iNR-Drug: Predicting the Interaction of Drugs with Nuclear Receptors in Cellular Networking

Abstract
:1. Introduction
2. Results and Discussion
2.1. Benchmark Dataset
 can be formulated as+ is the positive subset that consists of the interactive drug-NR pairs only, while
− the negative subset that contains of the non-interactive drug-NR pairs only, and the symbol ∪ represents the union in the set theory. The so-called “interactive” pair here means the pair whose two counterparts are interacting with each other in the drug-target networks as defined in the KEGG database [71]; while the “non-interactive” pair means that its two counterparts are not interacting with each other in the drug-target networks. The positive dataset
+ contains 86 drug-NR pairs, which were taken from He et al. [59]. The negative dataset
− contains 172 non-interactive drug-NR pairs, which were derived according to the following procedures: (a) separating each of the pairs in
+ into single drug and NR; (b) re-coupling each of the single drugs with each of the single NRs into pairs in a way that none of them occurred in
+ ; (c) randomly picking the pairs thus formed until reaching the number two times as many as the pairs in
+. The 86 interactive drug-NR pairs and 172 non-interactive drug-NR pairs are given in Supplementary Information S1, from which we can see that the 86 + 172 = 258 pairs in the current benchmark dataset
are actually formed by 25 different NRs and 53 different compounds.
can be formulated as+ is the positive subset that consists of the interactive drug-NR pairs only, while
− the negative subset that contains of the non-interactive drug-NR pairs only, and the symbol ∪ represents the union in the set theory. The so-called “interactive” pair here means the pair whose two counterparts are interacting with each other in the drug-target networks as defined in the KEGG database [71]; while the “non-interactive” pair means that its two counterparts are not interacting with each other in the drug-target networks. The positive dataset
+ contains 86 drug-NR pairs, which were taken from He et al. [59]. The negative dataset
− contains 172 non-interactive drug-NR pairs, which were derived according to the following procedures: (a) separating each of the pairs in
+ into single drug and NR; (b) re-coupling each of the single drugs with each of the single NRs into pairs in a way that none of them occurred in
+ ; (c) randomly picking the pairs thus formed until reaching the number two times as many as the pairs in
+. The 86 interactive drug-NR pairs and 172 non-interactive drug-NR pairs are given in Supplementary Information S1, from which we can see that the 86 + 172 = 258 pairs in the current benchmark dataset
are actually formed by 25 different NRs and 53 different compounds.2.2. Sample Representation
2.2.1. Use 2D Molecular Fingerprints to Represent Drugs
are, respectively, given in Supplementary Information S2.2.2.2. Use Pseudo Amino Acid Composition to Represent the Nuclear Receptors
are listed in Supplementary Information S3. Suppose the sequence of a nuclear receptor protein P with L residues is generally expressed by2.2.3. Formulate the Pair of Drugs with Nuclear Receptor
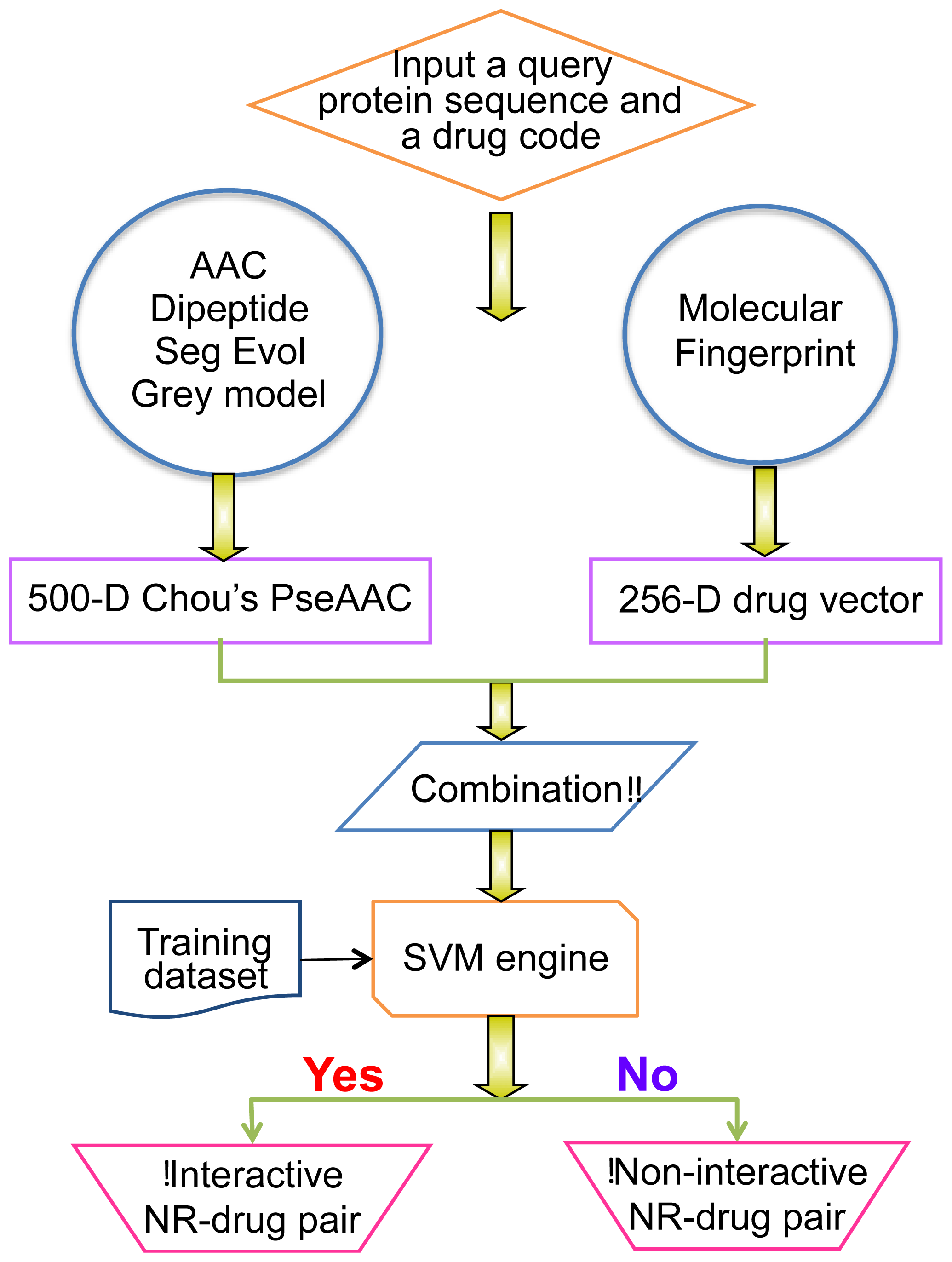
2.2.4. Operation Engine or Algorithm
3. Experimental Section
3.1. Metrics for Measuring Prediction Quality
+ and none of the non-interactive NR-drug pairs in
− was incorrectly predicted, we have the overall accuracy Acc = 1; while
and
meaning that all the interactive NR-drug pairs in the dataset
+ and all the non-interactive NR-drug pairs in
− were mispredicted, we have the overall accuracy Acc = 0. The Matthews correlation coefficient MCC is usually used for measuring the quality of binary (two-class) classifications. When
meaning that none of the interactive NR-drug pairs in the dataset
+ and none of the non-interactive NR-drug pairs in
− was mispredicted, we have MCC = 1; when
and
we have MCC = 0 meaning no better than random prediction; when
and
we have MCC = 0 meaning total disagreement between prediction and observation. As we can see from the above discussion, it is much more intuitive and easier to understand when using Equation (23) to examine a predictor for its four metrics, particularly for its Mathew’s correlation coefficient. It is instructive to point out that the metrics as defined in Equation (23) are valid for single label systems; for multi-label systems, a set of more complicated metrics should be used as given in [162].3.2. Jackknife Test Approach
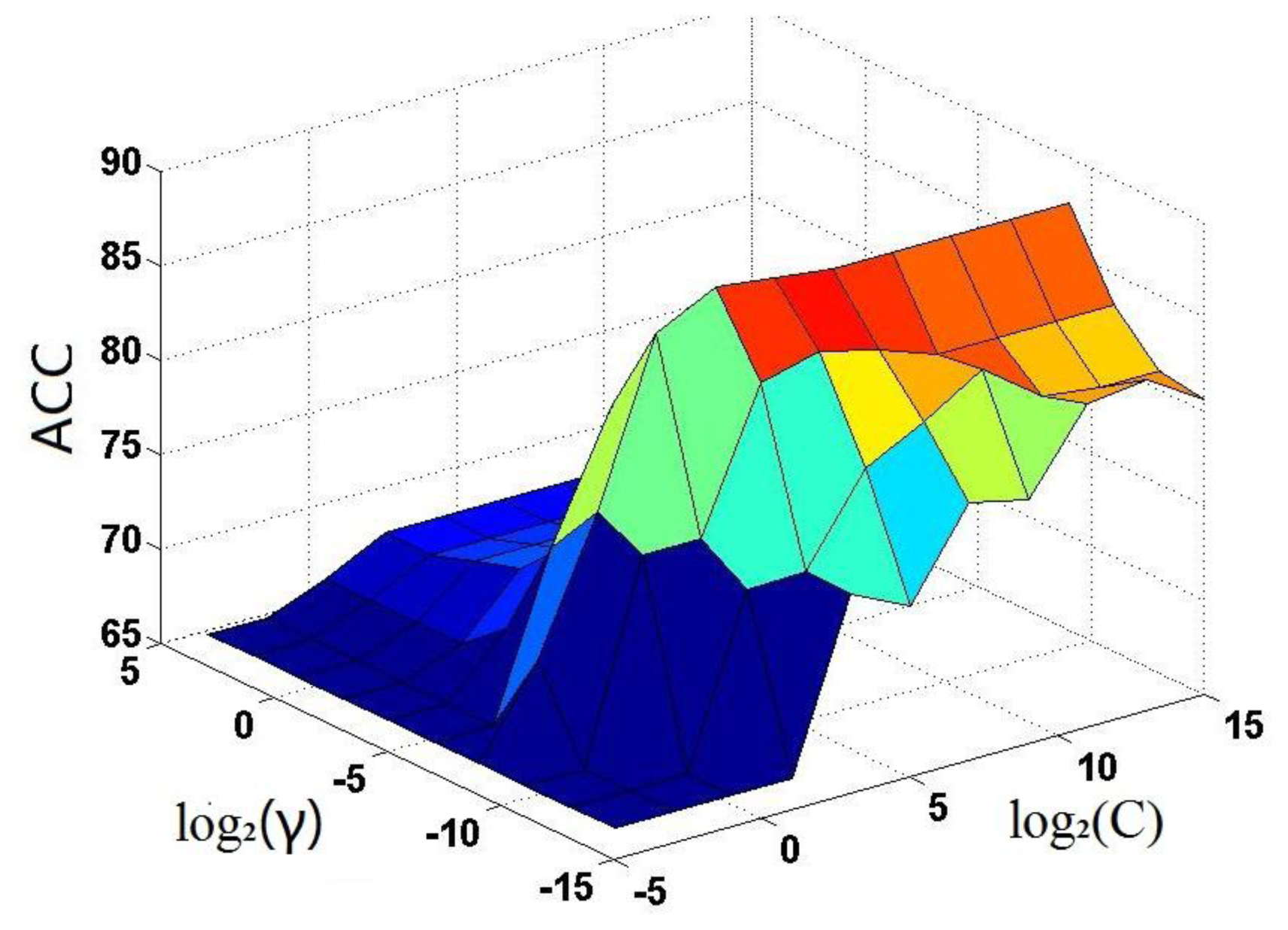
. The results thus obtained are shown in Figure 3, from which it can be seen that the iNR-Drug predictor reaches its optimal status when C = 23 and γ = 2−9. The corresponding rates for the four metrics (cf. Equation (23)) are given in Table 1, where for facilitating comparison, the overall accuracy Acc reported by He et al. [59] on the same benchmark dataset is also given although no results were reported by them for Sn, Sp and MCC. It can be observed from the table that the overall accuracy obtained by iNR-Drug is remarkably higher that of He et al. [59], and that the rates achieved by iNR-Drug for the other three metrics are also quite higher. These facts indicate that the current predictor not only can yield higher overall prediction accuracy but also is quite stable with low false prediction rates.3.3. Independent Dataset Test
via the jackknife test as reported in Table 1.4. Conclusions
Supplementary Information
ijms-15-04915-s001.pdfAcknowledgments
Conflicts of Interest
References
- Altucci, L.; Gronemeyer, H. Nuclear receptors in cell life and death. Trends Endocrinol. Metab. 2001, 12, 460–468. [Google Scholar]
- Bates, M.K.; Kerr, R.M. Nuclear Receptors; Nova Science: Hauppauge, NY, USA, 2011. [Google Scholar]
- Bunce, C.M.; Campbell, M.J. Nuclear Receptors: Current Concepts and Future Challenges; Springer: Dordrecht, The Netherlands; New York, NY, USA, 2010. [Google Scholar]
- Robinson-Rechavi, M.; Garcia, H.E.; Laudet, V. The nuclear receptor superfamily. J. Cell Sci. 2003, 116, 585–586. [Google Scholar]
- Kastner, P. Non-steroid nuclear receptors: What are genetic studies telling us their role in renal life? Cell 1995, 83, 859–869. [Google Scholar]
- Chen, T. Nuclear receptor drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 418–426. [Google Scholar]
- Tirona, R.G.; Kim, R.B. Nuclear receptors and drug disposition gene regulation. J. Pharm. Sci. 2005, 94, 1169–1186. [Google Scholar]
- Lin, W.Z.; Xiao, X.; Chou, K.C. GPCR-GIA: A web-server for identifying G-protein coupled receptors and their families with grey incidence analysis. Protein Eng. Des. Sel. 2009, 22, 699–705. [Google Scholar]
- Chou, K.C.; Elrod, D.W. Bioinformatical analysis of G-protein-coupled receptors. J. Proteome Res. 2002, 1, 429–433. [Google Scholar]
- Xiao, X.; Min, J.L.; Wang, P.; Chou, K.C. iGPCR-Drug: A web server for predicting interaction between GPCRs and drugs in cellular networking. PLoS One 2013, 8, e72234. [Google Scholar]
- Xiao, X.; Wang, P.; Chou, K.C. GPCR-CA: A cellular automaton image approach for predicting G-protein-coupled receptor functional classes. J. Comput. Chem. 2009, 30, 1414–1423. [Google Scholar]
- Xiao, X.; Wang, P.; Chou, K.C. GPCR-2L: Predicting G protein-coupled receptors and their types by hybridizing two different modes of pseudo amino acid compositions. Mol. Biosyst. 2011, 7, 911–919. [Google Scholar]
- Gu, Q.; Ding, Y.S.; Zhang, T.L. Prediction of G-protein-coupled receptor classes in low homology using Chou’s pseudo amino acid composition with approximate entropy and hydrophobicity patterns. Protein Pept. Lett. 2010, 17, 559–567. [Google Scholar]
- Qiu, J.D.; Huang, J.H.; Liang, R.P.; Lu, X.Q. Prediction of G-protein-coupled receptor classes based on the concept of Chou’s pseudo amino acid composition: An approach from discrete wavelet transform. Anal. Biochem. 2009, 390, 68–73. [Google Scholar]
- Xie, H.L.; Fu, L.; Nie, X.D. Using ensemble SVM to identify human GPCRs N-linked glycosylation sites based on the general form of Chou’s PseAAC. Protein Eng. Des. Sel. 2013, 26, 735–742. [Google Scholar]
- Zia Ur, R.; Khan, A. Identifying GPCRs and their types with Chou’s pseudo amino acid composition: An approach from multi-scale energy representation and position specific scoring matrix. Protein Pept. Lett. 2012, 19, 890–903. [Google Scholar]
- Chou, K.C. Prediction of G-protein-coupled receptor classes. J. Proteome Res. 2005, 4, 1413–1418. [Google Scholar]
- Xiao, X.; Min, J.L.; Wang, P.; Chou, K.C. iCDI-PseFpt: Identify the channel-drug interaction in cellular networking with PseAAC and molecular fingerprints. J. Theor. Biol. 2013, 337C, 71–79. [Google Scholar]
- Chou, K.C. Insights from modelling three-dimensional structures of the human potassium and sodium channels. J. Proteome Res. 2004, 3, 856–861. [Google Scholar]
- Pielak, R.M.; Chou, J.J. Influenza M2 proton channels. Biochim. Biophys. Acta 2011, 1808, 522–529. [Google Scholar]
- Chou, K.C.; Watenpaugh, K.D.; Heinrikson, R.L. A Model of the complex between cyclin-dependent kinase 5 (Cdk5) and the activation domain of neuronal Cdk5 activator. Biochem. Biophys. Res. Commun. 1999, 259, 420–428. [Google Scholar]
- Schnell, J.R.; Zhou, G.P.; Zweckstetter, M.; Rigby, A.C.; Chou, J.J. Rapid and accurate structure determination of coiled-coil domains using NMR dipolar couplings: Application to cGMP-dependent protein kinase Ialpha. Protein Sci. 2005, 14, 2421–2428. [Google Scholar]
- Zhou, G.P.; Surks, H.K.; Schnell, J.R.; Chou, J.J.; Mendelsohn, M.E.; Rigby, A.C. The three-dimensional structure of the cGMP-dependent protein kinase I-α leucine zipper domain and its interaction with the myosin binding subunit. Blood 2004, 104, 963a. [Google Scholar]
- Zweckstetter, M.; Schnell, J.R.; Chou, J.J. Determination of the packing mode of the coiled-coil domain of cGMP-dependent protein kinase Ialpha in solution using charge-predicted dipolar couplings. J. Am. Chem. Soc. 2005, 127, 11918–11919. [Google Scholar]
- Knowles, J.; Gromo, G. A guide to drug discovery: Target selection in drug discovery. Nat. Rev. Drug Discov. 2003, 2, 63–69. [Google Scholar]
- Lindsay, M.A. Target discovery. Nat. Rev. Drug Discov. 2003, 2, 831–838. [Google Scholar]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar]
- Chou, K.C.; Wei, D.Q.; Zhong, W.Z. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS (Erratum: ibid2003 Vol 310 675). Biochem. Biophys. Res. Commun. 2003, 308, 148–151. [Google Scholar]
- Zhou, G.P.; Troy, F.A. NMR studies on how the binding complex of polyisoprenol recognition sequence peptides and polyisoprenols can modulate membrane structure. Curr. Protein Pept. Sci. 2005, 6, 399–411. [Google Scholar]
- Chou, K.C.; Wei, D.Q.; Du, Q.S.; Sirois, S.; Zhong, W.Z. Review: Progress in computational approach to drug development against SARS. Curr. Med. Chem. 2006, 13, 3263–3270. [Google Scholar]
- Du, Q.S.; Wang, S.; Wei, D.Q.; Sirois, S.; Chou, K.C. Molecular modelling and chemical modification for finding peptide inhibitor against SARS CoV Mpro. Anal. Biochem. 2005, 337, 262–270. [Google Scholar]
- Huang, R.B.; Du, Q.S.; Wang, C.H.; Chou, K.C. An in-depth analysis of the biological functional studies based on the NMR M2 channel structure of influenza A virus. Biochem. Biophys. Res. Commun. 2008, 377, 1243–1247. [Google Scholar]
- Du, Q.S.; Huang, R.B.; Wang, C.H.; Li, X.M.; Chou, K.C. Energetic analysis of the two controversial drug binding sites of the M2 proton channel in influenza A virus. J. Theor. Biol. 2009, 259, 159–164. [Google Scholar]
- Wei, H.; Wang, C.H.; Du, Q.S.; Meng, J.; Chou, K.C. Investigation into adamantane-based M2 inhibitors with FB-QSAR. Med. Chem. 2009, 5, 305–317. [Google Scholar]
- Du, Q.S.; Huang, R.B.; Wang, S.Q.; Chou, K.C. Designing inhibitors of M2 proton channel against H1N1 swine influenza virus. PLoS One 2010, 5, e9388. [Google Scholar]
- Wang, S.Q.; Du, Q.S.; Huang, R.B.; Zhang, D.W.; Chou, K.C. Insights from investigating the interaction of oseltamivir (Tamiflu) with neuraminidase of the 2009 H1N1 swine flu virus. Biochem. Biophys. Res. Commun. 2009, 386, 432–436. [Google Scholar]
- Chou, K.C. Review: Structural bioinformatics and its impact to biomedical science. Curr. Med. Chem. 2004, 11, 2105–2134. [Google Scholar]
- Cai, L.; Wang, Y.; Wang, J.F.; Chou, K.C. Identification of proteins interacting with human SP110 during the process of viral infections. Med. Chem. 2011, 7, 121–126. [Google Scholar]
- Liao, Q.H.; Gao, Q.Z.; Wei, J.; Chou, K.C. Docking and molecular dynamics study on the inhibitory activity of novel inhibitors on epidermal growth factor receptor (EGFR). Med. Chem. 2011, 7, 24–31. [Google Scholar]
- Li, X.B.; Wang, S.Q.; Xu, W.R.; Wang, R.L.; Chou, K.C. Novel inhibitor design for hemagglutinin against H1N1 influenza virus by core hopping method. PLoS One 2011, 6, e28111. [Google Scholar]
- Ma, Y.; Wang, S.Q.; Xu, W.R.; Wang, R.L.; Chou, K.C. Design novel dual agonists for treating type-2 diabetes by targeting peroxisome proliferator-activated receptors with core hopping approach. PLoS One 2012, 7, e38546. [Google Scholar]
- Wang, J.F.; Chou, K.C. Insights from modeling the 3D structure of New Delhi metallo-beta-lactamase and its binding interactions with antibiotic drugs. PLoS One 2011, 6, e18414. [Google Scholar]
- Wang, J.F.; Chou, K.C. Insights into the mutation-induced HHH syndrome from modeling human mitochondrial ornithine transporter-1. PLoS One 2012, 7, e31048. [Google Scholar]
- Berardi, M.J.; Shih, W.M.; Harrison, S.C.; Chou, J.J. Mitochondrial uncoupling protein 2 structure determined by NMR molecular fragment searching. Nature 2011, 476, 109–113. [Google Scholar]
- Schnell, J.R.; Chou, J.J. Structure and mechanism of the M2 proton channel of influenza A virus. Nature 2008, 451, 591–595. [Google Scholar]
- OuYang, B.; Xie, S.; Berardi, M.J.; Zhao, X.M.; Dev, J.; Yu, W.; Sun, B.; Chou, J.J. Unusual architecture of the p7 channel from hepatitis C virus. Nature 2013, 498, 521–525. [Google Scholar]
- Oxenoid, K.; Chou, J.J. The structure of phospholamban pentamer reveals a channel-like architecture in membranes. Proc. Natl. Acad. Sci. USA 2005, 102, 10870–10875. [Google Scholar]
- Call, M.E.; Wucherpfennig, K.W.; Chou, J.J. The structural basis for intramembrane assembly of an activating immunoreceptor complex. Nat. Immunol. 2010, 11, 1023–1029. [Google Scholar]
- Pielak, R.M.; Chou, J.J. Solution NMR structure of the V27A drug resistant mutant of influenza A M2 channel. Biochem. Biophys. Res. Commun. 2010, 401, 58–63. [Google Scholar]
- Pielak, R.M.; Jason, R.; Schnell, J.R.; Chou, J.J. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc. Natl. Acad. Sci. USA 2009, 106, 7379–7384. [Google Scholar]
- Wang, J.; Pielak, R.M.; McClintock, M.A.; Chou, J.J. Solution structure and functional analysis of the influenza B proton channel. Nat. Struct. Mol. Biol. 2009, 16, 1267–1271. [Google Scholar]
- Chou, K.C.; Jones, D.; Heinrikson, R.L. Prediction of the tertiary structure and substrate binding site of caspase-8. FEBS Lett. 1997, 419, 49–54. [Google Scholar]
- Chou, K.C.; Tomasselli, A.G.; Heinrikson, R.L. Prediction of the tertiary structure of a caspase-9/inhibitor complex. FEBS Lett. 2000, 470, 249–256. [Google Scholar]
- Chou, K.C.; Howe, W.J. Prediction of the tertiary structure of the beta-secretase zymogen. Biochem. Biophys. Res. Commun. 2002, 292, 702–708. [Google Scholar]
- Chou, K.C. Coupling interaction between thromboxane A2 receptor and alpha-13 subunit of guanine nucleotide-binding protein. J. Proteome Res. 2005, 4, 1681–1686. [Google Scholar]
- Chou, K.C. Insights from modeling the 3D structure of DNA-CBF3b complex. J. Proteome Res. 2005, 4, 1657–1660. [Google Scholar]
- Chou, K.C. Modeling the tertiary structure of human cathepsin-E. Biochem. Biophys. Res. Commun. 2005, 331, 56–60. [Google Scholar]
- Sirois, S.; Hatzakis, G.E.; Wei, D.Q.; Du, Q.S.; Chou, K.C. Assessment of chemical libraries for their druggability. Comput. Biol. Chem. 2005, 29, 55–67. [Google Scholar]
- He, Z.; Zhang, J.; Shi, X.H.; Hu, L.L.; Kong, X.; Cai, Y.D.; Chou, K.C. Predicting drug-target interaction networks based on functional groups and biological features. PLoS One 2010, 5, e9603. [Google Scholar]
- Chou, K.C. Some remarks on protein attribute prediction and pseudo amino acid composition (50th Anniversary Year Review). J. Theor. Biol. 2011, 273, 236–247. [Google Scholar]
- Qiu, W.R.; Xiao, X.; Chou, K.C. iRSpot-TNCPseAAC: Identify recombination spots with trinucleotide composition and pseudo amino acid components. Int. J. Mol. Sci. 2014, 15, 1746–1766. [Google Scholar]
- Chen, W.; Feng, P.M.; Lin, H.; Chou, K.C. iRSpot-PseDNC: Identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res. 2013, 41, e69. [Google Scholar]
- Feng, P.M.; Chen, W.; Lin, H.; Chou, K.C. iHSP-PseRAAAC: Identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal. Biochem. 2013, 442, 118–125. [Google Scholar]
- Liu, B.; Zhang, D.; Xu, R.; Xu, J.; Wang, X.; Chen, Q.; Dong, Q.; Chou, K.C. Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection. Bioinformatics 2013. [Google Scholar] [CrossRef]
- Chen, W.; Lin, H.; Feng, P.M.; Ding, C.; Zuo, Y.C.; Chou, K.C. iNuc-PhysChem: A sequence-based predictor for identifying nucleosomes via physicochemical properties. PLoS One 2012, 7, e47843. [Google Scholar]
- Xu, Y.; Ding, J.; Wu, L.Y.; Chou, K.C. iSNO-PseAAC: Predict cysteine S-nitrosylation sites in proteins by incorporating position specific amino acid propensity into pseudo amino acid composition. PLoS One 2013, 8, e55844. [Google Scholar]
- Xu, Y.; Shao, X.J.; Wu, L.Y.; Deng, N.Y.; Chou, K.C. iSNO-AAPair: Incorporating amino acid pairwise coupling into PseAAC for predicting cysteine S-nitrosylation sites in proteins. Peer J. 2013, 1, e171. [Google Scholar]
- Min, J.L.; Xiao, X.; Chou, K.C. iEzy-Drug: A web server for identifying the interaction between enzymes and drugs in cellular networking. BioMed Res. Int. 2013, 2013, 701317. [Google Scholar]
- Xiao, X.; Wang, P.; Lin, W.Z.; Jia, J.H.; Chou, K.C. iAMP-2L: A two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem. 2013, 436, 168–177. [Google Scholar]
- Guo, S.H.; Deng, E.Z.; Xu, L.Q.; Ding, H.; Lin, H.; Chen, W.; Chou, K.C. iNuc-PseKNC: A sequence-based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition. Bioinformatics 2014. [Google Scholar] [CrossRef]
- Kotera, M.; Hirakawa, M.; Tokimatsu, T.; Goto, S.; Kanehisa, M. The KEGG databases and tools facilitating omics analysis: Latest developments involving human diseases and pharmaceuticals. Methods Mol. Biol. 2012, 802, 19–39. [Google Scholar]
- Chou, K.C.; Shen, H.B. Review: Recent progresses in protein subcellular location prediction. Anal. Biochem. 2007, 370, 1–16. [Google Scholar]
- Zhou, G.P. An intriguing controversy over protein structural class prediction. J. Protein Chem. 1998, 17, 729–738. [Google Scholar]
- Zhou, G.P.; Assa-Munt, N. Some insights into protein structural class prediction. Proteins: Struct. Funct. Genet. 2001, 44, 57–59. [Google Scholar]
- Chou, K.C.; Elrod, D.W. Prediction of enzyme family classes. J. Proteome Res. 2003, 2, 183–190. [Google Scholar]
- Wang, M.; Yang, J.; Xu, Z.J.; Chou, K.C. SLLE for predicting membrane protein types. J. Theor. Biol. 2005, 232, 7–15. [Google Scholar]
- Xiao, X.; Wang, P.; Chou, K.C. Predicting protein structural classes with pseudo amino acid composition: An approach using geometric moments of cellular automaton image. J. Theor. Biol. 2008, 254, 691–696. [Google Scholar]
- Chou, K.C. A novel approach to predicting protein structural classes in a (20–1)-D amino acid composition space. Proteins: Struct. Funct. Genet. 1995, 21, 319–344. [Google Scholar]
- Zhou, G.P.; Doctor, K. Subcellular location prediction of apoptosis proteins. Proteins: Struct. Funct. Genet. 2003, 50, 44–48. [Google Scholar]
- Feng, K.Y.; Cai, Y.D.; Chou, K.C. Boosting classifier for predicting protein domain structural class. Biochem. Biophys. Res. Commun. 2005, 334, 213–217. [Google Scholar]
- Cai, Y.D.; Chou, K.C. Artificial neural network for predicting alpha-turn types. Anal. Biochem. 1999, 268, 407–409. [Google Scholar]
- Thompson, T.B.; Chou, K.C.; Zheng, C. Neural network prediction of the HIV-1 protease cleavage sites. J. Theor. Biol. 1995, 177, 369–379. [Google Scholar]
- Xiao, X.; Wang, P.; Chou, K.C. iNR-PhysChem: A sequence-based predictor for identifying nuclear receptors and their subfamilies via physical-chemical property matrix. PLoS One 2012, 7, e30869. [Google Scholar]
- Lin, W.Z.; Fang, J.A.; Xiao, X.; Chou, K.C. iDNA-Prot: Identification of DNA binding proteins using random forest with grey model. PLoS One 2011, 6, e24756. [Google Scholar]
- Kandaswamy, K.K.; Chou, K.C.; Martinetz, T.; Moller, S.; Suganthan, P.N.; Sridharan, S.; Pugalenthi, G. AFP-Pred: A random forest approach for predicting antifreeze proteins from sequence-derived properties. J. Theor. Biol. 2011, 270, 56–62. [Google Scholar]
- Cai, Y.D.; Chou, K.C. Predicting subcellular localization of proteins in a hybridization space. Bioinformatics 2004, 20, 1151–1156. [Google Scholar]
- Chou, K.C.; Cai, Y.D. Prediction of protease types in a hybridization space. Biochem. Biophys. Res. Commun. 2006, 339, 1015–1020. [Google Scholar]
- Chou, K.C.; Shen, H.B. Predicting eukaryotic protein subcellular location by fusing optimized evidence-theoretic K-nearest neighbor classifiers. J. Proteome Res. 2006, 5, 1888–1897. [Google Scholar]
- Chou, K.C.; Shen, H.B. Hum-PLoc: A novel ensemble classifier for predicting human protein subcellular localization. Biochem. Biophys. Res. Commun. 2006, 347, 150–157. [Google Scholar]
- Chou, K.C.; Shen, H.B. Large-scale predictions of Gram-negative bacterial protein subcellular locations. J. Proteome Res. 2006, 5, 3420–3428. [Google Scholar]
- Chou, K.C.; Shen, H.B. Euk-mPLoc: A fusion classifier for large-scale eukaryotic protein subcellular location prediction by incorporating multiple sites. J. Proteome Res. 2007, 6, 1728–1734. [Google Scholar]
- Chou, K.C.; Shen, H.B. Signal-CF: A subsite-coupled and window-fusing approach for predicting signal peptides. Biochem. Biophys. Res. Commun. 2007, 357, 633–640. [Google Scholar]
- Shen, H.B.; Chou, K.C. Using optimized evidence-theoretic K-nearest neighbor classifier and pseudo amino acid composition to predict membrane protein types. Biochem. Biophys. Res. Commun. 2005, 334, 288–292. [Google Scholar]
- Shen, H.B.; Chou, K.C. A top-down approach to enhance the power of predicting human protein subcellular localization: Hum-mPLoc 20. Anal. Biochem. 2009, 394, 269–274. [Google Scholar]
- Shen, H.B.; Yang, J.; Chou, K.C. Fuzzy KNN for predicting membrane protein types from pseudo amino acid composition. J. Theor. Biol. 2006, 240, 9–13. [Google Scholar]
- Chou, K.C. Prediction of protein cellular attributes using pseudo amino acid composition. Proteins: Struct. Funct. Genet. 2001, 43, 246–255. [Google Scholar]
- Chou, K.C. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics 2005, 21, 10–19. [Google Scholar]
- Lin, S.X.; Lapointe, J. Theoretical and experimental biology in one. J. Biomed. Sci. Eng. (JBiSE) 2013, 6, 435–442. [Google Scholar]
- Hajisharifi, Z.; Piryaiee, M.; Mohammad Beigi, M.; Behbahani, M.; Mohabatkar, H. Predicting anticancer peptides with Chou’s pseudo amino acid composition and investigating their mutagenicity via Ames test. J. Theor. Biol. 2014, 341, 34–40. [Google Scholar]
- Mei, S. Predicting plant protein subcellular multi-localization by Chou’s PseAAC formulation based multi-label homolog knowledge transfer learning. J. Theor. Biol. 2012, 310, 80–87. [Google Scholar]
- Chang, T.H.; Wu, L.C.; Lee, T.Y.; Chen, S.P.; Huang, H.D.; Horng, J.T. EuLoc: A web-server for accurately predict protein subcellular localization in eukaryotes by incorporating various features of sequence segments into the general form of Chou’s PseAAC. J. Comput.-Aided Mol. Des. 2013, 27, 91–103. [Google Scholar]
- Fan, G.L.; Li, Q.Z. Predict mycobacterial proteins subcellular locations by incorporating pseudo-average chemical shift into the general form of Chou’s pseudo amino acid composition. J. Theor. Biol. 2012, 304, 88–95. [Google Scholar]
- Huang, C.; Yuan, J. Using radial basis function on the general form of Chou’s pseudo amino acid composition and PSSM to predict subcellular locations of proteins with both single and multiple sites. Biosystems 2013, 113, 50–57. [Google Scholar]
- Lin, H.; Wang, H.; Ding, H.; Chen, Y.L.; Li, Q.Z. Prediction of subcellular localization of apoptosis protein using Chou’s pseudo amino acid composition. Acta Biotheor. 2009, 57, 321–330. [Google Scholar]
- Wan, S.; Mak, M.W.; Kung, S.Y. GOASVM: A subcellular location predictor by incorporating term-frequency gene ontology into the general form of Chou’s pseudo-amino acid composition. J. Theor. Biol. 2013, 323, 40–48. [Google Scholar]
- Huang, C.; Yuan, J.Q. Predicting protein subchloroplast locations with both single and multiple sites via three different modes of Chou’s pseudo amino acid compositions. J. Theor. Biol. 2013, 335, 205–212. [Google Scholar]
- Chen, Y.K.; Li, K.B. Predicting membrane protein types by incorporating protein topology domains signal peptides and physicochemical properties into the general form of Chou’s pseudo amino acid composition. J. Theor. Biol. 2013, 318, 1–12. [Google Scholar]
- Huang, C.; Yuan, J.Q. A multilabel model based on Chou’s pseudo-amino acid composition for identifying membrane proteins with both single and multiple functional types. J. Membr. Biol. 2013, 246, 327–334. [Google Scholar]
- Nanni, L.; Lumini, A. Genetic programming for creating Chou’s pseudo amino acid based features for submitochondria localization. Amino Acids 2008, 34, 653–660. [Google Scholar]
- Fan, G.L.; Li, Q.Z. Predicting protein submitochondria locations by combining different descriptors into the general form of Chou’s pseudo amino acid composition. Amino Acids 2012, 43, 545–555. [Google Scholar]
- Mei, S. Multi-kernel transfer learning based on Chou’s PseAAC formulation for protein submitochondria localization. J. Theor. Biol. 2012, 293, 121–130. [Google Scholar]
- Zeng, Y.H.; Guo, Y.Z.; Xiao, R.Q.; Yang, L.; Yu, L.Z.; Li, M.L. Using the augmented Chou’s pseudo amino acid composition for predicting protein submitochondria locations based on auto covariance approach. J. Theor. Biol. 2009, 259, 366–372. [Google Scholar]
- Mohabatkar, H.; Mohammad Beigi, M.; Esmaeili, A. Prediction of GABA(A) receptor proteins using the concept of Chou’s pseudo-amino acid composition and support vector machine. J. Theor. Biol. 2011, 281, 18–23. [Google Scholar]
- Zhou, X.B.; Chen, C.; Li, Z.C.; Zou, X.Y. Using Chou’s amphiphilic pseudo-amino acid composition and support vector machine for prediction of enzyme subfamily classes. J. Theor. Biol. 2007, 248, 546–551. [Google Scholar]
- Khosravian, M.; Faramarzi, F.K.; Beigi, M.M.; Behbahani, M.; Mohabatkar, H. Predicting antibacterial peptides by the concept of Chou;s pseudo-amino acid composition and machine learning methods. Protein Pept. Lett. 2013, 20, 180–186. [Google Scholar]
- Zou, D.; He, Z.; He, J.; Xia, Y. Supersecondary structure prediction using Chou’s pseudo amino acid composition. J. Comput. Chem. 2011, 32, 271–278. [Google Scholar]
- Nanni, L.; Lumini, A.; Gupta, D.; Garg, A. Identifying bacterial virulent proteins by fusing a set of classifiers based on variants of Chou’s pseudo amino acid composition and on evolutionary information. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 467–475. [Google Scholar]
- Sahu, S.S.; Panda, G. A novel feature representation method based on Chou’s pseudo amino acid composition for protein structural class prediction. Comput. Biol. Chem. 2010, 34, 320–327. [Google Scholar]
- Zhang, G.Y.; Fang, B.S. Predicting the cofactors of oxidoreductases based on amino acid composition distribution and Chou’s amphiphilic pseudo amino acid composition. J. Theor. Biol. 2008, 253, 310–315. [Google Scholar]
- Mohammad Beigi, M.; Behjati, M.; Mohabatkar, H. Prediction of metalloproteinase family based on the concept of Chou’s pseudo amino acid composition using a machine learning approach. J. Struct. Funct. Genomics 2011, 12, 191–197. [Google Scholar]
- Yu, L.; Guo, Y.; Li, Y.; Li, G.; Li, M.; Luo, J.; Xiong, W.; Qin, W. SecretP: Identifying bacterial secreted proteins by fusing new features into Chou’s pseudo-amino acid composition. J. Theor. Biol. 2010, 267, 1–6. [Google Scholar]
- Mohabatkar, H.; Beigi, M.M.; Abdolahi, K.; Mohsenzadeh, S. Prediction of allergenic proteins by means of the concept of Chou’s pseudo amino acid composition and a machine learning approach. Med. Chem. 2013, 9, 133–137. [Google Scholar]
- Zhang, S.W.; Chen, W.; Yang, F.; Pan, Q. Using Chou’s pseudo amino acid composition to predict protein quaternary structure: A sequence-segmented PseAAC approach. Amino Acids 2008, 35, 591–598. [Google Scholar]
- Sun, X.Y.; Shi, S.P.; Qiu, J.D.; Suo, S.B.; Huang, S.Y.; Liang, R.P. Identifying protein quaternary structural attributes by incorporating physicochemical properties into the general form of Chou’s PseAAC via discrete wavelet transform. Mol. BioSyst. 2012, 8, 3178–3184. [Google Scholar]
- Esmaeili, M.; Mohabatkar, H.; Mohsenzadeh, S. Using the concept of Chou’s pseudo amino acid composition for risk type prediction of human papillomaviruses. J. Theor. Biol. 2010, 263, 203–209. [Google Scholar]
- Mohabatkar, H. Prediction of cyclin proteins using Chou’s pseudo amino acid composition. Protein Pept. Lett. 2010, 17, 1207–1214. [Google Scholar]
- Hayat, M.; Khan, A. Discriminating outer membrane proteins with fuzzy K-nearest neighbor algorithms based on the general form of Chou’s PseAAC. Protein Pept. Lett. 2012, 19, 411–421. [Google Scholar]
- Georgiou, D.N.; Karakasidis, T.E.; Nieto, J.J.; Torres, A. Use of fuzzy clustering technique and matrices to classify amino acids and its impact to Chou’s pseudo amino acid composition. J. Theor. Biol. 2009, 257, 17–26. [Google Scholar]
- Liu, B.; Wang, X.; Zou, Q.; Dong, Q.; Chen, Q. Protein remote homology detection by combining Chou’s pseudo amino acid composition and profile-based protein representation. Mol. Inform. 2013, 32, 775–782. [Google Scholar]
- Li, B.Q.; Huang, T.; Liu, L.; Cai, Y.D.; Chou, K.C. Identification of colorectal cancer related genes with mRMR and shortest path in protein-protein interaction network. PLoS One 2012, 7, e33393. [Google Scholar]
- Huang, T.; Wang, J.; Cai, Y.D.; Yu, H.; Chou, K.C. Hepatitis C virus network based classification of hepatocellular cirrhosis and carcinoma. PLoS One 2012, 7, e34460. [Google Scholar]
- Jiang, Y.; Huang, T.; Lei, C.; Gao, Y.F.; Cai, Y.D.; Chou, K.C. Signal propagation in protein interaction network during colorectal cancer progression. BioMed Res. Int. 2013, 2013, 287019. [Google Scholar]
- Du, P.; Wang, X.; Xu, C.; Gao, Y. PseAAC-Builder: A cross-platform stand-alone program for generating various special Chou’s pseudo-amino acid compositions. Anal. Biochem. 2012, 425, 117–119. [Google Scholar]
- Cao, D.S.; Xu, Q.S.; Liang, Y.Z. Propy: A tool to generate various modes of Chou’s PseAAC. Bioinformatics 2013, 29, 960–962. [Google Scholar]
- Shen, H.B.; Chou, K.C. PseAAC: A flexible web-server for generating various kinds of protein pseudo amino acid composition. Anal. Biochem. 2008, 373, 386–388. [Google Scholar]
- Nakashima, H.; Nishikawa, K.; Ooi, T. The folding type of a protein is relevant to the amino acid composition. J. Biochem. 1986, 99, 153–162. [Google Scholar]
- Zhang, C.T.; Chou, K.C. An optimization approach to predicting protein structural class from amino acid composition. Protein Sci. 1992, 1, 401–408. [Google Scholar]
- Zhang, C.T.; Chou, K.C. Monte Carlo simulation studies on the prediction of protein folding types from amino acid composition. Biophys. J. 1992, 63, 1523–1529. [Google Scholar]
- Chou, K.C.; Zhang, C.T. Predicting protein folding types by distance functions that make allowances for amino acid interactions. J. Biol. Chem. 1994, 269, 22014–22020. [Google Scholar]
- Zhang, C.T.; Chou, K.C. Monte Carlo simulation studies on the prediction of protein folding types from amino acid composition II correlative effect. J. Protein Chem. 1995, 14, 251–258. [Google Scholar]
- Chou, K.C. Does the folding type of a protein depend on its amino acid composition? FEBS Lett. 1995, 363, 127–131. [Google Scholar]
- Liu, W.; Chou, K.C. Protein secondary structural content prediction. Protein Eng. 1999, 12, 1041–1050. [Google Scholar]
- Chou, K.C. The convergence-divergence duality in lectin domains of the selectin family and its implications. FEBS Lett. 1995, 363, 123–126. [Google Scholar]
- Chou, K.C.; Wu, Z.C.; Xiao, X. iLoc-Euk: A multi-label classifier for predicting the subcellular localization of singleplex and multiplex eukaryotic proteins. PLoS One 2011, 6, e18258. [Google Scholar]
- Chou, K.C.; Wu, Z.C.; Xiao, X. iLoc-Hum: Using accumulation-label scale to predict subcellular locations of human proteins with both single and multiple sites. Mol. Biosyst. 2012, 8, 629–641. [Google Scholar]
- Schaffer, A.A.; Aravind, L.; Madden, T.L.; Shavirin, S.; Spouge, J.L.; Wolf, Y.I.; Koonin, E.V.; Altschul, S.F. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 2001, 29, 2994–3005. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar]
- Schadt, E.E.; Li, C.; Ellis, B.; Wong, W.H. Feature extraction and normalization algorithms for high-density oligonucleotide gene expression array data. J. Cell Biochem. Suppl. 2001, 37, 120–125. [Google Scholar]
- Shi, J.Y.; Zhang, S.W.; Pan, Q.; Cheng, Y.M.; Xie, J. Prediction of protein subcellular localization by support vector machines using multi-scale energy and pseudo amino acid composition. Amino Acids 2007, 33, 69–74. [Google Scholar]
- Liu, H.; Wang, M.; Chou, K.C. Low-frequency Fourier spectrum for predicting membrane protein types. Biochem. Biophys. Res. Commun. 2005, 336, 737–739. [Google Scholar]
- Wang, S.Q.; Yang, J.; Chou, K.C. Using stacked generalization to predict membrane protein types based on pseudo amino acid composition. J. Theor. Biol. 2006, 242, 941–946. [Google Scholar]
- Chen, J.; Liu, H.; Yang, J.; Chou, K.C. Prediction of linear B-cell epitopes using amino acid pair antigenicity scale. Amino Acids 2007, 33, 423–428. [Google Scholar]
- Lin, W.Z.; Fang, J.A.; Xiao, X.; Chou, K.C. Predicting secretory proteins of malaria parasite by incorporating sequence evolution information into pseudo amino acid composition via grey system model. PLoS One 2012, 7, e49040. [Google Scholar]
- Chou, K.C.; Cai, Y.D. Using functional domain composition and support vector machines for prediction of protein subcellular location. J. Biol. Chem. 2002, 277, 45765–45769. [Google Scholar]
- Cai, Y.D.; Zhou, G.P.; Chou, K.C. Support vector machines for predicting membrane protein types by using functional domain composition. Biophys. J. 2003, 84, 3257–3263. [Google Scholar]
- Cristianini, N.; Shawe-Taylor, J. An Introduction of Support Vector Machines and Other Kernel-Based Learning Methodds; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar]
- Chang, C.; Lin, C. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. (TIST) 2011, 2. [Google Scholar] [CrossRef]
- Chou, K.C. Prediction of protein signal sequences and their cleavage sites. Proteins: Struct. Funct. Genet. 2001, 42, 136–139. [Google Scholar]
- Chou, K.C. Using subsite coupling to predict signal peptides. Protein Eng. 2001, 14, 75–79. [Google Scholar]
- Chou, K.C. Prediction of signal peptides using scaled window. Peptides 2001, 22, 1973–1979. [Google Scholar]
- Chou, K.C. Some remarks on predicting multi-label attributes in molecular biosystems. Mol. Biosyst. 2013, 9, 1092–1100. [Google Scholar]
- Chou, K.C.; Zhang, C.T. Review: Prediction of protein structural classes. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 275–349. [Google Scholar]
- Chou, K.C.; Shen, H.B. Cell-PLoc: A package of Web servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc. 2008, 3, 153–162. [Google Scholar]
- Chou, K.C.; Shen, H.B. Cell-PLoc 20: An improved package of web-servers for predicting subcellular localization of proteins in various organisms. Nat. Sci. 2010, 2, 1090–1103. [Google Scholar]
- Cai, Y.D.; Zhou, G.P.; Chou, K.C. Predicting enzyme family classes by hybridizing gene product composition and pseudo-amino acid composition. J. Theor. Biol. 2005, 234, 145–149. [Google Scholar]
- Cai, Y.D.; Zhou, G.P.; Jen, C.H.; Lin, S.L.; Chou, K.C. Identify catalytic triads of serine hydrolases by support vector machines. J. Theor. Biol. 2004, 228, 551–557. [Google Scholar]
- Shi, J.Y.; Zhang, S.W.; Pan, Q.; Zhou, G.P. Using pseudo amino acid composition to predict protein subcellular location: Approached with amino acid composition distribution. Amino Acids 2008, 35, 321–327. [Google Scholar]
- Fan, G.L.; Li, Q.Z. Discriminating bioluminescent proteins by incorporating average chemical shift and evolutionary information into the general form of Chou’s pseudo amino acid composition. J. Theor. Biol. 2013, 334, 45–51. [Google Scholar]
- Wu, Z.C.; Xiao, X.; Chou, K.C. iLoc-Gpos: A multi-layer classifier for predicting the subcellular localization of singleplex and multiplex gram-positive bacterial proteins. Protein Pept. Lett. 2012, 19, 4–14. [Google Scholar]
- Yamanishi, Y.; Kotera, M.; Kanehisa, M.; Goto, S. Drug-target interaction prediction from chemical genomic and pharmacological data in an integrated framework. Bioinformatics 2010, 26, i246–i254. [Google Scholar]
- Chou, K.C.; Shen, H.B. Review: Recent advances in developing web-servers for predicting protein attributes. Nat. Sci. 2009, 2, 63–92. [Google Scholar]
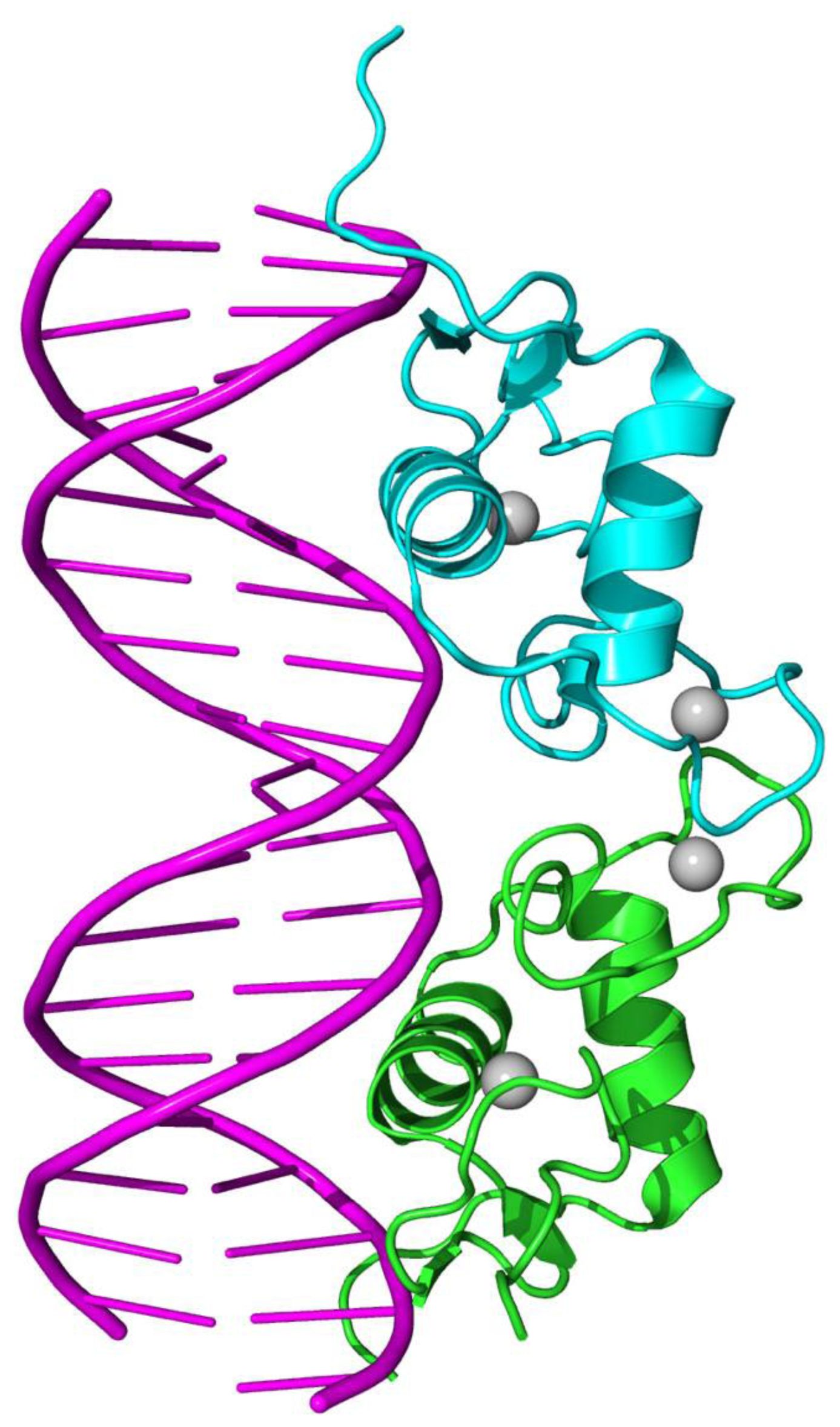



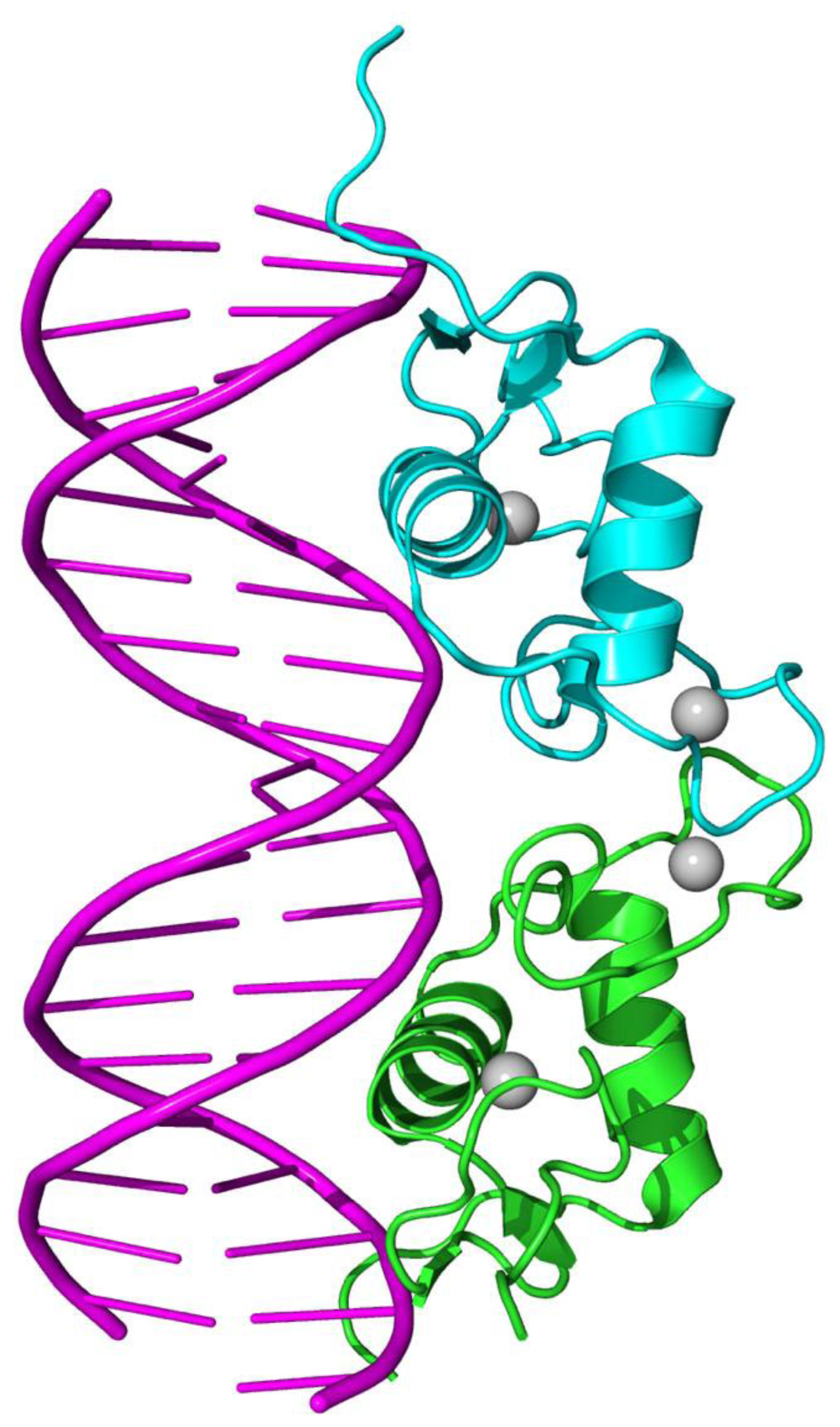{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link} (cf. Supplementary Information S1).
(cf. Supplementary Information S1).
| Metrics used for measuring prediction quality (cf. Equation (23)) | iNR-Drug a | Method by He et al. b |
|---|---|---|
| Sn | N/A | |
| Sp | N/A | |
| Acc | 85.66% | |
| MCC | 75.19% | N/A |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fan, Y.-N.; Xiao, X.; Min, J.-L.; Chou, K.-C. iNR-Drug: Predicting the Interaction of Drugs with Nuclear Receptors in Cellular Networking. Int. J. Mol. Sci. 2014, 15, 4915-4937. https://doi.org/10.3390/ijms15034915
Fan Y-N, Xiao X, Min J-L, Chou K-C. iNR-Drug: Predicting the Interaction of Drugs with Nuclear Receptors in Cellular Networking. International Journal of Molecular Sciences. 2014; 15(3):4915-4937. https://doi.org/10.3390/ijms15034915
Chicago/Turabian StyleFan, Yue-Nong, Xuan Xiao, Jian-Liang Min, and Kuo-Chen Chou. 2014. "iNR-Drug: Predicting the Interaction of Drugs with Nuclear Receptors in Cellular Networking" International Journal of Molecular Sciences 15, no. 3: 4915-4937. https://doi.org/10.3390/ijms15034915
APA StyleFan, Y.-N., Xiao, X., Min, J.-L., & Chou, K.-C. (2014). iNR-Drug: Predicting the Interaction of Drugs with Nuclear Receptors in Cellular Networking. International Journal of Molecular Sciences, 15(3), 4915-4937. https://doi.org/10.3390/ijms15034915