Application of Computational Methods for the Design of BACE-1 Inhibitors: Validation of in Silico Modelling

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Analysis of Selected Crystal Structures
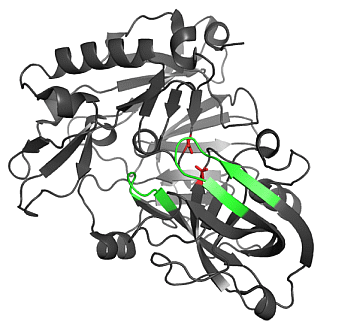
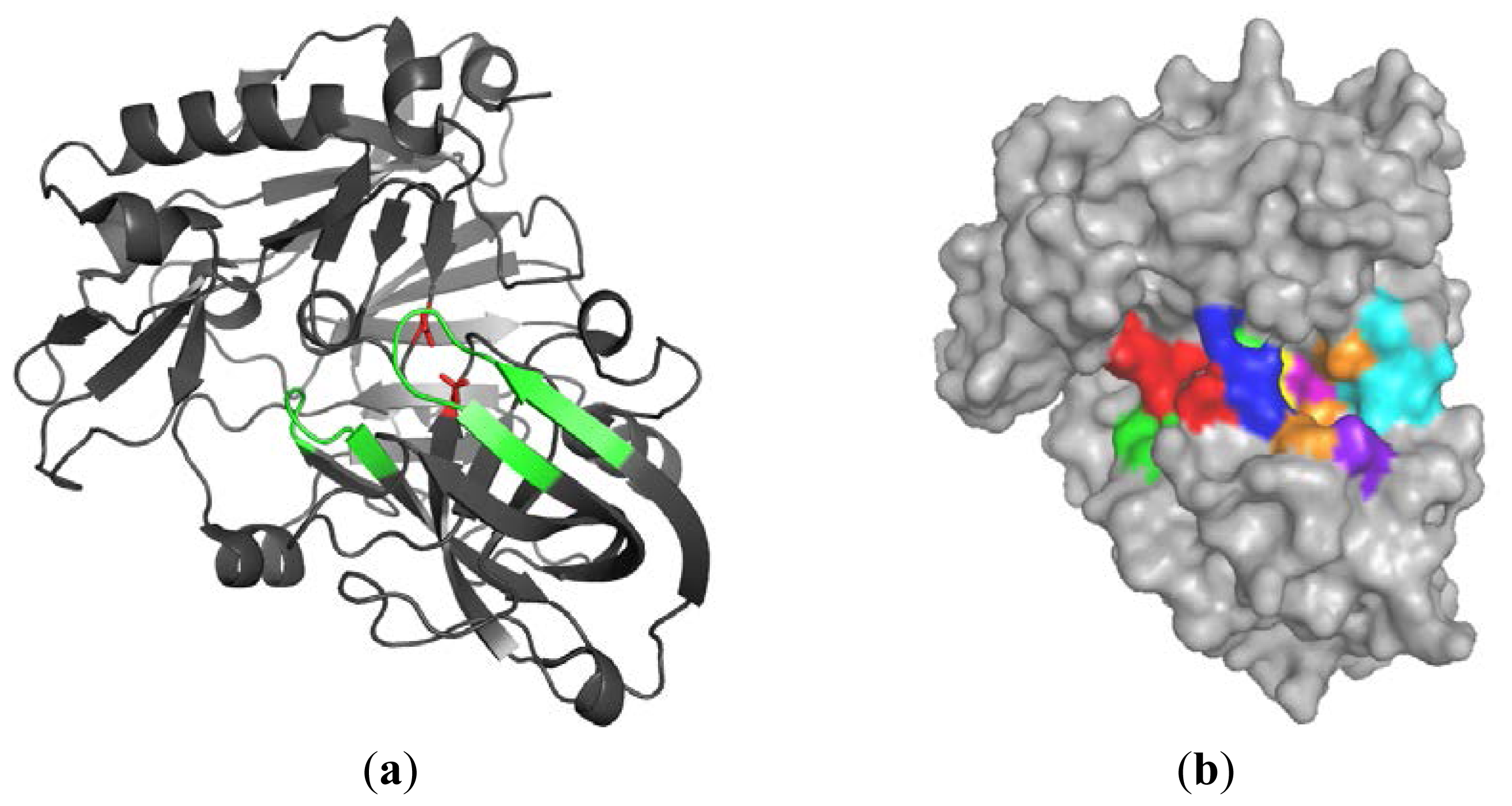
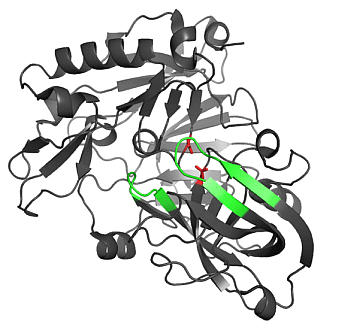
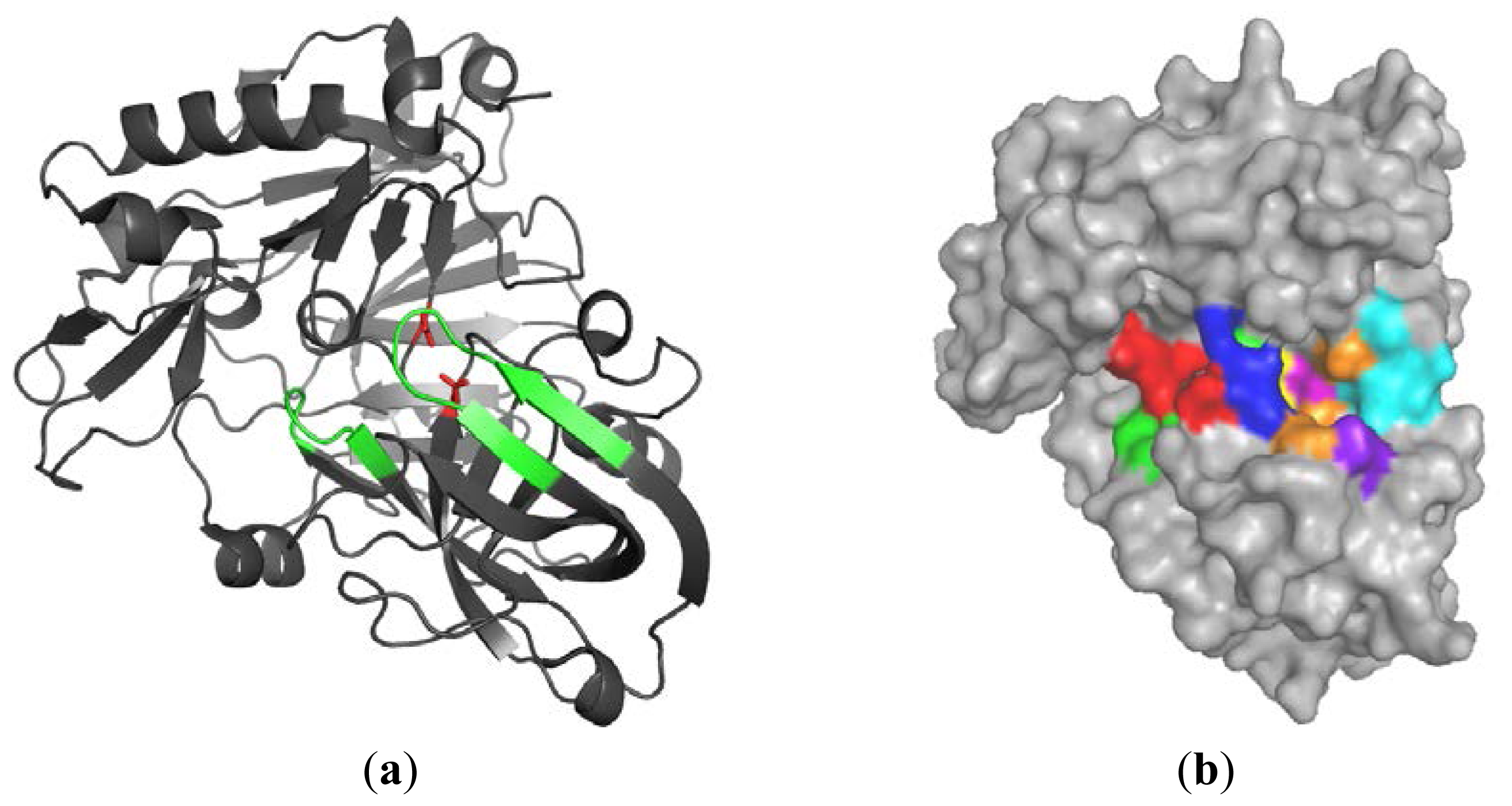
2.1.1. β-Secretase (BACE-1)
2.1.2. Water Molecules in Crystal Structures
2.2. Validation of Docking with Gold Suite
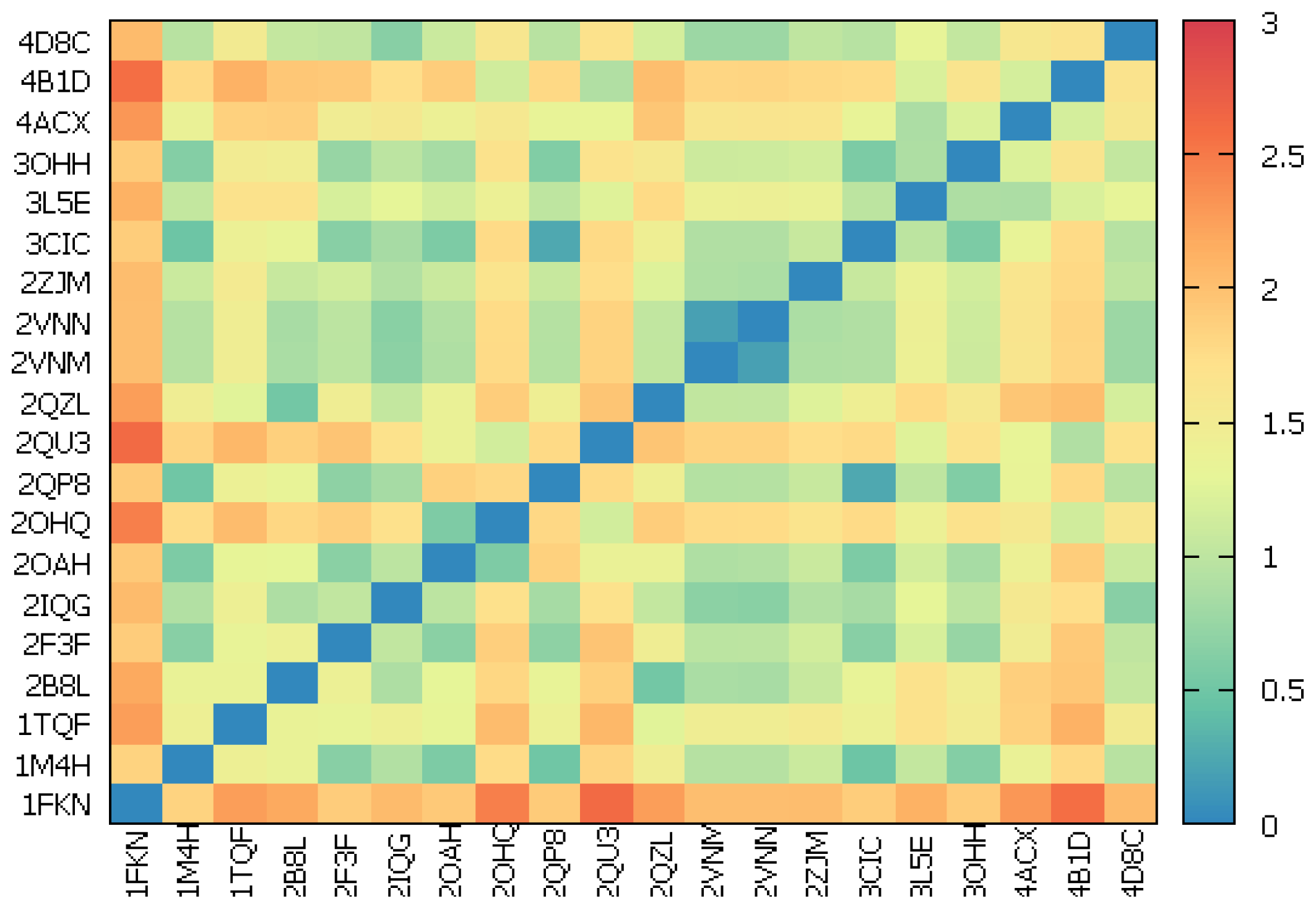
2.2.1. Redocking
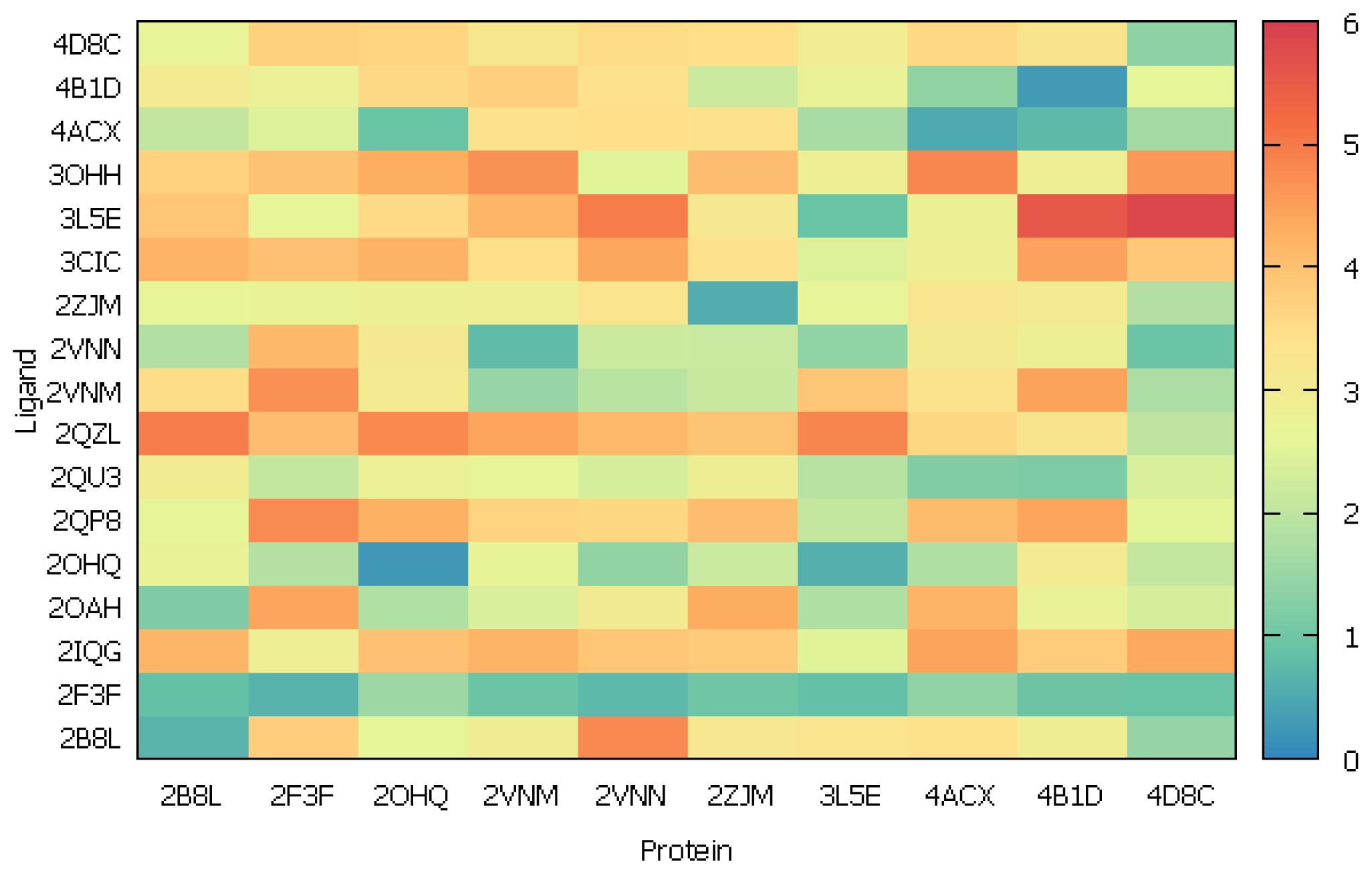
2.2.2. Cross-Docking
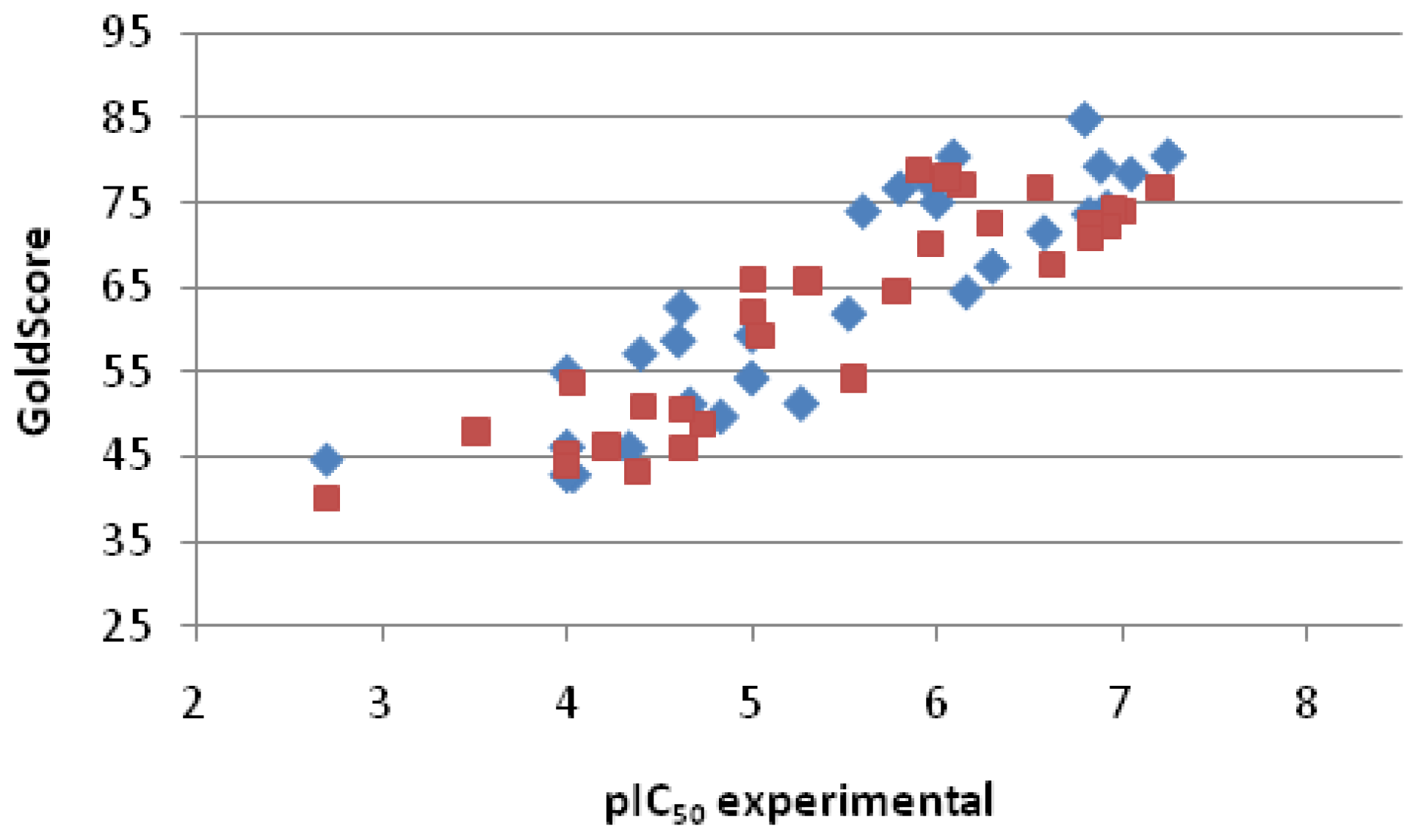
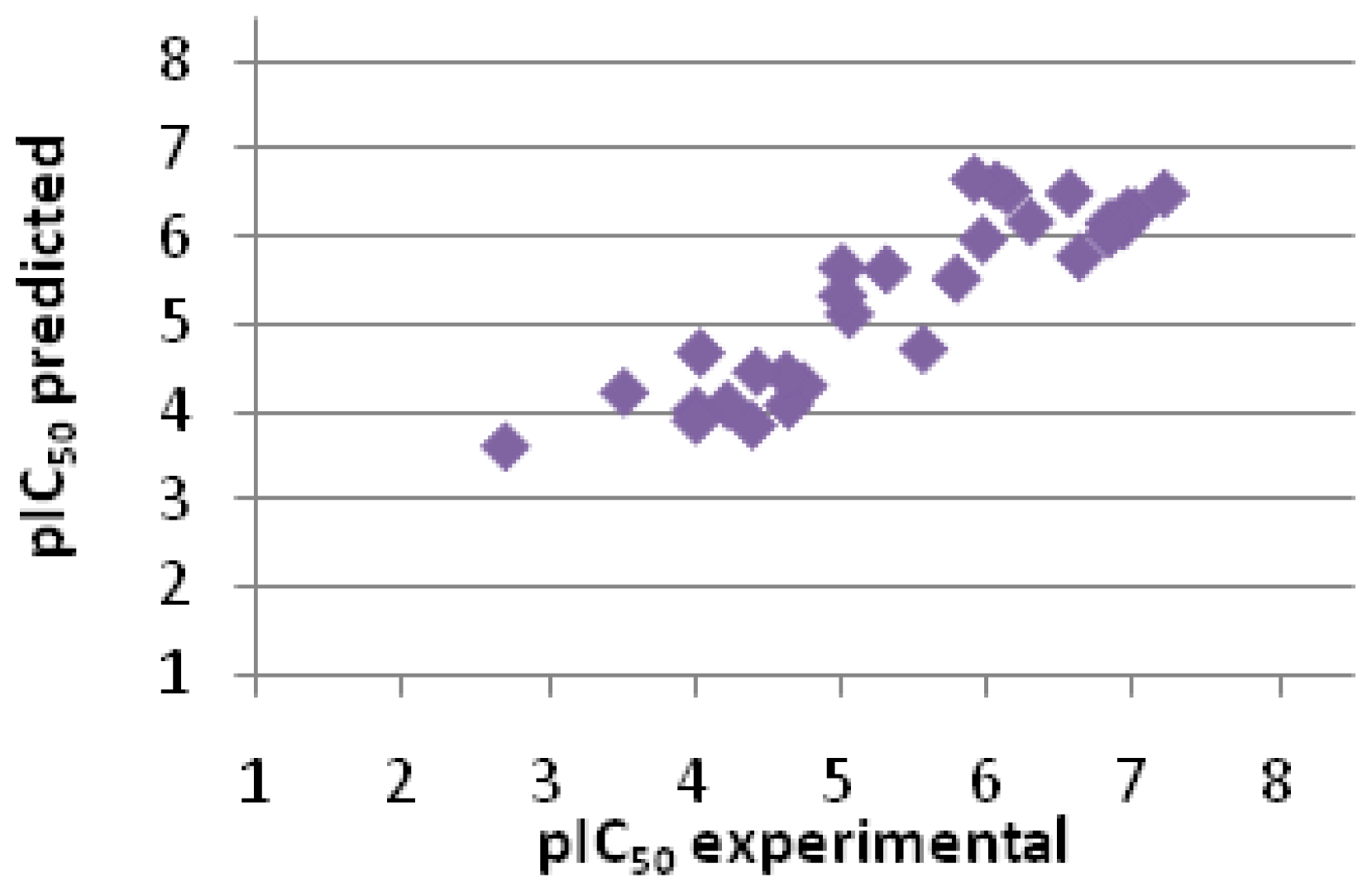
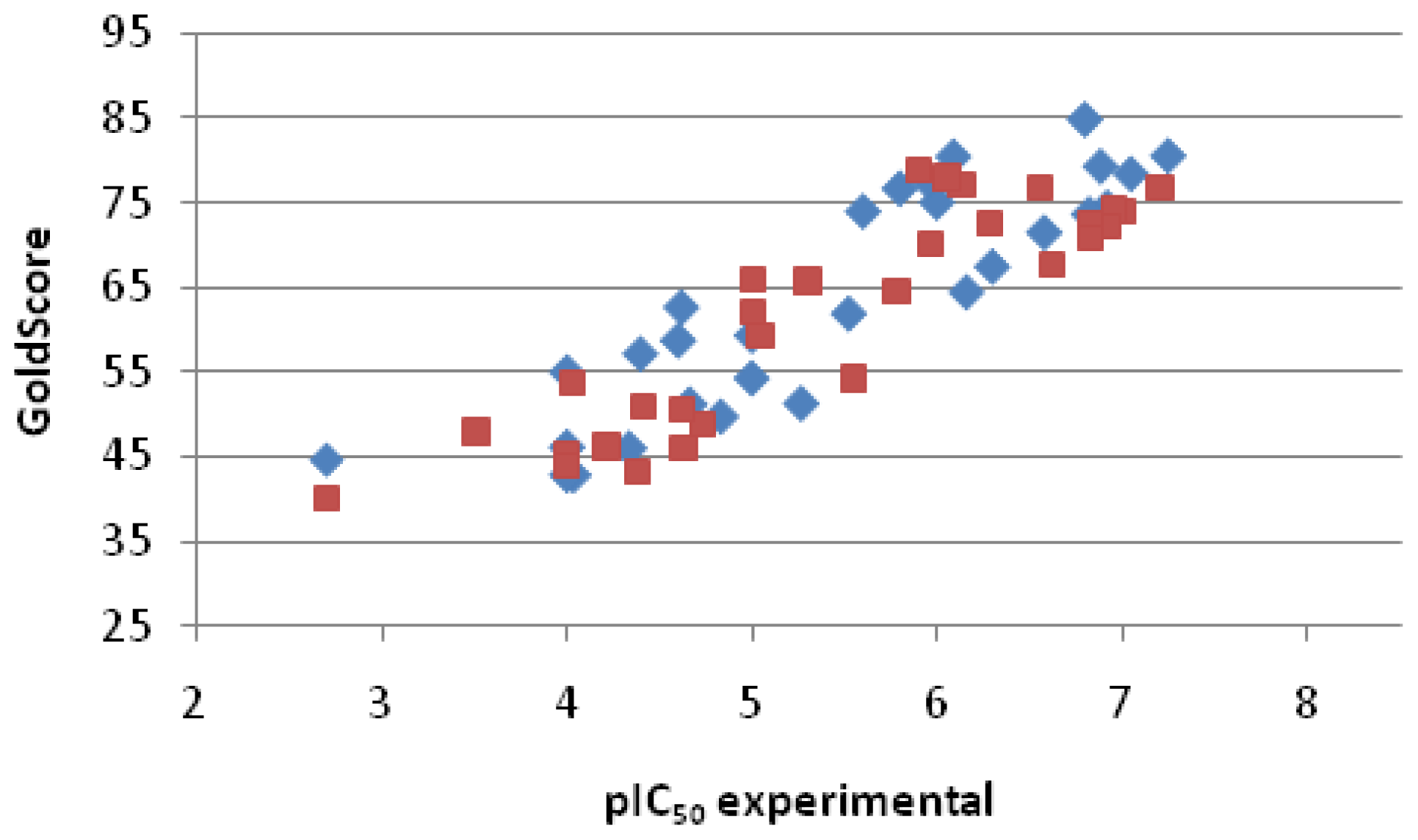
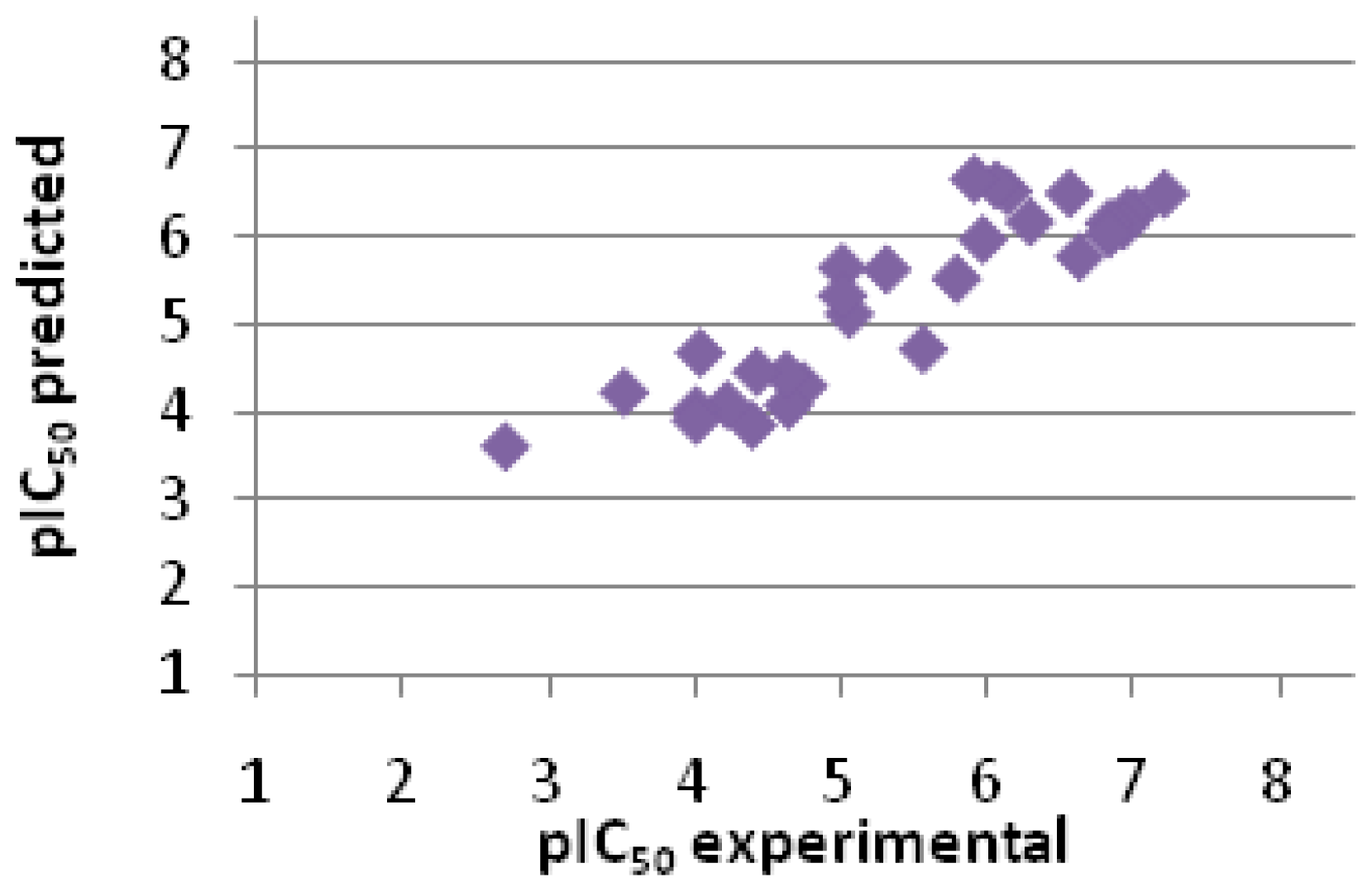
2.2.3. Scoring
3. Experimental Section
3.1. Analysis of Selected Crystal Structures
3.2. Docking and Scoring
4. Conclusions
Supplementary Information
ijms-15-05128-s001.pdfAcknowledgments
Conflicts of Interest
References
- Yuan, J.; Venkatraman, S.; Zheng, Y.; McKeever, B.M.; Dillard, L.W.; Singh, S.B. Structure-based design of β-site APP cleaving enzyme 1 (BACE1) inhibitors for the treatment of Alzheimer’s disease. J. Med. Chem. 2013, 56, 4156–4180. [Google Scholar]
- Stamford, A.; Strickland, C. Inhibitors of BACE for treating Alzheimer’s disease: A fragment-based drug discovery story. Curr. Opin. Chem. Biol. 2013, 17, 320–328. [Google Scholar]
- Mullane, K.; Williams, M. Alzheimer’s therapeutics: Continued clinical failures question the validity of the amyloid hypothesis-but what lies beyond? Biochem. Pharmacol. 2013, 85, 289–305. [Google Scholar]
- Marks, N.; Berg, M.J. BACE and γ-secretase characterization and their sorting as therapeutic targets to reduce amyloidogenesis. Neurochem. Res. 2010, 35, 181–210. [Google Scholar]
- De Strooper, B.; Iwatsubo, T.; Wolfe, M.S. Presenilins and γ-secretase: Structure function and role in Alzheimer’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006304. [Google Scholar]
- Wolfe, K.J.; Cyr, D.M. Amyloid in neurodegenerative diseases: Friend or foe? Semin. Cell Dev. Biol. 2011, 22, 476–481. [Google Scholar]
- Tamagno, E.; Guglielmotto, M.; Monteleone, D.; Tabaton, M. Amyloid-β production: Major link between oxidative stress and BACE1. Neurotox. Res. 2012, 22, 208–219. [Google Scholar]
- Zhang, X.M.; Cai, Y.; Xiong, K.; Cai, H.; Luo, X.G.; Feng, J.C.; Clough, R.W.; Struble, R.G.; Patrylo, P.R.; Yan, X.X. β-Secretase-1 elevation in transgenic mouse models of Alzheimer’s disease is associated with synaptic/axonal pathology and amyloidogenesis: Implications for neuritic plaque development. Eur. J. Neurosci. 2009, 30, 2271–2283. [Google Scholar]
- Vassar, R.; Kovacs, D.M.; Yan, R.; Wong, P.C. The β-secretase enzyme BACE in health and Alzheimer’s disease: Regulation cell biology function and therapeutic potential. J. Neurosci. 2009, 29, 12787–12794. [Google Scholar]
- Treiber, H.; Hagemeyer, N.; Ehrenreich, H.; Simons, M. BACE1 in central nervous system myelination revisited. Mol. Psychiatry 2012, 17, 237–239. [Google Scholar]
- Sathya, M.; Premkumar, P.; Karthick, C.; Moorthi, P.; Jayachandran, K.S.; Anusuyadevi, M. BACE1 in Alzheimer’s disease. Clin. Chim. Acta 2012, 414, 171–178. [Google Scholar]
- Sankaranarayanan, S.; Price, E.A.; Wu, G.; Crouthamel, M.C.; Shi, X.P.; Tugusheva, K.; Tyler, K.X.; Kahana, J.; Ellis, J.; Jin, L.; et al. In vivo β-secretase 1 inhibition leads to brain Abeta lowering and increased α-secretase processing of amyloid precursor protein without effect on neuregulin-1. J. Pharmacol. Exp. Ther. 2008, 324, 957–969. [Google Scholar]
- Charrier, N.; Clarke, B.; Cutler, L.; Demont, E.; Dingwall, C.; Dunsdon, R.; Hawkins, J.; Howes, C.; Hubbard, J.; Hussain, I.; et al. Second generation of BACE-1 inhibitors Part 1: The need for improved pharmacokinetics. Bioorg. Med. Chem. Lett. 2009, 19, 3664–3668. [Google Scholar]
- Charrier, N.; Clarke, B.; Cutler, L.; Demont, E.; Dingwall, C.; Dunsdon, R.; Hawkins, J.; Howes, C.; Hubbard, J.; Hussain, I.; et al. Second generation of BACE-1 inhibitors part 3: Towards non hydroxyethylamine transition state mimetics. Bioorg. Med. Chem. Lett. 2009, 19, 3674–3678. [Google Scholar]
- Charrier, N.; Clarke, B.; Demont, E.; Dingwall, C.; Dunsdon, R.; Hawkins, J.; Hubbard, J.; Hussain, I.; Maile, G.; Matico, R.; et al. Second generation of BACE-1 inhibitors part 2: Optimisation of the non-prime side substituent. Bioorg. Med. Chem. Lett. 2009, 19, 3669–3673. [Google Scholar]
- Clarke, B.; Demont, E.; Dingwall, C.; Dunsdon, R.; Faller, A.; Hawkins, J.; Hussain, I.; MacPherson, D.; Maile, G.; Matico, R.; et al. BACE-1 inhibitors part 1: Identification of novel hydroxy ethylamines (HEAs). Bioorg. Med. Chem. Lett. 2008, 18, 1011–1016. [Google Scholar]
- Clarke, B.; Demont, E.; Dingwall, C.; Dunsdon, R.; Faller, A.; Hawkins, J.; Hussain, I.; MacPherson, D.; Maile, G.; Matico, R.; et al. BACE-1 inhibitors part 2: Identification of hydroxy ethylamines (HEAs) with reduced peptidic character. Bioorg. Med. Chem. Lett. 2008, 18, 1017–1021. [Google Scholar]
- Ghosh, A.K.; Brindisi, M.; Tang, J. Developing β-secretase inhibitors for treatment of Alzheimer’s disease. J. Neurochem. 2012, 120, 71–83. [Google Scholar]
- Venugopal, C.; Demos, C.M.; Rao, K.S.; Pappolla, M.A.; Sambamurti, K. β-Secretase: Structure function and evolution. CNS Neurol. Disord. Drug Targets 2008, 7, 278–294. [Google Scholar]
- Evin, G.; Barakat, A.; Masters, C.L. BACE: Therapeutic target and potential biomarker for Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2010, 42, 1923–1926. [Google Scholar]
- Kim, D.Y.; Gersbacher, M.T.; Inquimbert, P.; Kovacs, D.M. Reduced sodium channel Nav11 levels in BACE1-null mice. J. Biol. Chem. 2011, 286, 8106–8116. [Google Scholar]
- Panza, F.; Solfrizzi, V.; Frisardi, V.; Capurso, C.; D’Introno, A.; Colacicco, A.M.; Vendemiale, G.; Capurso, A.; Imbimbo, B.P. Disease-modifying approach to the treatment of Alzheimer’s disease: From α-Secretase activators to γ-secretase inhibitors and modulators. Drugs Aging 2009, 26, 537–555. [Google Scholar]
- Guo, J.H.; Cheng, H.P.; Yu, L.; Zhao, S. Natural antisense transcripts of Alzheimer’s disease associated genes. DNA Seq. 2006, 17, 170–173. [Google Scholar]
- Limongelli, V.; Marinelli, L.; Cosconati, S.; Braun, H.A.; Schmidt, B.; Novellino, E. Ensemble-docking approach on BACE-1: Pharmacophore perception and guidelines for drug design. ChemMedChem 2007, 2, 667–678. [Google Scholar]
- Hong, L. Structure of the protease domain of memapsin 2 (β-secretase) complexed with inhibitor. Science 2000, 290, 150–153. [Google Scholar]
- Wang, H.; Li, R.; Shen, Y. β-Secretase: Its biology as a therapeutic target in diseases. Trends Pharmacol. Sci. 2013, 34, 215–225. [Google Scholar]
- Kandalepas, P.C.; Vassar, R. Identification and biology of β-secretase. J. Neurochem. 2012, 120, 55–61. [Google Scholar]
- Patel, S.; Vuillard, L.; Cleasby, A.; Murray, C.W.; Yon, J. Apo and inhibitor complex structures of BACE (β-secretase). J. Mol. Biol. 2004, 343, 407–416. [Google Scholar]
- Xu, Y.; Li, M.J.; Greenblatt, H.; Chen, W.; Paz, A.; Dym, O.; Peleg, Y.; Chen, T.; Shen, X.; He, J.; et al. Flexibility of the flap in the active site of BACE1 as revealed by crystal structures and molecular dynamics simulations. Acta Crystallogr. 2012, 68, 13–25. [Google Scholar]
- Chakraborty, S.; Kumar, S.; Basu, S. Conformational transition in the substrate binding domain of β-secretase exploited by nma and its implication in inhibitor recognition: BACE1-myricetin a case study. Neurochem. Int. 2011, 58, 914–923. [Google Scholar]
- Hunt, C.E.; Turner, A.J. Cell biology regulation and inhibition of β-secretase (BACE-1). FEBS J. 2009, 276, 1845–1859. [Google Scholar]
- Guo, T.; Hobbs, D.W. Development of BACE1 inhibitors for Alzheimer’s disease. Curr. Med. Chem. 2006, 13, 1811–1829. [Google Scholar]
- Cole, S.L.; Vassar, R. The Alzheimer’s disease β-secretase enzyme BACE-1. Mol. Neurodegener. 2007, 2, 22:1–22:25. [Google Scholar]
- Turner, R.T., III; Hong, L.; Koelsch, G.; Ghosh, A.K.; Tang, J. Structural locations and functional roles of new subsites S5 S6 and S7 in memapsin 2 (β-secretase). Biochemistry 2005, 44, 105–112. [Google Scholar]
- Bajda, M.; Guzior, N.; Ignasik, M.; Malawska, B. Multi-target-directed ligands in Alzheimer’s disease treatment. Curr. Med.Chem. 2011, 18, 4949–4975. [Google Scholar]
- Jeppsson, F.; Eketjall, S.; Janson, J.; Karlstrom, S.; Gustavsson, S.; Olsson, L.L.; Radesater, A.C.; Ploeger, B.; Cebers, G.; Kolmodin, K.; et al. Discovery of AZD3839 a potent and selective BACE1 inhibitor clinical candidate for the treatment of alzheimer disease. J. Biol. Chem. 2012, 287, 41245–41257. [Google Scholar]
- Silvestri, R. Boom in the development of non-peptidic β-secretase (BACE1) inhibitors for the treatment of Alzheimer’s disease. Med. Res. Rev. 2009, 29, 295–338. [Google Scholar]
- Protein Data Bank. Available online: http://www.rcsb.org (accessed on 1 March 2013).
- Gold 5.1; The Cambridge Crystallographic Data Centre: Cambridge, UK, 2011.
- Congreve, M.; Aharony, D.; Albert, J.; Callaghan, O.; Campbell, J.; Carr, R.A.; Chessari, G.; Cowan, S.; Edwards, P.D.; Frederickson, M.; et al. Application of fragment screening by X-ray crystallography to the discovery of aminopyridines as inhibitors of β-secretase. J. Med. Chem. 2007, 50, 1124–1132. [Google Scholar]
- Piazzi, L.; Cavalli, A.; Colizzi, F.; Belluti, F.; Bartolini, M.; Mancini, F.; Recanatini, M.; Andrisano, V.; Rampa, A. Multi-target-directed coumarin derivatives: hAChE and BACE1 inhibitors as potential anti-Alzheimer compounds. Bioorg. Med. Chem. Lett. 2008, 18, 423–426. [Google Scholar]
- Shimmyo, Y.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. Flavonols and flavones as BACE-1 inhibitors: Structure-activity relationship in cell-free cell-based and in silico studies reveal novel pharmacophore features. Biochim. Biophys. Acta 2008, 1780, 819–825. [Google Scholar]
- Jung, H.A.; Lee, E.J.; Kim, J.S.; Kang, S.S.; Lee, J.H.; Min, B.S.; Choi, J.S. Cholinesterase and BACE1 inhibitory diterpenoids from Aralia cordata. Arch. Pharm. Res. 2009, 32, 1399–1408. [Google Scholar]
- Asso, V.; Ghilardi, E.; Bertini, S.; Digiacomo, M.; Granchi, C.; Minutolo, F.; Rapposelli, S.; Bortolato, A.; Moro, S.; Macchia, M. α-Naphthylaminopropan-2-ol derivatives as BACE1 inhibitors. ChemMedChem 2008, 3, 1530–1534. [Google Scholar]
- Chirapu, S.R.; Pachaiyappan, B.; Nural, H.F.; Cheng, X.; Yuan, H.; Lankin, D.C.; Abdul-Hay, S.O.; Thatcher, G.R.; Shen, Y.; Kozikowski, A.P.; et al. Molecular modeling synthesis and activity studies of novel biaryl and fused-ring BACE1 inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 264–274. [Google Scholar]
- PyMOL 0.99rc6; DeLano Scientific LLC: Palo Alto, CA, USA, 2006.
- Sybyl X 1.2; Tripos: St. Louis, MO, USA, 2010.
- MOE 2009.10; Chemical Computing Group: Montreal, QC, Canada, 2009.
- Online Demo—Fast 3D Structure Generation with CORINA. Available online: http://www.molecular-networks.com/online_demos/corina_demo (accessed on 1 March 2013).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Ligand * | Monomer (chain) | Binding site radius (Å) | Scoring function | Score value | RMSD | Water |
|---|---|---|---|---|---|---|---|
| 1FKN | OM99-2 | A | 12 | ChemPLP | 101.86 | 3.94 | no |
| 1M4H | OM00-3 | A | 8 | GoldScore | 113.00 | 4.84 | no |
| 1TQF | 32P | A | 10 | GoldScore | 92.02 | 3.60 | no |
| 2B8L | 5HA | A | 12 | GoldScore | 133.76 | 0.65 | no |
| 2F3F | BDF488 | C | 12 | ASP | 54.23 | 0.31 | no |
| 2IQG | 2FI | A | 10 | ASP | 54.87 | 1.13 | no |
| 2OAH | QIN | A | 8 | ChemPLP | 129.36 | 1.22 | no |
| 2OHQ | 7IP | A | 12 | GoldScore | 72.57 | 0.20 | no |
| 2QP8 | SCH734723 | A | 8 | GoldScore | 97.51 | 1.43 | no |
| 2QU3 | 462 | A | 12 | GoldScore | 67.69 | 1.01 | no |
| 2QZL | IXS | A | 10 | GoldScore | 116.81 | 1.15 | no |
| 2VNM | CM8 | A | 12 | GoldScore | 113.13 | 0.81 | no |
| 2VNN | CM7 | A | 8 | ChemScore | 52.00 | 0.47 | yes |
| 2ZJM | F1M | A | 8 | GoldScore | 96.86 | 0.64 | no |
| 3CIC | SCH709583 | A | 10 | GoldScore | 114.20 | 1.27 | no |
| 3L5E | SCH736062 | A | 8 | GoldScore | 77.80 | 1.00 | yes |
| 3OHH | BMS681889 | A | 10 | GoldScore | 99.81 | 1.99 | no |
| 4ACX | S82 | A | 10 | ChemPLP | 99.61 | 0.51 | yes |
| 4B1D | 6TG | A | 10 | ChemScore | 32.00 | 0.27 | no |
| 4D8C | NVP-BXD552 | C | 10 | GoldScore | 102.69 | 0.95 | no |
| Entry No. | GoldScore | pIC50 pred. | pIC50 exp. | Error |
|---|---|---|---|---|
| 1 | 76.74 | 6.48 | 7.20 | −0.72 |
| 2 | 74.07 | 6.27 | 7.00 | −0.73 |
| 3 | 74.15 | 6.28 | 6.95 | −0.68 |
| 4 | 72.20 | 6.12 | 6.92 | −0.80 |
| 5 | 72.49 | 6.14 | 6.82 | −0.68 |
| 6 | 70.94 | 6.02 | 6.82 | −0.80 |
| 7 | 67.61 | 5.76 | 6.62 | −0.86 |
| 8 | 76.82 | 6.49 | 6.55 | −0.06 |
| 9 | 72.70 | 6.16 | 6.28 | −0.12 |
| 10 | 77.07 | 6.51 | 6.13 | 0.38 |
| 11 | 78.03 | 6.58 | 6.05 | 0.53 |
| 12 | 70.10 | 5.96 | 5.96 | 0.00 |
| 13 | 78.88 | 6.65 | 5.90 | 0.75 |
| 14 | 64.45 | 5.51 | 5.78 | −0.27 |
| 15 | 54.23 | 4.70 | 5.55 | −0.85 |
| 16 | 65.78 | 5.61 | 5.30 | 0.31 |
| 17 | 59.32 | 5.10 | 5.05 | 0.06 |
| 18 | 66.03 | 5.63 | 5.00 | 0.63 |
| 19 | 61.99 | 5.31 | 5.00 | 0.31 |
| 20 | 48.90 | 4.28 | 4.73 | −0.45 |
| 21 | 46.12 | 4.06 | 4.63 | −0.57 |
| 22 | 50.47 | 4.40 | 4.62 | −0.22 |
| 23 | 50.96 | 4.44 | 4.41 | 0.03 |
| 24 | 43.23 | 3.83 | 4.38 | −0.55 |
| 25 | 46.15 | 4.06 | 4.21 | −0.15 |
| 26 | 53.70 | 4.66 | 4.03 | 0.63 |
| 27 | 45.37 | 4.00 | 4.00 | 0.00 |
| 28 | 43.96 | 3.89 | 4.00 | −0.11 |
| 29 | 47.96 | 4.20 | 3.51 | 0.70 |
| 30 | 40.10 | 3.58 | 2.70 | 0.88 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bajda, M.; Jończyk, J.; Malawska, B.; Filipek, S. Application of Computational Methods for the Design of BACE-1 Inhibitors: Validation of in Silico Modelling. Int. J. Mol. Sci. 2014, 15, 5128-5139. https://doi.org/10.3390/ijms15035128
Bajda M, Jończyk J, Malawska B, Filipek S. Application of Computational Methods for the Design of BACE-1 Inhibitors: Validation of in Silico Modelling. International Journal of Molecular Sciences. 2014; 15(3):5128-5139. https://doi.org/10.3390/ijms15035128
Chicago/Turabian StyleBajda, Marek, Jakub Jończyk, Barbara Malawska, and Sławomir Filipek. 2014. "Application of Computational Methods for the Design of BACE-1 Inhibitors: Validation of in Silico Modelling" International Journal of Molecular Sciences 15, no. 3: 5128-5139. https://doi.org/10.3390/ijms15035128