Sphingolipids: Key Regulators of Apoptosis and Pivotal Players in Cancer Drug Resistance



{kind=link}
{kind=link}
{kind=link}
{kind=link}
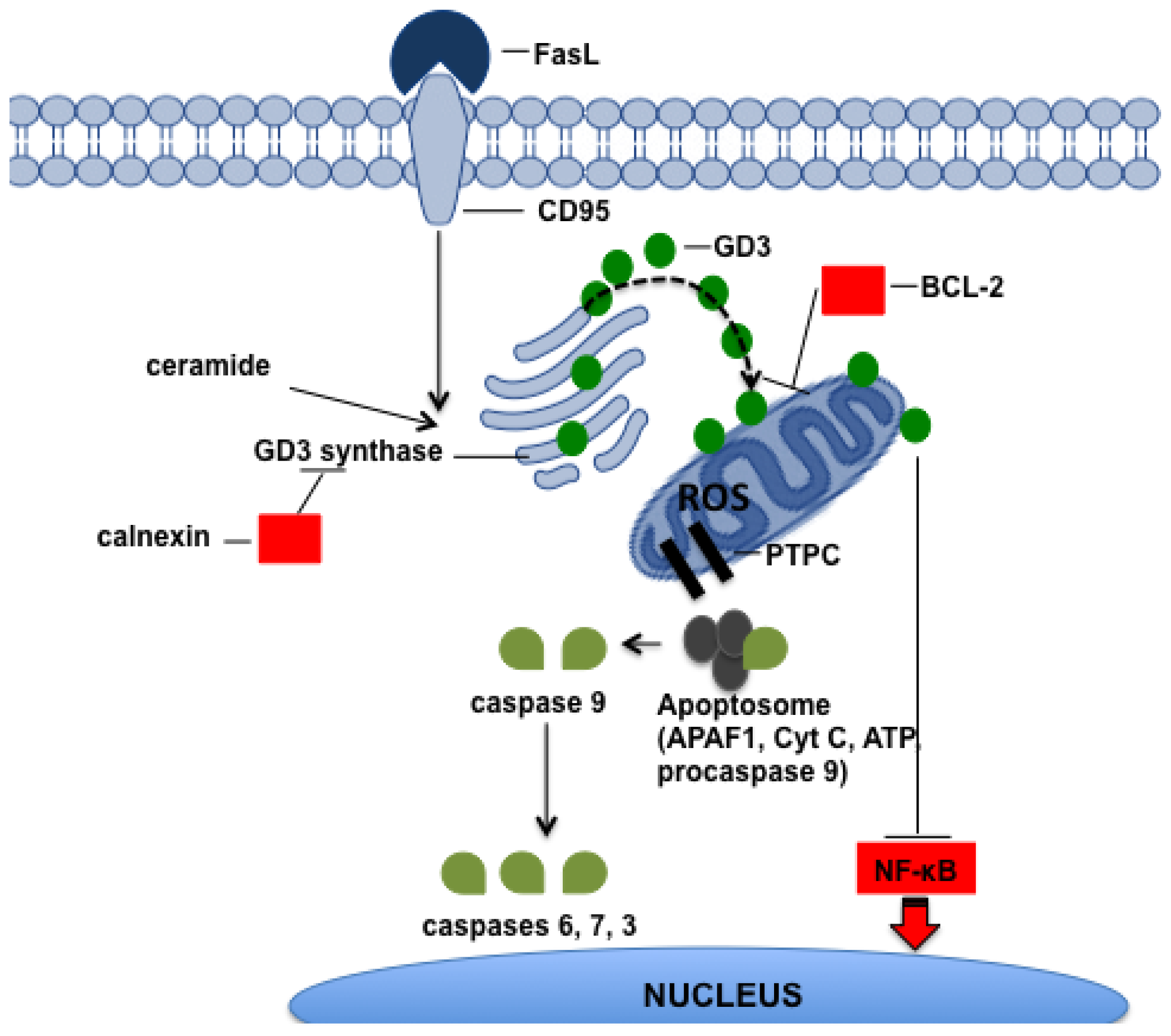{kind=link}
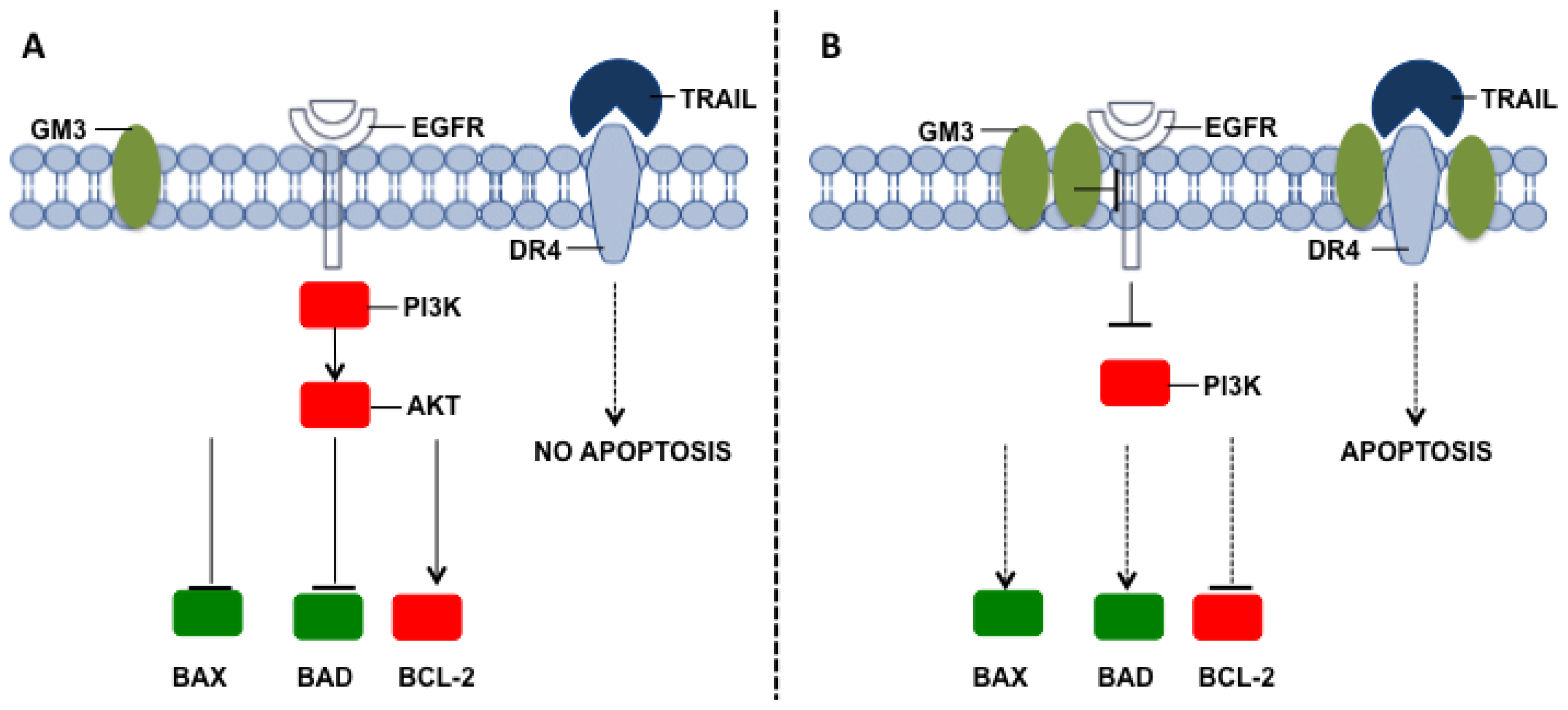{kind=link}
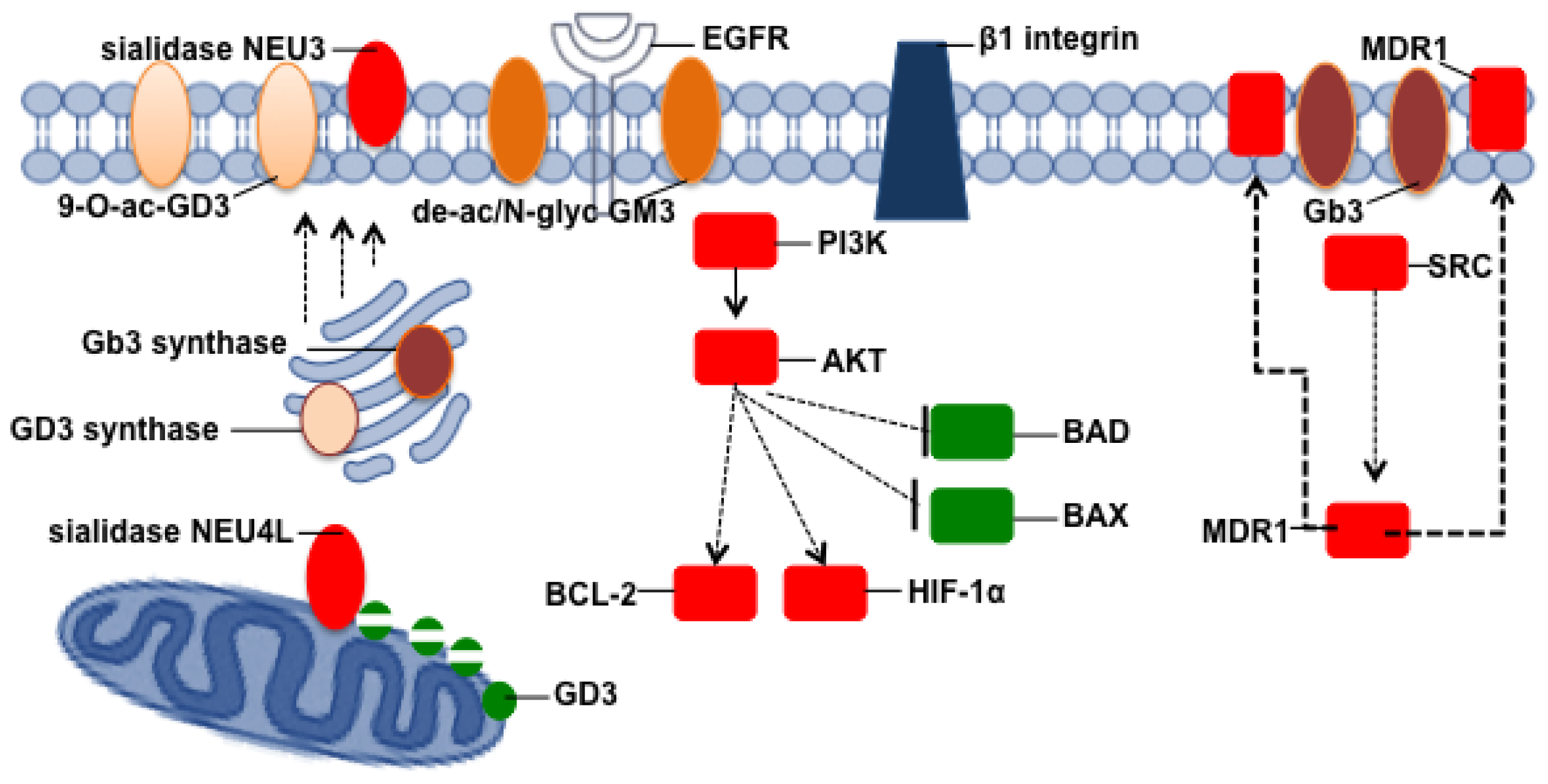{kind=link}
Abstract
:1. Introduction
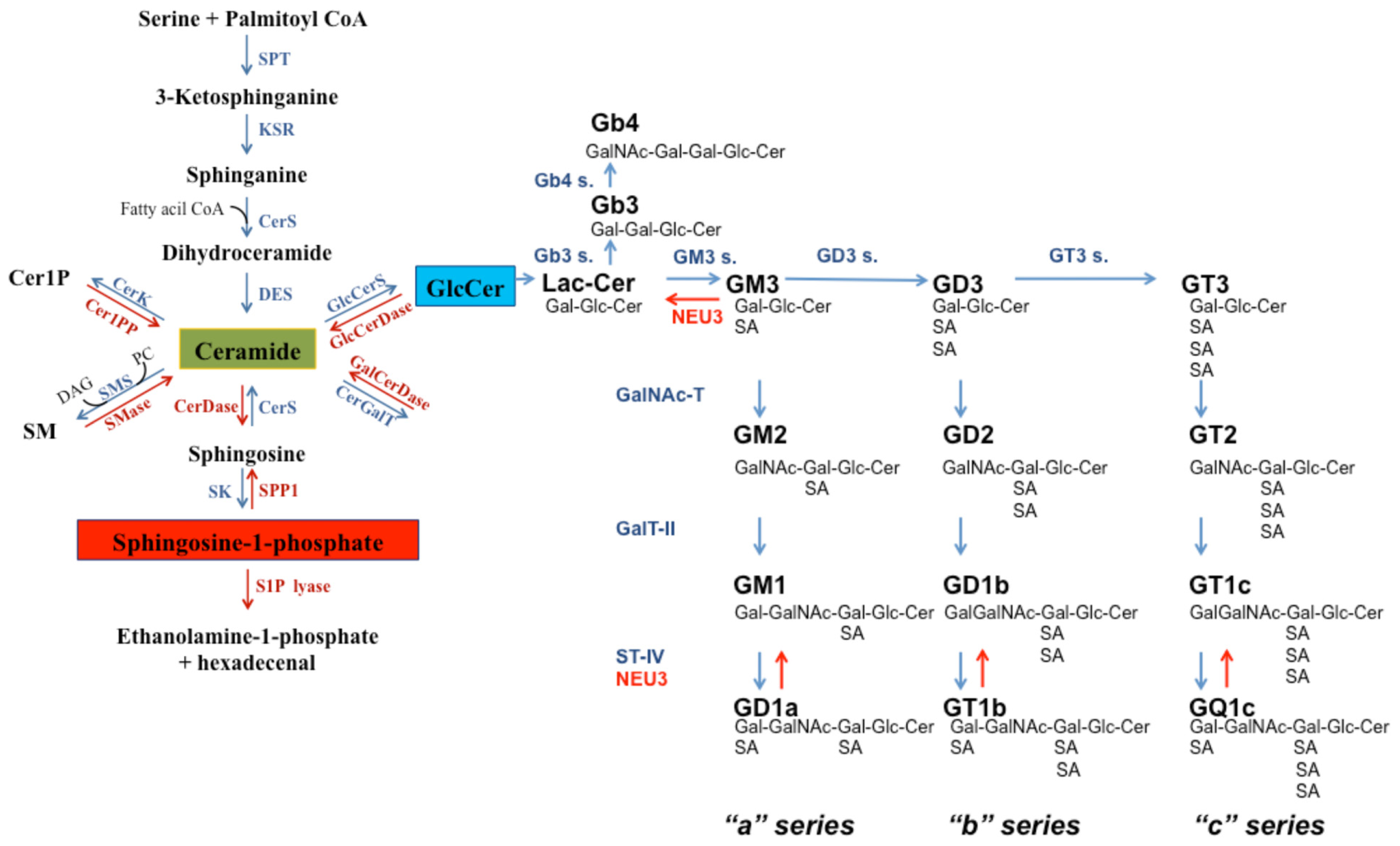
2. Sphingolipids: Insight into Their Structure and Metabolism
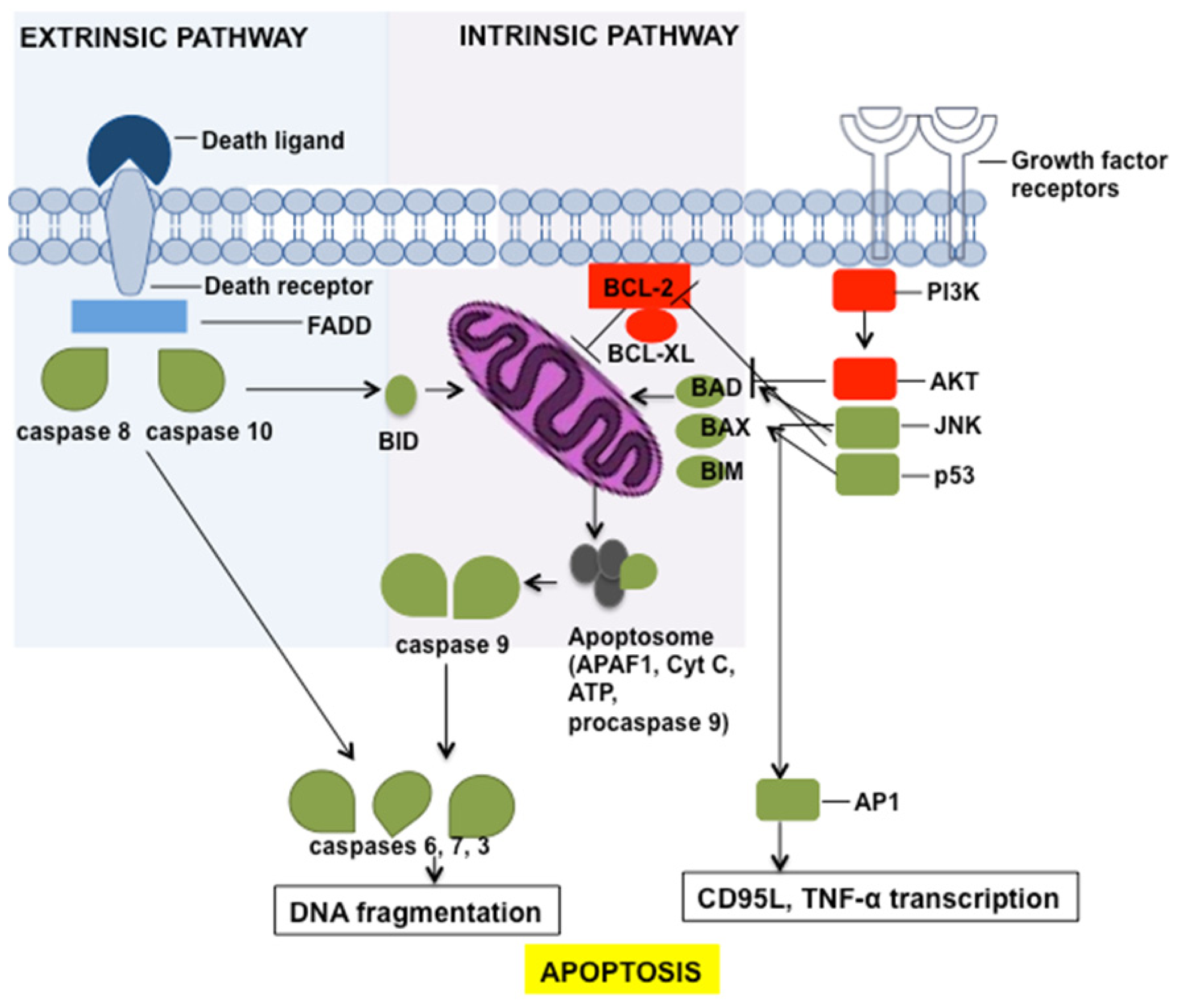
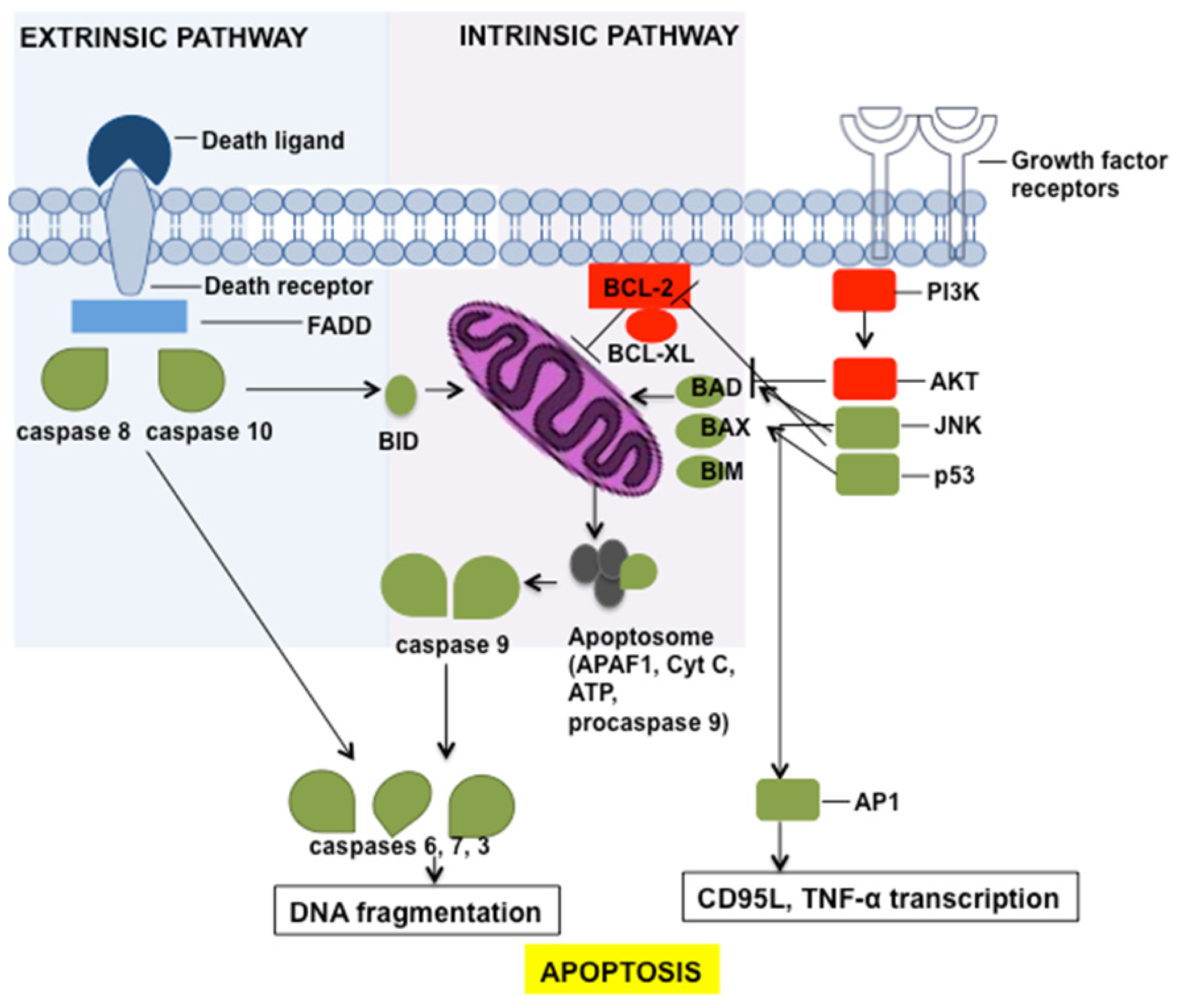
3. Apoptosis Induction by Chemotherapeutic Drugs
4. The Role of Sphingolipids in Apoptosis and Apoptosis Resistance
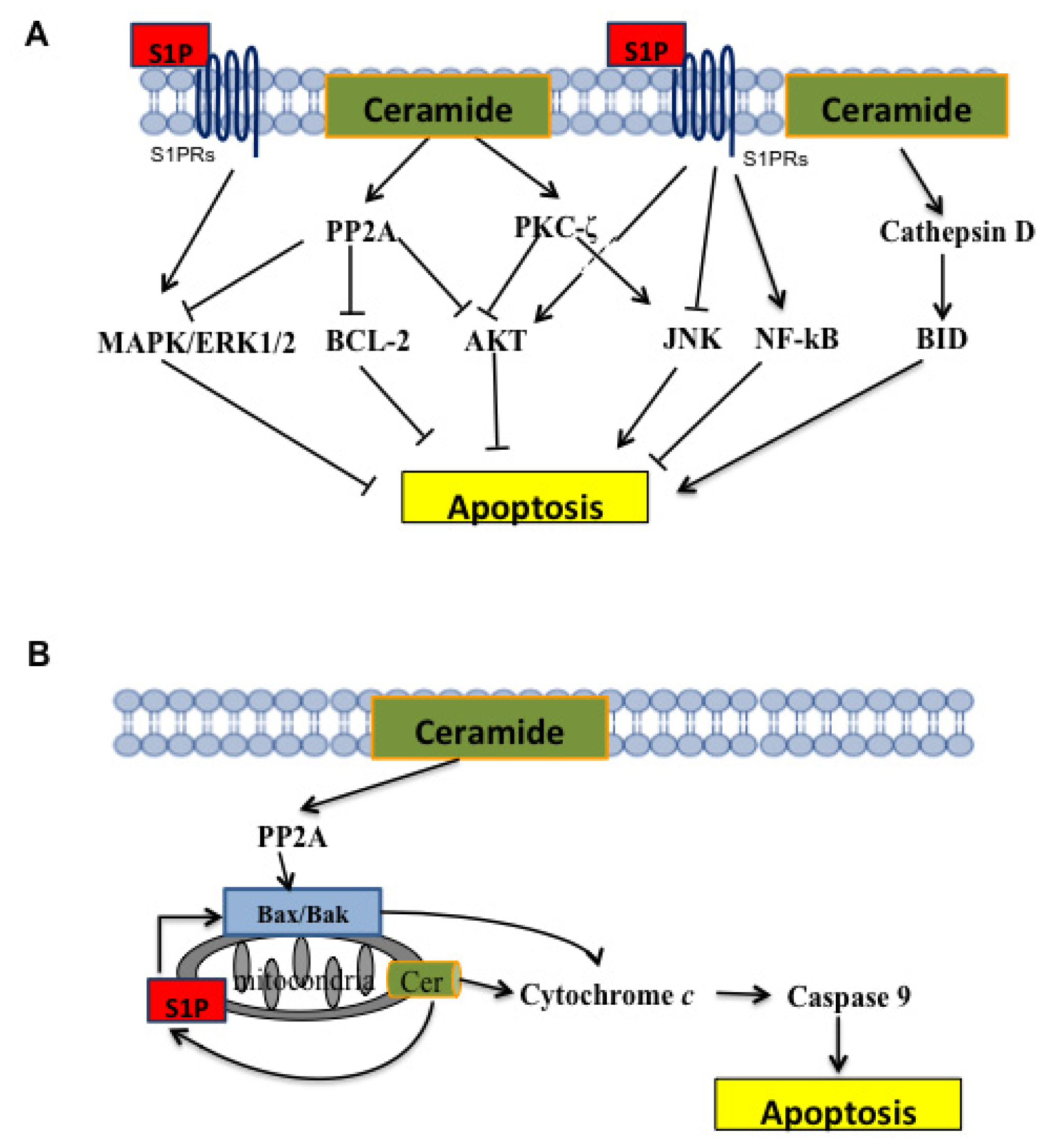
4.1. Ceramide
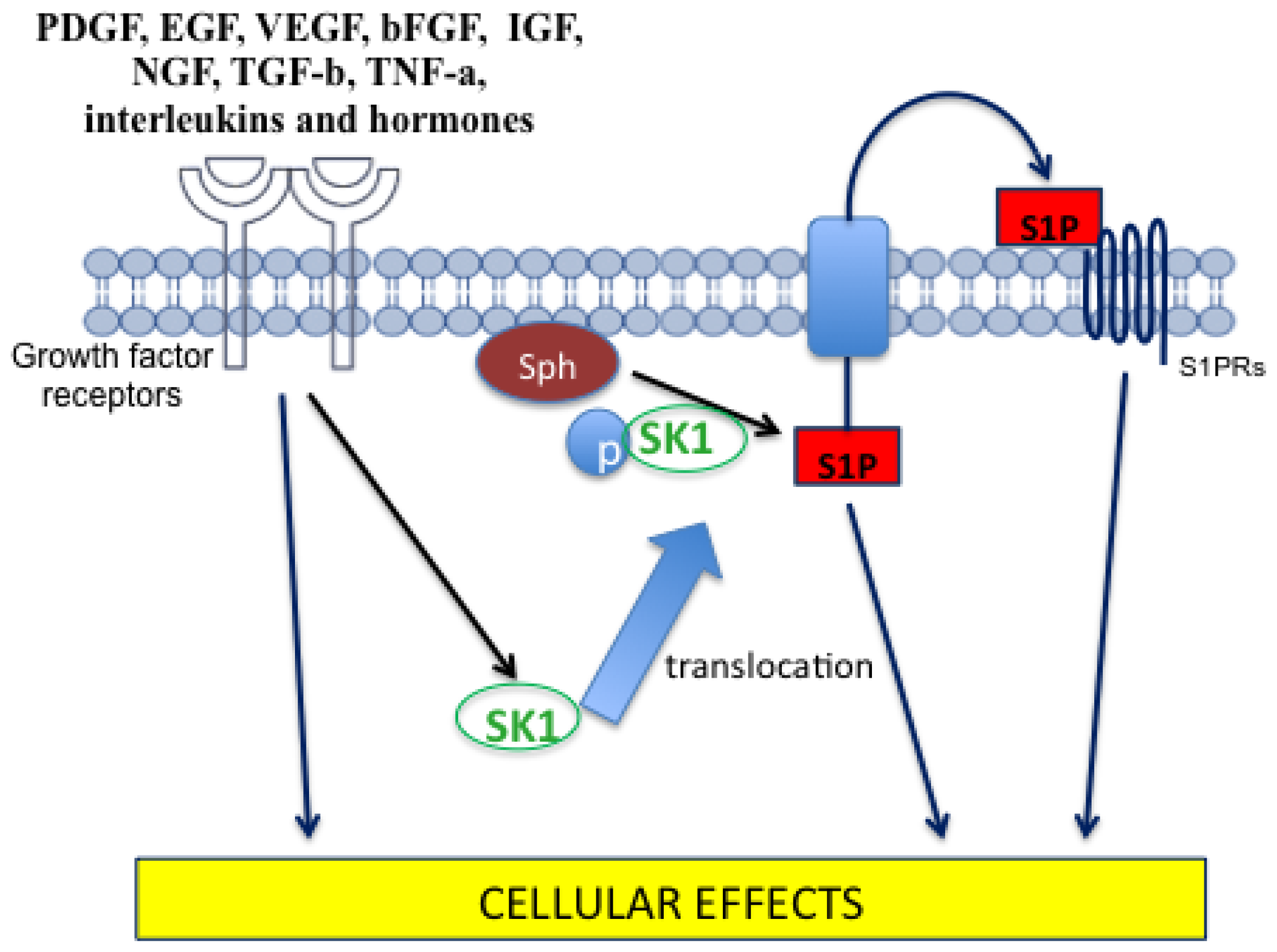
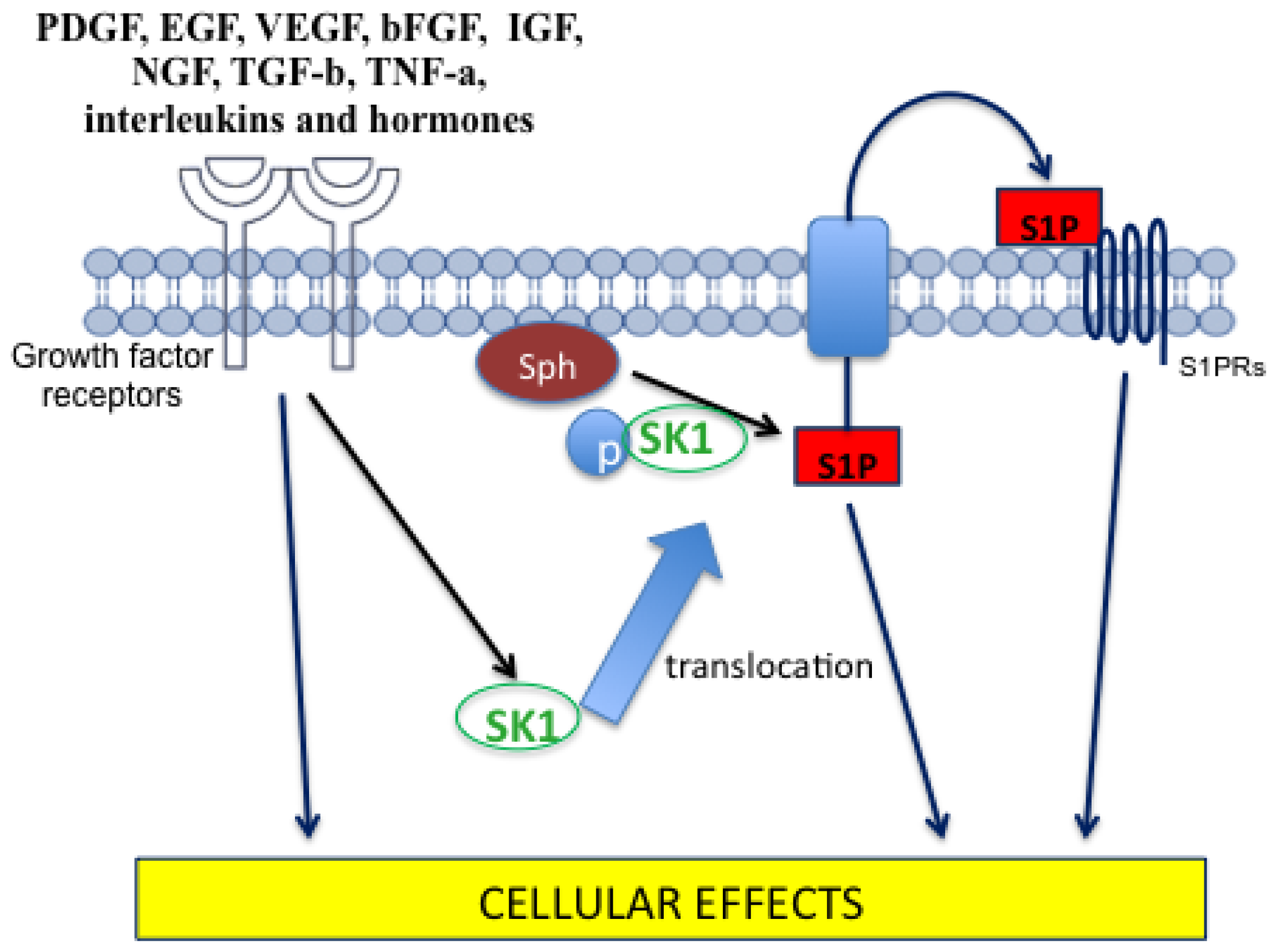
4.2. Sphingosine-1-Phosphate
5. The Role of Complex Glycosphingolipids in Apoptosis and Apoptosis Resistance
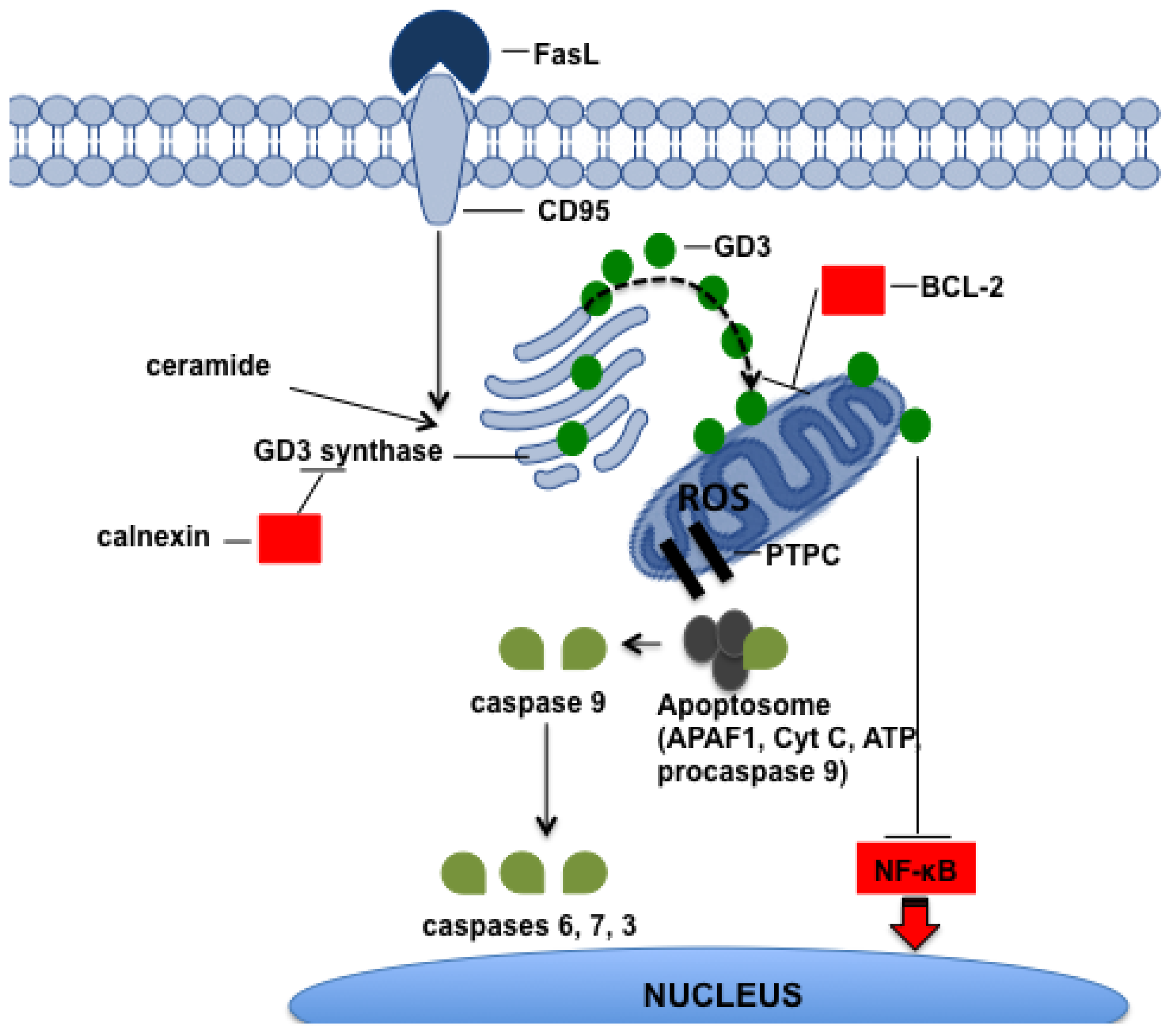
5.1. Gangliosides
5.2. Globosides
6. Pivotal Enzymes Involved in Glycosphingolipid Metabolism and in Apoptosis Resistance
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar]
- Borst, P.; Elferink, R.O. Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 2002, 71, 537–592. [Google Scholar]
- Debatin, K.M.; Krammer, P.H. Death receptors in chemotherapy and cancer. Oncogene 2004, 23, 2950–2966. [Google Scholar]
- Valent, P.; Bonnet, D.; de Maria, R.; Lapidot, T.; Copland, M.; Melo, J.V.; Chomienne, C.; Ishikawa, F.; Schuringa, J.J.; Stassi, G.; et al. Cancer stem cell definitions and terminology: The devil is in the details. Nat. Rev. Cancer 2012, 12, 767–775. [Google Scholar]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumour suppression. Nature 2004, 432, 307–315. [Google Scholar]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar]
- Ryland, L.K.; Fox, T.E.; Liu, X.; Loughran, T.P.; Kester, M. Dysregulation of sphingolipid metabolism in cancer. Cancer Biol. Ther. 2011, 11, 138–149. [Google Scholar]
- Maurer, B.J.; Metelitsa, L.S.; Seeger, R.C.; Cabot, M.C.; Reynolds, C.P. Increase of ceramide and induction of mixed apoptosis/necrosis by N-(4-hydroxyphenyl)-retinamide in neuroblastoma cell lines. J. Natl. Cancer Inst. 1999, 91, 1138–1146. [Google Scholar]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar]
- Hakomori, S.I. Glycosynaptic microdomains controlling tumor cell phenotype through alteration of cell growth adhesion and motility. FEBS Lett. 2010, 584, 1901–1906. [Google Scholar]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar]
- Tettamanti, G.; Bassi, R.; Viani, P.; Riboni, L. Salvage pathways in glycosphingolipid metabolism. Biochimie 2003, 85, 423–437. [Google Scholar]
- Levy, M.; Futerman, A.H. Mammalian ceramide synthases. IUBMB Life 2010, 62, 347–356. [Google Scholar]
- Pewzner-Jung, Y.; Ben-Dor, S.; Futerman, A.H. When do Lasses (longevity assurance genes) become CerS (ceramide synthases)?: Insights into the regulation of ceramide synthesis. J. Biol. Chem. 2006, 281, 25001–25005. [Google Scholar]
- Michel, C.; van Echten-Deckert, G. Conversion of dihydroceramide to ceramide occurs at the cytosolic face of the endoplasmic reticulum. FEBS Lett. 1997, 416, 153–155. [Google Scholar]
- Bornancin, F. Ceramide kinase: The first decade. Cell Signal. 2011, 23, 999–1008. [Google Scholar]
- Huitema, K.; van den Dikkenberg, J.; Brouwers, J.F.; Holthuis, J.C. Identification of a family of animal sphingomyelin synthases. EMBO J. 2004, 23, 33–44. [Google Scholar]
- Riboni, L.; Giussani, P.; Viani, P. Sphingolipid transport. Adv. Exp. Med. Biol. 2010, 688, 24–45. [Google Scholar]
- Jeckel, D.; Karrenbauer, A.; Burger, K.N.; van Meer, G.; Wieland, F. Glucosylceramide is synthesized at the cytosolic surface of various Golgi subfractions. J. Cell Biol. 1992, 117, 259–267. [Google Scholar]
- Ichikawa, S.; Hirabayashi, Y. Glucosylceramide synthase and glycosphingolipid synthesis. Trends Cell Biol. 1998, 8, 198–202. [Google Scholar]
- Degroote, S.; Wolthoorn, J.; van Meer, G. The cell biology of glycosphingolipids. Semin. Cell Dev. Biol. 2004, 15, 375–387. [Google Scholar]
- Durrant, L.G.; Noble, P.; Spendlove, I. Immunology in the clinic review series; focus on cancer: Glycolipids as targets for tumour immunotherapy. Clin. Exp. Immunol. 2012, 167, 206–215. [Google Scholar]
- Tettamanti, G. Ganglioside/glycosphingolipid turnover: New concepts. Glycoconj. J. 2004, 20, 301–317. [Google Scholar]
- Papini, N.; Anastasia, L.; Tringali, C.; Croci, G.; Bresciani, R.; Yamaguchi, K.; Miyagi, T.; Preti, A.; Prinetti, A.; Prioni, S.; et al. The plasma membrane-associated sialidase MmNEU3 modifies the ganglioside pattern of adjacent cells supporting its involvement in cell-to-cell interactions. J. Biol. Chem. 2004, 279, 16989–16995. [Google Scholar]
- Ledeen, R.; Wu, G. New findings on nuclear gangliosides: Overview on metabolism and function. J. Neurochem. 2011, 116, 714–720. [Google Scholar]
- Hasegawa, T.; Sugeno, N.; Takeda, A.; Matsuzaki-Kobayashi, M.; Kikuchi, A.; Furukawa, K.; Miyagi, T.; Itoyama, Y. Role of Neu4L sialidase and its substrate ganglioside GD3 in neuronal apoptosis induced by catechol metabolites. FEBS Lett. 2007, 581, 406–412. [Google Scholar]
- Tringali, C.; Papini, N.; Fusi, P.; Croci, G.; Borsani, G.; Preti, A.; Tortora, P.; Tettamanti, G.; Venerando, B.; Monti, E. Properties of recombinant human cytosolic sialidase HsNEU2 The enzyme hydrolyzes monomerically dispersed GM1 ganglioside molecules. J. Biol. Chem. 2004, 279, 3169–3179. [Google Scholar]
- Marchesini, N.; Hannun, Y.A. Acid and neutral sphingomyelinases: Roles and mechanisms of regulation. Biochem. Cell Biol. 2004, 82, 27–44. [Google Scholar]
- Romiti, E.; Vasta, V.; Meacci, E.; Farnararo, M.; Linke, T.; Ferlinz, K.; Sandhoff, K.; Bruni, P. Characterization of sphingomyelinase activity released by thrombin-stimulated platelets. Mol. Cell Biochem. 2000, 205, 75–81. [Google Scholar]
- Clarke, C.J.; Wu, B.X.; Hannun, Y.A. The neutral sphingomyelinase family: Identifying biochemical connections. Adv. Enzyme Regul. 2011, 51, 51–58. [Google Scholar]
- Duan, R.D. Alkaline sphingomyelinase: An old enzyme with novel implications. Biochim. Biophys. Acta 2006, 1761, 281–291. [Google Scholar]
- Mao, C.; Obeid, L.M. Ceramidases: Regulators of cellular responses mediated by ceramide sphingosine and sphingosine-1-phosphate. Biochim. Biophys. Acta 2008, 1781, 424–434. [Google Scholar]
- Ferlinz, K.; Kopal, G.; Bernardo, K.; Linke, T.; Bar, J.; Breiden, B.; Neumann, U.; Lang, F.; Schuchman, E.H.; Sandhoff, K. Human acid ceramidase: Processing glycosylation and lysosomal targeting. J. Biol. Chem. 2001, 276, 35352–35360. [Google Scholar]
- Romiti, E.; Meacci, E.; Tani, M.; Nuti, F.; Farnararo, M.; Ito, M.; Bruni, P. Neutral/alkaline and acid ceramidase activities are actively released by murine endothelial cells. Biochem. Biophys. Res. Commun. 2000, 275, 746–751. [Google Scholar]
- Hwang, Y.H.; Tani, M.; Nakagawa, T.; Okino, N.; Ito, M. Subcellular localization of human neutral ceramidase expressed in HEK293 cells. Biochem. Biophys. Res. Commun. 2005, 331, 37–42. [Google Scholar]
- Mao, C.; Xu, R.; Szulc, Z.M.; Bielawska, A.; Galadari, S.H.; Obeid, L.M. Cloning and characterization of a novel human alkaline ceramidase A mammalian enzyme that hydrolyzes phytoceramide. J. Biol. Chem. 2001, 276, 26577–26588. [Google Scholar]
- Sandhoff, K.; Kolter, T. Topology of glycosphingolipid degradation. Trends Cell Biol. 1996, 6, 98–103. [Google Scholar]
- Nava, V.E.; Lacana, E.; Poulton, S.; Liu, H.; Sugiura, M.; Kono, K.; Milstien, S.; Kohama, T.; Spiegel, S. Functional characterization of human sphingosine kinase-1. FEBS Lett. 2000, 473, 81–84. [Google Scholar]
- Liu, H.; Chakravarty, D.; Maceyka, M.; Milstien, S.; Spiegel, S. Sphingosine kinases: A novel family of lipid kinases. Prog. Nucleic Acid Res. Mol. Biol. 2002, 71, 493–511. [Google Scholar]
- Liu, H.; Sugiura, M.; Nava, V.E.; Edsall, L.C.; Kono, K.; Poulton, S.; Milstien, S.; Kohama, T.; Spiegel, S. Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J. Biol. Chem. 2000, 275, 19513–19520. [Google Scholar]
- Serra, M.; Saba, J.D. Sphingosine 1-phosphate lyase a key regulator of sphingosine 1-phosphate signaling and function. Adv. Enzyme Regul. 2010, 50, 349–362. [Google Scholar]
- Le Stunff, H.; Peterson, C.; Thornton, R.; Milstien, S.; Mandala, S.M.; Spiegel, S. Characterization of murine sphingosine-1-phosphate phosphohydrolase. J. Biol. Chem. 2002, 277, 8920–8927. [Google Scholar]
- Le Stunff, H.; Giussani, P.; Maceyka, M.; Lepine, S.; Milstien, S.; Spiegel, S. Recycling of sphingosine is regulated by the concerted actions of sphingosine-1-phosphate phosphohydrolase 1 and sphingosine kinase 2. J. Biol. Chem. 2007, 282, 34372–34380. [Google Scholar]
- Pyne, S.; Lee, S.C.; Long, J.; Pyne, N.J. Role of sphingosine kinases and lipid phosphate phosphatases in regulating spatial sphingosine 1-phosphate signalling in health and disease. Cell Signal. 2009, 21, 14–21. [Google Scholar]
- Krammer, P.H. CD95(APO-1/Fas)-mediated apoptosis: Live and let die. Adv. Immunol. 1999, 71, 163–210. [Google Scholar]
- Schmitz, I.; Kirchhoff, S.; Krammer, P.H. Regulation of death receptor-mediated apoptosis pathways. Int. J. Biochem. Cell Biol. 2000, 32, 1123–1136. [Google Scholar]
- Pitti, R.M.; Marsters, S.A.; Lawrence, D.A.; Roy, M.; Kischkel, F.C.; Dowd, P.; Huang, A.; Donahue, C.J.; Sherwood, S.W.; Baldwin, D.T.; et al. Genomic amplification of a decoy receptor for Fas ligand in lung and colon cancer. Nature 1998, 396, 699–703. [Google Scholar]
- Yu, K.Y.; Kwon, B.; Ni, J.; Zhai, Y.; Ebner, R.; Kwon, B.S. A newly identified member of tumor necrosis factor receptor superfamily (TR6) suppresses LIGHT-mediated apoptosis. J. Biol. Chem. 1999, 274, 13733–13736. [Google Scholar]
- Ashkenazi, A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat. Rev. Cancer 2002, 2, 420–430. [Google Scholar]
- Sprick, M.R.; Weigand, M.A.; Rieser, E.; Rauch, C.T.; Juo, P.; Blenis, J.; Krammer, P.H.; Walczak, H. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity 2000, 12, 599–609. [Google Scholar]
- Kischkel, F.C.; Hellbardt, S.; Behrmann, I.; Germer, M.; Pawlita, M.; Krammer, P.H.; Peter, M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995, 14, 5579–5588. [Google Scholar]
- Zamzami, N.; Kroemer, G. The mitochondrion in apoptosis: How Pandora’s box opens. Nat. Rev. Mol. Cell Biol. 2001, 2, 67–71. [Google Scholar]
- Martinou, J.C.; Green, D.R. Breaking the mitochondrial barrier. Nat. Rev. Mol. Cell Biol. 2001, 2, 63–67. [Google Scholar]
- Rathmell, J.C.; Thompson, C.B. The central effectors of cell death in the immune system. Annu. Rev. Immunol. 1999, 17, 781–828. [Google Scholar]
- Nakano, K.; Vousden, K.H. PUMA a novel proapoptotic gene is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar]
- Oda, K.; Arakawa, H.; Tanaka, T.; Matsuda, K.; Tanikawa, C.; Mori, T.; Nishimori, H.; Tamai, K.; Tokino, T.; Nakamura, Y.; et al. p53AIP1 a potential mediator of p53-dependent apoptosis and its regulation by Ser-46-phosphorylated p53. Cell 2000, 102, 849–862. [Google Scholar]
- Muller, M.; Wilder, S.; Bannasch, D.; Israeli, D.; Lehlbach, K.; Li-Weber, M.; Friedman, S.L.; Galle, P.R.; Stremmel, W.; Oren, M.; et al. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J. Exp. Med. 1998, 188, 2033–2045. [Google Scholar]
- Wu, G.S.; Burns, T.F.; McDonald, E.R., 3rd; Jiang, W.; Meng, R.; Krantz, I.D.; Kao, G.; Gan, D.D.; Zhou, J.Y.; Muschel, R.; et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat. Genet. 1997, 17, 141–143. [Google Scholar]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar]
- Holohan, C.; van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar]
- Sharma, S.V.; Haber, D.A.; Settleman, J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat. Rev. Cancer 2010, 10, 241–253. [Google Scholar]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar]
- Frisch, S.M.; Screaton, R.A. Anoikis mechanisms. Curr. Opin. Cell Biol. 2001, 13, 555–562. [Google Scholar]
- Herr, I.; Debatin, K.M. Cellular stress response and apoptosis in cancer therapy. Blood 2001, 98, 2603–2614. [Google Scholar]
- Hannun, Y.A. Functions of ceramide in coordinating cellular responses to stress. Science 1996, 274, 1855–1859. [Google Scholar]
- Hannun, Y.A.; Obeid, L.M. The Ceramide-centric universe of lipid-mediated cell regulation: Stress encounters of the lipid kind. J. Biol. Chem. 2002, 277, 25847–25850. [Google Scholar]
- Ogretmen, B.; Hannun, Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004, 4, 604–616. [Google Scholar]
- Gouaze-Andersson, V.; Cabot, M.C. Glycosphingolipids and drug resistance. Biochim. Biophys. Acta 2006, 1758, 2096–2103. [Google Scholar]
- Reynolds, C.P.; Maurer, B.J.; Kolesnick, R.N. Ceramide synthesis and metabolism as a target for cancer therapy. Cancer Lett. 2004, 206, 169–180. [Google Scholar]
- Morad, S.A.; Cabot, M.C. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 2013, 13, 51–65. [Google Scholar]
- Bose, R.; Verheij, M.; Haimovitz-Friedman, A.; Scotto, K.; Fuks, Z.; Kolesnick, R. Ceramide synthase mediates daunorubicin-induced apoptosis: An alternative mechanism for generating death signals. Cell 1995, 82, 405–414. [Google Scholar]
- Gomez del Pulgar, T.; Velasco, G.; Sanchez, C.; Haro, A.; Guzman, M. De novo-synthesized ceramide is involved in cannabinoid-induced apoptosis. Biochem. J. 2002, 363, 183–188. [Google Scholar]
- Perry, D.K.; Carton, J.; Shah, A.K.; Meredith, F.; Uhlinger, D.J.; Hannun, Y.A. Serine palmitoyltransferase regulates de novo ceramide generation during etoposide-induced apoptosis. J. Biol. Chem. 2000, 275, 9078–9084. [Google Scholar]
- Chalfant, C.E.; Ogretmen, B.; Galadari, S.; Kroesen, B.J.; Pettus, B.J.; Hannun, Y.A. FAS activation induces dephosphorylation of SR proteins; dependence on the de novo generation of ceramide and activation of protein phosphatase 1. J. Biol. Chem. 2001, 276, 44848–44855. [Google Scholar]
- Luberto, C.; Hassler, D.F.; Signorelli, P.; Okamoto, Y.; Sawai, H.; Boros, E.; Hazen-Martin, D.J.; Obeid, L.M.; Hannun, Y.A.; Smith, G.K. Inhibition of tumor necrosis factor-induced cell death in MCF7 by a novel inhibitor of neutral sphingomyelinase. J. Biol. Chem. 2002, 277, 41128–41139. [Google Scholar]
- Liu, J.J.; Wang, J.Y.; Hertervig, E.; Cheng, Y.; Nilsson, A.; Duan, R.D. Activation of neutral sphingomyelinase participates in ethanol-induced apoptosis in Hep G2 cells. Alcohol. Alcohol. 2000, 35, 569–573. [Google Scholar]
- Sorli, S.C.; Colie, S.; Albinet, V.; Dubrac, A.; Touriol, C.; Guilbaud, N.; Bedia, C.; Fabrias, G.; Casas, J.; Segui, B.; et al. The nonlysosomal β-glucosidase GBA2 promotes endoplasmic reticulum stress and impairs tumorigenicity of human melanoma cells. FASEB J. 2013, 27, 489–498. [Google Scholar]
- Liu, Y.Y.; Han, T.Y.; Giuliano, A.E.; Cabot, M.C. Ceramide glycosylation potentiates cellular multidrug resistance. FASEB J. 2001, 15, 719–730. [Google Scholar]
- Kok, J.W.; Sietsma, H. Sphingolipid metabolism enzymes as targets for anticancer therapy. Curr. Drug Targets 2004, 5, 375–382. [Google Scholar]
- Giussani, P.; Bassi, R.; Anelli, V.; Brioschi, L.; de Zen, F.; Riccitelli, E.; Caroli, M.; Campanella, R.; Gaini, S.M.; Viani, P.; et al. Glucosylceramide synthase protects glioblastoma cells against autophagic and apoptotic death induced by temozolomide and Paclitaxel. Cancer Investig. 2012, 30, 27–37. [Google Scholar]
- Senchenkov, A.; Litvak, D.A.; Cabot, M.C. Targeting ceramide metabolism—A strategy for overcoming drug resistance. J. Natl. Cancer Inst. 2001, 93, 347–357. [Google Scholar]
- Grazide, S.; Terrisse, A.D.; Lerouge, S.; Laurent, G.; Jaffrezou, J.P. Cytoprotective effect of glucosylceramide synthase inhibition against daunorubicin-induced apoptosis in human leukemic cell lines. J. Biol. Chem. 2004, 279, 18256–18261. [Google Scholar]
- Owczarek, T.B.; Suchanski, J.; Pula, B.; Kmiecik, A.M.; Chadalski, M.; Jethon, A.; Dziegiel, P.; Ugorski, M. Galactosylceramide affects tumorigenic and metastatic properties of breast cancer cells as an anti-apoptotic molecule. PLoS One 2013, 8, e84191. [Google Scholar]
- Itoh, M.; Kitano, T.; Watanabe, M.; Kondo, T.; Yabu, T.; Taguchi, Y.; Iwai, K.; Tashima, M.; Uchiyama, T.; Okazaki, T. Possible role of ceramide as an indicator of chemoresistance: Decrease of the ceramide content via activation of glucosylceramide synthase and sphingomyelin synthase in chemoresistant leukemia. Clin. Cancer Res. 2003, 9, 415–423. [Google Scholar]
- Lafont, E.; Milhas, D.; Carpentier, S.; Garcia, V.; Jin, Z.X.; Umehara, H.; Okazaki, T.; Schulze-Osthoff, K.; Levade, T.; Benoist, H.; et al. Caspase-mediated inhibition of sphingomyelin synthesis is involved in FasL-triggered cell death. Cell Death Differ. 2010, 17, 642–654. [Google Scholar]
- Separovic, D.; Hanada, K.; Maitah, M.Y.; Nagy, B.; Hang, I.; Tainsky, M.A.; Kraniak, J.M.; Bielawski, J. Sphingomyelin synthase 1 suppresses ceramide production and apoptosis post-photodamage. Biochem. Biophys. Res. Commun. 2007, 358, 196–202. [Google Scholar]
- Dolgachev, V.; Farooqui, M.S.; Kulaeva, O.I.; Tainsky, M.A.; Nagy, B.; Hanada, K.; Separovic, D. De novo ceramide accumulation due to inhibition of its conversion to complex sphingolipids in apoptotic photosensitized cells. J. Biol. Chem. 2004, 279, 23238–23249. [Google Scholar]
- Barth, B.M.; Cabot, M.C.; Kester, M. Ceramide-based therapeutics for the treatment of cancer. Anti-Cancer Agents Med. Chem. 2011, 11, 911–919. [Google Scholar]
- Hanada, K.; Kumagai, K.; Yasuda, S.; Miura, Y.; Kawano, M.; Fukasawa, M.; Nishijima, M. Molecular machinery for non-vesicular trafficking of ceramide. Nature 2003, 426, 803–809. [Google Scholar]
- Swanton, C.; Marani, M.; Pardo, O.; Warne, P.H.; Kelly, G.; Sahai, E.; Elustondo, F.; Chang, J.; Temple, J.; Ahmed, A.A.; et al. Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer Cell 2007, 11, 498–512. [Google Scholar]
- Lee, A.J.; Roylance, R.; Sander, J.; Gorman, P.; Endesfelder, D.; Kschischo, M.; Jones, N.P.; East, P.; Nicke, B.; Spassieva, S.; et al. CERT depletion predicts chemotherapy benefit and mediates cytotoxic and polyploid-specific cancer cell death through autophagy induction. J. Pathol. 2012, 226, 482–494. [Google Scholar]
- Heering, J.; Weis, N.; Holeiter, M.; Neugart, F.; Staebler, A.; Fehm, T.N.; Bischoff, A.; Schiller, J.; Duss, S.; Schmid, S.; et al. Loss of the ceramide transfer protein augments EGF receptor signaling in breast cancer. Cancer Res. 2012, 72, 2855–2866. [Google Scholar]
- Van Brocklyn, J.R. Sphingolipid signaling pathways as potential therapeutic targets in gliomas. Mini Rev. Med. Chem. 2007, 7, 984–990. [Google Scholar]
- Hara, S.; Nakashima, S.; Kiyono, T.; Sawada, M.; Yoshimura, S.; Iwama, T.; Sakai, N. Ceramide triggers caspase activation during gamma-radiation-induced apoptosis of human glioma cells lacking functional p53. Oncol. Rep. 2004, 12, 119–123. [Google Scholar]
- Hara, S.; Nakashima, S.; Kiyono, T.; Sawada, M.; Yoshimura, S.; Iwama, T.; Banno, Y.; Shinoda, J.; Sakai, N. p53-Independent ceramide formation in human glioma cells during gamma-radiation-induced apoptosis. Cell Death Differ. 2004, 11, 853–861. [Google Scholar]
- Velasco, G.; Galve-Roperh, I.; Sanchez, C.; Blazquez, C.; Haro, A.; Guzman, M. Cannabinoids and ceramide: Two lipids acting hand-by-hand. Life Sci. 2005, 77, 1723–1731. [Google Scholar]
- Viani, P.; Giussani, P.; Brioschi, L.; Bassi, R.; Anelli, V.; Tettamanti, G.; Riboni, L. Ceramide in nitric oxide inhibition of glioma cell growth Evidence for the involvement of ceramide traffic. J. Biol. Chem. 2003, 278, 9592–9601. [Google Scholar]
- Gramatzki, D.; Herrmann, C.; Happold, C.; Becker, K.A.; Gulbins, E.; Weller, M.; Tabatabai, G. Glioma cell death induced by irradiation or alkylating agent chemotherapy is independent of the intrinsic ceramide pathway. PLoS One 2013, 8, e63527. [Google Scholar]
- Barcelo-Coblijn, G.; Martin, M.L.; de Almeida, R.F.; Noguera-Salva, M.A.; Marcilla-Etxenike, A.; Guardiola-Serrano, F.; Luth, A.; Kleuser, B.; Halver, J.E.; Escriba, P.V. Sphingomyelin and sphingomyelin synthase (SMS) in the malignant transformation of glioma cells and in 2-hydroxyoleic acid therapy. Proc. Natl. Acad. Sci. USA 2011, 108, 19569–19574. [Google Scholar]
- Giussani, P.; Colleoni, T.; Brioschi, L.; Bassi, R.; Hanada, K.; Tettamanti, G.; Riboni, L.; Viani, P. Ceramide traffic in C6 glioma cells: Evidence for CERT-dependent and independent transport from ER to the Golgi apparatus. Biochim. Biophys. Acta 2008, 1781, 40–51. [Google Scholar]
- Giussani, P.; Maceyka, M.; le Stunff, H.; Mikami, A.; Lepine, S.; Wang, E.; Kelly, S.; Merrill, A.H., Jr; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate phosphohydrolase regulates endoplasmic reticulum-to-golgi trafficking of ceramide. Mol. Cell. Biol. 2006, 26, 5055–5069. [Google Scholar]
- Veret, J.; Coant, N.; Gorshkova, I.A.; Giussani, P.; Fradet, M.; Riccitelli, E.; Skobeleva, A.; Goya, J.; Kassis, N.; Natarajan, V.; et al. Role of palmitate-induced sphingoid base-1-phosphate biosynthesis in INS-1 β-cell survival. Biochim. Biophys. Acta 2013, 1831, 251–262. [Google Scholar]
- Sami, A.; Karsy, M. Targeting the PI3K/AKT/mTOR signaling pathway in glioblastoma: Novel therapeutic agents and advances in understanding. Tumour Biol. 2013, 34, 1991–2002. [Google Scholar]
- Giussani, P.; Brioschi, L.; Bassi, R.; Riboni, L.; Viani, P. Phosphatidylinositol 3-kinase/AKT pathway regulates the endoplasmic reticulum to golgi traffic of ceramide in glioma cells: A link between lipid signaling pathways involved in the control of cell survival. J. Biol. Chem. 2009, 284, 5088–5096. [Google Scholar]
- Hannun, Y.A.; Obeid, L.M. Many ceramides. J. Biol. Chem. 2011, 286, 27855–27862. [Google Scholar]
- Dbaibo, G.S.; Pushkareva, M.Y.; Jayadev, S.; Schwarz, J.K.; Horowitz, J.M.; Obeid, L.M.; Hannun, Y.A. Retinoblastoma gene product as a downstream target for a ceramide-dependent pathway of growth arrest. Proc. Natl. Acad. Sci. USA 1995, 92, 1347–1351. [Google Scholar]
- Chalfant, C.E.; Rathman, K.; Pinkerman, R.L.; Wood, R.E.; Obeid, L.M.; Ogretmen, B.; Hannun, Y.A. De novo ceramide regulates the alternative splicing of caspase 9 and Bcl-x in A549 lung adenocarcinoma cells Dependence on protein phosphatase-1. J. Biol. Chem. 2002, 277, 12587–12595. [Google Scholar]
- Ruvolo, P.P.; Deng, X.; Ito, T.; Carr, B.K.; May, W.S. Ceramide induces Bcl2 dephosphorylation via a mechanism involving mitochondrial PP2A. J. Biol. Chem. 1999, 274, 20296–20300. [Google Scholar]
- Young, M.M.; Kester, M.; Wang, H.G. Sphingolipids: Regulators of crosstalk between apoptosis and autophagy. J. Lipid Res. 2013, 54, 5–19. [Google Scholar]
- Riboni, L.; Viani, P.; Bassi, R.; Stabilini, A.; Tettamanti, G. Biomodulatory role of ceramide in basic fibroblast growth factor-induced proliferation of cerebellar astrocytes in primary culture. Glia 2000, 32, 137–145. [Google Scholar]
- Meloche, S.; Pouyssegur, J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 2007, 26, 3227–3239. [Google Scholar]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar]
- Chalfant, C.E.; Szulc, Z.; Roddy, P.; Bielawska, A.; Hannun, Y.A. The structural requirements for ceramide activation of serine-threonine protein phosphatases. J. Lipid Res. 2004, 45, 496–506. [Google Scholar]
- Lee, W.J.; Kim, D.U.; Lee, M.Y.; Choi, K.Y. Identification of proteins interacting with the catalytic subunit of PP2A by proteomics. Proteomics 2007, 7, 206–214. [Google Scholar]
- Adams, D.G.; Coffee, R.L., Jr; Zhang, H.; Pelech, S.; Strack, S.; Wadzinski, B.E. Positive regulation of Raf1-MEK1/2-ERK1/2 signaling by protein serine/threonine phosphatase 2A holoenzymes. J. Biol. Chem. 2005, 280, 42644–42654. [Google Scholar]
- Lahiri, S.; Futerman, A.H. The metabolism and function of sphingolipids and glycosphingolipids. Cell. Mol. Life Sci. 2007, 64, 2270–2284. [Google Scholar]
- Bollinger, C.R.; Teichgraber, V.; Gulbins, E. Ceramide-enriched membrane domains. Biochim. Biophys. Acta 2005, 1746, 284–294. [Google Scholar]
- Grassme, H.; Cremesti, A.; Kolesnick, R.; Gulbins, E. Ceramide-mediated clustering is required for CD95-DISC formation. Oncogene 2003, 22, 5457–5470. [Google Scholar]
- Matsko, C.M.; Hunter, O.C.; Rabinowich, H.; Lotze, M.T.; Amoscato, A.A. Mitochondrial lipid alterations during Fas- and radiation-induced apoptosis. Biochem. Biophys. Res. Commun. 2001, 287, 1112–1120. [Google Scholar]
- Birbes, H.; El Bawab, S.; Hannun, Y.A.; Obeid, L.M. Selective hydrolysis of a mitochondrial pool of sphingomyelin induces apoptosis. FASEB J. 2001, 15, 2669–2679. [Google Scholar]
- Siskind, L.J.; Kolesnick, R.N.; Colombini, M. Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J. Biol. Chem. 2002, 277, 26796–26803. [Google Scholar]
- Sawada, M.; Nakashima, S.; Banno, Y.; Yamakawa, H.; Takenaka, K.; Shinoda, J.; Nishimura, Y.; Sakai, N.; Nozawa, Y. Influence of Bax or Bcl-2 overexpression on the ceramide-dependent apoptotic pathway in glioma cells. Oncogene 2000, 19, 3508–3520. [Google Scholar]
- Birbes, H.; Luberto, C.; Hsu, Y.T.; El Bawab, S.; Hannun, Y.A.; Obeid, L.M. A mitochondrial pool of sphingomyelin is involved in TNFα-induced Bax translocation to mitochondria. Biochem. J. 2005, 386, 445–451. [Google Scholar]
- Von Haefen, C.; Wieder, T.; Gillissen, B.; Starck, L.; Graupner, V.; Dorken, B.; Daniel, P.T. Ceramide induces mitochondrial activation and apoptosis via a Bax-dependent pathway in human carcinoma cells. Oncogene 2002, 21, 4009–4019. [Google Scholar]
- Kim, H.J.; Mun, J.Y.; Chun, Y.J.; Choi, K.H.; Kim, M.Y. Bax-dependent apoptosis induced by ceramide in HL-60 cells. FEBS Lett. 2001, 505, 264–268. [Google Scholar]
- Chipuk, J.E.; McStay, G.P.; Bharti, A.; Kuwana, T.; Clarke, C.J.; Siskind, L.J.; Obeid, L.M.; Green, D.R. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 2012, 148, 988–1000. [Google Scholar]
- Young, M.; Wheaton, B. The impact of neighborhood composition on work-family conflict and distress. J. Health Soc. Behav. 2013, 54, 481–497. [Google Scholar]
- Xin, M.; Deng, X. Protein phosphatase 2A enhances the proapoptotic function of Bax through dephosphorylation. J. Biol. Chem. 2006, 281, 18859–18867. [Google Scholar]
- Bryan, L.; Kordula, T.; Spiegel, S.; Milstien, S. Regulation and functions of sphingosine kinases in the brain. Biochim. Biophys. Acta 2008, 1781, 459–466. [Google Scholar]
- Shida, D.; Takabe, K.; Kapitonov, D.; Milstien, S.; Spiegel, S. Targeting SphK1 as a new strategy against cancer. Curr. Drug Targets 2008, 9, 662–673. [Google Scholar]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar]
- Pchejetski, D.; Golzio, M.; Bonhoure, E.; Calvet, C.; Doumerc, N.; Garcia, V.; Mazerolles, C.; Rischmann, P.; Teissie, J.; Malavaud, B.; et al. Sphingosine kinase-1 as a chemotherapy sensor in prostate adenocarcinoma cell and mouse models. Cancer Res. 2005, 65, 11667–11675. [Google Scholar]
- Bektas, M.; Jolly, P.S.; Muller, C.; Eberle, J.; Spiegel, S.; Geilen, C.C. Sphingosine kinase activity counteracts ceramide-mediated cell death in human melanoma cells: Role of Bcl-2 expression. Oncogene 2005, 24, 178–187. [Google Scholar]
- Sukocheva, O.; Wang, L.; Verrier, E.; Vadas, M.A.; Xia, P. Restoring endocrine response in breast cancer cells by inhibition of the sphingosine kinase-1 signaling pathway. Endocrinology 2009, 150, 4484–4492. [Google Scholar]
- Taha, T.A.; Kitatani, K.; Bielawski, J.; Cho, W.; Hannun, Y.A.; Obeid, L.M. Tumor necrosis factor induces the loss of sphingosine kinase-1 by a cathepsin B-dependent mechanism. J. Biol. Chem. 2005, 280, 17196–17202. [Google Scholar]
- Okada, T.; Ding, G.; Sonoda, H.; Kajimoto, T.; Haga, Y.; Khosrowbeygi, A.; Gao, S.; Miwa, N.; Jahangeer, S.; Nakamura, S. Involvement of N-terminal-extended form of sphingosine kinase 2 in serum-dependent regulation of cell proliferation and apoptosis. J. Biol. Chem. 2005, 280, 36318–36325. [Google Scholar]
- Liu, H.; Toman, R.E.; Goparaju, S.K.; Maceyka, M.; Nava, V.E.; Sankala, H.; Payne, S.G.; Bektas, M.; Ishii, I.; Chun, J.; et al. Sphingosine kinase type 2 is a putative BH3-only protein that induces apoptosis. J. Biol. Chem. 2003, 278, 40330–40336. [Google Scholar]
- Igarashi, N.; Okada, T.; Hayashi, S.; Fujita, T.; Jahangeer, S.; Nakamura, S. Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J. Biol. Chem. 2003, 278, 46832–46839. [Google Scholar]
- Van Brocklyn, J.R.; Jackson, C.A.; Pearl, D.K.; Kotur, M.S.; Snyder, P.J.; Prior, T.W. Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: Roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J. Neuropathol. Exp. Neurol. 2005, 64, 695–705. [Google Scholar]
- Min, J.; Mesika, A.; Sivaguru, M.; van Veldhoven, P.P.; Alexander, H.; Futerman, A.H.; Alexander, S. (Dihydro)ceramide synthase 1 regulated sensitivity to cisplatin is associated with the activation of p38 mitogen-activated protein kinase and is abrogated by sphingosine kinase 1. Mol. Cancer Res. 2007, 5, 801–812. [Google Scholar]
- Zhang, H.; Li, W.; Sun, S.; Yu, S.; Zhang, M.; Zou, F. Inhibition of sphingosine kinase 1 suppresses proliferation of glioma cells under hypoxia by attenuating activity of extracellular signal-regulated kinase. Cell Prolif. 2012, 45, 167–175. [Google Scholar]
- Edsall, L.C.; Cuvillier, O.; Twitty, S.; Spiegel, S.; Milstien, S. Sphingosine kinase expression regulates apoptosis and caspase activation in PC12 cells. J. Neurochem. 2001, 76, 1573–1584. [Google Scholar]
- Riccitelli, E.; Giussani, P.; di Vito, C.; Condomitti, G.; Tringali, C.; Caroli, M.; Galli, R.; Viani, P.; Riboni, L. Extracellular sphingosine-1-phosphate: A novel actor in human glioblastoma stem cell survival. PLoS One 2013, 8, e68229. [Google Scholar]
- Maceyka, M.; Sankala, H.; Hait, N.C.; le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H., Jr; et al. SphK1 and SphK2 sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 2005, 280, 37118–37129. [Google Scholar]
- Payne, S.G.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate: Dual messenger functions. FEBS Lett. 2002, 531, 54–57. [Google Scholar]
- Bassi, R.; Anelli, V.; Giussani, P.; Tettamanti, G.; Viani, P.; Riboni, L. Sphingosine-1-phosphate is released by cerebellar astrocytes in response to bFGF and induces astrocyte proliferation through Gi-protein-coupled receptors. Glia 2006, 53, 621–630. [Google Scholar]
- Le Stunff, H.; Mikami, A.; Giussani, P.; Hobson, J.P.; Jolly, P.S.; Milstien, S.; Spiegel, S. Role of sphingosine-1-phosphate phosphatase 1 in epidermal growth factor-induced chemotaxis. J. Biol. Chem. 2004, 279, 34290–34297. [Google Scholar]
- Igarashi, J.; Bernier, S.G.; Michel, T. Sphingosine 1-phosphate and activation of endothelial nitric-oxide synthase differential regulation of Akt and MAP kinase pathways by EDG and bradykinin receptors in vascular endothelial cells. J. Biol. Chem. 2001, 276, 12420–12426. [Google Scholar]
- Siehler, S.; Wang, Y.; Fan, X.; Windh, R.T.; Manning, D.R. Sphingosine 1-phosphate activates nuclear factor-kappa B through Edg receptors Activation through Edg-3 and Edg-5 but not Edg-1 in human embryonic kidney 293 cells. J. Biol. Chem. 2001, 276, 48733–48739. [Google Scholar]
- Zondag, G.C.; Postma, F.R.; Etten, I.V.; Verlaan, I.; Moolenaar, W.H. Sphingosine 1-phosphate signalling through the G-protein-coupled receptor Edg-1. Biochem. J. 1998, 330, 605–609. [Google Scholar]
- Meyer zu Heringdorf, D.; Liliom, K.; Schaefer, M.; Danneberg, K.; Jaggar, J.H.; Tigyi, G.; Jakobs, K.H. Photolysis of intracellular caged sphingosine-1-phosphate causes Ca2+ mobilization independently of G-protein-coupled receptors. FEBS Lett. 2003, 554, 443–449. [Google Scholar]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar]
- French, K.J.; Schrecengost, R.S.; Lee, B.D.; Zhuang, Y.; Smith, S.N.; Eberly, J.L.; Yun, J.K.; Smith, C.D. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003, 63, 5962–5969. [Google Scholar]
- Sweeney, E.A.; Sakakura, C.; Shirahama, T.; Masamune, A.; Ohta, H.; Hakomori, S.; Igarashi, Y. Sphingosine and its methylated derivative NN-dimethylsphingosine (DMS) induce apoptosis in a variety of human cancer cell lines. Int. J. Cancer. J. Int. Cancer 1996, 66, 358–366. [Google Scholar]
- Shirahama, T.; Sweeney, E.A.; Sakakura, C.; Singhal, A.K.; Nishiyama, K.; Akiyama, S.; Hakomori, S.; Igarashi, Y. In vitro and in vivo induction of apoptosis by sphingosine and NN-dimethylsphingosine in human epidermoid carcinoma KB-3-1 and its multidrug-resistant cells. Clin. Cancer Res. 1997, 3, 257–264. [Google Scholar]
- Cuvillier, O.; Levade, T. Sphingosine 1-phosphate antagonizes apoptosis of human leukemia cells by inhibiting release of cytochrome c and Smac/DIABLO from mitochondria. Blood 2001, 98, 2828–2836. [Google Scholar]
- Adan-Gokbulut, A.; Kartal-Yandim, M.; Iskender, G.; Baran, Y. Novel agents targeting bioactive sphingolipids for the treatment of cancer. Curr. Med. Chem. 2013, 20, 108–122. [Google Scholar]
- Dickson, M.A.; Carvajal, R.D.; Merrill, A.H., Jr; Gonen, M.; Cane, L.M.; Schwartz, G.K. A phase I clinical trial of safingol in combination with cisplatin in advanced solid tumors. Clin. Cancer Res. 2011, 17, 2484–2492. [Google Scholar]
- Kapitonov, D.; Allegood, J.C.; Mitchell, C.; Hait, N.C.; Almenara, J.A.; Adams, J.K.; Zipkin, R.E.; Dent, P.; Kordula, T.; Milstien, S.; et al. Targeting sphingosine kinase 1 inhibits Akt signaling induces apoptosis and suppresses growth of human glioblastoma cells and xenografts. Cancer Res. 2009, 69, 6915–6923. [Google Scholar]
- Beljanski, V.; Knaak, C.; Smith, C.D. A novel sphingosine kinase inhibitor induces autophagy in tumor cells. J. Pharmacol. Exp. Ther. 2010, 333, 454–464. [Google Scholar]
- Matsuoka, Y.; Nagahara, Y.; Ikekita, M.; Shinomiya, T. A novel immunosuppressive agent FTY720 induced Akt dephosphorylation in leukemia cells. Br. J. Pharmacol. 2003, 138, 1303–1312. [Google Scholar]
- Azuma, H.; Takahara, S.; Horie, S.; Muto, S.; Otsuki, Y.; Katsuoka, Y. Induction of apoptosis in human bladder cancer cells in vitro and in vivo caused by FTY720 treatment. J. Urol. 2003, 169, 2372–2377. [Google Scholar]
- Sonoda, Y.; Yamamoto, D.; Sakurai, S.; Hasegawa, M.; Aizu-Yokota, E.; Momoi, T.; Kasahara, T. FTY720 a novel immunosuppressive agent induces apoptosis in human glioma cells. Biochem. Biophys. Res. Commun. 2001, 281, 282–288. [Google Scholar]
- Wang, J.D.; Takahara, S.; Nonomura, N.; Ichimaru, N.; Toki, K.; Azuma, H.; Matsumiya, K.; Okuyama, A.; Suzuki, S. Early induction of apoptosis in androgen-independent prostate cancer cell line by FTY720 requires caspase-3 activation. Prostate 1999, 40, 50–55. [Google Scholar]
- Yasui, H.; Hideshima, T.; Raje, N.; Roccaro, A.M.; Shiraishi, N.; Kumar, S.; Hamasaki, M.; Ishitsuka, K.; Tai, Y.T.; Podar, K.; et al. FTY720 induces apoptosis in multiple myeloma cells and overcomes drug resistance. Cancer Res. 2005, 65, 7478–7484. [Google Scholar]
- Azuma, H.; Takahara, S.; Ichimaru, N.; Wang, J.D.; Itoh, Y.; Otsuki, Y.; Morimoto, J.; Fukui, R.; Hoshiga, M.; Ishihara, T.; et al. Marked prevention of tumor growth and metastasis by a novel immunosuppressive agent FTY720 in mouse breast cancer models. Cancer Res. 2002, 62, 1410–1419. [Google Scholar]
- Lepine, S.; Allegood, J.C.; Edmonds, Y.; Milstien, S.; Spiegel, S. Autophagy induced by deficiency of sphingosine-1-phosphate phosphohydrolase 1 is switched to apoptosis by calpain-mediated autophagy-related gene 5 (Atg5) cleavage. J. Biol. Chem. 2011, 286, 44380–44390. [Google Scholar]
- Young, M.M.; Takahashi, Y.; Khan, O.; Park, S.; Hori, T.; Yun, J.; Sharma, A.K.; Amin, S.; Hu, C.D.; Zhang, J.; et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J. Biol. Chem. 2012, 287, 12455–12468. [Google Scholar]
- Scarlatti, F.; Bauvy, C.; Ventruti, A.; Sala, G.; Cluzeaud, F.; Vandewalle, A.; Ghidoni, R.; Codogno, P. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J. Biol. Chem. 2004, 279, 18384–18391. [Google Scholar]
- Prinetti, A.; Basso, L.; Appierto, V.; Villani, M.G.; Valsecchi, M.; Loberto, N.; Prioni, S.; Chigorno, V.; Cavadini, E.; Formelli, F.; et al. Altered sphingolipid metabolism in N-(4-hydroxyphenyl)-retinamide-resistant A2780 human ovarian carcinoma cells. J. Biol. Chem. 2003, 278, 5574–5583. [Google Scholar]
- Prinetti, A.; Millimaggi, D.; D’Ascenzo, S.; Clarkson, M.; Bettiga, A.; Chigorno, V.; Sonnino, S.; Pavan, A.; Dolo, V. Lack of ceramide generation and altered sphingolipid composition are associated with drug resistance in human ovarian carcinoma cells. Biochem. J 2006, 395, 311–318. [Google Scholar]
- Veldman, R.J.; Klappe, K.; Hinrichs, J.; Hummel, I.; van der Schaaf, G.; Sietsma, H.; Kok, J.W. Altered sphingolipid metabolism in multidrug-resistant ovarian cancer cells is due to uncoupling of glycolipid biosynthesis in the Golgi apparatus. FASEB J. 2002, 16, 1111–1113. [Google Scholar]
- Dijkhuis, A.J.; Douwes, J.; Kamps, W.; Sietsma, H.; Kok, J.W. Differential expression of sphingolipids in P-glycoprotein or multidrug resistance-related protein 1 expressing human neuroblastoma cell lines. FEBS Lett. 2003, 548, 28–32. [Google Scholar]
- Plo, I.; Lehne, G.; Beckstrom, K.J.; Maestre, N.; Bettaieb, A.; Laurent, G.; Lautier, D. Influence of ceramide metabolism on P-glycoprotein function in immature acute myeloid leukemia KG1a cells. Mol. Pharmacol. 2002, 62, 304–312. [Google Scholar]
- Hakomori, S.I. Cell adhesion/recognition and signal transduction through glycosphingolipid microdomain. Glycoconj. J. 2000, 17, 143–151. [Google Scholar]
- Anastasia, L.; Papini, N.; Colazzo, F.; Palazzolo, G.; Tringali, C.; Dileo, L.; Piccoli, M.; Conforti, E.; Sitzia, C.; Monti, E.; et al. NEU3 sialidase strictly modulates GM3 levels in skeletal myoblasts C2C12 thus favoring their differentiation and protecting them from apoptosis. J. Biol. Chem. 2008, 283, 36265–36271. [Google Scholar]
- Kang, S.K.; Kim, Y.S.; Kong, Y.J.; Song, K.H.; Chang, Y.C.; Park, Y.G.; Ko, J.H.; Lee, Y.C.; Kim, C.H. isialoganglioside GD3 synthase expression recruits membrane transglutaminase 2 during erythroid differentiation of the human chronic myelogenous leukemia K562 cells. Proteomics 2008, 8, 3317–3328. [Google Scholar]
- Hettmer, S.; McCarter, R.; Ladisch, S.; Kaucic, K. Alterations in neuroblastoma ganglioside synthesis by induction of GD1b synthase by retinoic acid. Br. J. Cancer 2004, 91, 389–397. [Google Scholar]
- Nojiri, H.; Yamana, H.; Shirouzu, G.; Suzuki, T.; Isono, H. Glycotherapy for cancer: Remodeling of ganglioside pattern as an effective approach for cancer therapy. Cancer Detect. Prev. 2002, 26, 114–120. [Google Scholar]
- Tringali, C.; Lupo, B.; Cirillo, F.; Papini, N.; Anastasia, L.; Lamorte, G.; Colombi, P.; Bresciani, R.; Monti, E.; Tettamanti, G.; et al. Silencing of membrane-associated sialidase Neu3 diminishes apoptosis resistance and triggers megakaryocytic differentiation of chronic myeloid leukemic cells K562 through the increase of ganglioside GM3. Cell Death Differ. 2009, 16, 164–174. [Google Scholar]
- Guan, F.; Handa, K.; Hakomori, S.I. Specific glycosphingolipids mediate epithelial-to-mesenchymal transition of human and mouse epithelial cell lines. Proc. Natl. Acad. Sci. USA 2009, 106, 7461–7466. [Google Scholar]
- Savagner, P. The epithelial-mesenchymal transition (EMT) phenomenon. Ann. Oncol. 2010, 21, vii89–vii92. [Google Scholar]
- Min, Y.; Shi, J.; Zhang, Y.; Liu, S.; Liu, Y.; Zheng, D. Death receptor 5-recruited raft components contributes to the sensitivity of Jurkat leukemia cell lines to TRAIL-induced cell death. IUBMB Life 2009, 61, 261–267. [Google Scholar]
- Falschlehner, C.; Emmerich, C.H.; Gerlach, B.; Walczak, H. TRAIL signalling: Decisions between life and death. Int. J. Biochem. Cell Biol. 2007, 39, 1462–1475. [Google Scholar]
- Lim, S.C.; Duong, H.Q.; Choi, J.E.; Lee, T.B.; Kang, J.H.; Oh, S.H.; Han, S.I. Lipid raft-dependent death receptor 5 (DR5) expression and activation are critical for ursodeoxycholic acid-induced apoptosis in gastric cancer cells. Carcinogenesis 2011, 32, 723–731. [Google Scholar]
- Ouyang, W.; Yang, C.; Zhang, S.; Liu, Y.; Yang, B.; Zhang, J.; Zhou, F.; Zhou, Y.; Xie, C. Absence of death receptor translocation into lipid rafts in acquired TRAIL-resistant NSCLC cells. Int. J. Oncol. 2013, 42, 699–711. [Google Scholar]
- Xiao, W.; Ishdorj, G.; Sun, J.; Johnston, J.B.; Gibson, S.B. Death receptor 4 is preferentially recruited to lipid rafts in chronic lymphocytic leukemia cells contributing to tumor necrosis related apoptosis inducing ligand-induced synergistic apoptotic responses. Leuk. Lymphoma 2011, 52, 1290–1301. [Google Scholar]
- Marconi, M.; Ascione, B.; Ciarlo, L.; Vona, R.; Garofalo, T.; Sorice, M.; Gianni, A.M.; Locatelli, S.L.; Carlo-Stella, C.; Malorni, W.; et al. Constitutive localization of DR4 in lipid rafts is mandatory for TRAIL-induced apoptosis in B-cell hematologic malignancies. Cell Death Dis. 2013, 4, e863. [Google Scholar]
- Tringali, C.; Lupo, B.; Silvestri, I.; Papini, N.; Anastasia, L.; Tettamanti, G.; Venerando, B. The plasma membrane sialidase NEU3 regulates the malignancy of renal carcinoma cells by controlling β1 integrin internalization and recycling. J. Biol. Chem. 2012, 287, 42835–42845. [Google Scholar]
- Dyatlovitskaya, E.V.; Bergelson, L.D. Glycosphingolipids and antitumor immunity. Biochim. Biophys. Acta 1987, 907, 125–143. [Google Scholar]
- Fredman, P. Gangliosides associated with primary brain tumors and their expression in cell lines established from these tumors. Prog. Brain Res. 1994, 101, 225–240. [Google Scholar]
- Berra, B.; Gaini, S.M.; Riboni, L. Correlation between ganglioside distribution and histological grading of human astrocytomas. Int. J. Cancer 1985, 36, 363–366. [Google Scholar]
- Riboni, L.; Campanella, R.; Bassi, R.; Villani, R.; Gaini, S.M.; Martinelli-Boneschi, F.; Viani, P.; Tettamanti, G. Ceramide levels are inversely associated with malignant progression of human glial tumors. Glia 2002, 39, 105–113. [Google Scholar]
- Malisan, F.; Testi, R. GD3 ganglioside and apoptosis. Biochim. Biophys. Acta 2002, 1585, 179–187. [Google Scholar]
- Garcia-Ruiz, C.; Colell, A.; Paris, R.; Fernandez-Checa, J.C. Direct interaction of GD3 ganglioside with mitochondria generates reactive oxygen species followed by mitochondrial permeability transition cytochrome c release and caspase activation. FASEB J. 2000, 14, 847–858. [Google Scholar]
- Kroemer, G.; Reed, J.C. Mitochondrial control of cell death. Nat. Med. 2000, 6, 513–519. [Google Scholar]
- Rippo, M.R.; Malisan, F.; Ravagnan, L.; Tomassini, B.; Condo, I.; Costantini, P.; Susin, S.A.; Rufini, A.; Todaro, M.; Kroemer, G.; et al. GD3 ganglioside directly targets mitochondria in a bcl-2-controlled fashion. FASEB J. 2000, 14, 2047–2054. [Google Scholar]
- De Maria, R.; Rippo, M.R.; Schuchman, E.H.; Testi, R. Acidic sphingomyelinase (ASM) is necessary for fas-induced GD3 ganglioside accumulation and efficient apoptosis of lymphoid cells. J. Exp. Med. 1998, 187, 897–902. [Google Scholar]
- Colell, A.; Morales, A.; Fernandez-Checa, J.C.; Garcia-Ruiz, C. Ceramide generated by acidic sphingomyelinase contributes to tumor necrosis factor-α-mediated apoptosis in human colon HT-29 cells through glycosphingolipids formation Possible role of ganglioside GD3. FEBS Lett. 2002, 526, 135–141. [Google Scholar]
- Lovat, P.E.; di Sano, F.; Corazzari, M.; Fazi, B.; Donnorso, R.P.; Pearson, A.D.; Hall, A.G.; Redfern, C.P.; Piacentini, M. Gangliosides link the acidic sphingomyelinase-mediated induction of ceramide to 12-lipoxygenase-dependent apoptosis of neuroblastoma in response to fenretinide. J. Natl. Cancer Inst. 2004, 96, 1288–1299. [Google Scholar]
- Birkle, S.; Zeng, G.; Gao, L.; Yu, R.K.; Aubry, J. Role of tumor-associated gangliosides in cancer progression. Biochimie 2003, 85, 455–463. [Google Scholar]
- Koochekpour, S.; Merzak, A.; Pilkington, G.J. Vascular endothelial growth factor production is stimulated by gangliosides and TGF-β isoforms in human glioma cells in vitro. Cancer Lett. 1996, 102, 209–215. [Google Scholar]
- Ziche, M.; Morbidelli, L.; Alessandri, G.; Gullino, P.M. Angiogenesis can be stimulated or repressed in vivo by a change in GM3:GD3 ganglioside ratio. Lab. Investig. 1992, 67, 711–715. [Google Scholar]
- Zeng, G.; Gao, L.; Birkle, S.; Yu, R.K. Suppression of ganglioside GD3 expression in a rat F-11 tumor cell line reduces tumor growth angiogenesis and vascular endothelial growth factor production. Cancer Res. 2000, 60, 6670–6676. [Google Scholar]
- Malisan, F.; Franchi, L.; Tomassini, B.; Ventura, N.; Condo, I.; Rippo, M.R.; Rufini, A.; Liberati, L.; Nachtigall, C.; Kniep, B.; et al. Acetylation suppresses the proapoptotic activity of GD3 ganglioside. J. Exp. Med. 2002, 196, 1535–1541. [Google Scholar]
- Mukherjee, K.; Chava, A.K.; Mandal, C.; Dey, S.N.; Kniep, B.; Chandra, S.; Mandal, C. O-acetylation of GD3 prevents its apoptotic effect and promotes survival of lymphoblasts in childhood acute lymphoblastic leukaemia. J. Cell. Biochem. 2008, 105, 724–734. [Google Scholar]
- Birks, S.M.; Danquah, J.O.; King, L.; Vlasak, R.; Gorecki, D.C.; Pilkington, G.J. Targeting the GD3 acetylation pathway selectively induces apoptosis in glioblastoma. Neuro-Oncol. 2011, 13, 950–960. [Google Scholar]
- Mandal, C.; Tringali, C.; Mondal, S.; Anastasia, L.; Chandra, S.; Venerando, B.; Mandal, C. Down regulation of membrane-bound Neu3 constitutes a new potential marker for childhood acute lymphoblastic leukemia and induces apoptosis suppression of neoplastic cells. Int. J. Cancer J. Int. Cancer 2010, 126, 337–349. [Google Scholar]
- Chen, H.Y.; Varki, A. O-acetylation of GD3: An enigmatic modification regulating apoptosis? J. Exp. Med. 2002, 196, 1529–1533. [Google Scholar]
- Tomassini, B.; Malisan, F.; Franchi, L.; Nicolo, C.; Calvo, G.B.; Saito, T.; Testi, R. Calnexin suppresses GD3 synthase-induced apoptosis. FASEB J. 2004, 18, 1553–1555. [Google Scholar]
- Malisan, F.; Testi, R. The ganglioside GD3 as the Greek goddess Hecate: Several faces turned towards as many directions. IUBMB Life 2005, 57, 477–482. [Google Scholar]
- Watanabe, R.; Ohyama, C.; Aoki, H.; Takahashi, T.; Satoh, M.; Saito, S.; Hoshi, S.; Ishii, A.; Saito, M.; Arai, Y. Ganglioside G(M3) overexpression induces apoptosis and reduces malignant potential in murine bladder cancer. Cancer Res. 2002, 62, 3850–3854. [Google Scholar]
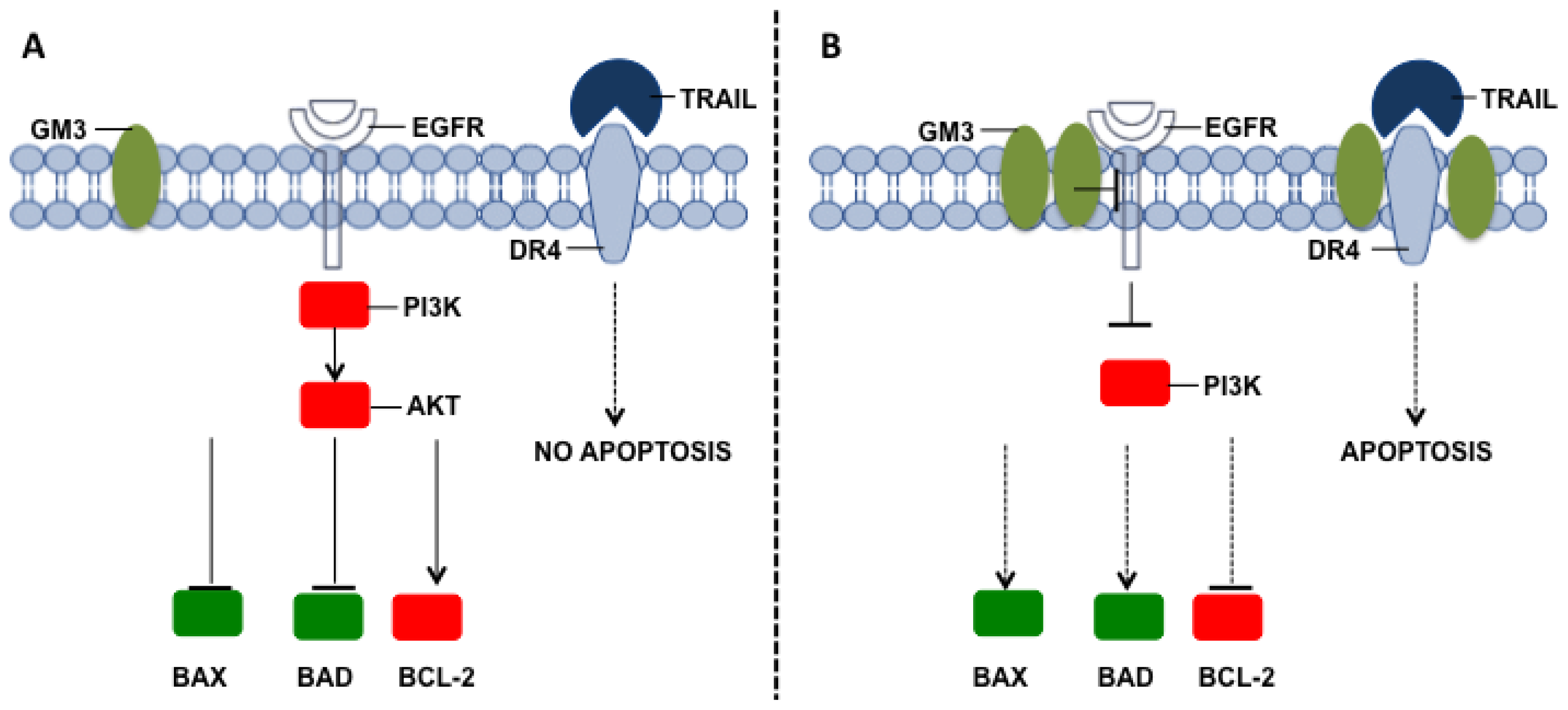
- Noguchi, M.; Suzuki, T.; Kabayama, K.; Takahashi, H.; Chiba, H.; Shiratori, M.; Abe, S.; Watanabe, A.; Satoh, M.; Hasegawa, T.; et al. GM3 synthase gene is a novel biomarker for histological classification and drug sensitivity against epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Cancer Sci. 2007, 98, 1625–1632. [Google Scholar]
- Kiura, K.; Watarai, S.; Ueoka, H.; Tabata, M.; Gemba, K.; Aoe, K.; Yamane, H.; Yasuda, T.; Harada, M. An alteration of ganglioside composition in cisplatin-resistant lung cancer cell line. Anticancer Res. 1998, 18, 2957–2960. [Google Scholar]
- Uemura, S.; Kabayama, K.; Noguchi, M.; Igarashi, Y.; Inokuchi, J. Sialylation and sulfation of lactosylceramide distinctly regulate anchorage-independent growth apoptosis and gene expression in 3LL Lewis lung carcinoma cells. Glycobiology 2003, 13, 207–216. [Google Scholar]
- Noguchi, M.; Kabayama, K.; Uemura, S.; Kang, B.W.; Saito, M.; Igarashi, Y.; Inokuchi, J. Endogenously produced ganglioside GM3 endows etoposide and doxorubicin resistance by up-regulating Bcl-2 expression in 3LL Lewis lung carcinoma cells. Glycobiology 2006, 16, 641–650. [Google Scholar]
- Liu, J.W.; Sun, P.; Yan, Q.; Paller, A.S.; Gerami, P.; Ho, N.; Vashi, N.; le Poole, I.C.; Wang, X.Q. De-N-acetyl GM3 promotes melanoma cell migration and invasion through urokinase plasminogen activator receptor signaling-dependent MMP-2 activation. Cancer Res. 2009, 69, 8662–8669. [Google Scholar]
- Blanco, R.; Rengifo, C.E.; Cedeno, M.; Frometa, M.; Rengifo, E.; Carr, A. Immunoreactivity of the 14F7 Mab (raised against N-glycolyl GM3 Ganglioside) as a positive prognostic factor in non-small-cell lung cancer. Pathol. Res. Int. 2012, 2012, 235418. [Google Scholar]
- Blanco, R.; Rengifo, E.; Cedeno, M.; Rengifo, C.E.; Alonso, D.F.; Carr, A. Immunoreactivity of the 14F7 mab raised against N-glycolyl GM3 ganglioside in epithelial malignant tumors from digestive system. ISRN Gastroenterol. 2011, 2011, 645641. [Google Scholar]
- Carr, A.; Mullet, A.; Mazorra, Z.; Vazquez, A.M.; Alfonso, M.; Mesa, C.; Rengifo, E.; Perez, R.; Fernandez, L.E. A mouse IgG1 monoclonal antibody specific for N-glycolyl GM3 ganglioside recognized breast and melanoma tumors. Hybridoma 2000, 19, 241–247. [Google Scholar]
- Bremer, E.G.; Schlessinger, J.; Hakomori, S. Ganglioside-mediated modulation of cell growth Specific effects of GM3 on tyrosine phosphorylation of the epidermal growth factor receptor. J. Biol. Chem. 1986, 261, 2434–2440. [Google Scholar]
- Hayashi, N.; Chiba, H.; Kuronuma, K.; Go, S.; Hasegawa, Y.; Takahashi, M.; Gasa, S.; Watanabe, A.; Hasegawa, T.; Kuroki, Y.; et al. Detection of N-glycolyated gangliosides in non-small-cell lung cancer using GMR8 monoclonal antibody. Cancer Sci. 2013, 104, 43–47. [Google Scholar]
- Hanai, N.; Dohi, T.; Nores, G.A.; Hakomori, S. A novel ganglioside de-N-acetyl-GM3 (II3NeuNH2LacCer) acting as a strong promoter for epidermal growth factor receptor kinase and as a stimulator for cell growth. J. Biol. Chem. 1988, 263, 6296–6301. [Google Scholar]
- Goel, S.; Hidalgo, M.; Perez-Soler, R. EGFR inhibitor-mediated apoptosis in solid tumors. J. Exp. Ther. Oncol. 2007, 6, 305–320. [Google Scholar]
- Lingwood, C.A. Role of verotoxin receptors in pathogenesis. Trends Microbiol. 1996, 4, 147–153. [Google Scholar]
- Wiels, J.; Fellous, M.; Tursz, T. Monoclonal antibody against a Burkitt lymphoma-associated antigen. Proc. Natl. Acad. Sci. USA 1981, 78, 6485–6488. [Google Scholar]
- Distler, U.; Souady, J.; Hulsewig, M.; Drmic-Hofman, I.; Haier, J.; Friedrich, A.W.; Karch, H.; Senninger, N.; Dreisewerd, K.; Berkenkamp, S.; et al. Shiga toxin receptor Gb3Cer/CD77: Tumor-association and promising therapeutic target in pancreas and colon cancer. PLoS One 2009, 4, e6813. [Google Scholar]
- Johansson, D.; Kosovac, E.; Moharer, J.; Ljuslinder, I.; Brannstrom, T.; Johansson, A.; Behnam-Motlagh, P. Expression of verotoxin-1 receptor Gb3 in breast cancer tissue and verotoxin-1 signal transduction to apoptosis. BMC Cancer 2009, 9, 67. [Google Scholar]
- Liu, Y.Y.; Gupta, V.; Patwardhan, G.A.; Bhinge, K.; Zhao, Y.; Bao, J.; Mehendale, H.; Cabot, M.C.; Li, Y.T.; Jazwinski, S.M. Glucosylceramide synthase upregulates MDR1 expression in the regulation of cancer drug resistance through cSrc and β-catenin signaling. Mol. Cancer 2010, 9, 145. [Google Scholar]
- Willison, K.R.; Karol, R.A.; Suzuki, A.; Kundu, S.K.; Marcus, D.M. Neutral glycolipid antigens as developmental markers of mouse teratocarcinoma and early embryos: An immunologic and chemical analysis. J. Immunol. 1982, 129, 603–609. [Google Scholar]
- Park, S.Y.; Kwak, C.Y.; Shayman, J.A.; Kim, J.H. Globoside promotes activation of ERK by interaction with the epidermal growth factor receptor. Biochim. Biophys. Acta 2012, 1820, 1141–1148. [Google Scholar]
- Ruckhaberle, E.; Karn, T.; Rody, A.; Hanker, L.; Gatje, R.; Metzler, D.; Holtrich, U.; Kaufmann, M. Gene expression of ceramide kinase galactosyl ceramide synthase and ganglioside GD3 synthase is associated with prognosis in breast cancer. J. Cancer Res. Clin. Oncol. 2009, 135, 1005–1013. [Google Scholar]
- Ko, K.; Furukawa, K.; Takahashi, T.; Urano, T.; Sanai, Y.; Nagino, M.; Nimura, Y.; Furukawa, K. Fundamental study of small interfering RNAs for ganglioside GD3 synthase gene as a therapeutic target of lung cancers. Oncogene 2006, 25, 6924–6935. [Google Scholar]
- Wada, T.; Hata, K.; Yamaguchi, K.; Shiozaki, K.; Koseki, K.; Moriya, S.; Miyagi, T. A crucial role of plasma membrane-associated sialidase in the survival of human cancer cells. Oncogene 2007, 26, 2483–2490. [Google Scholar]
- Mikhaylova, O.; Stratton, Y.; Hall, D.; Kellner, E.; Ehmer, B.; Drew, A.F.; Gallo, C.A.; Plas, D.R.; Biesiada, J.; Meller, J.; et al. VHL-regulated MiR-204 suppresses tumor growth through inhibition of LC3B-mediated autophagy in renal clear cell carcinoma. Cancer Cell 2012, 21, 532–546. [Google Scholar]
- Kakugawa, Y.; Wada, T.; Yamaguchi, K.; Yamanami, H.; Ouchi, K.; Sato, I.; Miyagi, T. Up-regulation of plasma membrane-associated ganglioside sialidase (Neu3) in human colon cancer and its involvement in apoptosis suppression. Proc. Natl. Acad. Sci. USA 2002, 99, 10718–10723. [Google Scholar]
- Miyata, M.; Kambe, M.; Tajima, O.; Moriya, S.; Sawaki, H.; Hotta, H.; Kondo, Y.; Narimatsu, H.; Miyagi, T.; Furukawa, K.; et al. Membrane sialidase NEU3 is highly expressed in human melanoma cells promoting cell growth with minimal changes in the composition of gangliosides. Cancer Sci. 2011, 102, 2139–2149. [Google Scholar]
- Kawamura, S.; Sato, I.; Wada, T.; Yamaguchi, K.; Li, Y.; Li, D.; Zhao, X.; Ueno, S.; Aoki, H.; Tochigi, T.; et al. Plasma membrane-associated sialidase (NEU3) regulates progression of prostate cancer to androgen-independent growth through modulation of androgen receptor signaling. Cell Death Differ. 2012, 19, 170–179. [Google Scholar]
- Scaringi, R.; Piccoli, M.; Papini, N.; Cirillo, F.; Conforti, E.; Bergante, S.; Tringali, C.; Garatti, A.; Gelfi, C.; Venerando, B.; et al. NEU3 sialidase is activated under hypoxia and protects skeletal muscle cells from apoptosis through the activation of the epidermal growth factor receptor signaling pathway and the hypoxia-inducible factor (HIF)-1α. J. Biol. Chem. 2013, 288, 3153–3162. [Google Scholar]
- Miyagi, T.; Wada, T.; Yamaguchi, K. Roles of plasma membrane-associated sialidase NEU3 in human cancers. Biochim. Biophys. Acta 2008, 1780, 532–537. [Google Scholar]
- Yamaguchi, K.; Hata, K.; Koseki, K.; Shiozaki, K.; Akita, H.; Wada, T.; Moriya, S.; Miyagi, T. Evidence for mitochondrial localization of a novel human sialidase (NEU4). Biochem. J. 2005, 390, 85–93. [Google Scholar]
- Basu, S.; Ma, R.; Mikulla, B.; Bradley, M.; Moulton, C.; Basu, M.; Banerjee, S.; Inokuchi, J. Apoptosis of human carcinoma cells in the presence of inhibitors of glycosphingolipid biosynthesis: I Treatment of Colo-205 and SKBR3 cells with isomers of PDMP and PPMP. Glycoconj. J. 2004, 20, 157–168. [Google Scholar]
- Chai, L.; McLaren, R.P.; Byrne, A.; Chuang, W.L.; Huang, Y.; Dufault, M.R.; Pacheco, J.; Madhiwalla, S.; Zhang, X.; Zhang, M.; et al. The chemosensitizing activity of inhibitors of glucosylceramide synthase is mediated primarily through modulation of P-gp function. Int. J. Oncol. 2011, 38, 701–711. [Google Scholar]
- Sun, Y.L.; Zhou, G.Y.; Li, K.N.; Gao, P.; Zhang, Q.H.; Zhen, J.H.; Bai, Y.H.; Zhang, X.F. Suppression of glucosylceramide synthase by RNA interference reverses multidrug resistance in human breast cancer cells. Neoplasma 2006, 53, 1–8. [Google Scholar]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar]

© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Giussani, P.; Tringali, C.; Riboni, L.; Viani, P.; Venerando, B. Sphingolipids: Key Regulators of Apoptosis and Pivotal Players in Cancer Drug Resistance. Int. J. Mol. Sci. 2014, 15, 4356-4392. https://doi.org/10.3390/ijms15034356
Giussani P, Tringali C, Riboni L, Viani P, Venerando B. Sphingolipids: Key Regulators of Apoptosis and Pivotal Players in Cancer Drug Resistance. International Journal of Molecular Sciences. 2014; 15(3):4356-4392. https://doi.org/10.3390/ijms15034356
Chicago/Turabian StyleGiussani, Paola, Cristina Tringali, Laura Riboni, Paola Viani, and Bruno Venerando. 2014. "Sphingolipids: Key Regulators of Apoptosis and Pivotal Players in Cancer Drug Resistance" International Journal of Molecular Sciences 15, no. 3: 4356-4392. https://doi.org/10.3390/ijms15034356