide synthase. Hepatology 2002, 35, 289–298. [Google Scholar]
- Novick, D.; Kim, S.H.; Fantuzzi, G.; Reznikov, L.L.; Dinarello, C.A.; Rubinstein, M. Interleukin-18 binding protein: A novel modulator of the Th1 cytokine response. Immunity 1999, 10, 127–136. [Google Scholar]



© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tsutsui, H.; Nishiguchi, S. Importance of Kupffer Cells in the Development of Acute Liver Injuries in Mice. Int. J. Mol. Sci. 2014, 15, 7711-7730. https://doi.org/10.3390/ijms15057711
Tsutsui H, Nishiguchi S. Importance of Kupffer Cells in the Development of Acute Liver Injuries in Mice. International Journal of Molecular Sciences. 2014; 15(5):7711-7730. https://doi.org/10.3390/ijms15057711
Chicago/Turabian StyleTsutsui, Hiroko, and Shuhei Nishiguchi. 2014. "Importance of Kupffer Cells in the Development of Acute Liver Injuries in Mice" International Journal of Molecular Sciences 15, no. 5: 7711-7730. https://doi.org/10.3390/ijms15057711