PseAAC-General: Fast Building Various Modes of General Form of Chou’s Pseudo-Amino Acid Composition for Large-Scale Protein Datasets

Abstract
:1. Introduction
2. Results and Discussion
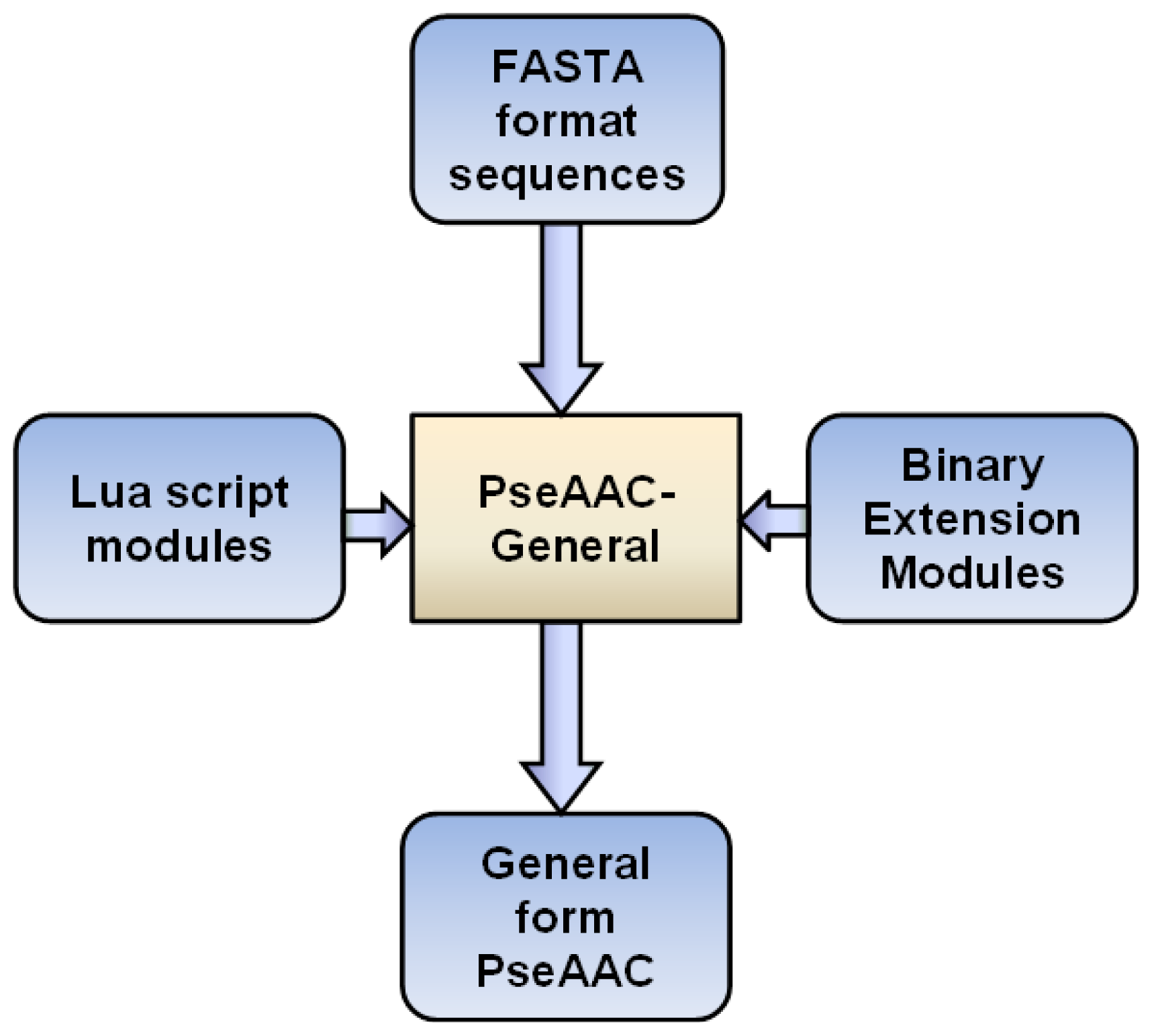
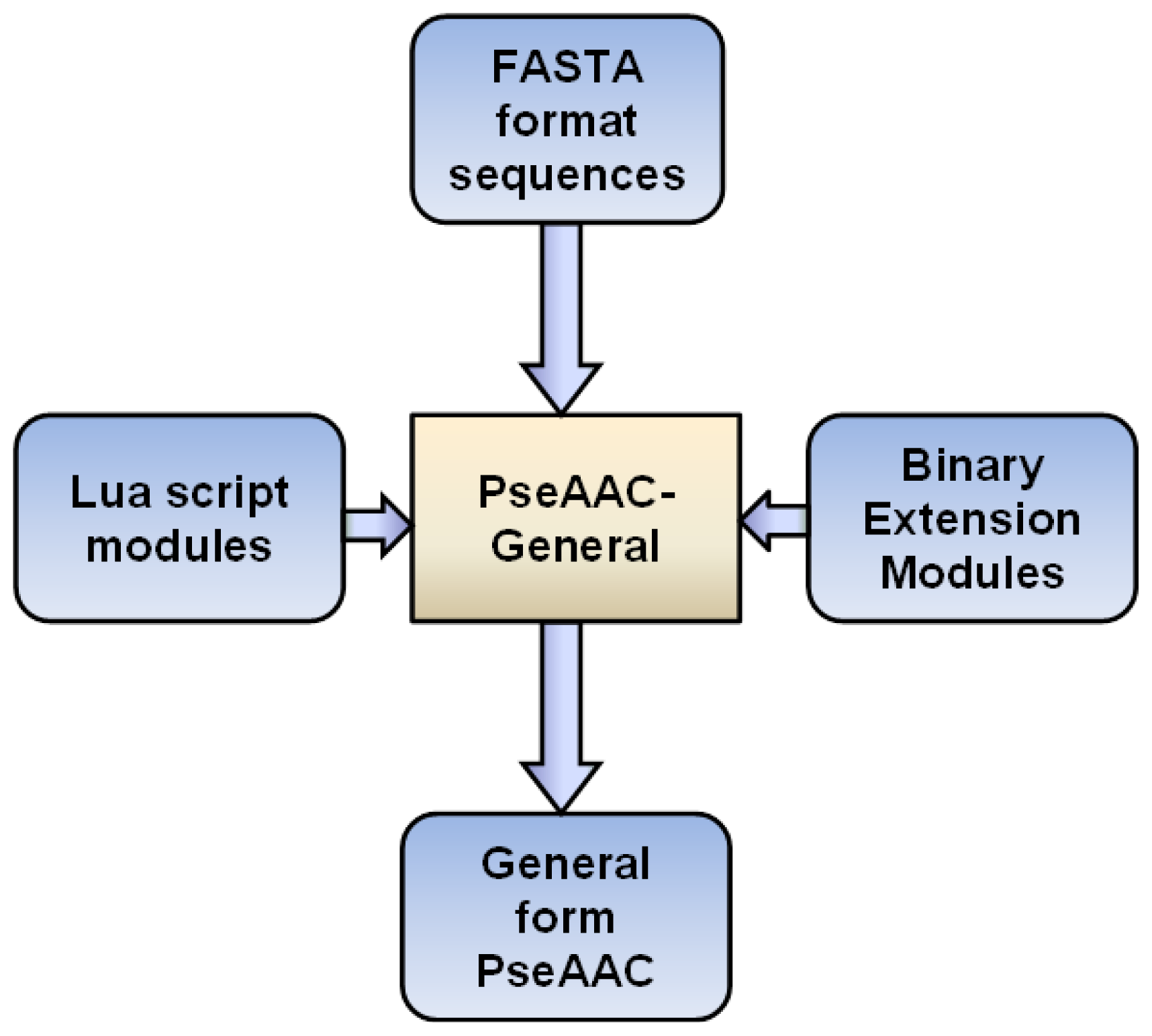
3. Implementations
4. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsP.D. designed the software, partially wrote the code and wrote the manuscript. S.G. and Y.J. partially wrote the code, carried out testing experiments and partially wrote the manuscript.
References
- Chou, K.C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins 2001, 43, 246–255. [Google Scholar]
- Lee, K.; Chuang, H.-Y.; Beyer, A.; Sung, M.-K.; Huh, W.-K.; Lee, B.; Ideker, T. Protein networks markedly improve prediction of subcellular localization in multiple eukaryotic species. Nucleic Acids Res. 2008, 36, e136. [Google Scholar]
- Chou, K.-C.; Shen, H.-B. Cell-PLoc: A package of web servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc. 2008, 3, 153–162. [Google Scholar]
- Huang, C.; Yuan, J. Using radial basis function on the general form of Chou’s pseudo amino acid composition and PSSM to predict subcellular locations of proteins with both single and multiple sites. BioSystems 2013, 113, 50–57. [Google Scholar]
- Jiang, X.; Wei, R.; Zhang, T.; Gu, Q. Using the concept of Chou’s pseudo amino acid composition to predict apoptosis proteins subcellular location: An approach by approximate entropy. Protein Pept. Lett. 2008, 15, 392–396. [Google Scholar]
- Lin, H.; Wang, H.; Ding, H.; Chen, Y.-L.; Li, Q.-Z. Prediction of subcellular localization of apoptosis protein using Chou’s pseudo amino acid composition. Acta Biotheor. 2009, 57, 321–330. [Google Scholar]
- Lin, J.; Wang, Y. Using a novel AdaBoost algorithm and Chou’s Pseudo amino acid composition for predicting protein subcellular localization. Protein Pept. Lett. 2011, 18, 1219–1225. [Google Scholar]
- Mei, S. Predicting plant protein subcellular multi-localization by Chou’s PseAAC formulation based multi-label homolog knowledge transfer learning. J. Theor. Biol. 2012, 310, 80–87. [Google Scholar]
- Pacharawongsakda, E.; Theeramunkong, T. Predict subcellular locations of singleplex and multiplex proteins by semi-supervised learning and dimension-reducing general mode of Chou’s PseAAC. NanoBioscience 2013, 12, 311–320. [Google Scholar]
- Wan, S.; Mak, M.-W.; Kung, S.-Y. GOASVM: A subcellular location predictor by incorporating term-frequency gene ontology into the general form of Chou’s pseudo-amino acid composition. J. Theor. Biol. 2013, 323, 40–48. [Google Scholar]
- Wang, X.; Li, G.-Z.; Lu, W.-C. Virus-ECC-mPLoc: A multi-label predictor for predicting the subcellular localization of virus proteins with both single and multiple sites based on a general form of Chou’s pseudo amino acid composition. Protein Pept. Lett. 2013, 20, 309–317. [Google Scholar]
- Du, P.; Li, Y. Prediction of protein submitochondria locations by hybridizing pseudo-amino acid composition with various physicochemical features of segmented sequence. BMC Bioinforma. 2006, 7, 518. [Google Scholar]
- Du, P.; Yu, Y. SubMito-PSPCP: Predicting protein submitochondrial locations by hybridizing positional specific physicochemical properties with pseudoamino acid compositions. BioMed Res. Int. 2013, 2013, 263829–263836. [Google Scholar]
- Fan, G.-L.; Li, Q.-Z. Predicting protein submitochondria locations by combining different descriptors into the general form of Chou’s pseudo amino acid composition. Amino Acids 2012, 43, 545–555. [Google Scholar]
- Mei, S. Multi-kernel transfer learning based on Chou’s PseAAC formulation for protein submitochondria localization. J. Theor. Biol. 2012, 293, 121–130. [Google Scholar]
- Huang, C.; Yuan, J.-Q. Predicting protein subchloroplast locations with both single and multiple sites via three different modes of Chou’s pseudo amino acid compositions. J. Theor. Biol. 2013, 335, 205–212. [Google Scholar]
- Jiang, X.; Wei, R.; Zhao, Y.; Zhang, T. Using Chou’s pseudo amino acid composition based on approximate entropy and an ensemble of AdaBoost classifiers to predict protein subnuclear location. Amino Acids 2008, 34, 669–675. [Google Scholar]
- Shen, H.-B.; Chou, K.-C. Nuc-PLoc: A new web-server for predicting protein subnuclear localization by fusing PseAA composition and PsePSSM. Protein Eng. Des. Sel. 2007, 20, 561–567. [Google Scholar]
- Li, F.-M.; Li, Q.-Z. Predicting protein subcellular location using Chou’s pseudo amino acid composition and improved hybrid approach. Protein Pept. Lett. 2008, 15, 612–616. [Google Scholar]
- Li, L.-Q.; Zhang, Y.; Zou, L.-Y.; Zhou, Y.; Zheng, X.-Q. Prediction of protein subcellular multi-localization based on the general form of Chou’s pseudo amino acid composition. Protein Pept. Lett. 2012, 19, 375–387. [Google Scholar]
- Nanni, L.; Lumini, A. Genetic programming for creating Chou’s pseudo amino acid based features for submitochondria localization. Amino Acids 2008, 34, 653–660. [Google Scholar]
- Zeng, Y.; Guo, Y.; Xiao, R.; Yang, L.; Yu, L.; Li, M. Using the augmented Chou’s pseudo amino acid composition for predicting protein submitochondria locations based on auto covariance approach. J. Theor. Biol. 2009, 259, 366–372. [Google Scholar]
- Pierleoni, A.; Martelli, P.L.; Casadio, R. MemLoci: Predicting subcellular localization of membrane proteins in eukaryotes. Bioinformatics 2011, 27, 1224–1230. [Google Scholar]
- Du, P.; Tian, Y.; Yan, Y. Subcellular localization prediction for human internal and organelle membrane proteins with projected gene ontology scores. J. Theor. Biol. 2012, 313, 61–67. [Google Scholar]
- Huang, C.; Yuan, J.-Q. A multilabel model based on Chou’s pseudo-amino acid composition for identifying membrane proteins with both single and multiple functional types. J. Membr. Biol. 2013, 246, 327–334. [Google Scholar]
- Zhang, S.-W.; Zhang, Y.-L.; Yang, H.-F.; Zhao, C.-H.; Pan, Q. Using the concept of Chou’s pseudo amino acid composition to predict protein subcellular localization: An approach by incorporating evolutionary information and von Neumann entropies. Amino Acids 2008, 34, 565–572. [Google Scholar]
- Cao, J.-Z.; Liu, W.-Q.; Gu, H. Predicting viral protein subcellular localization with Chou’s pseudo amino acid composition and imbalance-weighted multi-label K-nearest neighbor algorithm. Protein Pept. Lett. 2012, 19, 1163–1169. [Google Scholar]
- Shen, H.-B.; Chou, K.-C. Virus-mPLoc: A fusion classifier for viral protein subcellular location prediction by incorporating multiple sites. J. Biomol. Struct. Dyn. 2010, 28, 175–186. [Google Scholar]
- Sahu, S.S.; Panda, G. A novel feature representation method based on Chou’s pseudo amino acid composition for protein structural class prediction. Comput. Biol. Chem. 2010, 34, 320–327. [Google Scholar]
- Chen, C.; Shen, Z.-B.; Zou, X.-Y. Dual-layer wavelet SVM for predicting protein structural class via the general form of Chou’s pseudo amino acid composition. Protein Pept. Lett. 2012, 19, 422–429. [Google Scholar]
- Kong, L.; Zhang, L.; Lv, J. Accurate prediction of protein structural classes by incorporating predicted secondary structure information into the general form of Chou’s pseudo amino acid composition. J. Theor. Biol. 2013, 344, 12–18. [Google Scholar]
- Li, Z.-C.; Zhou, X.-B.; Dai, Z.; Zou, X.-Y. Prediction of protein structural classes by Chou’s pseudo amino acid composition: Approached using continuous wavelet transform and principal component analysis. Amino Acids 2009, 37, 415–425. [Google Scholar]
- Liao, B.; Xiang, Q.; Li, D. Incorporating secondary features into the general form of Chou’s PseAAC for predicting protein structural class. Protein Pept. Lett. 2012, 19, 1133–1138. [Google Scholar]
- Liu, L.; Hu, X.-Z.; Liu, X.-X.; Wang, Y.; Li, S.-B. Predicting protein fold types by the general form of Chou’s pseudo amino acid composition: Approached from optimal feature extractions. Protein Pept. Lett. 2012, 19, 439–449. [Google Scholar]
- Qin, Y.-F.; Wang, C.-H.; Yu, X.-Q.; Zhu, J.; Liu, T.-G.; Zheng, X.-Q. Predicting protein structural class by incorporating patterns of over-represented k-mers into the general form of Chou’s PseAAC. Protein Pept. Lett. 2012, 19, 388–397. [Google Scholar]
- Chen, C.; Chen, L.; Zou, X.; Cai, P. Prediction of protein secondary structure content by using the concept of Chou’s pseudo amino acid composition and support vector machine. Protein Pept. Lett. 2009, 16, 27–31. [Google Scholar]
- Zou, D.; He, Z.; He, J.; Xia, Y. Supersecondary structure prediction using Chou’s pseudo amino acid composition. J. Comput. Chem. 2011, 32, 271–278. [Google Scholar]
- Sun, X.-Y.; Shi, S.-P.; Qiu, J.-D.; Suo, S.-B.; Huang, S.-Y.; Liang, R.-P. Identifying protein quaternary structural attributes by incorporating physicochemical properties into the general form of Chou’s PseAAC via discrete wavelet transform. Mol. Biosyst. 2012, 8, 3178–3184. [Google Scholar]
- Zhang, S.-W.; Chen, W.; Yang, F.; Pan, Q. Using Chou’s pseudo amino acid composition to predict protein quaternary structure: A sequence-segmented PseAAC approach. Amino Acids 2008, 35, 591–598. [Google Scholar]
- Gu, Q.; Ding, Y.S.; Zhang, T.L. Prediction of G-protein-coupled receptor classes in low homology using Chou’s pseudo amino acid composition with approximate entropy and hydrophobicity patterns. Protein Pept. Lett. 2010, 17, 559–567. [Google Scholar]
- Qiu, J.-D.; Huang, J.-H.; Liang, R.-P.; Lu, X.-Q. Prediction of G-protein-coupled receptor classes based on the concept of Chou’s pseudo amino acid composition: An approach from discrete wavelet transform. Anal. Biochem. 2009, 390, 68–73. [Google Scholar]
- Zia-Ur-Rehman Khan, A. Identifying GPCRs and their types with Chou’s pseudo amino acid composition: An approach from multi-scale energy representation and position specific scoring matrix. Protein Pept. Lett. 2012, 19, 890–903. [Google Scholar]
- Qiu, J.-D.; Huang, J.-H.; Shi, S.-P.; Liang, R.-P. Using the concept of Chou’s pseudo amino acid composition to predict enzyme family classes: An approach with support vector machine based on discrete wavelet transform. Protein Pept. Lett. 2010, 17, 715–722. [Google Scholar]
- Zhou, X.-B.; Chen, C.; Li, Z.-C.; Zou, X.-Y. Using Chou’s amphiphilic pseudo-amino acid composition and support vector machine for prediction of enzyme subfamily classes. J. Theor. Biol. 2007, 248, 546–551. [Google Scholar]
- Chen, Y.-K.; Li, K.-B. Predicting membrane protein types by incorporating protein topology domains signal peptides and physicochemical properties into the general form of Chou’s pseudo amino acid composition. J. Theor. Biol. 2013, 318, 1–12. [Google Scholar]
- Han, G.-S.; Yu, Z.-G.; Anh, V. A two-stage SVM method to predict membrane protein types by incorporating amino acid classifications and physicochemical properties into a general form of Chou’s PseAAC. J. Theor. Biol. 2013, 344, 31–39. [Google Scholar]
- Hayat, M.; Khan, A. Discriminating outer membrane proteins with fuzzy K-nearest neighbor algorithms based on the general form of Chou’s PseAAC. Protein Pept. Lett. 2012, 19, 411–421. [Google Scholar]
- Mohammad Beigi, M.; Behjati, M.; Mohabatkar, H. Prediction of metalloproteinase family based on the concept of Chou’s pseudo amino acid composition using a machine learning approach. J. Struct. Funct. Genomics 2011, 12, 191–197. [Google Scholar]
- Esmaeili, M.; Mohabatkar, H.; Mohsenzadeh, S. Using the concept of Chou’s pseudo amino acid composition for risk type prediction of human papillomaviruses. J. Theor. Biol. 2010, 263, 203–209. [Google Scholar]
- Ding, H.; Luo, L.; Lin, H. Prediction of cell wall lytic enzymes using Chou’s amphiphilic pseudo amino acid composition. Protein Pept. Lett. 2009, 16, 351–355. [Google Scholar]
- Mohabatkar, H. Prediction of cyclin proteins using Chou’s pseudo amino acid composition. Protein Pept. Lett. 2010, 17, 1207–1214. [Google Scholar]
- Mohabatkar, H.; Mohammad Beigi, M.; Abdolahi, K.; Mohsenzadeh, S. Prediction of allergenic proteins by means of the concept of Chou’s pseudo amino acid composition and a machine learning approach. Med. Chem. 2013, 9, 133–137. [Google Scholar]
- Fan, G.-L.; Li, Q.-Z. Discriminating bioluminescent proteins by incorporating average chemical shift and evolutionary information into the general form of Chou’s pseudo amino acid composition. J. Theor. Biol. 2013, 334, 45–51. [Google Scholar]
- Fang, Y.; Guo, Y.; Feng, Y.; Li, M. Predicting DNA-binding proteins: Approached from Chou’s pseudo amino acid composition and other specific sequence features. Amino Acids 2008, 34, 103–109. [Google Scholar]
- Mohabatkar, H.; Mohammad Beigi, M.; Esmaeili, A. Prediction of GABAA receptor proteins using the concept of Chou’s pseudo-amino acid composition and support vector machine. J. Theor. Biol. 2011, 281, 18–23. [Google Scholar]
- Nanni, L.; Lumini, A.; Gupta, D.; Garg, A. Identifying bacterial virulent proteins by fusing a set of classifiers based on variants of Chou’s pseudo amino acid composition and on evolutionary information. IEEE/ACM Trans. Comput. Biol. Bioinforma. 2012, 9, 467–475. [Google Scholar]
- Sarangi, A.N.; Lohani, M.; Aggarwal, R. Prediction of essential proteins in prokaryotes by incorporating various physico-chemical features into the general form of Chou’s pseudo amino acid composition. Protein Pept. Lett. 2013, 20, 781–795. [Google Scholar]
- Hajisharifi, Z.; Piryaiee, M.; Mohammad Beigi, M.; Behbahani, M.; Mohabatkar, H. Predicting anticancer peptides with Chou’s pseudo amino acid composition and investigating their mutagenicity via Ames test. J. Theor. Biol. 2014, 341, 34–40. [Google Scholar]
- Khosravian, M.; Faramarzi, F.K.; Beigi, M.M.; Behbahani, M.; Mohabatkar, H. Predicting antibacterial peptides by the concept of Chou’s pseudo-amino acid composition and machine learning methods. Protein Pept. Lett. 2013, 20, 180–186. [Google Scholar]
- Zhao, X.-W.; Ma, Z.-Q.; Yin, M.-H. Predicting protein–protein interactions by combing various sequence-derived features into the general form of Chou’s pseudo amino acid composition. Protein Pept. Lett. 2012, 19, 492–500. [Google Scholar]
- Niu, X.-H.; Hu, X.-H.; Shi, F.; Xia, J.-B. Predicting protein solubility by the general form of Chou’s pseudo amino acid composition: Approached from chaos game representation and fractal dimension. Protein Pept. Lett. 2012, 19, 940–948. [Google Scholar]
- Yu, H.; Chen, J.; Xu, X.; Li, Y.; Zhao, H.; Fang, Y.; Li, X.; Zhou, W.; Wang, W.; Wang, Y.A. Systematic prediction of multiple drug-target interactions from chemical genomic and pharmacological data. PLoS One 2012, 7, e37608. [Google Scholar]
- Georgiou, D.N.; Karakasidis, T.E.; Nieto, J.J.; Torres, A. Use of fuzzy clustering technique and matrices to classify amino acids and its impact to Chou’s pseudo amino acid composition. J. Theor. Biol. 2009, 257, 17–26. [Google Scholar]
- Gupta, M.K.; Niyogi, R.; Misra, M. An alignment-free method to find similarity among protein sequences via the general form of Chou’s pseudo amino acid composition. SAR QSAR Environ. Res. 2013, 24, 597–609. [Google Scholar]
- Lin, H. The modified Mahalanobis discriminant for predicting outer membrane proteins by using Chou’s pseudo amino acid composition. J. Theor. Biol. 2008, 252, 350–356. [Google Scholar]
- Nanni, L.; Brahnam, S.; Lumini, A. Wavelet images and Chou’s pseudo amino acid composition for protein classification. Amino Acids 2012, 43, 657–665. [Google Scholar]
- Qiu, J.-D.; Suo, S.-B.; Sun, X.-Y.; Shi, S.-P.; Liang, R.-P. OligoPred: A web-server for predicting homo-oligomeric proteins by incorporating discrete wavelet transform into Chou’s pseudo amino acid composition. J. Mol. Graph. Model. 2011, 30, 129–134. [Google Scholar]
- Ren, L.-Y.; Zhang, Y.-S.; Gutman, I. Predicting the classification of transcription factors by incorporating their binding site properties into a novel mode of Chou’s pseudo amino acid composition. Protein Pept. Lett. 2012, 19, 1170–1176. [Google Scholar]
- Xiaohui, N.; Nana, L.; Jingbo, X.; Dingyan, C.; Yuehua, P.; Yang, X.; Weiquan, W.; Dongming, W.; Zengzhen, W. Using the concept of Chou’s pseudo amino acid composition to predict protein solubility: An approach with entropies in information theory. J. Theor. Biol. 2013, 332, 211–217. [Google Scholar]
- Xie, H.-L.; Fu, L.; Nie, X.-D. Using ensemble SVM to identify human GPCRs N-linked glycosylation sites based on the general form of Chou’s PseAAC. Protein Eng. Des. Sel. 2013, 26, 735–742. [Google Scholar]
- Yu, L.; Guo, Y.; Li, Y.; Li, G.; Li, M.; Luo, J.; Xiong, W.; Qin, W. SecretP: Identifying bacterial secreted proteins by fusing new features into Chou’s pseudo-amino acid composition. J. Theor. Biol. 2010, 267, 1–6. [Google Scholar]
- Zhang, G.-Y.; Fang, B.-S. Predicting the cofactors of oxidoreductases based on amino acid composition distribution and Chou’s amphiphilic pseudo-amino acid composition. J. Theor. Biol. 2008, 253, 310–315. [Google Scholar]
- Zhang, G.-Y.; Li, H.-C.; Gao, J.-Q.; Fang, B.-S. Predicting lipase types by improved Chou’s pseudo-amino acid composition. Protein Pept. Lett. 2008, 15, 1132–1137. [Google Scholar]
- Liu, B.; Wang, X.; Zou, Q.; Dong, Q.; Chen, Q. Protein remote homology detection by combining Chou’s pseudo amino acid composition and profile-based protein representation. Mol. Inform. 2013, 32, 775–782. [Google Scholar]
- Georgiou, D.N.; Karakasidis, T.E.; Nieto, J.J.; Torres, A. A study of entropy/clarity of genetic sequences using metric spaces and fuzzy sets. J. Theor. Biol. 2010, 267, 95–105. [Google Scholar]
- Georgiou, T.N.; Karakasidis, T.E.; Megaritis, A.C. A short survey on genetic sequences Chou’s pseudo amino acid composition and its combination with fuzzy set theory. Open Bioinforma. J. 2013, 7, 41–48. [Google Scholar]
- Chen, W.; Feng, P.-M.; Lin, H.; Chou, K.-C. iRSpot-PseDNC: Identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res. 2013, 41, e68. [Google Scholar]
- Chou, K.-C. Some remarks on protein attribute prediction and pseudo amino acid composition. J. Theor. Biol. 2011, 273, 236–247. [Google Scholar]
- Chou, K.-C. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics 2005, 21, 10–19. [Google Scholar]
- Shen, H.-B.; Chou, K.-C. PseAAC: A flexible web server for generating various kinds of protein pseudo amino acid composition. Anal. Biochem. 2008, 373, 386–388. [Google Scholar]
- Du, P.; Wang, X.; Xu, C.; Gao, Y. PseAAC-Builder: A cross-platform stand-alone program for generating various special Chou’s pseudo-amino acid compositions. Anal. Biochem. 2012, 425, 117–119. [Google Scholar]
- Cao, D.-S.; Xu, Q.-S.; Liang, Y.-Z. Propy: A tool to generate various modes of Chou’s PseAAC. Bioinformatics 2013, 29, 960–962. [Google Scholar]
- Chou, K.-C.; Cai, Y.-D. Prediction of protein subcellular locations by GO-FunD-PseAA predictor. Biochem. Biophys. Res. Commun. 2004, 320, 1236–1239. [Google Scholar]
- Feng, Z.P.; Zhang, C.T. Prediction of membrane protein types based on the hydrophobic index of amino acids. J. Protein Chem. 2000, 19, 269–275. [Google Scholar]
- Lin, Z.; Pan, X.M. Accurate prediction of protein secondary structural content. J. Protein Chem. 2001, 20, 217–220. [Google Scholar]
- Horne, D.S. Prediction of protein helix content from an autocorrelation analysis of sequence hydrophobicities. Biopolymers 1988, 27, 451–477. [Google Scholar]
- Sokal, R.R.; Thomson, B.A. Population structure inferred by local spatial autocorrelation: An example from an Amerindian tribal population. Am. J. Phys. Anthropol. 2006, 129, 121–131. [Google Scholar]
- Dubchak, I.; Muchnik, I.; Mayor, C.; Dralyuk, I.; Kim, S.H. Recognition of a protein fold in the context of the Structural Classification of Proteins (SCOP) classification. Proteins 1999, 35, 401–407. [Google Scholar]
- Chou, K.-C. Prediction of protein subcellular locations by incorporating quasi-sequence-order effect. Biochem. Biophys. Res. Commun. 2000, 27, 477–483. [Google Scholar]
- Steinbiss, S.; Gremme, G.; Schärfer, C.; Mader, M.; Kurtz, S. AnnotationSketch: A genome annotation drawing library. Bioinformatics 2009, 25, 533–534. [Google Scholar]
- PseAAC-General. Available online: http://pseb.sf.net (accessed on 19 February 2014).
- PseAAC-General SourceForge Site. Available online: http://sourceforge.net/projects/pseb/files (accessed on 19 February 2014).

{kind=link}
| Program Functions a | PseAAC-General | PseAAC-Builder | Propy | PseAAC Server |
|---|---|---|---|---|
| Physicochemical Properties | 544 | 544 | 8 | 6 |
| Output Features | ||||
| Type I PseAAC [1] | Y | Y | Y | Y |
| Type II PseAAC [79] | Y | Y | Y | Y |
| Amino acid composition | Y | Y | Y | Y |
| di-Peptide composition | Y | Y | Y | Y |
| tri-Peptide composition | Y | N | Y | N |
| Normalized Moreau-Broto autocorrelation [84,85] | Y | N | Y | N |
| Moran autocorrelation [86] | Y | N | Y | N |
| Geary autocorrelation [87] | Y | N | Y | N |
| Composition-Transition-Distribution (CTD) [88] | Y | N | Y | N |
| Quasi-sequence order [89] | Y | N | Y | N |
| Gene ontology mode [83] | Y | N | N | N |
| Functional domain mode [83] | Y | N | N | N |
| Sequential evolution mode [18] | Y | N | N | N |
| Other functions | ||||
| User defined | Y | N | N | N |
| Online updates | Y | N | N | N |
| Graphical User Interface (GUI) | Y | Y | N | Y |
| Execution efficiency b | ~17,000 seqs/s | ~170 seqs/s | N.A. | ~15 seqs/s |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Du, P.; Gu, S.; Jiao, Y. PseAAC-General: Fast Building Various Modes of General Form of Chou’s Pseudo-Amino Acid Composition for Large-Scale Protein Datasets. Int. J. Mol. Sci. 2014, 15, 3495-3506. https://doi.org/10.3390/ijms15033495
Du P, Gu S, Jiao Y. PseAAC-General: Fast Building Various Modes of General Form of Chou’s Pseudo-Amino Acid Composition for Large-Scale Protein Datasets. International Journal of Molecular Sciences. 2014; 15(3):3495-3506. https://doi.org/10.3390/ijms15033495
Chicago/Turabian StyleDu, Pufeng, Shuwang Gu, and Yasen Jiao. 2014. "PseAAC-General: Fast Building Various Modes of General Form of Chou’s Pseudo-Amino Acid Composition for Large-Scale Protein Datasets" International Journal of Molecular Sciences 15, no. 3: 3495-3506. https://doi.org/10.3390/ijms15033495