Alcohol Induced Hepatic Degeneration in a Hepatitis C Virus Core Protein Transgenic Mouse Model

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
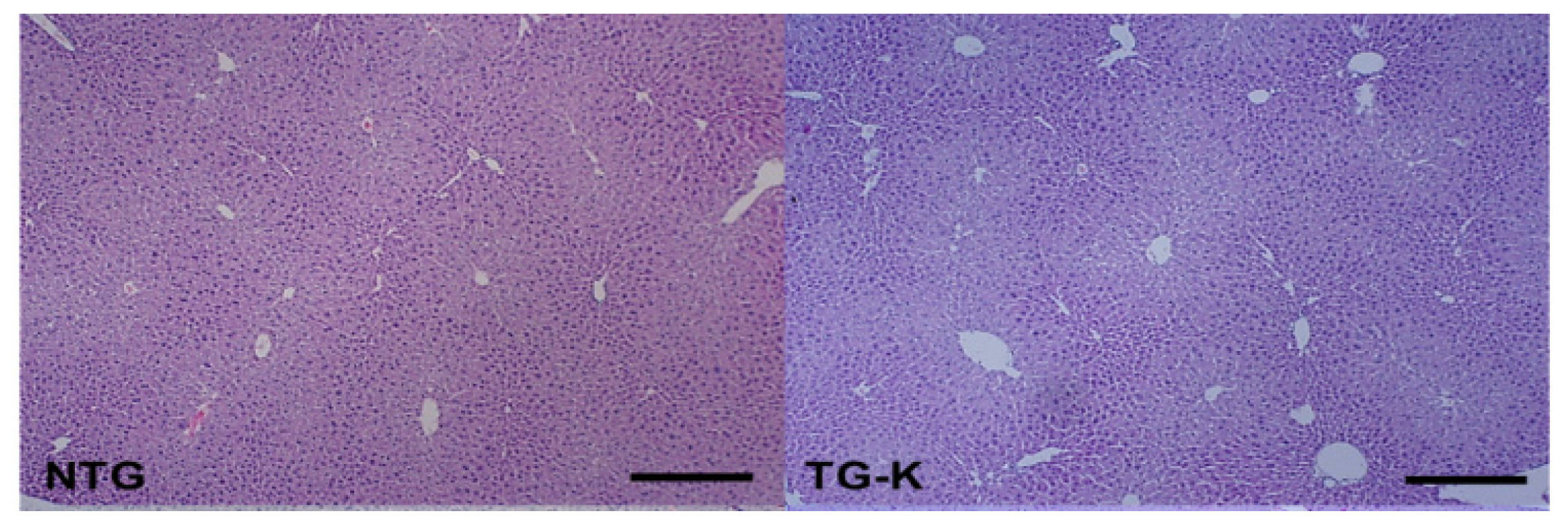
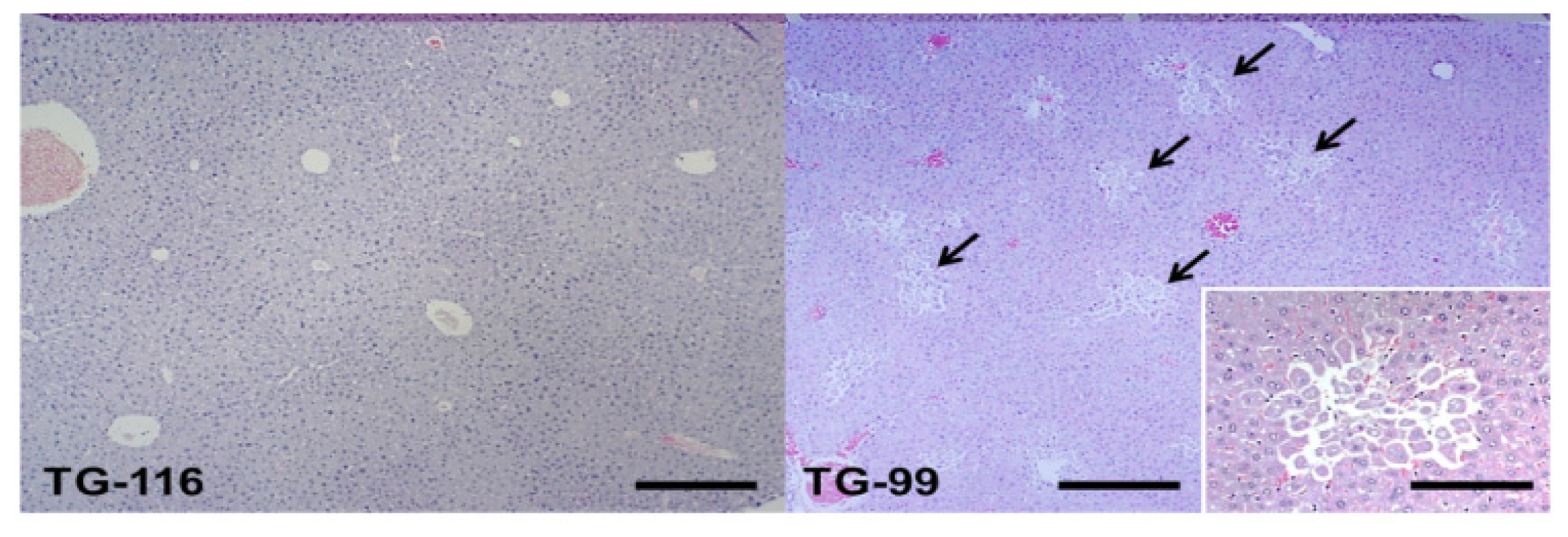
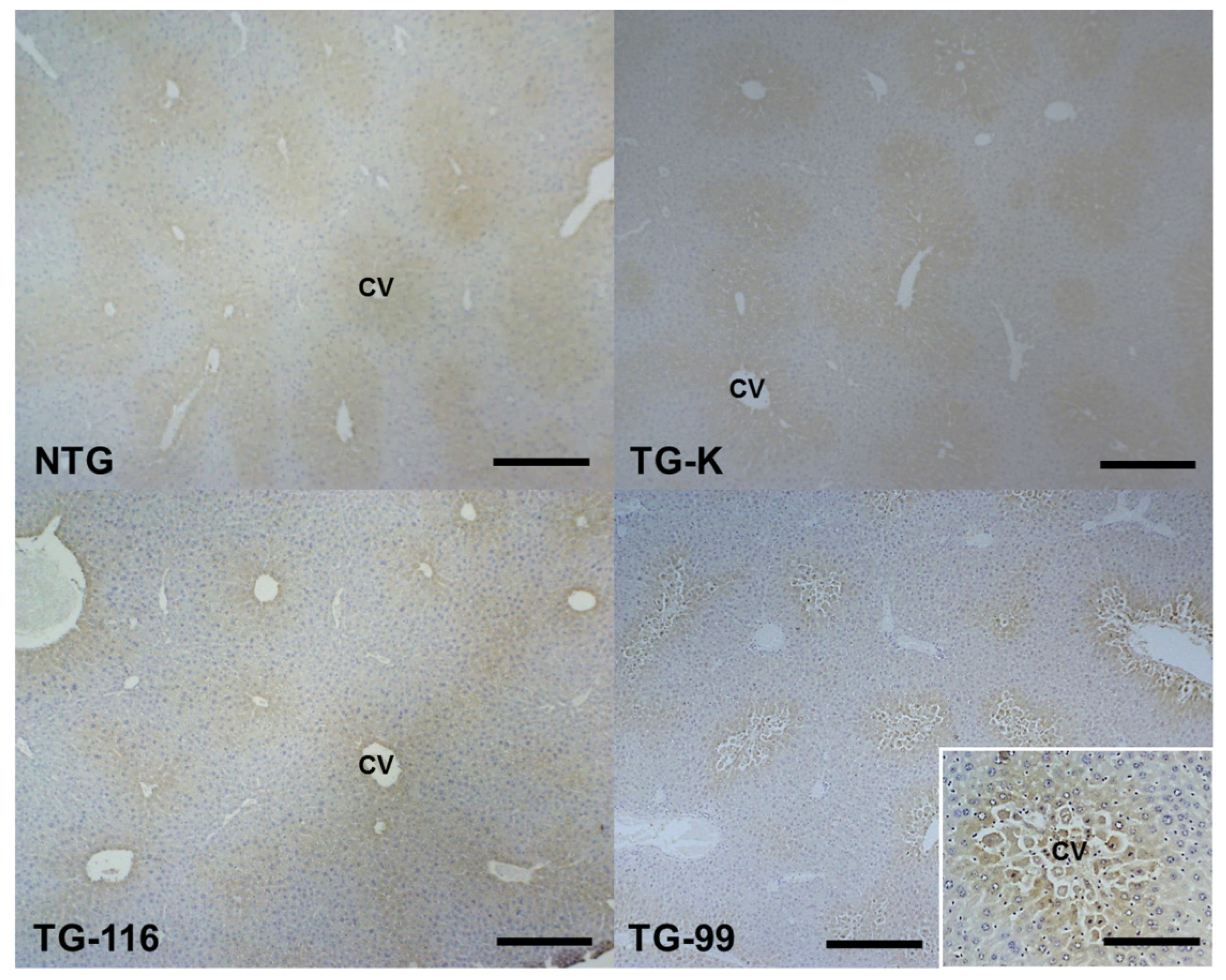
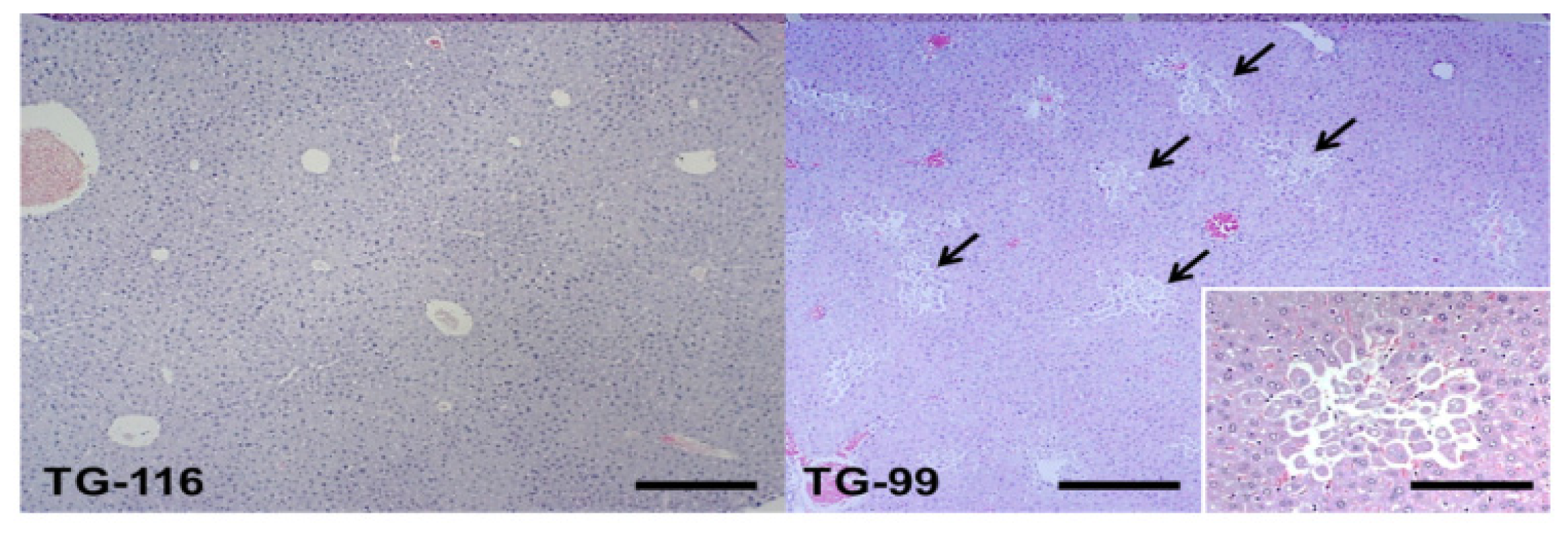
2.1. Histopathological Changes in the HCV Core Tg-Mice
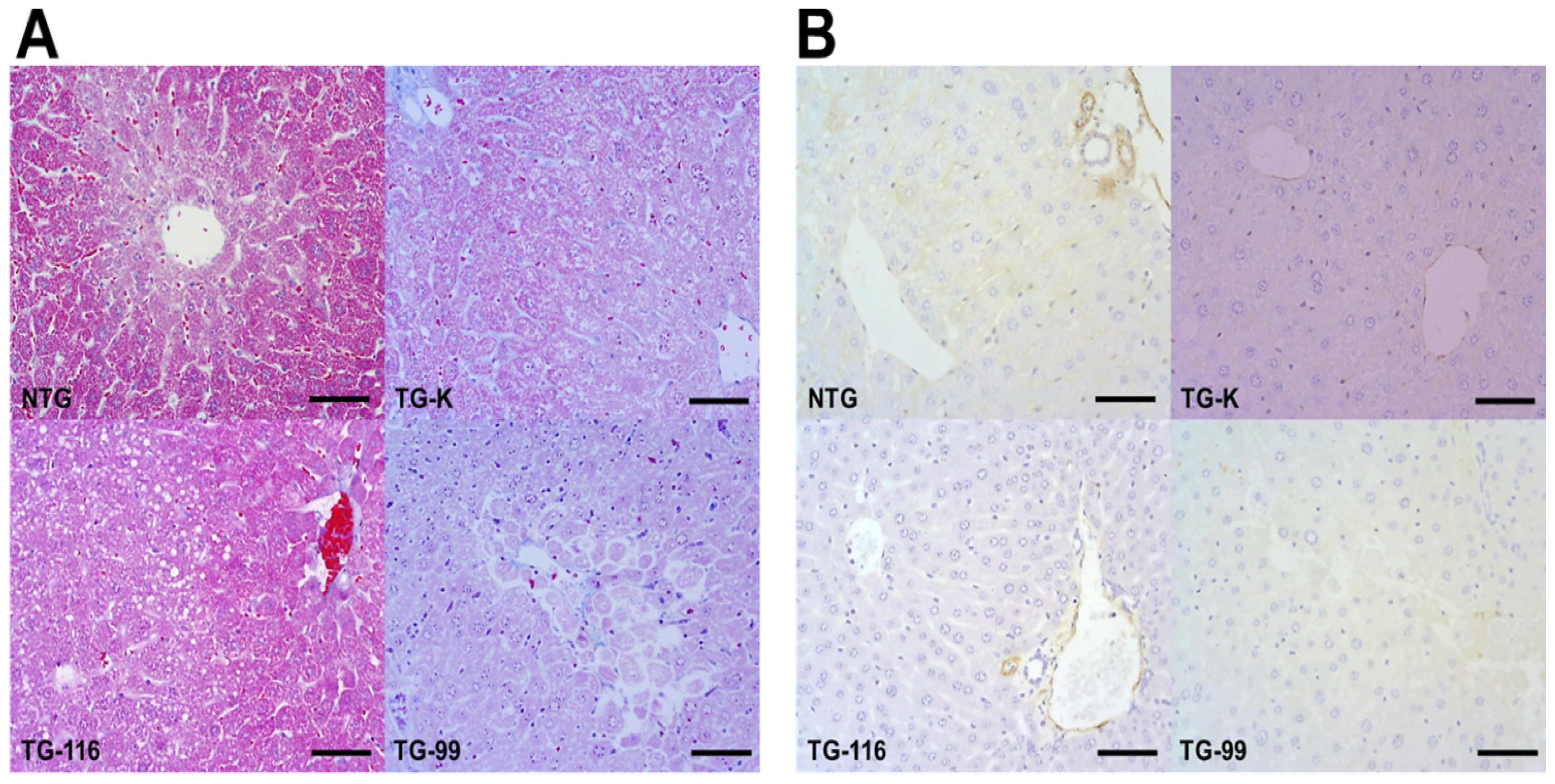
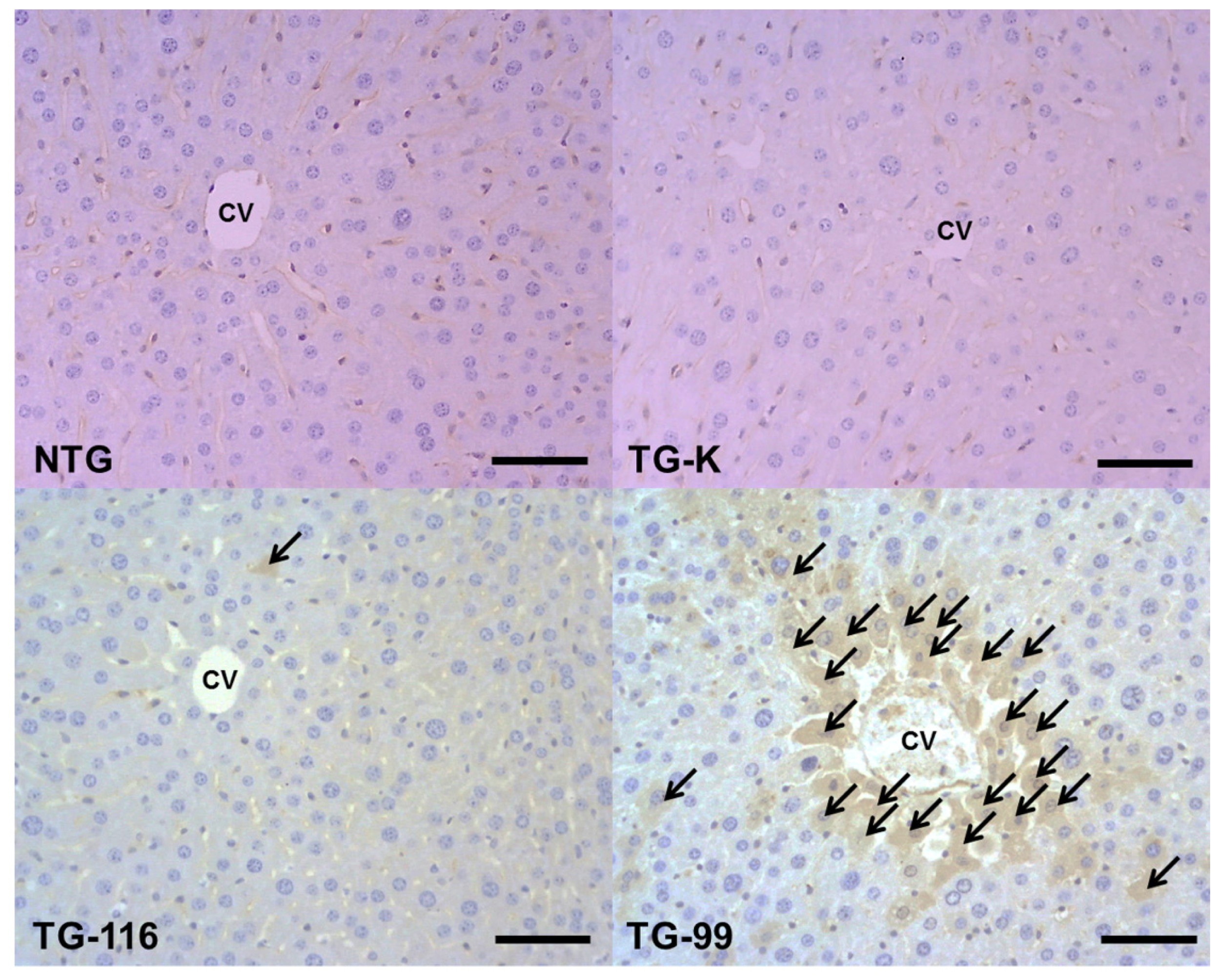
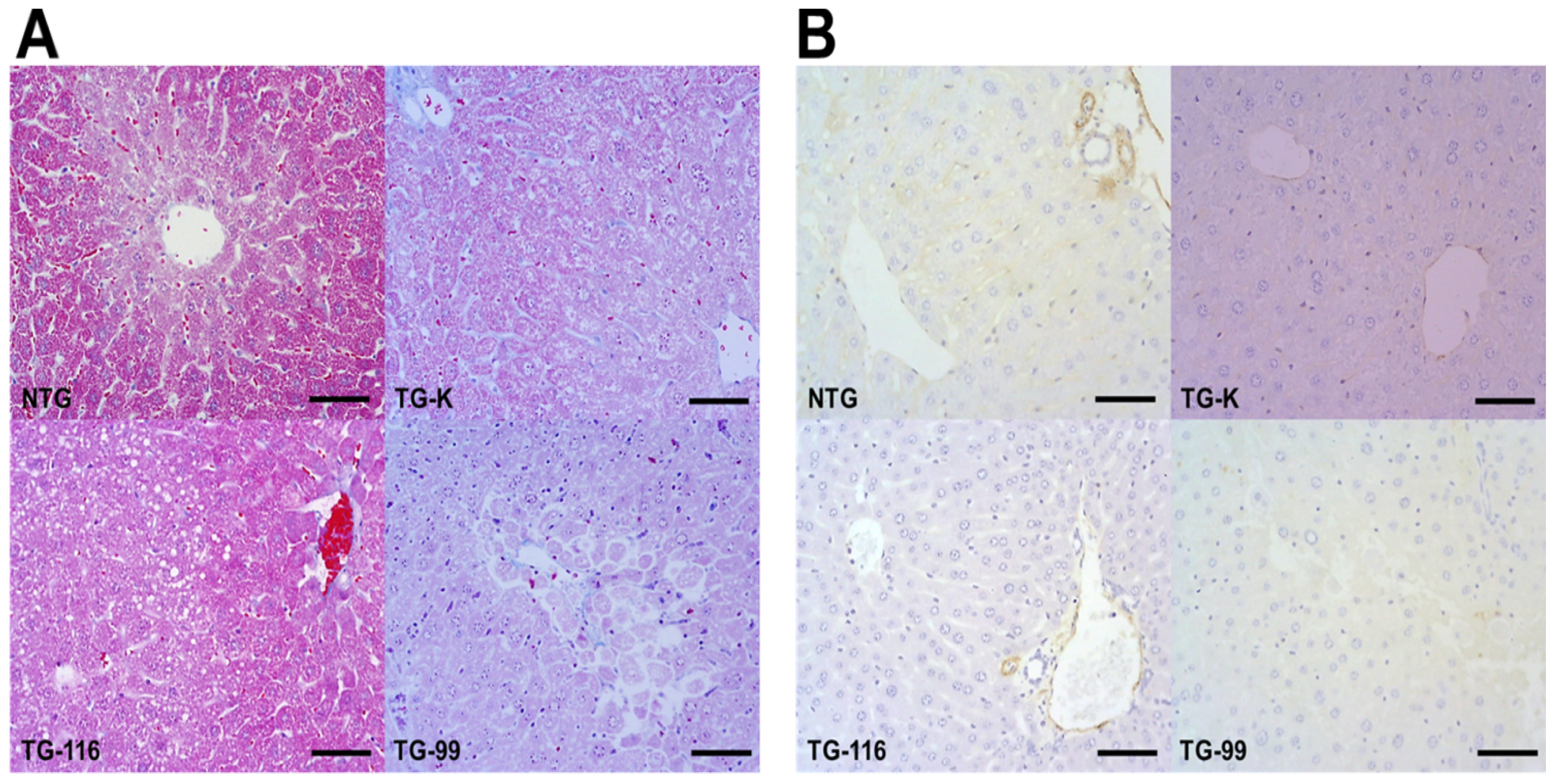
2.2. Evaluation of Collagen Production
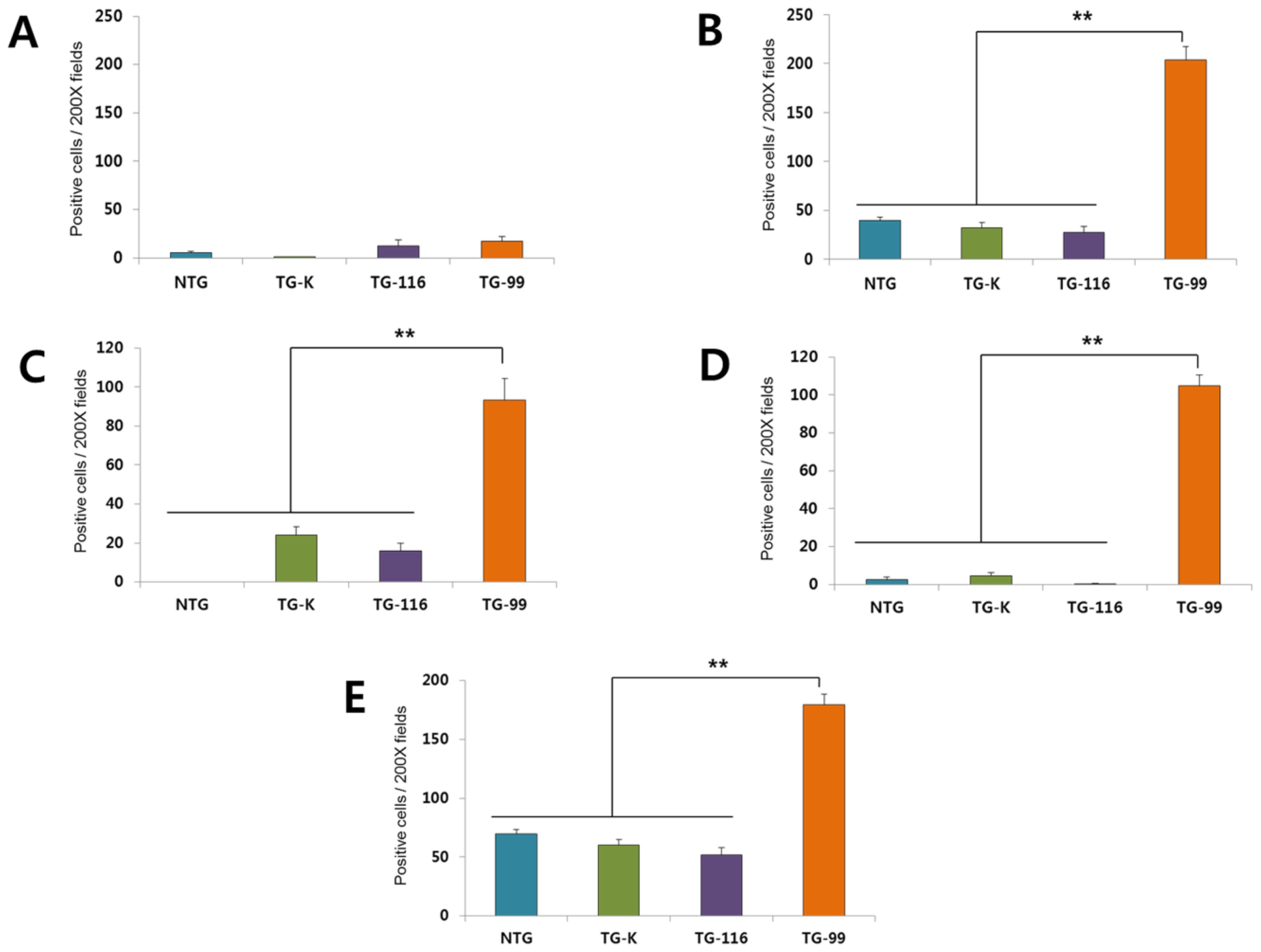
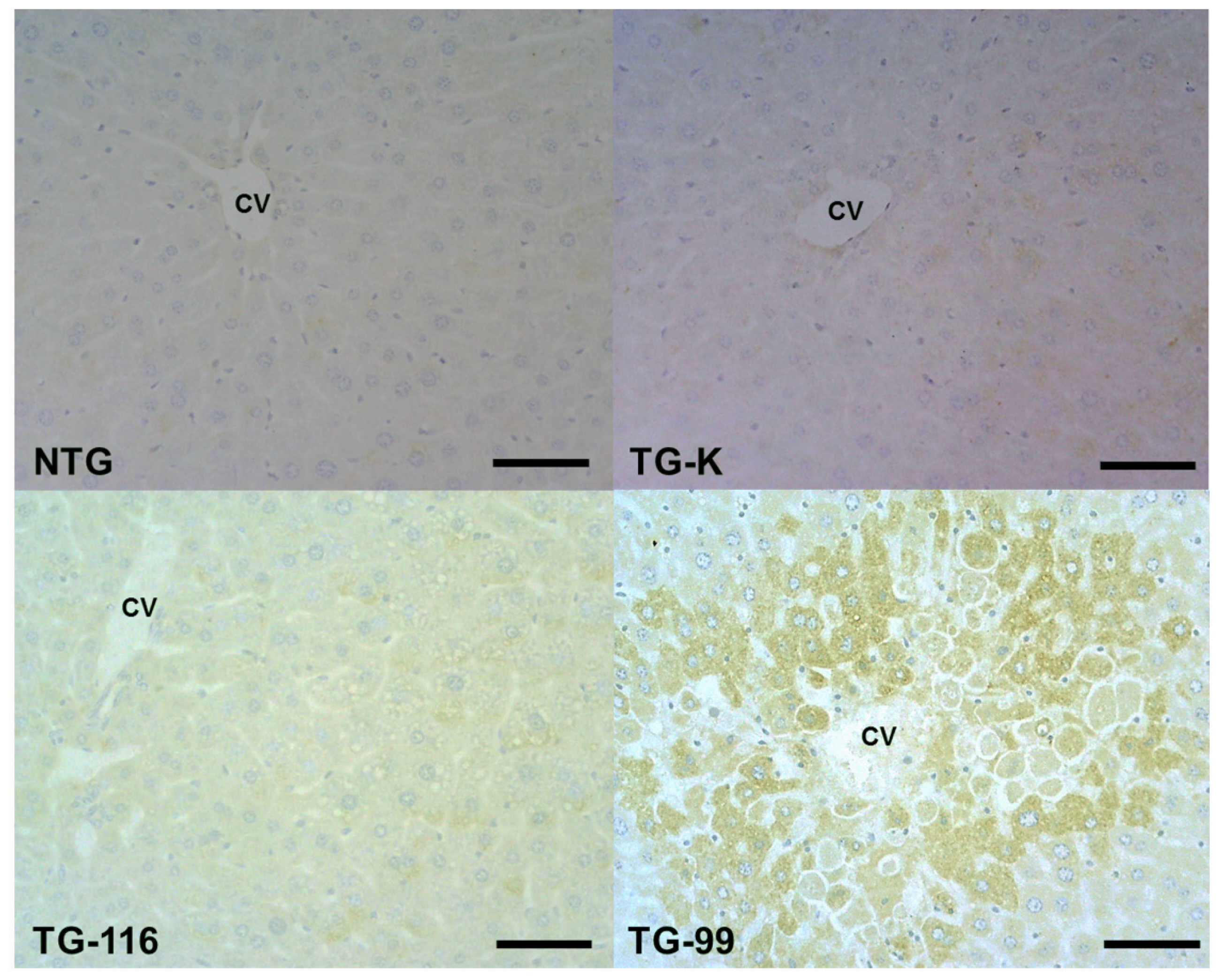
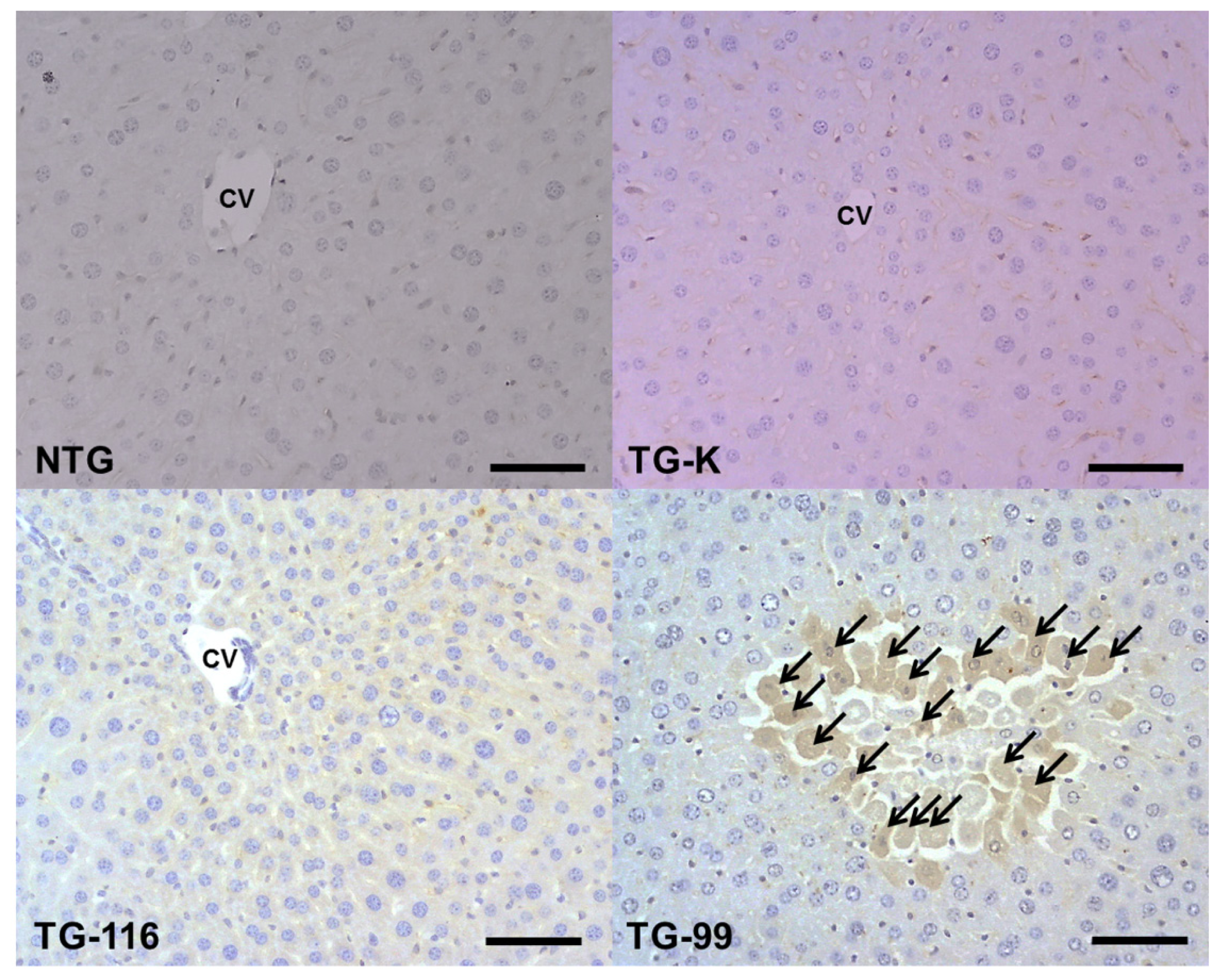
2.3. Identification of Marker for Alcoholic Hepatitis in HCV Core Tg-Mice
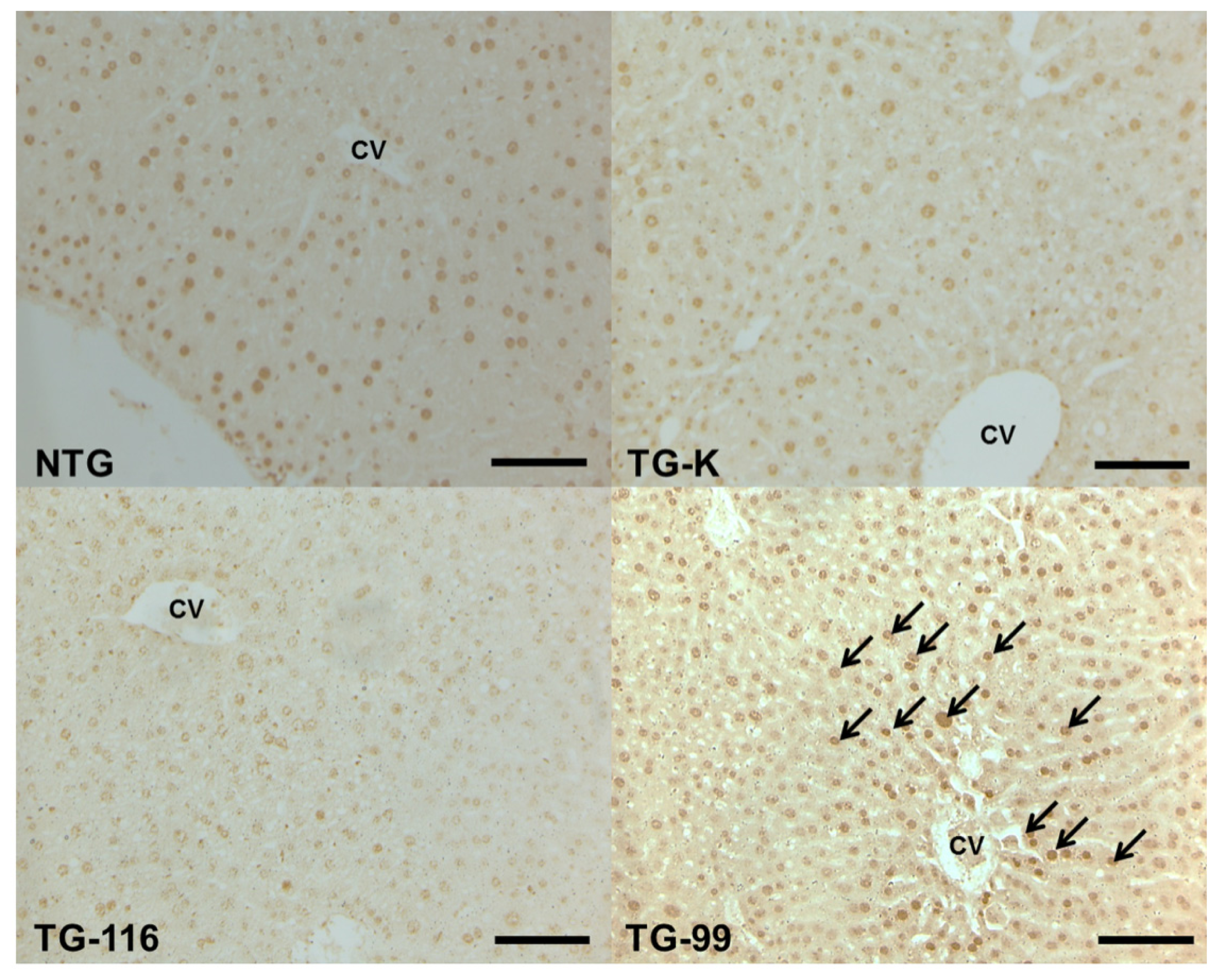
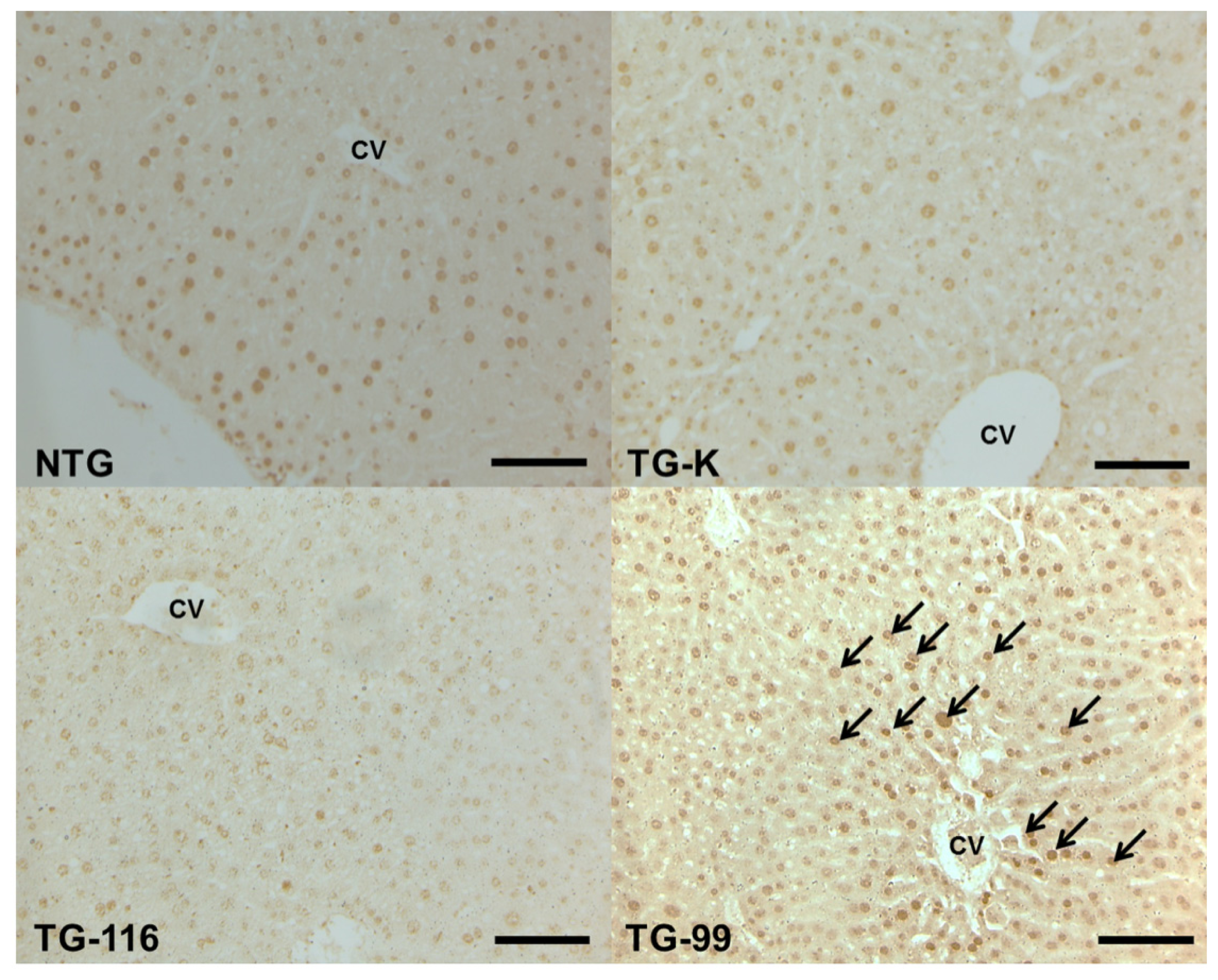
2.4. TGF-β1 Signaling Cascade in HCV Core Tg-Mice
3. Discussion
4. Experimental Section
4.1. Animals and Experimental Design
4.2. Histopathologic and Immunohistochemistry Analyses
4.3. Statistical Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsDong-Hyung Noh performed animal experiment and wrote manuscript. Eun-Joo Lee interpreted data and wrote manuscript. Ah-Young Kim and Eun-Mi Lee performed the statistical analysis. Chang-Woo Min and Kyung-Ku Kang carried out immunohistochemistry. Myeong-Mi Lee, Sang-Hyeob Kim and Soo-Eun Sung helped the animal experiment and immunohistochemistry. Meeyul Hwang, Dae-Yeul Yu and Kyu-Shik Jeong helped to draft the manuscript.
References
- Irshad, M.; Dhar, I. Hepatitis C virus core protein: An update on its molecular biology cellular functions and clinical implications. Med. Princ. Pract 2006, 15, 405–416. [Google Scholar]
- Shimotohno, K. Hepatitis C virus and its pathogenesis. Semin. Cancer Biol 2000, 10, 233–240. [Google Scholar]
- Irshad, M.; Mankotia, D.S.; Irshad, K. An insight into the diagnosis and pathogenesis of hepatitis C virus infection. World J. Gastroenterol 2013, 19, 7896–7909. [Google Scholar]
- Moudgil, V.; Redhu, D.; Dhanda, S.; Singh, J. A review of molecular mechanisms in the development of hepatocellular carcinoma by aflatoxin and hepatitis B and C viruses. J. Environ. Pathol. Toxicol. Oncol 2013, 32, 165–175. [Google Scholar]
- Koike, K. Hepatitis C virus contributes to hepatocarcinogenesis by modulating metabolic and intracellular signaling pathways. J. Gastroenterol. Hepatol 2007, 22, S108–S111. [Google Scholar]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med 1998, 4, 1065–1067. [Google Scholar]
- Dubourdeau, M.; Miyamura, T.; Matsuura, Y.; Alric, L.; Pipy, B.; Rousseau, D. Infection of HepG2 cells with recombinant adenovirus encoding the HCV core protein induces p21(WAF1) down-regulation–Effect of transforming growth factor beta. J. Hepatol 2002, 37, 486–492. [Google Scholar]
- Honda, A.; Hatano, M.; Kohara, M.; Arai, Y.; Hartatik, T.; Moriyama, T.; Imawari, M.; Koike, K.; Yokosuka, O.; Shimotohno, K.; et al. HCV-core protein accelerates recovery from the insensitivity of liver cells to fas-mediated apoptosis induced by an injection of anti-fas antibody in mice. J. Hepatol 2000, 33, 440–447. [Google Scholar]
- Morgan, T.R.; Brenner, D.; Everhart, J.; French, S.W.; Fried, M.W.; Gretch, D.R.; Koziel, M.J.; McClain, C.J.; Peters, M.G.; Weinman, S.A.; et al. Hepatitis C and alcohol: Fundamental and translational research directions. Alcohol. Clin. Exp. Res 2003, 27, 726–731. [Google Scholar]
- Cardin, R.; D’Errico, A.; Fiorentino, M.; Cecchetto, A.; Naccarato, R.; Farinati, F. Hepatocyte proliferation and apoptosis in relation to oxidative damage in alcohol-related liver disease. Alcohol Alcohol 2002, 37, 43–48. [Google Scholar]
- Niemela, O. Distribution of ethanol-induced protein adducts in vivo: Relationship to tissue injury. Free Radic. Biol. Med 2001, 31, 1533–1538. [Google Scholar]
- Comporti, M.; Signorini, C.; Leoncini, S.; Gardi, C.; Ciccoli, L.; Giardini, A.; Vecchio, D.; Arezzini, B. Ethanol-induced oxidative stress: Basic knowledge. Genes Nutr 2010, 5, 101–109. [Google Scholar]
- Federico, A.; Morgillo, F.; Tuccillo, C.; Ciardiello, F.; Loguercio, C. Chronic inflammation and oxidative stress in human carcinogenesis. Int. J. Cancer 2007, 121, 2381–2386. [Google Scholar]
- Gitto, S.; Micco, L.; Conti, F.; Andreone, P.; Bernardi, M. Alcohol and viral hepatitis: A mini-review. Dig. Liver Dis 2009, 41, 67–70. [Google Scholar]
- Cooksley, W. Chronic liver disease: Do alcohol and hepatitis C virus. Interact. J. Gastroenterol. Hepatol 1996, 11, 187–192. [Google Scholar]
- Moriya, K.; Nakagawa, K.; Santa, T.; Shintani, Y.; Fujie, H.; Miyoshi, H.; Tsutsumi, T.; Miyazawa, T.; Ishibashi, K.; Horie, T.; et al. Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res 2001, 61, 4365–4370. [Google Scholar]
- Donato, F.; Tagger, A.; Chiesa, R.; Ribero, M.L.; Tomasoni, V.; Fasola, M.; Gelatti, U.; Portera, G.; Boffetta, P.; Nardi, G. Hepatitis B and C virus infection alcohol drinking and hepatocellular carcinoma: A case-control study in Italy Brescia HCC study. Hepatology 1997, 26, 579–584. [Google Scholar]
- Mercer, D.F. Animal models for studying hepatitis C and alcohol effects on liver. World J. Gastroenterol 2011, 17, 2515–2519. [Google Scholar]
- Wang, A.G.; Moon, H.B.; Lee, Y.H.; Yu, S.L.; Kwon, H.J.; Han, Y.H.; Fang, W.; Lee, T.H.; Jang, K.L.; Kim, S.K.; et al. Transgenic expression of Korean type hepatitis C virus core protein and related mutants in mice. Exp. Mol. Med 2004, 36, 588–593. [Google Scholar]
- Shih, C.M.; Chen, C.M.; Chen, S.Y.; Lee, Y.H. Modulation of the trans-suppression activity of hepatitis C virus core protein by phosphorylation. J. Virol 1995, 69, 1160–1171. [Google Scholar]
- Ekstrom, G.; Ingelman-Sundberg, M. Rat liver microsomal NADPH-supported oxidase activity and lipid peroxidation dependent on ethanol-inducible cytochrome P-450 (P-450IIE1). Biochem. Pharmacol 1989, 38, 1313–1319. [Google Scholar]
- Zatloukal, K.; Stumptner, C.; Lehner, M.; Denk, H.; Baribault, H.; Eshkind, L.G.; Franke, W.W. Cytokeratin 8 protects from hepatotoxicity and its ratio to cytokeratin 18 determines the ability of hepatocytes to form mallory bodies. Am. J. Pathol 2000, 156, 1263–1274. [Google Scholar]
- Shen, H.; Huang, G.; Hadi, M.; Choy, P.; Zhang, M.; Minuk, G.Y.; Chen, Y.; Gong, Y. Transforming growth factor-beta1 downregulation of smad1 gene expression in rat hepatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol 2003, 285, G539–G546. [Google Scholar]
- Matsuzaki, K. Modulation of TGF-beta signaling during progression of chronic liver diseases. Front. Biosci. (Landmark Ed.) 2009, 14, 2923–2934. [Google Scholar]
- Kessova, I.; Cederbaum, A.I. CYP2E1: Biochemistry toxicology regulation and function in ethanol-induced liver injury. Curr. Mol. Med 2003, 3, 509–518. [Google Scholar]
- Lai, M.M. Hepatitis C virus proteins: Direct link to hepatic oxidative stress steatosis carcinogenesis and more. Gastroenterology 2002, 122, 568–571. [Google Scholar]
- Okuda, M.; Li, K.; Beard, M.R.; Showalter, L.A.; Scholle, F.; Lemon, S.M.; Weinman, S.A. Mitochondrial injury oxidative stress and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology 2002, 122, 366–375. [Google Scholar]
- De la, M.; Hall, P.; Lieber, C.S.; DeCarli, L.M.; French, S.W.; Lindros, K.O.; Jarvelainen, H.; Bode, C.; Parlesak, A.; Bode, J.C. Models of alcoholic liver disease in rodents: A critical evaluation. Alcohol. Clin. Exp. Res 2001, 25, 254S–261S. [Google Scholar]
- Jensen, K.; Gluud, C. The mallory body: Morphological clinical and experimental studies (part 1 of a literature survey). Hepatology 1994, 20, 1061–1077. [Google Scholar]
- Jensen, K.; Gluud, C. The mallory body: Theories on development and pathological significance (part 2 of a literature survey). Hepatology 1994, 20, 1330–1342. [Google Scholar]
- Wang, M.X.; Morgan, T.; Lungo, W.; Wang, L.; Sze, G.Z.; French, S.W. “Piecemeal” necrosis: Renamed troxis necrosis. Exp. Mol. Pathol 2001, 71, 137–146. [Google Scholar]
- Matsuzaki, K.; Seki, T.; Okazaki, K. TGF-beta signal shifting between tumor suppression and fibro-carcinogenesis in human chronic liver diseases. J. Gastroenterol 2013. [Google Scholar] [CrossRef]
- Wagayama, H.; Shiraki, K.; Sugimoto, K.; Ito, T.; Fujikawa, K.; Yamanaka, T.; Takase, K.; Nakano, T. High expression of p21WAF1/CIP1 is correlated with human hepatocellular carcinoma in patients with hepatitis C virus-associated chronic liver diseases. Hum. Pathol 2002, 33, 429–434. [Google Scholar]
- Elbendary, A.A.; Cirisano, F.D.; Evans, A.C., Jr; Davis, P.L.; Iglehart, J.D.; Marks, J.R.; Berchuck, A. Relationship between p21 expression and mutation of the p53 tumor suppressor gene in normal and malignant ovarian epithelial cells. Clin. Cancer Res 1996, 2, 1571–1575. [Google Scholar]
- Suzuki, A.; Tsutomi, Y.; Akahane, K.; Araki, T.; Miura, M. Resistance to fas-mediated apoptosis: activation of caspase 3 is regulated by cell cycle regulator p21WAF1 and IAP gene family ILP. Oncogene 1998, 17, 931–939. [Google Scholar]
- Pines, J. Cell cycle p21 inhibits cyclin shock. Nature 1994, 369, 520–521. [Google Scholar]
- Datto, M.B.; Li, Y.; Panus, J.F.; Howe, D.J.; Xiong, Y.; Wang, X.F. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 5545–5549. [Google Scholar]
- Russo, T.; Zambrano, N.; Esposito, F.; Ammendola, R.; Cimino, F.; Fiscella, M.; Jackman, J.; O’Connor, P.M.; Anderson, C.W.; Appella, E. A p53-independent pathway for activation of WAF1/CIP1 expression following oxidative stress. J. Biol. Chem 1995, 270, 29386–29391. [Google Scholar]
- Qiu, X.; Forman, H.J.; Schonthal, A.H.; Cadenas, E. Induction of p21 mediated by reactive oxygen species formed during the metabolism of aziridinylbenzoquinones by HCT116 cells. J. Biol. Chem 1996, 271, 31915–31921. [Google Scholar]
- Marx, J. Cell biology Cell cycle inhibitors may help brake growth as cells develop. Science 1995, 267, 963–964. [Google Scholar]
- Gorospe, M.; Shack, S.; Guyton, K.Z.; Samid, D.; Holbrook, N.J. Up-regulation and functional role of p21Waf1/Cip1 during growth arrest of human breast carcinoma MCF-7 cells by phenylacetate. Cell Growth Differ 1996, 7, 1609–1615. [Google Scholar]
- Guo, W.; Baluda, M.A.; Park, N.H. Ethanol upregulates the expression of p21 WAF1/CIP1 and prolongs G1 transition via a p53-independent pathway in human epithelial cells. Oncogene 1997, 15, 1143–1149. [Google Scholar]
- Kobayashi, S.; Matsushita, K.; Saigo, K.; Urashima, T.; Asano, T.; Hayashi, H.; Ochiai, T. P21WAF1/CIP1 messenger RNA expression in hepatitis B, C virus-infected human hepatocellular carcinoma tissues. Cancer 2001, 91, 2096–2103. [Google Scholar]
- Ozcelik, H.; Mousses, S.; Andrulis, I.L. Low levels of expression of an inhibitor of cyclin-dependent kinases (CIP1/WAF1) in primary breast carcinomas with p53 mutations. Clin. Cancer Res 1995, 1, 907–912. [Google Scholar]
- Kondo, T.; Suda, T.; Fukuyama, H.; Adachi, M.; Nagata, S. Essential roles of the fas ligand in the development of hepatitis. Nat. Med 1997, 3, 409–413. [Google Scholar]
- Qin, L.F.; Ng, I.O. Expression of p27(KIP1) and p21(WAF1/CIP1) in primary hepatocellular carcinoma: Clinicopathologic correlation and survival analysis. Hum. Pathol 2001, 32, 778–784. [Google Scholar]
- Yamanaka, T.; Kodama, T.; Doi, T. Subcellular localization of HCV core protein regulates its ability for p53 activation and p21 suppression. Biochem. Biophys. Res. Commun 2002, 294, 528–534. [Google Scholar]
- Yamanaka, T.; Uchida, M.; Doi, T. Innate form of HCV core protein plays an important role in the localization and the function of HCV core protein. Biochem. Biophys. Res. Commun 2002, 294, 521–527. [Google Scholar]
- Matsuzaki, K. Smad phosphoisoform signals in acute and chronic liver injury: Similarities and differences between epithelial and mesenchymal cells. Cell Tissue Res 2012, 347, 225–243. [Google Scholar]
- Hayashi, H.; Sakai, T. Animal models for the study of liver fibrosis: New insights from knockout mouse models. Am. J. Physiol. Gastrointest. Liver Physiol 2011, 300, G729–G738. [Google Scholar]
- Brandon-Warner, E.; Schrum, L.W.; Schmidt, C.M.; McKillop, I.H. Rodent models of alcoholic liver disease: Of mice and men. Alcohol 2012, 46, 715–725. [Google Scholar]
- Arteel, G.E. Animal models of alcoholic liver disease. Dig. Dis 2010, 28, 729–736. [Google Scholar]






© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Noh, D.-H.; Lee, E.-J.; Kim, A.-Y.; Lee, E.-M.; Min, C.-W.; Kang, K.-K.; Lee, M.-M.; Kim, S.-H.; Sung, S.-E.; Hwang, M.; et al. Alcohol Induced Hepatic Degeneration in a Hepatitis C Virus Core Protein Transgenic Mouse Model. Int. J. Mol. Sci. 2014, 15, 4126-4141. https://doi.org/10.3390/ijms15034126
Noh D-H, Lee E-J, Kim A-Y, Lee E-M, Min C-W, Kang K-K, Lee M-M, Kim S-H, Sung S-E, Hwang M, et al. Alcohol Induced Hepatic Degeneration in a Hepatitis C Virus Core Protein Transgenic Mouse Model. International Journal of Molecular Sciences. 2014; 15(3):4126-4141. https://doi.org/10.3390/ijms15034126
Chicago/Turabian StyleNoh, Dong-Hyung, Eun-Joo Lee, Ah-Young Kim, Eun-Mi Lee, Chang-Woo Min, Kyung-Ku Kang, Myeong-Mi Lee, Sang-Hyeob Kim, Soo-Eun Sung, Meeyul Hwang, and et al. 2014. "Alcohol Induced Hepatic Degeneration in a Hepatitis C Virus Core Protein Transgenic Mouse Model" International Journal of Molecular Sciences 15, no. 3: 4126-4141. https://doi.org/10.3390/ijms15034126