Aerobic Interval Training Attenuates Mitochondrial Dysfunction in Rats Post-Myocardial Infarction: Roles of Mitochondrial Network Dynamics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
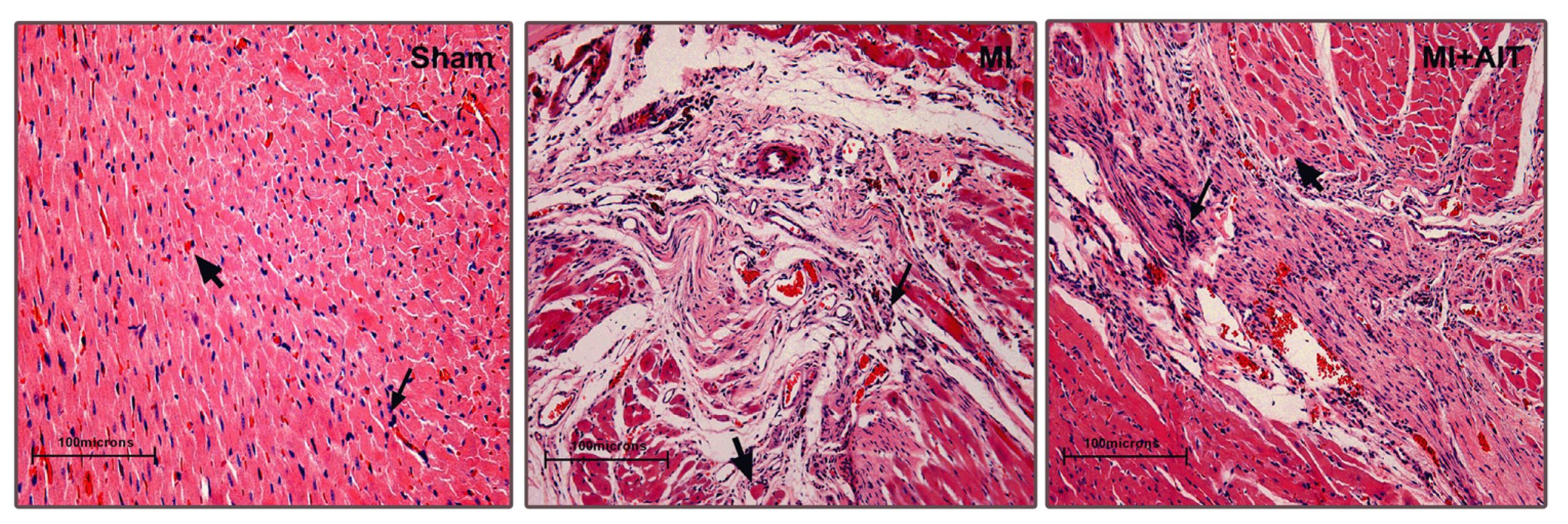
2.1. Effects of MI and AIT on Left Ventricular Tissue Microstructure
2.2. Effects of MI and AIT on Membrane Potential and Cytochrome C Leakage
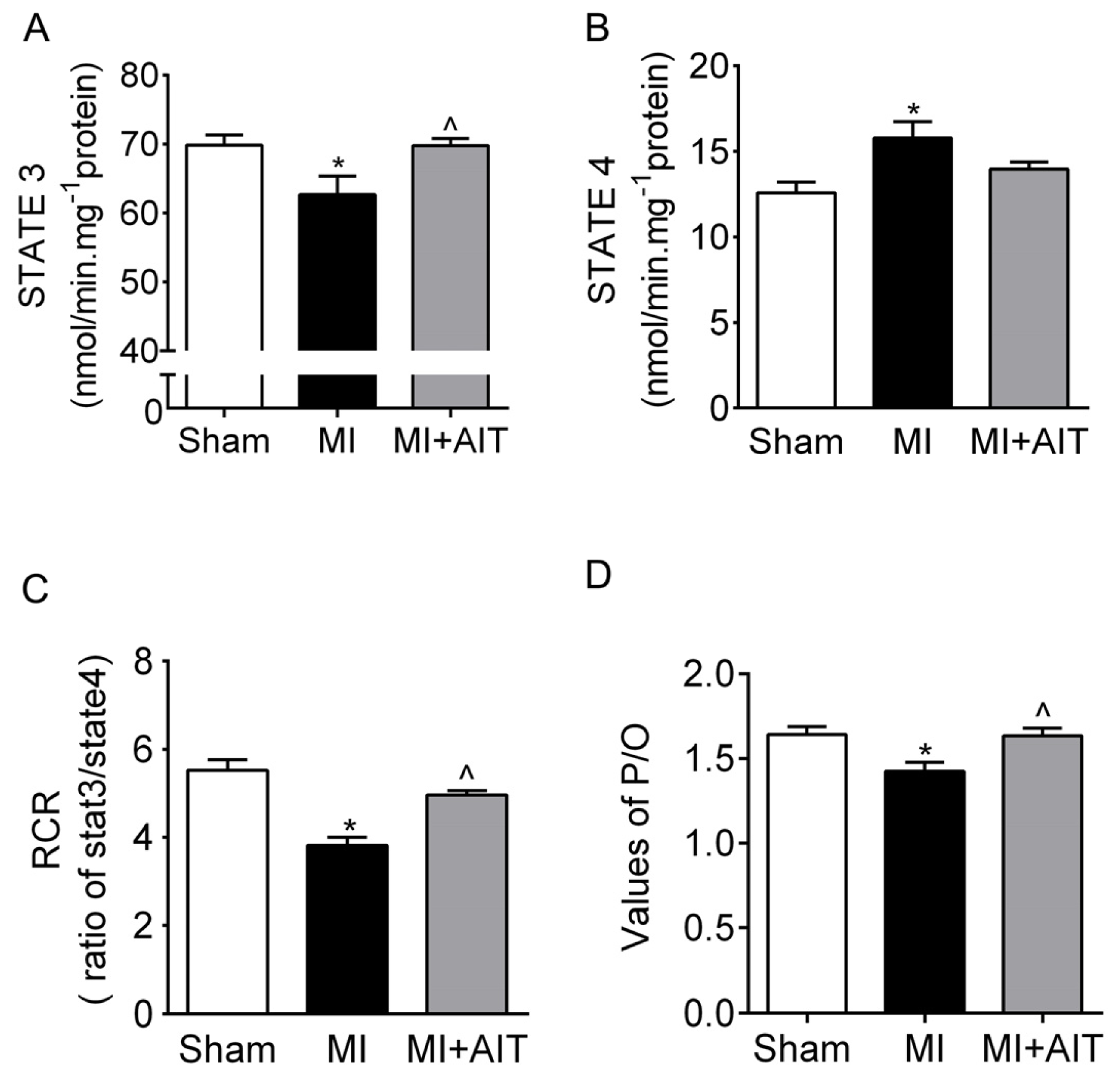
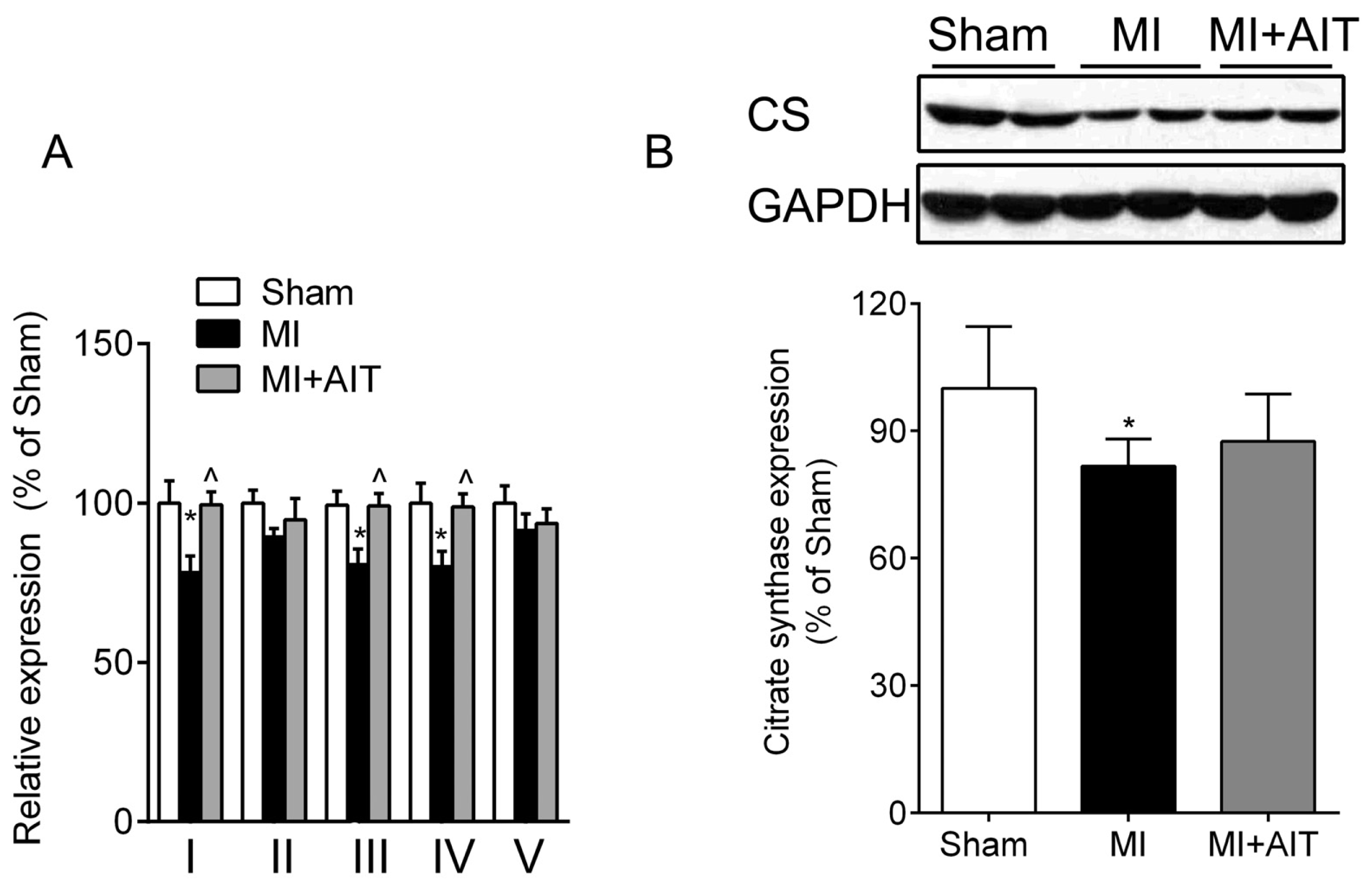
2.3. Effects of MI and AIT on Mitochondrial Respiratory Functions, Electron Transport Chain (ETC) Complex Activities and Protein Expression of Citrate Synthase (CS)
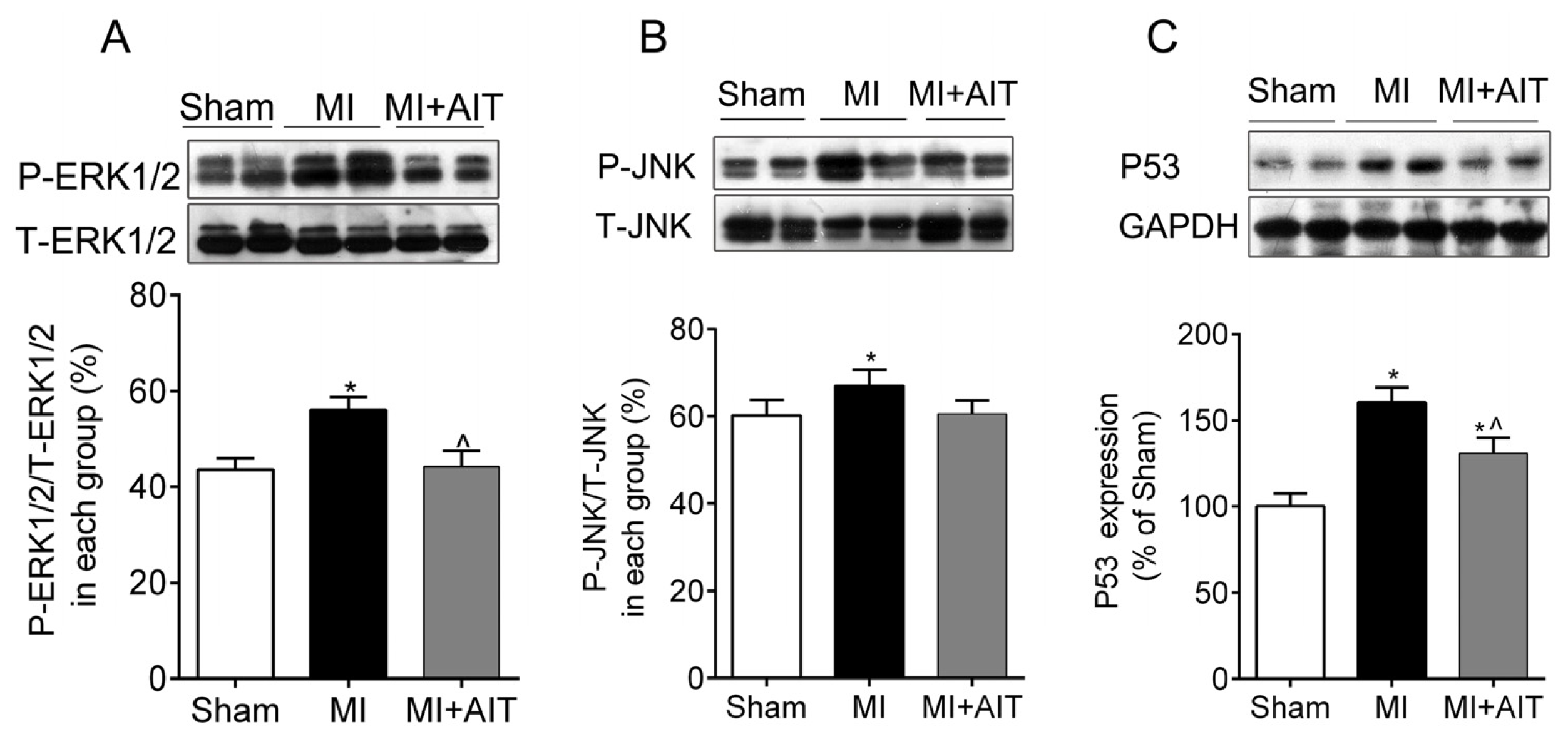
2.4. Effects of MI and AIT on Remodeling of Mitochondrial Dynamics and Oxidative Signaling
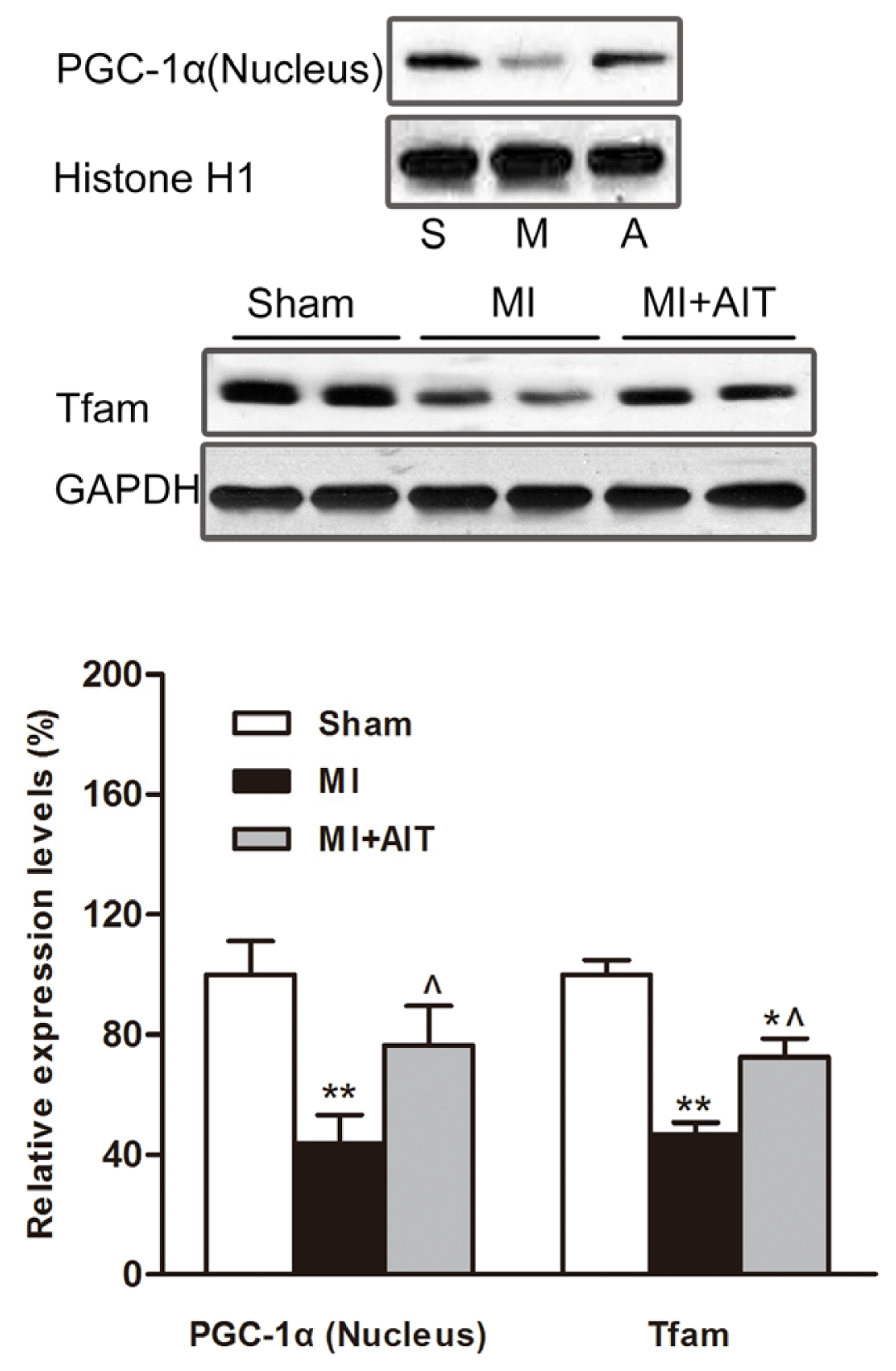
2.5. Effects of MI and AIT on PGC-1α and Tfam
3. Discussion
4. Experimental Section
4.1. Animals
4.2. In Vivo Myocardial Infarction Model
4.3. Experimental Grouping and Animal Use
4.4. Treadmill AIT Protocol
4.5. HE Staining
4.6. Myocardial Mitochondria Preparations
4.7. Mitochondrial Enzyme Activity Assay
4.8. Mitochondrial Respiratory Activity Determination
4.9. Measuring Mitochondrial Membrane Potential
4.10. Nuclear Protein Extraction
4.11. Western Blot Analysis
4.12. Statistical Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Leistner, D.M.; Zeiher, A.M. Novel avenues for cell therapy in acute myocardial infarction. Circ. Res 2012, 110, 195–197. [Google Scholar]
- Hori, M.; Nishida, K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc. Res 2009, 81, 457–464. [Google Scholar]
- Sena, S.; Hu, P.; Zhang, D.; Wang, X.; Wayment, B.; Olsen, C.; Avelar, E.; Abel, E.D.; Litwin, S.E. Impaired insulin signaling accelerates cardiac mitochondrial dysfunction after myocardial infarction. J. Mol. Cell. Cardiol 2009, 46, 910–918. [Google Scholar]
- Heather, L.C.; Carr, C.A.; Stuckey, D.J.; Pope, S.; Morten, K.J.; Carter, E.E.; Edwards, L.M.; Clarke, K. Critical role of complex III in the early metabolic changes following myocardial infarction. Cardiovasc. Res 2010, 85, 127–136. [Google Scholar]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ. Res 2001, 88, 529–535. [Google Scholar]
- Rosca, M.G.; Vazquez, E.J.; Kerner, J.; Parland, W.; Chandler, M.P.; Stanley, W.; Sabbah, H.N.; Hoppel, C.L. Cardiac mitochondria in heart failure: Decrease in respirasomes and oxidative phosphorylation. Cardiovasc. Res 2008, 80, 30–39. [Google Scholar]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc. Res 2009, 81, 449–456. [Google Scholar]
- Ong, S.B.; Hausenloy, D.J. Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res 2010, 88, 16–29. [Google Scholar]
- Bo, H.; Zhang, Y.; Ji, L.L. Redefining the role of mitochondria in exercise: A dynamic remodeling. Ann. N. Y. Acad. Sci 2010, 1201, 121–128. [Google Scholar]
- Palaniyandi, S.S.; Qi, X.; Yogalingam, G.; Ferreira, J.C.; Mochly-Rosen, D. Regulation of mitochondrial processes: A target for heart failure. Drug Discov. Today Dis. Mech 2010, 7, e95–e102. [Google Scholar]
- Frederico, M.J.; Justo, S.L.; Da Luz, G.; Da Silva, S.; Medeiros, C.; Barbosa, V.A.; Silva, L.A.; Boeck, C.R.; de Pinho, R.A.; de Souza, C.T. Exercise training provides cardioprotection via a reduction in reactive oxygen species in rats submitted to myocardial infarction induced by isoproterenol. Free Radic. Res 2009, 43, 957–964. [Google Scholar]
- Kavazis, A.N.; Alvarez, S.; Talbert, E.; Lee, Y.; Powers, S.K. Exercise training induces a cardioprotective phenotype and alterations in cardiac subsarcolemmal and intermyofibrillar mitochondrial proteins. Am. J. Physiol. Heart Circ. Physiol 2009, 297, H144–H152. [Google Scholar]
- Campos, J.C.; Queliconi, B.B.; Dourado, P.M.; Cunha, T.F.; Zambelli, V.O.; Bechara, L.R.; Kowaltowski, A.J.; Brum, P.C.; Mochly-Rosen, D.; Ferreira, J.C. Exercise training restores cardiac protein quality control in heart failure. PLoS One 2012, 7, e52764. [Google Scholar]
- Little, J.P.; Cochran, A.J. Regulating the regulators: The role of transcriptional regulatory proteins in the adaptive response to exercise in human skeletal muscle. J. Physiol 2011, 589, 1511–1512. [Google Scholar]
- Yan, Z.; Lira, V.A.; Greene, N.P. Exercise training-induced regulation of mitochondrial quality. Exerc. Sport Sci. Rev 2012, 40, 159–164. [Google Scholar]
- Wisloff, U.; Stoylen, A.; Loennechen, J.P.; Bruvold, M.; Rognmo, O.; Haram, P.M.; Tjonna, A.E.; Helgerud, J.; Slordahl, S.A.; Lee, S.J.; et al. Superior cardiovascular effect of aerobic interval training versus moderate continuous training in heart failure patients: A randomized study. Circulation 2007, 115, 3086–3094. [Google Scholar]
- Daussin, F.N.; Zoll, J.; Dufour, S.P.; Ponsot, E.; Lonsdorfer-Wolf, E.; Doutreleau, S.; Mettauer, B.; Piquard, F.; Geny, B.; Richard, R. Effect of interval versus continuous training on cardiorespiratory and mitochondrial functions: Relationship to aerobic performance improvements in sedentary subjects. Am. J. Physiol. Regul. Integr. Comp. Physiol 2008, 295, R264–R272. [Google Scholar]
- Haram, P.M.; Kemi, O.J.; Lee, S.J.; Bendheim, M.O.; Al-Share, Q.Y.; Waldum, H.L.; Gilligan, L.J.; Koch, L.G.; Britton, S.L.; Najjar, S.M.; et al. Aerobic interval training vs. continuous moderate exercise in the metabolic syndrome of rats artificially selected for low aerobic capacity. Cardiovasc. Res 2009, 81, 723–732. [Google Scholar]
- Gibala, M.J.; Little, J.P.; Macdonald, M.J.; Hawley, J.A. Physiological adaptations to low-volume, high-intensity interval training in health and disease. J. Physiol 2012, 590, 1077–1084. [Google Scholar]
- Beckers, P.J.; Denollet, J.; Possemiers, N.M.; Wuyts, F.L.; Vrints, C.J.; Conraads, V.M. Combined endurance-resistance training vs. endurance training in patients with chronic heart failure: A prospective randomized study. Eur. Heart J 2008, 29, 1858–1866. [Google Scholar]
- Vona, M.; Codeluppi, G.M.; Iannino, T.; Ferrari, E.; Bogousslavsky, J.; von Segesser, L.K. Effects of different types of exercise training followed by detraining on endothelium-dependent dilation in patients with recent myocardial infarction. Circulation 2009, 119, 1601–1608. [Google Scholar]
- Guimaraes, G.V.; Ciolac, E.G.; Carvalho, V.O.; D’Avila, V.M.; Bortolotto, L.A.; Bocchi, E.A. Effects of continuous vs. interval exercise training on blood pressure and arterial stiffness in treated hypertension. Hypertens. Res 2010, 33, 627–632. [Google Scholar]
- Guiraud, T.; Juneau, M.; Nigam, A.; Gayda, M.; Meyer, P.; Mekary, S.; Paillard, F.; Bosquet, L. Optimization of high intensity interval exercise in coronary heart disease. Eur. J. Appl. Physiol 2010, 108, 733–740. [Google Scholar]
- Stolen, T.O.; Hoydal, M.A.; Kemi, O.J.; Catalucci, D.; Ceci, M.; Aasum, E.; Larsen, T.; Rolim, N.; Condorelli, G.; Smith, G.L.; et al. Interval training normalizes cardiomyocyte function, diastolic Ca2+ control, and SR Ca2+ release synchronicity in a mouse model of diabetic cardiomyopathy. Circ. Res 2009, 105, 527–536. [Google Scholar]
- Anderson, R.M.; Barger, J.L.; Edwards, M.G.; Braun, K.H.; O’Connor, C.E.; Prolla, T.A.; Weindruch, R. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell 2008, 7, 101–111. [Google Scholar]
- Di Lisa, F.; Canton, M.; Carpi, A.; Kaludercic, N.; Menabo, R.; Menazza, S.; Semenzato, M. Mitochondrial injury and protection in ischemic pre- and postconditioning. Antioxid. Redox. Signal 2011, 14, 881–891. [Google Scholar]
- Cheng, Y.; Qiu, F.; Tashiro, S.; Onodera, S.; Ikejima, T. ERK and JNK mediate TNFalpha-induced p53 activation in apoptotic and autophagic L929 cell death. Biochem. Biophys. Res 2008, 376, 483–488. [Google Scholar]
- Johri, A.; Chandra, A.; Beal, M.F. PGC-1alpha, mitochondrial dysfunction, and Huntington’s disease. Free Radic. Res 2013, 62, 37–46. [Google Scholar]
- Ding, H.; Jiang, N.; Liu, H.; Liu, X.; Liu, D.; Zhao, F.; Wen, L.; Liu, S.; Ji, L.L.; Zhang, Y. Response of mitochondrial fusion and fission protein gene expression to exercise in rat skeletal muscle. Biochim. Biophys. Acta 2010, 1800, 250–256. [Google Scholar]
- Ma, Z.A.; Zhao, Z.; Turk, J. Mitochondrial dysfunction and beta-cell failure in type 2 diabetes mellitus. Exp. Diabetes Res 2012, 2012, 703538. [Google Scholar]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 2011, 124, 444–453. [Google Scholar]
- Dorn, G.W. Mitochondrial dynamics in heart disease. Biochim. Biophys. Acta 2013, 1833, 233–241. [Google Scholar]
- Hall, A.R.; Hausenloy, D.J. The shape of things to come: Mitochondrial fusion and fission in the adult heart. Cardiovasc. Res 2012, 94, 391–392. [Google Scholar]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res 2009, 84, 91–99. [Google Scholar]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar]
- Garnier, A.; Fortin, D.; Zoll, J.; N’Guessan, B.; Mettauer, B.; Lampert, E.; Veksler, V.; Ventura-Clapier, R. Coordinated changes in mitochondrial function and biogenesis in healthy and diseased human skeletal muscle. FASEB J 2005, 19, 43–52. [Google Scholar]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci 2008, 9, 505–518. [Google Scholar]
- Wu, S.; Zhou, F.; Zhang, Z.; Xing, D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J 2011, 278, 941–954. [Google Scholar]
- Pohjoismaki, J.L.; Boettger, T.; Liu, Z.; Goffart, S.; Szibor, M.; Braun, T. Oxidative stress during mitochondrial biogenesis compromises mtDNA integrity in growing hearts and induces a global DNA repair response. Nucleic. Acids. Res 2012, 40, 6595–6607. [Google Scholar]
- Xu, X.; Zhao, W.; Wan, W.; Ji, L.L.; Powers, A.S.; Erikson, J.M.; Zhang, J.Q. Exercise training combined with angiotensin II receptor blockade reduces oxidative stress after myocardial infarction in rats. Exp. Physiol 2010, 95, 1008–1015. [Google Scholar]
- Gounder, S.S.; Kannan, S.; Devadoss, D.; Miller, C.J.; Whitehead, K.S.; Odelberg, S.J.; Firpo, M.A.; Paine, R., 3rd; Hoidal, J.R.; Abel, E.D.; et al. Impaired transcriptional activity of Nrf2 in age-related myocardial oxidative stress is reversible by moderate exercise training. PLoS One 2012, 7, e45697. [Google Scholar]
- Muthusamy, V.R.; Kannan, S.; Sadhaasivam, K.; Gounder, S.S.; Davidson, C.J.; Boeheme, C.; Hoidal, J.R.; Wang, L.; Rajasekaran, N.S. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic. Biol. Med 2012, 52, 366–376. [Google Scholar]
- Schwartz, P.J. Vagal stimulation for the treatment of heart failure: A translational success story. Heart 2012, 98, 1687–1689. [Google Scholar]
- Liu, J.J.; Li, D.L.; Zhou, J.; Sun, L.; Zhao, M.; Kong, S.S.; Wang, Y.H.; Yu, X.J.; Zhou, J.; Zang, W.J. Acetylcholine prevents angiotensin II-induced oxidative stress and apoptosis in H9c2 cells. Apoptosis 2011, 16, 94–103. [Google Scholar]
- Gottlieb, R.A.; Gustafsson, A.B. Mitochondrial turnover in the heart. Biochim. Biophys. Acta 2011, 1813, 1295–1301. [Google Scholar]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1660. [Google Scholar]
- Ikeuchi, M.; Matsusaka, H.; Kang, D.; Matsushima, S.; Ide, T.; Kubota, T.; Fujiwara, T.; Hamasaki, N.; Takeshita, A.; Sunagawa, K.; et al. Overexpression of mitochondrial transcription factor a ameliorates mitochondrial deficiencies and cardiac failure after myocardial infarction. Circulation 2005, 112, 683–690. [Google Scholar]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional control of mitochondrial biogenesis: The central role of PGC-1alpha. Cardiovasc. Res 2008, 79, 208–217. [Google Scholar]
- Pillai, V.B.; Sundaresan, N.R.; Jeevanandam, V.; Gupta, M.P. Mitochondrial SIRT3 and heart disease. Cardiovasc. Res 2010, 88, 250–256. [Google Scholar]
- Lanza, I.R.; Short, D.K.; Short, K.R.; Raghavakaimal, S.; Basu, R.; Joyner, M.J.; McConnell, J.P.; Nair, K.S. Endurance exercise as a countermeasure for aging. Diabetes 2008, 57, 2933–2942. [Google Scholar]
- Liu, J.; Shen, W.; Zhao, B.; Wang, Y.; Wertz, K.; Weber, P.; Zhang, P. Targeting mitochondrial biogenesis for preventing and treating insulin resistance in diabetes and obesity: Hope from natural mitochondrial nutrients. Adv. Drug. Deliv. Rev 2009, 61, 1343–1352. [Google Scholar]
- Cheng, X.W.; Kuzuya, M.; Kim, W.; Song, H.; Hu, L.; Inoue, A.; Nakamura, K.; Di, Q.; Sasaki, T.; Tsuzuki, M.; et al. Exercise training stimulates ischemia-induced neovascularization via phosphatidylinositol 3-kinase/Akt-dependent hypoxia-induced factor-1 alpha reactivation in mice of advanced age. Circulation 2010, 122, 707–716. [Google Scholar]
- Wang, Y.; Wang, S.; Wier, W.G.; Zhang, Q.; Jiang, H.; Li, Q.; Chen, S.; Tian, Z.; Li, Y.; Yu, X.; et al. Exercise improves the dilatation function of mesenteric arteries in postmyocardial infarction rats via a PI3K/Akt/eNOS pathway-mediated mechanism. Am. J. Physiol. Heart Circ. Physiol 2010, 299, H2097–H2106. [Google Scholar]
- Wang, G.; Liem, D.A.; Vondriska, T.M.; Honda, H.M.; Korge, P.; Pantaleon, D.M.; Qiao, X.; Wang, Y.; Weiss, J.N.; Ping, P. Nitric oxide donors protect murine myocardium against infarction via modulation of mitochondrial permeability transition. Am. J. Physiol. Heart Circ. Physiol 2005, 288, H1290–H1295. [Google Scholar]
- Borniquel, S.; Valle, I.; Cadenas, S.; Lamas, S.; Monsalve, M. Nitric oxide regulates mitochondrial oxidative stress protection via the transcriptional coactivator PGC-1alpha. FASEB J 2006, 20, 1889–1891. [Google Scholar]
- Nisoli, E.; Clementi, E.; Paolucci, C.; Cozzi, V.; Tonello, C.; Sciorati, C.; Bracale, R.; Valerio, A.; Francolini, M.; Moncada, S.; et al. Mitochondrial biogenesis in mammals: The role of endogenous nitric oxide. Science 2003, 299, 896–899. [Google Scholar]
- De Palma, C.; Falcone, S.; Pisoni, S.; Cipolat, S.; Panzeri, C.; Pambianco, S.; Pisconti, A.; Allevi, R.; Bassi, M.T.; Cossu, G.; et al. Nitric oxide inhibition of Drp1-mediated mitochondrial fission is critical for myogenic differentiation. Cell Death Differ 2010, 17, 1684–1696. [Google Scholar]
- Jiang, H.; Miao, Y.; Wang, Y.; Zhao, M.; Feng, Z.; Yu, X.; Zang, W. Aerobic interval training protects against myocardial infarction-induced oxidative injury by enhancing antioxidase system and mitochondrial biosynthesis. Clin. Exp. Pharmacol. Physiol 2014. [Google Scholar] [CrossRef]
- Wisloff, U.; Helgerud, J.; Kemi, O.J.; Ellingsen, O. Intensity-controlled treadmill running in rats: VO(2 max) and cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol 2001, 280, H1301–H1310. [Google Scholar]
- Hao, J.; Shen, W.; Yu, G.; Jia, H.; Li, X.; Feng, Z.; Wang, Y.; Weber, P.; Wertz, K.; Sharman, E.; et al. Hydroxytyrosol promotes mitochondrial biogenesis and mitochondrial function in 3T3-L1 adipocytes. J. Nutr. Biochem 2010, 21, 634–644. [Google Scholar]
- Feng, Z.; Zou, X.; Jia, H.; Li, X.; Zhu, Z.; Liu, X.; Bucheli, P.; Ballevre, O.; Hou, Y.; Zhang, W.; et al. Maternal docosahexaenoic acid feeding protects against impairment of learning and memory and oxidative stress in prenatally stressed rats: Possible role of neuronal mitochondria metabolism. Antioxid. Redox Signal 2012, 16, 275–289. [Google Scholar]
- Bo, H.; Jiang, N.; Ma, G.; Qu, J.; Zhang, G.; Cao, D.; Wen, L.; Liu, S.; Ji, L.L.; Zhang, Y. Regulation of mitochondrial uncoupling respiration during exercise in rat heart: Role of reactive oxygen species (ROS) and uncoupling protein 2. Free Radic. Biol. Med 2008, 44, 1373–1381. [Google Scholar]
 indicates nuclei (blue);
indicates nuclei (blue);
 indicates cytoplasm (pink). Sham: sham-operation; MI: myocardial infarction; MI + AIT: myocardial infarcted rats subjected to aerobic interval training. Bar: 100 μm. n = 6 per group.
indicates nuclei (blue);
indicates cytoplasm (pink). Sham: sham-operation; MI: myocardial infarction; MI + AIT: myocardial infarcted rats subjected to aerobic interval training. Bar: 100 μm. n = 6 per group.
indicates cytoplasm (pink). Sham: sham-operation; MI: myocardial infarction; MI + AIT: myocardial infarcted rats subjected to aerobic interval training. Bar: 100 μm. n = 6 per group.
indicates nuclei (blue);
indicates cytoplasm (pink). Sham: sham-operation; MI: myocardial infarction; MI + AIT: myocardial infarcted rats subjected to aerobic interval training. Bar: 100 μm. n = 6 per group.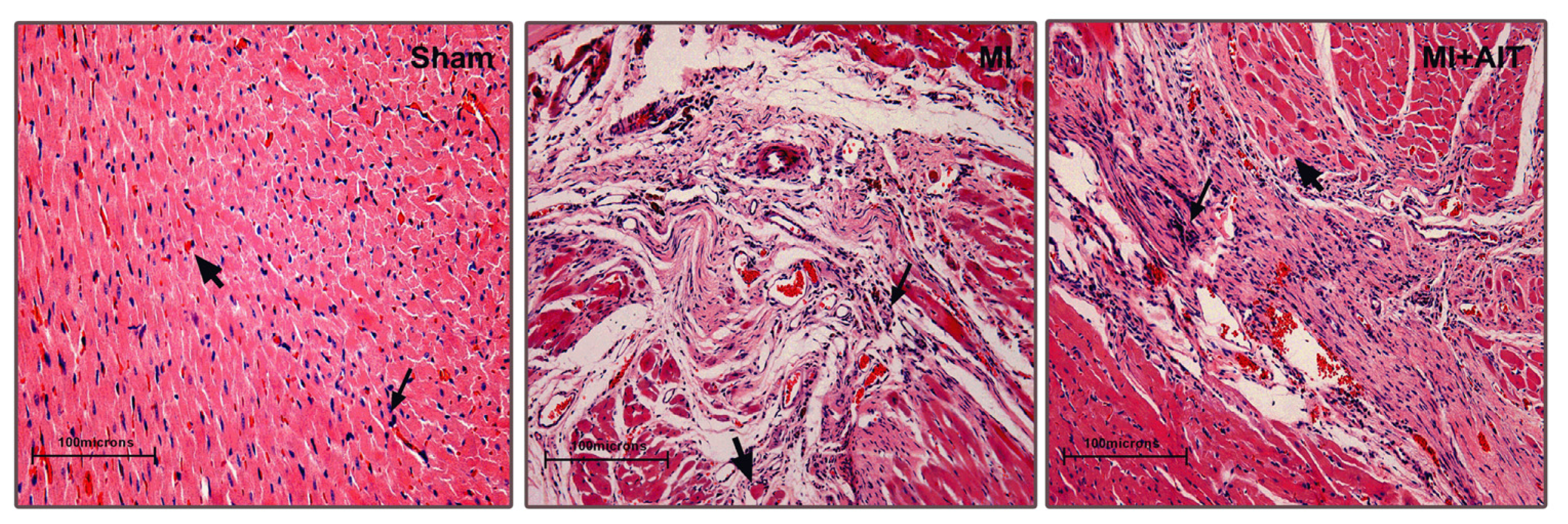





© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jiang, H.-K.; Wang, Y.-H.; Sun, L.; He, X.; Zhao, M.; Feng, Z.-H.; Yu, X.-J.; Zang, W.-J. Aerobic Interval Training Attenuates Mitochondrial Dysfunction in Rats Post-Myocardial Infarction: Roles of Mitochondrial Network Dynamics. Int. J. Mol. Sci. 2014, 15, 5304-5322. https://doi.org/10.3390/ijms15045304
Jiang H-K, Wang Y-H, Sun L, He X, Zhao M, Feng Z-H, Yu X-J, Zang W-J. Aerobic Interval Training Attenuates Mitochondrial Dysfunction in Rats Post-Myocardial Infarction: Roles of Mitochondrial Network Dynamics. International Journal of Molecular Sciences. 2014; 15(4):5304-5322. https://doi.org/10.3390/ijms15045304
Chicago/Turabian StyleJiang, Hong-Ke, You-Hua Wang, Lei Sun, Xi He, Mei Zhao, Zhi-Hui Feng, Xiao-Jiang Yu, and Wei-Jin Zang. 2014. "Aerobic Interval Training Attenuates Mitochondrial Dysfunction in Rats Post-Myocardial Infarction: Roles of Mitochondrial Network Dynamics" International Journal of Molecular Sciences 15, no. 4: 5304-5322. https://doi.org/10.3390/ijms15045304