A Genotoxic Stress-Responsive miRNA, miR-574-3p, Delays Cell Growth by Suppressing the Enhancer of Rudimentary Homolog Gene in Vitro

Abstract
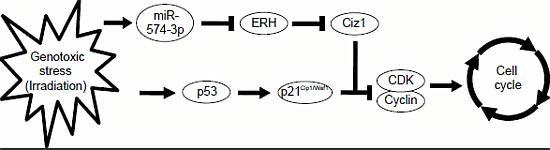
:
1. Introduction
2. Results and Discussion
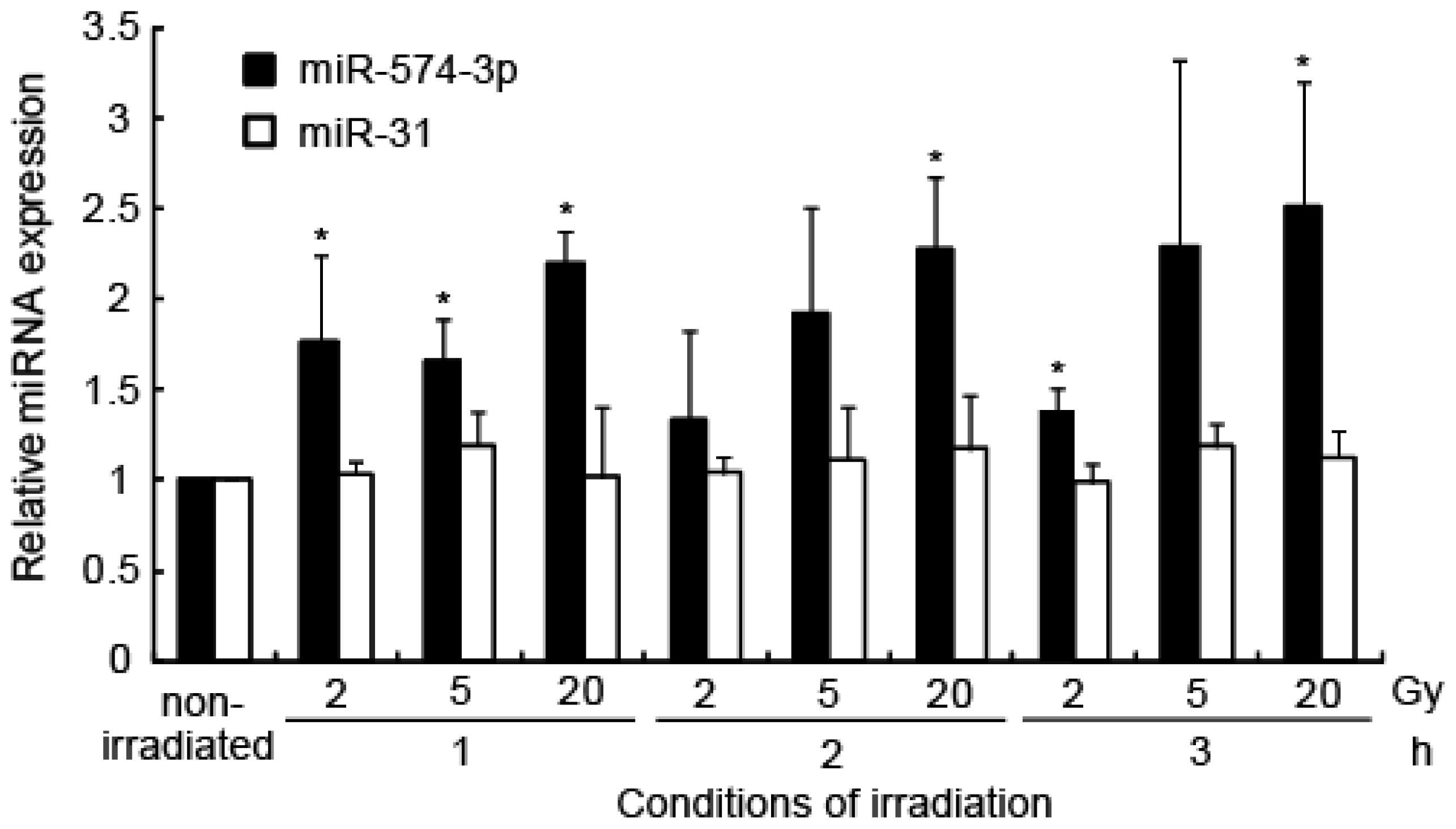
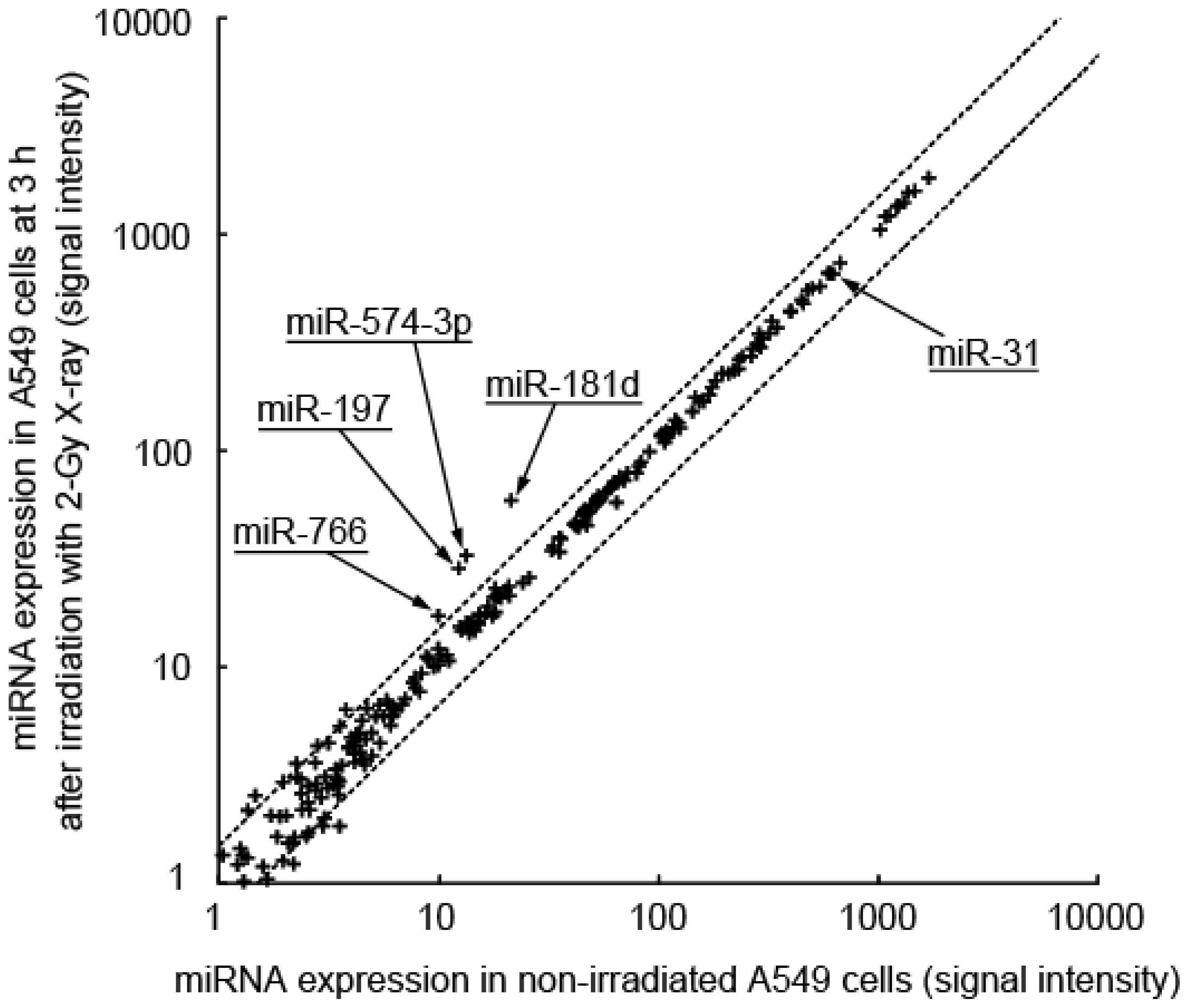
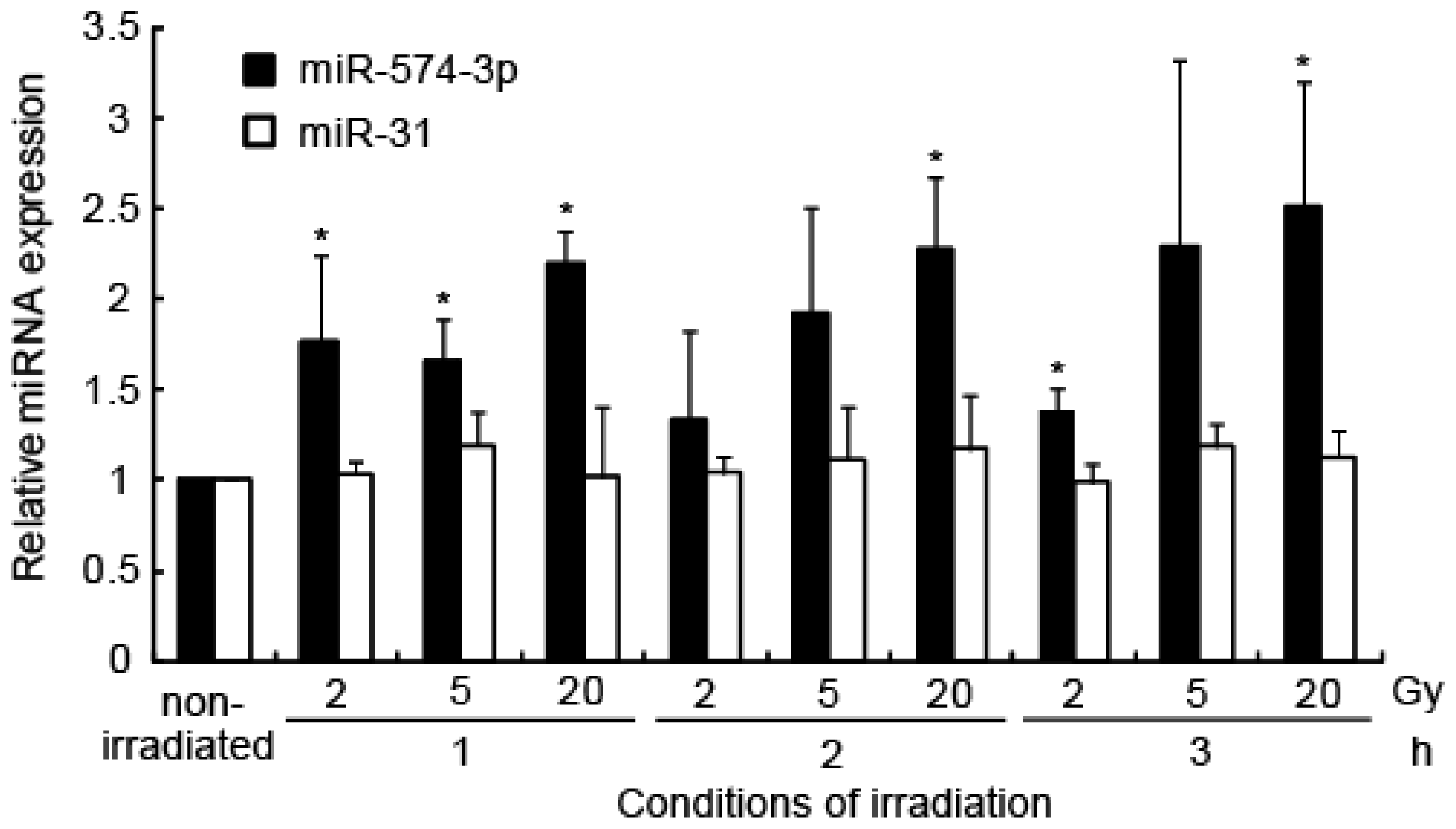
2.1. Induction of miRNA Expression after Irradiation
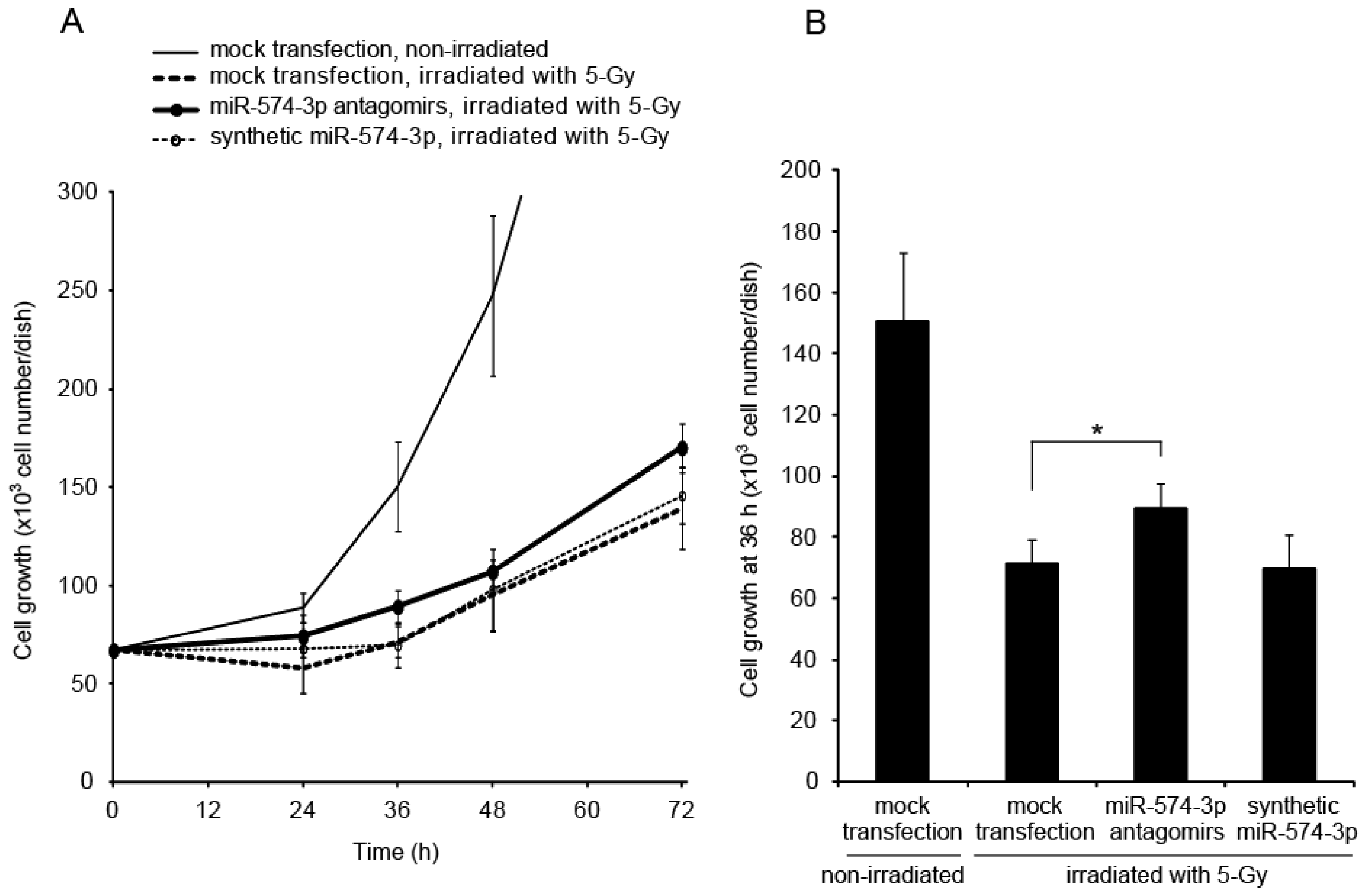
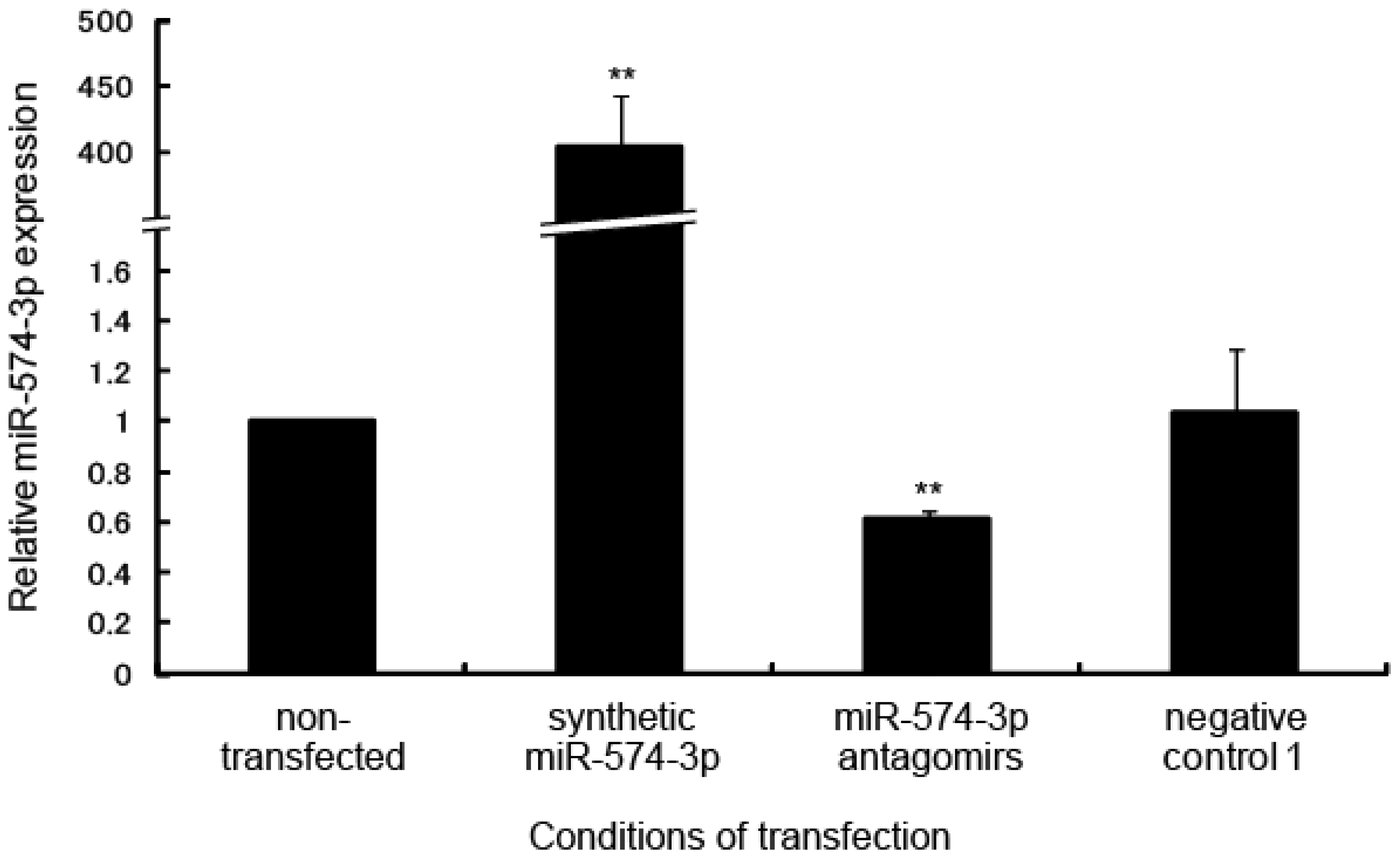
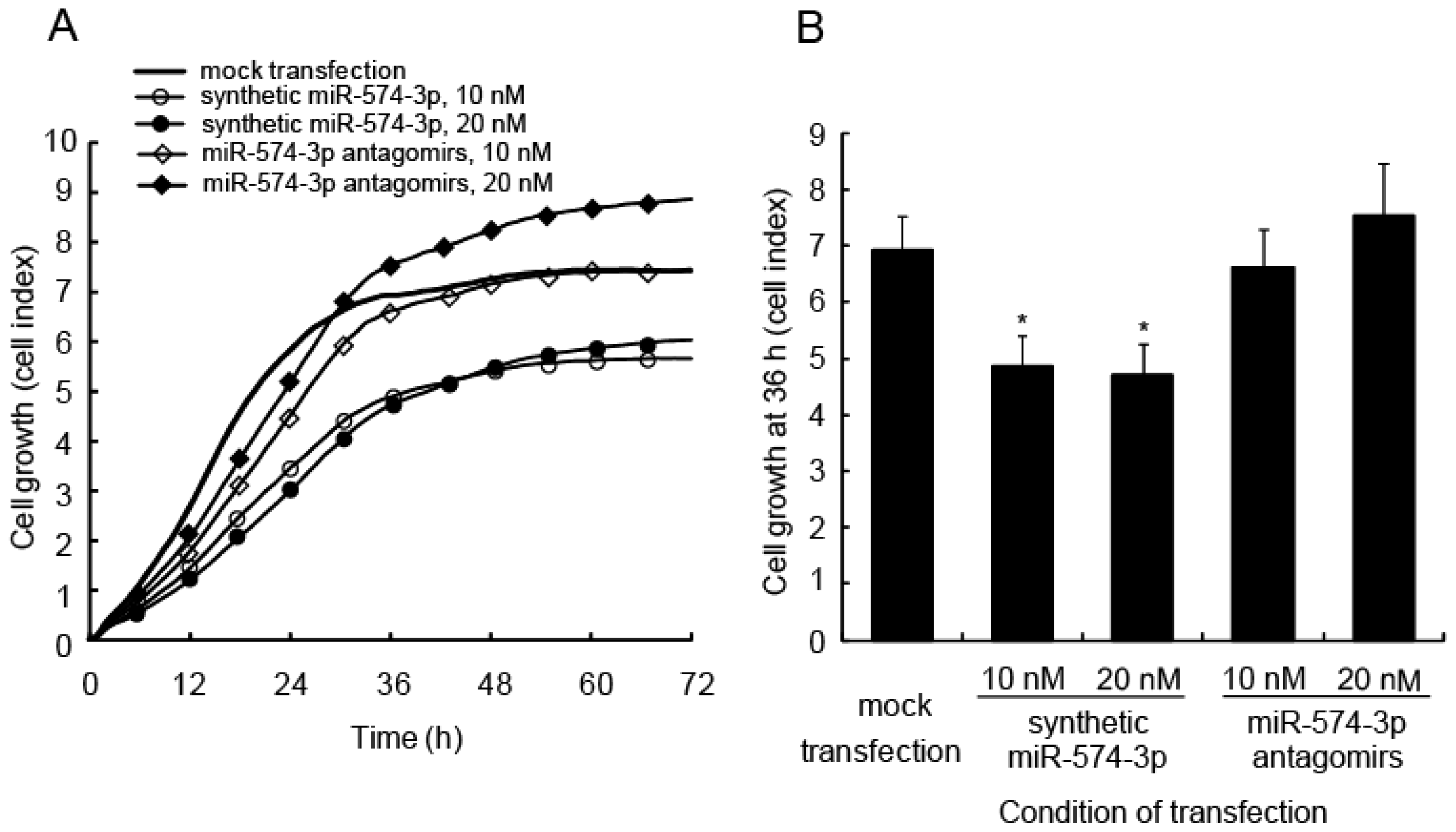
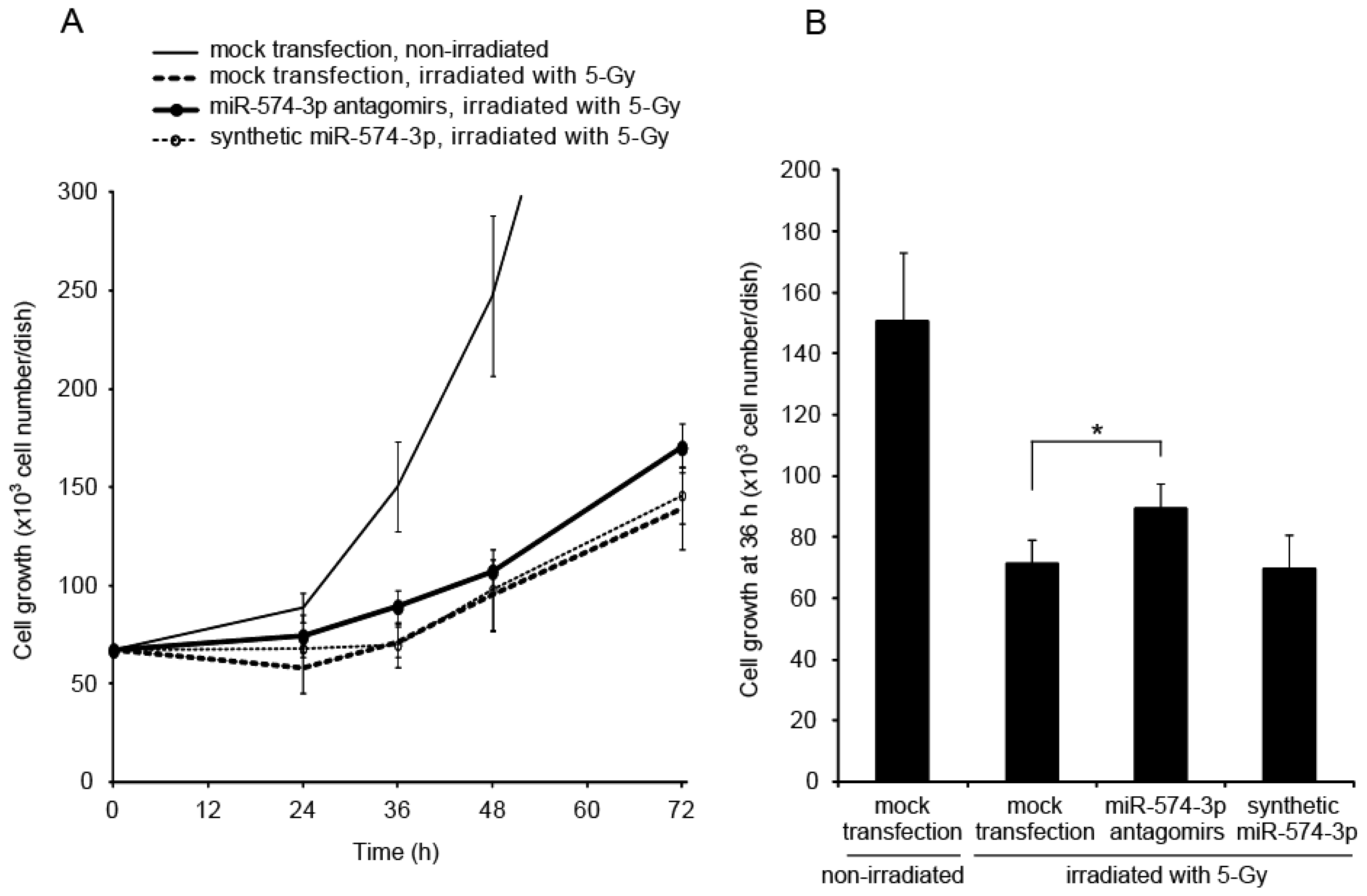
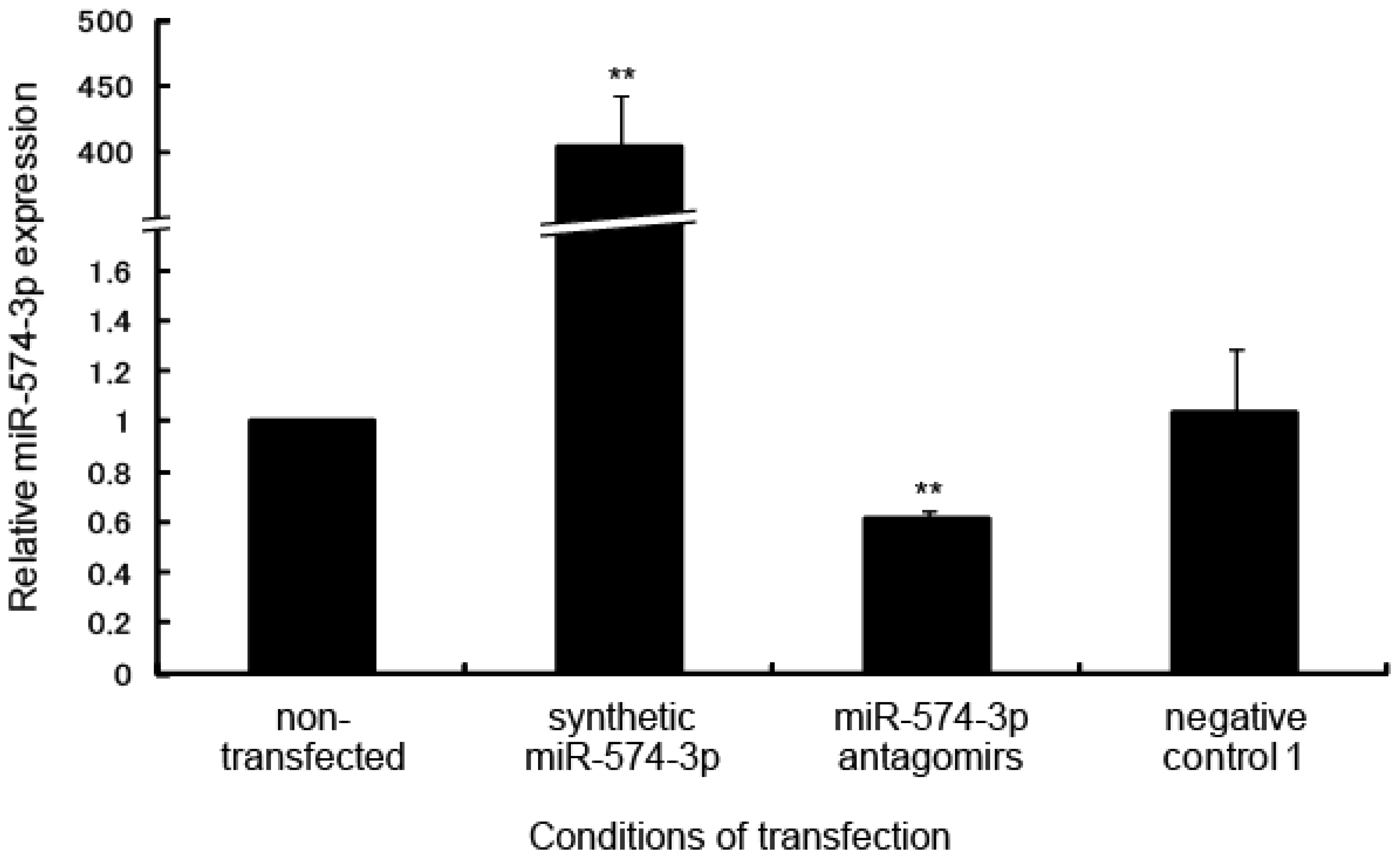
2.2. Delay in Cell Growth by Overexpression of miR-574-3p
2.3. Expression Analysis of Transcripts Regulated by miR-574-3p
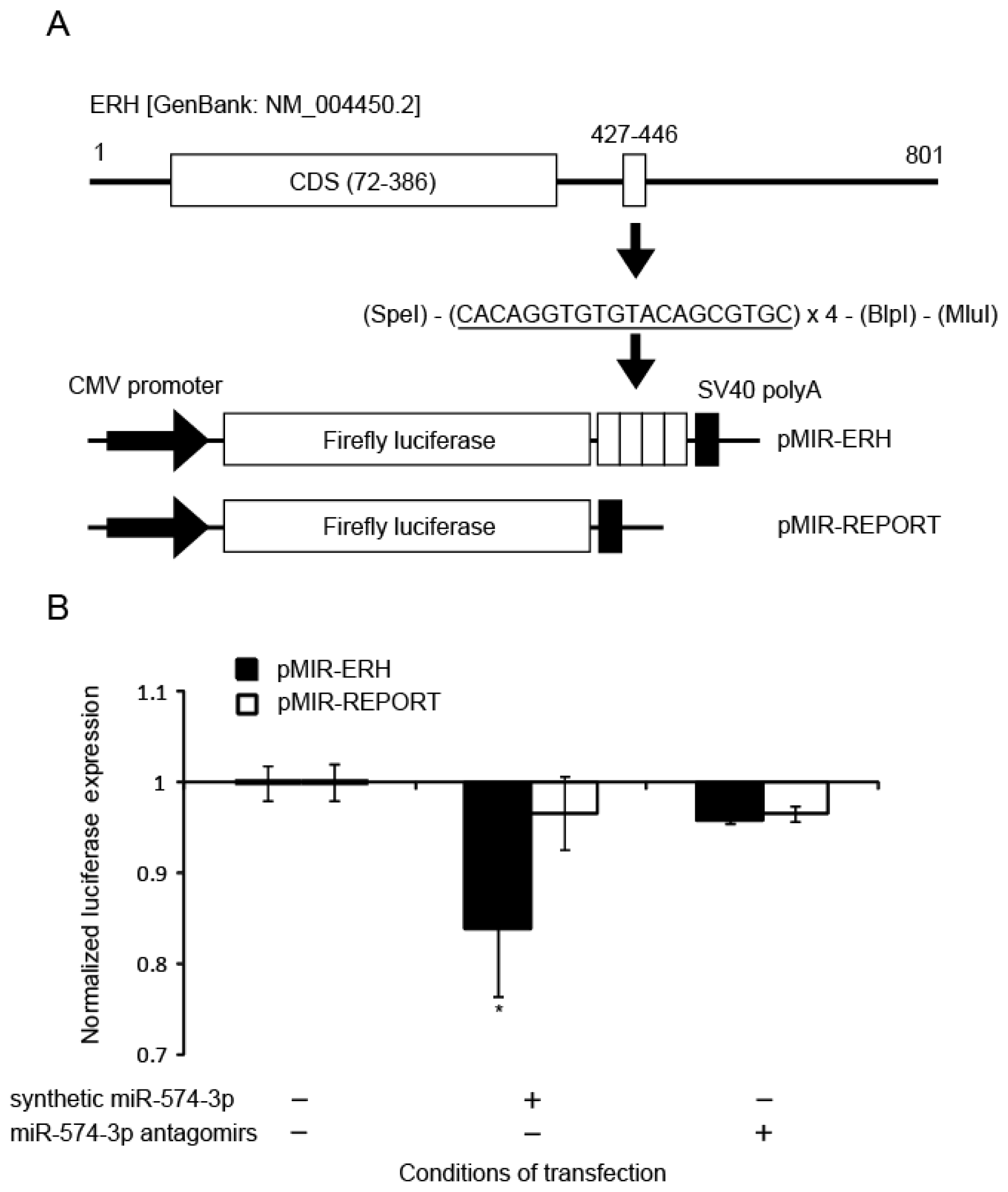
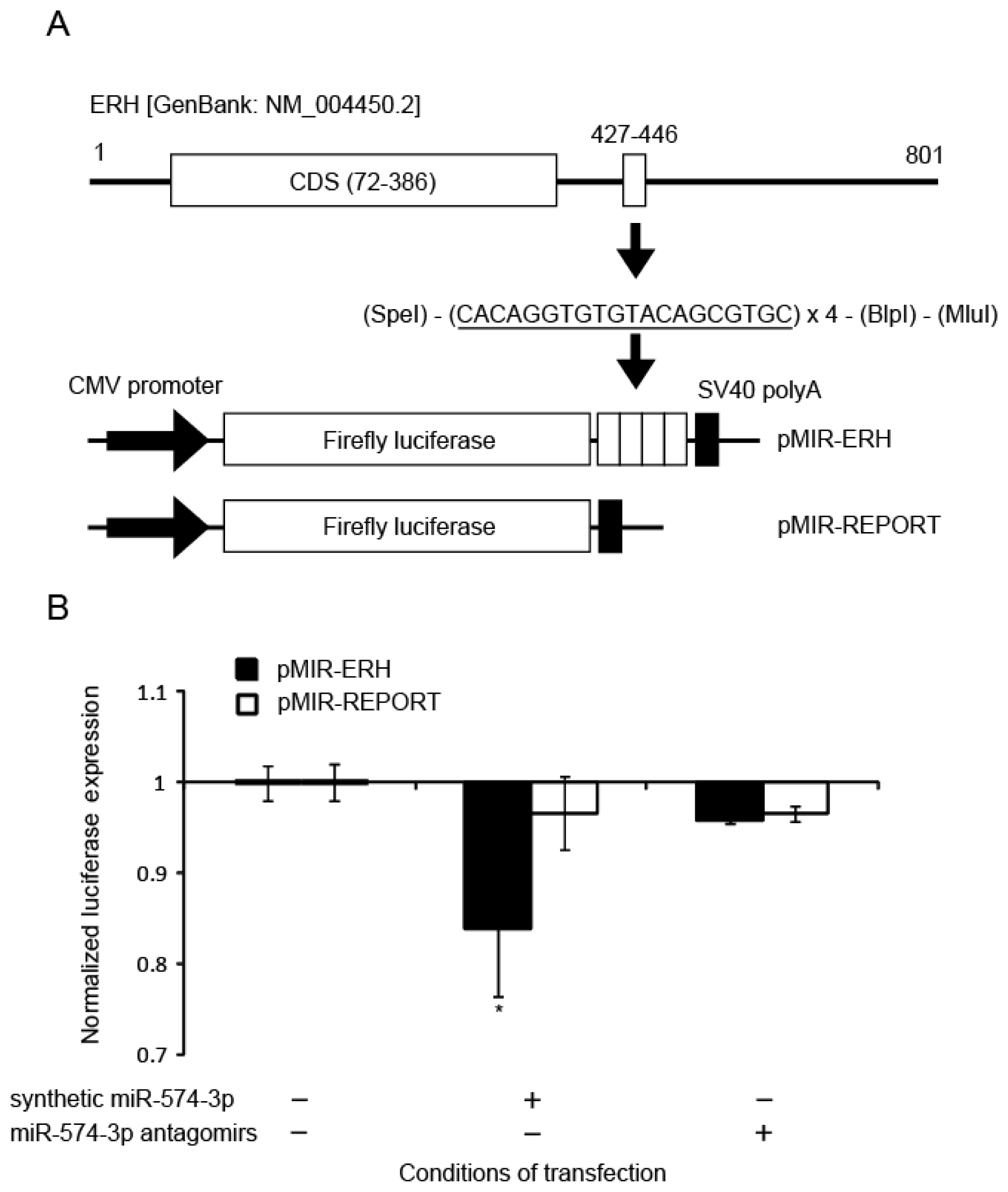
2.4. Binding of miR-574-3p to the 3′-UTR of ERH
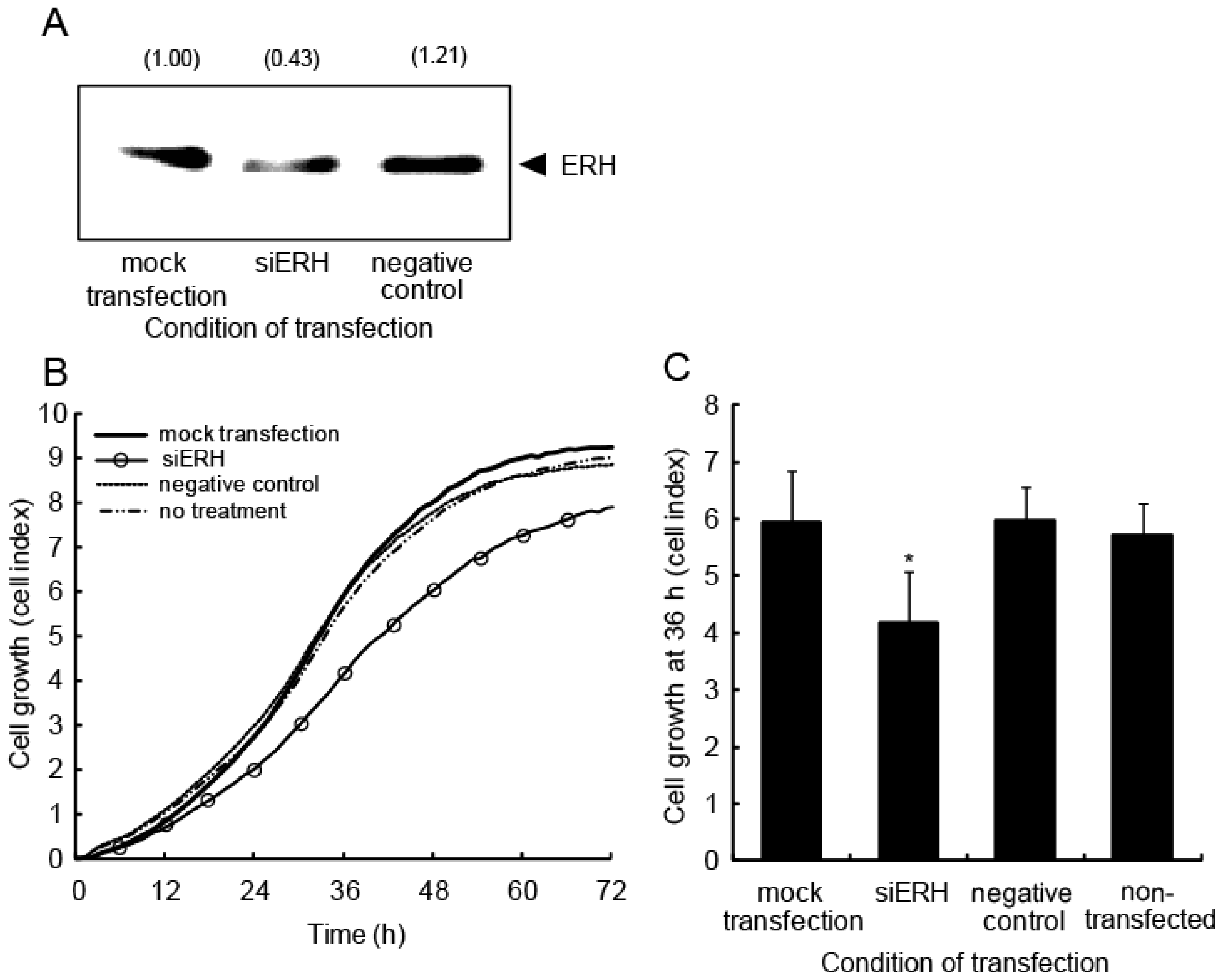
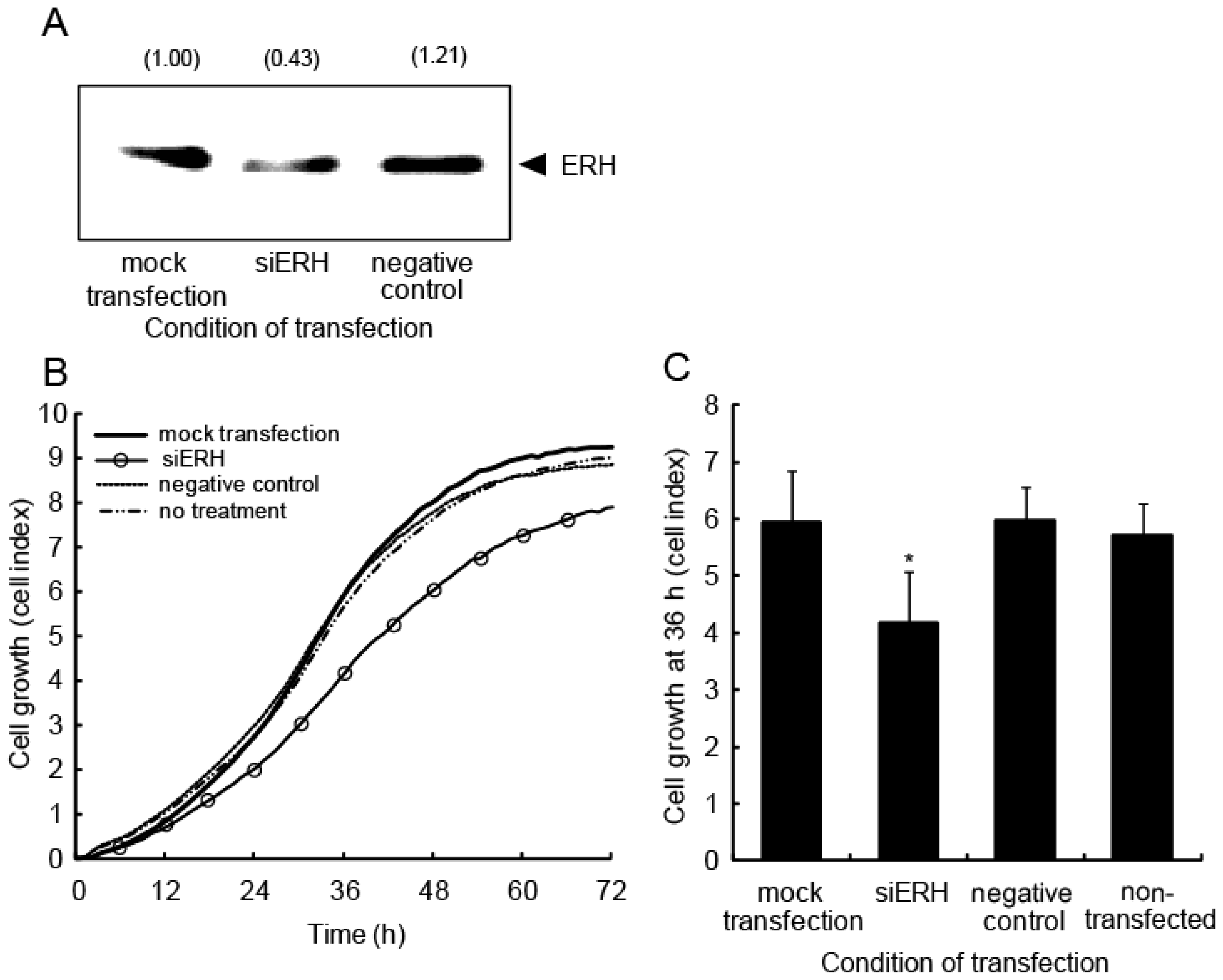
2.5. ERH Regulates Cell Cycle Progression
2.6. miR-574-3p Expression during Cellular Response to X-Ray Irradiation
3. Experimental Section
3.1. Cell Lines, Culture Conditions, and Irradiation
3.2. Isolation of Total RNA
3.3. Transfection
3.4. Microarray Analysis of miRNA Expression
3.5. Quantitative Real-Time Reverse Transcription (RT)-PCR Analysis of miRNA Expression
3.6. Microarray Analysis of mRNA Expression
3.7. Quantitative Real-Time RT-PCR Analysis of ERH Expression
3.8. Prediction of miR-574-3p Targets in Silico
3.9. Western Blotting
3.10. Reporter Constructs
3.11. Luciferase Assay
3.12. Real-Time Monitoring of Cell Proliferation
3.13. Statistical Analysis
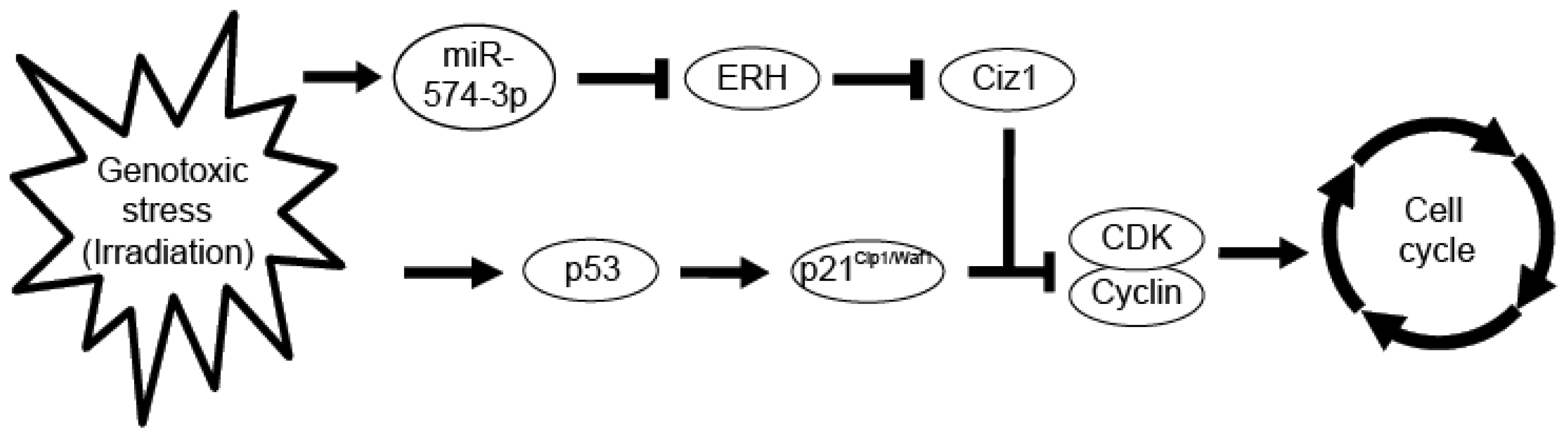
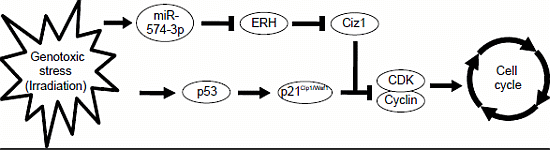
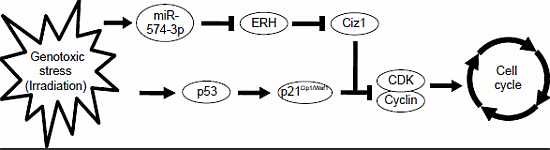
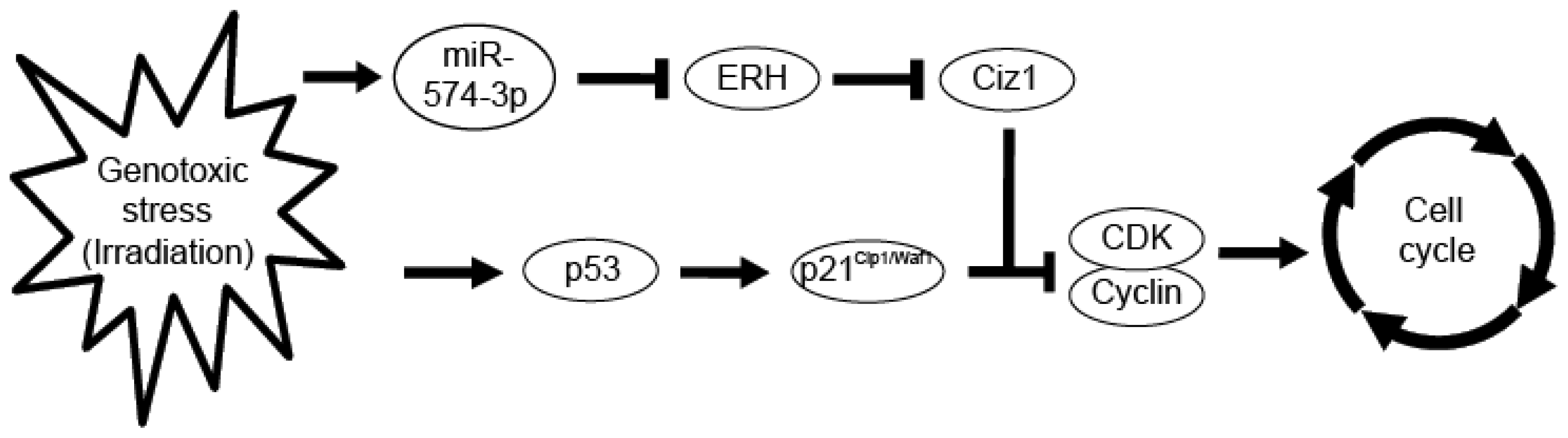
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Farrell, A.W.; Halliday, G.M.; Lyons, J.G. Chromatin structure following UV-induced DNA damage-repair or death? Int. J. Mol. Sci 2011, 12, 8063–8085. [Google Scholar]
- Ljungman, M. The DNA damage response—repair or despair? Environ. Mol. Mutagen 2010, 51, 879–889. [Google Scholar]
- Winter-Vann, A.M.; Johnson, G.L. Integrated activation of MAP3Ks balances cell fate in response to stress. J. Cell Biochem 2007, 102, 848–858. [Google Scholar]
- Hamada, N.; Imaoka, T.; Masunaga, S.; Ogata, T.; Okayasu, R.; Takahashi, A.; Kato, T.A.; Kobayashi, Y.; Ohnishi, T.; Ono, K.; et al. Recent advances in the biology of heavy-ion cancer therapy. J. Radiat. Res 2010, 51, 365–383. [Google Scholar]
- Ishikawa, K.; Koyama-Saegusa, K.; Otsuka, Y.; Ishikawa, A.; Kawai, S.; Yasuda, K.; Suga, T.; Michikawa, Y.; Suzuki, M.; Iwakawa, M.; et al. Gene expression profile changes correlating with radioresistance in human cell lines. Int. J. Radiat. Oncol 2006, 65, 234–245. [Google Scholar]
- Bonin, F.; Molina, M.; Malet, C.; Ginestet, C.; Berthier-Vergnes, O.; Martin, M.T.; Lamartine, J. GATA3 is a master regulator of the transcriptional response to low-dose ionizing radiation in human keratinocytes. BMC Genomics 2009, 10, 417. [Google Scholar]
- Luo, X.; Puig, O.; Hyun, J.; Bohmann, D.; Jasper, H. Foxo and Fos regulate the decision between cell death and survival in response to UV irradiation. EMBO J 2007, 26, 380–390. [Google Scholar]
- Thyss, R.; Virolle, V.; Imbert, V.; Peyron, J.F.; Aberdam, D.; Virolle, T. NF-kappaB/Egr-1/Gadd45 are sequentially activated upon UVB irradiation to mediate epidermal cell death. EMBO J 2005, 24, 128–137. [Google Scholar]
- Farazi, T.A.; Hoell, J.I.; Morozov, P.; Tuschl, T. MicroRNAs in human cancer. Adv. Exp. Med. Biol 2013, 774, 1–20. [Google Scholar]
- Ambros, V. MicroRNAs and developmental timing. Curr. Opin. Genet. Dev 2011, 21, 511–517. [Google Scholar]
- Van Kouwenhove, M.; Kedde, M.; Agami, R. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat. Rev. Cancer 2011, 11, 644–656. [Google Scholar]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 2001, 294, 853–858. [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar]
- Niemoeller, O.M.; Niyazi, M.; Corradini, S.; Zehentmayr, F.; Li, M.; Lauber, K.; Belka, C. MicroRNA expression profiles in human cancer cells after ionizing radiation. Radiat. Oncol 2011, 6, 29. [Google Scholar]
- Shin, S.; Cha, H.J.; Lee, E.M.; Lee, S.J.; Seo, S.K.; Jin, H.O.; Park, I.C.; Jin, Y.W.; An, S. Alteration of miRNA profiles by ionizing radiation in A549 human non-small cell lung cancer cells. Int. J. Oncol 2009, 35, 81–86. [Google Scholar]
- John-Aryankalayil, M.; Palayoor, S.T.; Makinde, A.Y.; Cerna, D.; Simone, C.B., 2nd; Falduto, M.T.; Magnuson, S.R.; Coleman, C.N. Fractionated radiation alters oncomir and tumor suppressor miRNAs in human prostate cancer cells. Radiat. Res 2012, 178, 105–117. [Google Scholar]
- Girardi, C.; De Pittà, C.; Casara, S.; Sales, G.; Lanfranchi, G.; Celotti, L.; Mognato, M. Analysis of miRNA and mRNA expression profiles highlights alterations in ionizing radiation response of human lymphocytes under modeled microgravity. PLoS One 2012, 7, e31293. [Google Scholar]
- Shin, S.; Cha, H.J.; Lee, E.M.; Jung, J.H.; Lee, S.J.; Park, I.C.; Jin, Y.W.; An, S. MicroRNAs are significantly influenced by p53 and radiation in HCT116 human colon carcinoma cells. Int. J. Oncol 2009, 34, 1645–1652. [Google Scholar]
- Joly-Tonetti, N.; Viñuelas, J.; Gandrillon, O.; Lamartine, J. Differential miRNA expression profiles in proliferating or differentiated keratinocytes in response to gamma irradiation. BMC Genomics 2013, 14, 184. [Google Scholar]
- Metheetrairut, C.; Slack, F.J. MicroRNAs in the ionizing radiation response and in radiotherapy. Curr. Opin. Genet. Dev 2013, 23, 12–19. [Google Scholar]
- Di Francesco, A.; De Pittà, C.; Moret, F.; Barbieri, V.; Celotti, L.; Mognato, M. The DNA-damage response to γ-radiation is affected by miR-27a in A549 cells. Int. J. Mol. Sci 2013, 14, 17881–17896. [Google Scholar]
- Kwon, J.E.; Kim, B.Y.; Kwak, S.Y.; Bae, I.H.; Han, Y.H. Ionizing radiation-inducible microRNA miR-193a-3p induces apoptosis by directly targeting Mcl-1. Apoptosis 2013, 18, 896–909. [Google Scholar]
- Cummins, J.M.; He, Y.; Leary, R.J.; Pagliarini, R.; Diaz, L.A., Jr.; Sjoblom, T.; Barad, O.; Bentwich, Z.; Szafranska, A.E.; Labourier, E.; et al. The colorectal microRNAome. Proc. Natl. Acad. Sci. USA 2006, 103, 3687–3692. [Google Scholar]
- Tatarano, S.; Chiyomaru, T.; Kawakami, K.; Enokida, H.; Yoshino, H.; Hidaka, H.; Nohata, N.; Yamasaki, T.; Gotanda, T.; Tachiwada, T.; et al. Novel oncogenic function of mesoderm development candidate 1 and its regulation by MiR-574-3p in bladder cancer cell lines. Int. J. Oncol 2012, 40, 951–959. [Google Scholar]
- Gui, J.; Tian, Y.; Wen, X.; Zhang, W.; Zhang, P.; Gao, J.; Run, W.; Tian, L.; Jia, X.; Gao, Y. Serum microRNA characterization identifies miR-885-5p as a potential marker for detecting liver pathologies. Clin. Sci 2011, 120, 183–193. [Google Scholar]
- Forbes, S.A.; Bhamra, G.; Bamford, S.; Dawson, E.; Kok, C.; Clements, J.; Menzies, A.; Teague, J.W.; Futreal, P.A.; Stratton, M.R. The Catalogue of Somatic Mutations in Cancer (COSMIC). In Current Protocols in Human Genetics; Haines, J.L., Korf, B.R., Morton, C.C., Seidman, C.E., Seidman, J.G., Smith, D.R., Sharer, J.D., Strong, T.B., Eds.; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2008. [Google Scholar] [CrossRef]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 2011, 39(Database issue), D945–D950. [Google Scholar]
- Shiau, C.K.; Gu, D.L.; Chen, C.F.; Lin, C.H.; Jou, Y.S. IGRhCellID: Integrated genomic resources of human cell lines for identification. Nucleic Acids Res 2011, 39(Database issue), D520–D524. [Google Scholar]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human MicroRNA targets. PLoS Biol 2005, 2, e363. [Google Scholar]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res 2008, 36, D149–D153. [Google Scholar]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar]
- Krek, A.; Grün, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet 2005, 37, 495–500. [Google Scholar]
- Rehmsmeier, M.; Steffen, P.; Hochsmann, M.; Giegerich, R. Fast and effective prediction of microRNA/target duplexes. RNA 2004, 10, 1507–1517. [Google Scholar]
- Witkos, T.M.; Koscianska, E.; Krzyzosiak, W.J. Practical aspects of microRNA target prediction. Curr. Mol. Med 2011, 11, 93–109. [Google Scholar]
- Wojcik, E.; Murphy, A.M.; Fares, H.; Dang-Vu, K.; Tsubota, S.I. Enhancer of rudimentaryp1, e(r)p1, a highly conserved enhancer of the rudimentary gene. Genetics 1994, 138, 1163–1170. [Google Scholar]
- Isomura, M.; Okui, K.; Fujiwara, T.; Shin, S.; Nakamura, Y. Cloning and mapping of a novel human cDNA homologous to DROER, the enhancer of the Drosophila melanogaster rudimentary gene. Genomics 1996, 32, 125–127. [Google Scholar]
- Gelsthorpe, M.; Pulumati, M.; McCallum, C.; Dang-Vu, K.; Tsubota, S.I. The putative cell cycle gene, enhancer of rudimentary, encodes a highly conserved protein found in plants and animals. Gene 1997, 186, 189–195. [Google Scholar]
- Pogge von Strandmann, E.; Senkel, S.; Ryffel, G.U. ERH (enhancer of rudimentary homologue), a conserved factor identical between frog and human, is a transcriptional repressor. Biol. Chem 2001, 382, 1379–1385. [Google Scholar]
- Lukasik, A.; Uniewicz, K.A.; Kulis, M.; Kozlowski, P. Ciz1, a p21 Cip1/Waf1-interacting zinc finger protein and DNA replication factor, is a novel molecular partner for human enhancer of rudimentary homolog. FEBS J 2008, 275, 332–340. [Google Scholar]
- Kwak, Y.T.; Guo, J.; Prajapati, S.; Park, K.J.; Surabhi, R.M.; Miller, B.; Gehrig, P.; Gaynor, R.B. Methylation of SPT5 regulates its interaction with RNA polymerase II and transcriptional elongation properties. Mol. Cell 2003, 11, 1055–1066. [Google Scholar]
- Fujimura, A.; Kishimoto, H.; Yanagisawa, J.; Kimura, K. Enhancer of rudimentary homolog (ERH) plays an essential role in the progression of mitosis by promoting mitotic chromosome alignment. Biochem. Biophys. Res. Commun 2012, 423, 588–592. [Google Scholar]
- Weng, M.T.; Lee, J.H.; Wei, S.C.; Li, Q.; Shahamatdar, S.; Hsu, D. Evolutionarily conserved protein ERH controls CENP-E mRNA splicing and is required for the survival of KRAS mutant cancer cells. Proc. Natl. Acad. Sci. USA 2012, 109, E3659–E3667. [Google Scholar]
- Pawlik, T.M.; Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol 2004, 59, 928–942. [Google Scholar]
- Mitsui, K.; Matsumoto, A.; Ohtsuka, S.; Ohtsubo, M.; Yoshimura, A. Cloning and characterization of a novel p21Cip1/Waf1-interacting zinc finger protein, ciz1. Biochem. Biophys. Res. Commun 1999, 264, 457–464. [Google Scholar]
- Higgins, G.; Roper, K.M.; Watson, I.J.; Blackhall, F.H.; Rom, W.N.; Pass, H.I.; Ainscough, J.F.; Coverley, D. Variant Ciz1 is a circulating biomarker for early-stage lung cancer. Proc. Natl. Acad. Sci. USA 2012, 109, E3128–E3135. [Google Scholar]
- den Hollander, P.; Rayala, S.K.; Coverley, D.; Kumar, R. Ciz1, a Novel DNA-binding coactivator of the estrogen receptor alpha, confers hypersensitivity to estrogen action. Cancer Res 2006, 66, 11021–11029. [Google Scholar]
- den Hollander, P.; Kumar, R. Dynein light chain 1 contributes to cell cycle progression by increasing cyclin-dependent kinase 2 activity in estrogen-stimulated cells. Cancer Res 2006, 66, 5941–5949. [Google Scholar]
- Rahman, F.A.; Aziz, N.; Coverley, D. Differential detection of alternatively spliced variants of Ciz1 in normal and cancer cells using a custom exon-junction microarray. BMC Cancer 2010, 10, 482. [Google Scholar]
- Nishibe, R.; Watanabe, W.; Ueda, T.; Yamasaki, N.; Koller, R.; Wolff, L.; Honda, Z.; Ohtsubo, M.; Honda, H. CIZ1, a p21Cip1/Waf1-interacting protein, functions as a tumor suppressor in vivo. FEBS Lett. 2013, 587, 1529–1535. [Google Scholar]
- Banko, M.I.; Krzyzanowski, M.K.; Turcza, P.; Maniecka, Z.; Kulis, M.; Kozlowski, P. Identification of Amino Acid Residues of ERH Required for Its Recruitment to Nuclear Speckles and Replication Foci in HeLa Cells. PLoS One 2013, 8, e74885. [Google Scholar]
- Rocco, J.W.; Sidransky, D. p16(MTS-1/CDKN2/INK4A) in cancer progression. Exp. Cell Res 2001, 264, 42–55. [Google Scholar]
- Sherr, C.J. The INK4a/ARF network in tumour suppression. Nat. Rev. Mol. Cell. Biol 2001, 2, 731–737. [Google Scholar]
- Nevins, J.R. E2F: A link between the Rb tumor suppressor protein and viral oncoproteins. Science 1992, 258, 424–429. [Google Scholar]
- Harbour, J.W.; Luo, R.X.; Dei Santi, A.; Postigo, A.A.; Dean, D.C. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999, 98, 859–869. [Google Scholar]
- Witkiewicz, A.K.; Knudsen, K.E.; Dicker, A.P.; Knudsen, E.S. The meaning of p16(ink4a) expression in tumors: Functional significance, clinical associations and future developments. Cell Cycle 2011, 10, 2497–2503. [Google Scholar]
- Eymin, B.; Karayan, L.; Séité, P.; Brambilla, C.; Brambilla, E.; Larsen, C.J.; Gazzéri, S. Human ARF binds E2F1 and inhibits its transcriptional activity. Oncogene 2001, 20, 1033–1041. [Google Scholar]
- Eymin, B.; Leduc, C.; Coll, J.L.; Brambilla, E.; Gazzeri, S. p14ARF induces G2 arrest and apoptosis independently of p53 leading to regression of tumours established in nude mice. Oncogene 2003, 22, 1822–1835. [Google Scholar]
- Hemmati, P.G.; Gillissen, B.; von Haefen, C.; Wendt, J.; Stärck, L.; Güner, D.; Dörken, B.; Daniel, P.T. Adenovirus-mediated overexpression of p14(ARF) induces p53 and Bax-independent apoptosis. Oncogene 2002, 21, 3149–3161. [Google Scholar]
- Tsuji, K.; Mizumoto, K.; Sudo, H.; Kouyama, K.; Ogata, E.; Matsuoka, M. p53-independent apoptosis is induced by the p19ARF tumor suppressor. Biochem. Biophys. Res. Commun 2002, 295, 621–629. [Google Scholar]
- Yarbrough, W.G.; Bessho, M.; Zanation, A.; Bisi, J.E.; Xiong, Y. Human tumor suppressor ARF impedes S-phase progression independent of p53. Cancer Res 2002, 62, 1171–1177. [Google Scholar]
- Matsumoto, Y.; Iwakawa, M.; Furusawa, Y.; Ishikawa, K.; Aoki, M.; Imadome, K.; Matsumoto, I.; Tsujii, H.; Ando, K.; Imai, T. Gene expression analysis in human malignant melanoma cell lines exposed to carbon beams. Int. J. Radiat. Biol 2008, 84, 299–314. [Google Scholar]
- Wang, H.; Ach, R.A.; Curry, B. Direct and sensitive miRNA profiling from low-input total RNA. RNA 2007, 13, 151–159. [Google Scholar]
- miRBase (release 9.1). Available online: http://microrna.sanger.ac.uk/sequences/ (accessed on 26 February 2007).
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 2006, 34, D140–D144. [Google Scholar]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 2011, 39, D152–D157. [Google Scholar]
- miRBase (release 10.1). Available online: http://microrna.sanger.ac.uk/sequences/ (accessed on 28 February 2008).
- NCBI/GENE. Available online: http://www.ncbi.nlm.nih.gov/Genebank/ (accessed on 28 February 2008).
- microRNA.org. Available online: http://www.microrna.org/microrna/ (accessed on 28 February 2008).
- NCBI/BLAST. Available online: http://blast.ncbi.nlm.nih.gov/Blast.cgi/ (accessed on 28 February 2008).
- Fujita, M.; Otsuka, Y.; Yamada, S.; Iwakawa, M.; Imai, T. X-ray irradiation and Rho-kinase inhibitor additively induce invasiveness of the cells of the pancreatic cancer line, MIAPaCa-2, which exhibits mesenchymal and amoeboid motility. Cancer Sci 2011, 102, 792–798. [Google Scholar]
- Cheng, A.M.; Byrom, M.W.; Shelton, J.; Ford, L.P. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res 2005, 33, 1290–1297. [Google Scholar]
- Solly, K.; Wang, X.; Xu, X.; Strulovici, B.; Zheng, W. Application of real-time cell electronic sensing (RT-CES) technology to cell-based assays. Assay Drug Dev. Technol 2004, 2, 363–372. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene symbol | Gene description | Accession no. | Expression level (signal intensity) | |||
|---|---|---|---|---|---|---|
| FC | KD | OE | p-value | |||
| ERH | Enhancer of rudimentary homolog (Drosophila) | NM_004450.1 | −2.1 | 31,602 | 14,698 | 3.8 × 10−36 |
| ZYG11A | zyg-11 homolog A (C. elegans) | XM_001133615.1 | −2.1 | 269 | 126 | 4.5 × 10−22 |
| GPR172B | G protein-coupled receptor 172B | NM_001104577.1 (var. 1) NM_017986.3 (var. 2) | −7.1 | 232 | 33 | 0 |
| ZMAT3 | Zinc finger, matrin type 3 | NM_022470.2 (var. 1) NM_152240.1 (var. 2) | −5.5 | 80 | 15 | 0 |
| ATPAF-AS1 | ATPAF1 antisense RNA 1 (non-protein coding) | NM_001145474.1 | −3.1 | 46 | 13 | 0.0006 |
| SLC34A1 | Solute carrier family 34 (sodium phosphate), member 1 | NM_003052.3 | −3.8 | 26 | 7 | 4.5 × 10−9 |
| AQP7 | Aquaporin 7 | NM_001170.1 | −2.0 | 24 | 12 | 9.7 × 10−7 |
| PRDM7 | PR domain containing 7 | NM_001098173.1 (var. 1) NM_052996.2 (var. 2) | −2.3 | 20 | 9 | 7.0 × 10−5 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ishikawa, K.-i.; Ishikawa, A.; Shoji, Y.; Imai, T. A Genotoxic Stress-Responsive miRNA, miR-574-3p, Delays Cell Growth by Suppressing the Enhancer of Rudimentary Homolog Gene in Vitro. Int. J. Mol. Sci. 2014, 15, 2971-2990. https://doi.org/10.3390/ijms15022971
Ishikawa K-i, Ishikawa A, Shoji Y, Imai T. A Genotoxic Stress-Responsive miRNA, miR-574-3p, Delays Cell Growth by Suppressing the Enhancer of Rudimentary Homolog Gene in Vitro. International Journal of Molecular Sciences. 2014; 15(2):2971-2990. https://doi.org/10.3390/ijms15022971
Chicago/Turabian StyleIshikawa, Ken-ichi, Atsuko Ishikawa, Yoshimi Shoji, and Takashi Imai. 2014. "A Genotoxic Stress-Responsive miRNA, miR-574-3p, Delays Cell Growth by Suppressing the Enhancer of Rudimentary Homolog Gene in Vitro" International Journal of Molecular Sciences 15, no. 2: 2971-2990. https://doi.org/10.3390/ijms15022971