1. Introduction
G protein-coupled receptors (GPCRs) are a large family of plasma membrane localized receptors with a characteristic seven transmembrane domain topology. Through activation of heterotrimeric G-proteins, GPCRs impact a multitude of normal cellular and disease processes and thus their modulation is of great interest. The regulation of receptor desensitization, internalization, and recycling back to the membrane has been extensively studied; less understood is the regulation of receptor biosynthesis and trafficking to the plasma membrane (reviewed in [
1,
2]).
GPCR progression through the endoplasmic reticulum (ER) and Golgi is mediated by interactions with several classes of proteins. Several of these, including many PDZ (
PSD-95,
Discs-large, and
ZO-1) containing proteins, interact with the
C terminus of their GPCR binding partners, while others show specific binding to the third intracellular loops [
2–
5]. Recently, a highly conserved leucine in the first intracellular loop was shown to be critical for the ER export of multiple GPCRs through an unknown mechanism [
6]. Receptor-activity-modifying proteins (RAMPs) not only promote ER exit of a subset of GPCRs, but can influence receptor specificity at the plasma membrane [
2,
7]. GPCRs also show different requirements for Ras-like small GTPases during their trafficking [
8]. In this paper we focus on the atypical role of golgin-160 in influencing the initial delivery of the beta-1 adrenergic receptor (β1AR) to the plasma membrane.
β1AR is the primary β adrenergic receptor in human cardiomyocytes and is responsible for the catecholamine-induced regulation of heart rate in the sympathetic “flight or fight” response. It also has been implicated in adipogenesis and memory formation; however, the majority of research has concentrated on its role in the heart as β1AR has been widely connected with heart disease [
9–
11]. Both the downregulation and overstimulation of β1AR have been connected to myocyte apoptosis [
12–
14]. Until relatively recently, the bulk of research on the surface expression of β1AR has focused on its cycle of receptor desensitization, internalization, and recycling to the plasma membrane. This started to shift in 2004 when He
et al. demonstrated that β1AR can be delayed at the Golgi when the Golgi resident PDZ-containing protein PIST (
PDZ domain protein
interacting
specifically with
TC10) is overexpressed [
3], and in 2006 Hicks
et al. found that golgin-160 promotes trafficking of β1AR from the Golgi complex to the plasma membrane [
15].
Golgins are a large family loosely characterized by their Golgi localization and extended coiled coil motifs and have traditionally been associated with maintenance of Golgi structure and assisting bulk flow of proteins through the organelle [
16]. Golgin-160 is a vertebrate-specific golgin that is one of only three golgins implicated in trafficking of specific cargoes [
15,
17–
20]. Hicks
et al. demonstrated
in vitro that the third intracellular loop of β1AR can interact with the N-terminal head domain of golgin-160 in a PIST-independent manner, and that depletion of golgin-160 from HeLa cells decreases the steady state surface levels of exogenously expressed β1AR without affecting internalization rates. Surprisingly, given the steady state localization of golgin-160 to the
cis-Golgi, this effect on trafficking occurs at or after the
trans-Golgi network [
15]. While we initially proposed that golgin-160 might promote palmitoylation of β1AR, we later demonstrated that palmitoylation of β1AR was independent of golgin-160 and instead impacts receptor internalization, leaving the mechanism by which golgin-160 facilitates trafficking of β1AR unknown [
21].
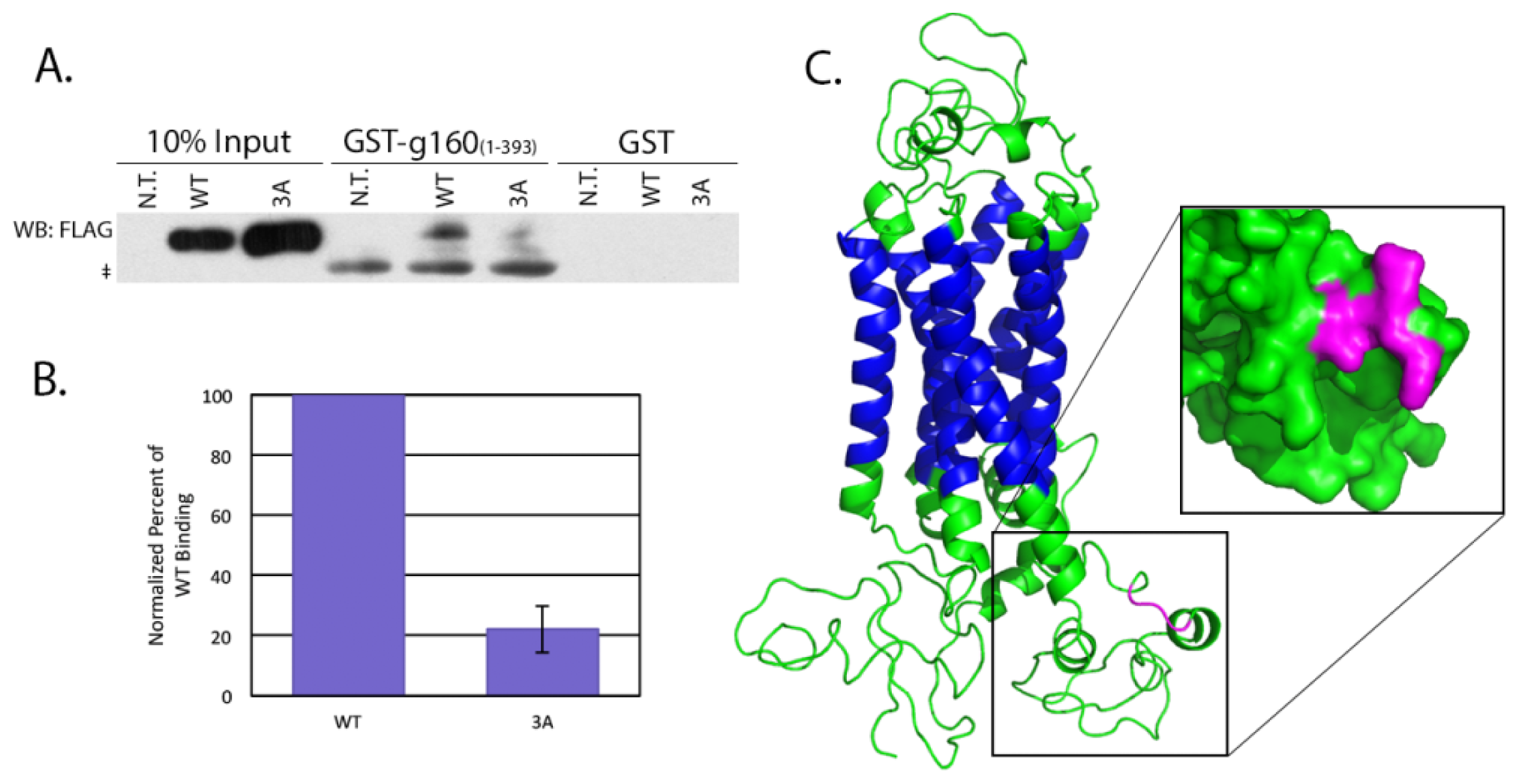
The specific interaction between golgin-160 and β1AR, in addition to being an unusual role for a golgin, suggests a novel mechanism of β1AR regulation independent of receptor desensitization and recycling. In this report, we further dissect the interaction between these two proteins and demonstrate that three basic residues in the third intracellular loop of β1AR are critical for binding to golgin-160. We further show that this binding is directly responsible for the efficient delivery of exogenously expressed β1AR from the trans-Golgi network to the cell surface.
3. Discussion
Golgin-160 was the first protein described to positively impact β1AR trafficking from the Golgi to the plasma membrane. We previously demonstrated
in vitro binding between these two proteins [
15], but the interaction was largely uncharacterized. In this report, we have shown that golgin-160 can directly bind β1AR, that three basic residues in the third intracellular loop of β1AR are critical for the interaction, and that these residues are necessary for efficient β1AR trafficking from the Golgi to the plasma membrane in cells.
The third intracellular loop and
C-terminus are the regions of greatest variance between the β1 and β2 adrenergic receptors. It is thought that the differences in protein interactions in these two domains account for many of the functional differences between the two receptors [
5]. The majority of proteins known to interact with β1AR bind to its
C-terminal cytoplasmic tail via their PDZ domains and are involved in signal transduction and recycling from the plasma membrane [
5]. Previously, only endophilins SH3p4 and SH3p13 have been shown to bind to the third intracellular loop of β1AR. This binding depends on the Src homology SH3 domain and mediates receptor internalization and coupling with G
s [
26]. Golgin-160 binding to the third intracellular loop may influence the interactions of these other proteins with β1AR. He
et al. found that PSD-95 can compete for a binding site with PIST via their PDZ domains in order to modulate β1AR trafficking [
3]; golgin-160 could be occluding a binding site in intracellular loop 3 in a similar manner. Golgin-160 could also affect proteins binding to the
C-terminal tail of β1AR. In 2007, Gardner
et al. found that binding of a SAP97-AKAP79 protein scaffold to the
C-terminus of β1AR was required for protein kinase A to phosphorylate a residue in loop 3, which is necessary for β1AR recycling to the plasma membrane [
27]. In an analogous fashion, golgin-160 may work cooperatively with other proteins that interact with the
C-terminus of β1AR. At present, the influence of golgin-160 on other β1AR binding partners has not been studied.
To date, golgin-160 is the only protein demonstrated to interact directly with β1AR to regulate its Golgi to plasma membrane trafficking. This is an atypical role for a golgin: most golgins maintain Golgi structure or assist in global trafficking, for example by acting as vesicle tethers. Only two other golgins,
trans-Golgi-localized golgin-97 and p230/golgin-245, have been implicated in trafficking of specific cargoes. Golgin-97 colocalizes with E-cadherin in post-Golgi transport carriers and golgin-97 depletion inhibits E-cadherin trafficking [
17]. Similarly, golgin-245 colocalizes with tumor necrosis factor-α (TNF) in TGN tubules and depletion of golgin-245 prevents TNF localization to the plasma membrane [
20]. Golgin-160, in addition to its role in facilitating plasma membrane delivery of β1AR, can bind to the apical potassium channel ROMK and promote its delivery to the cell surface [
18], and it is required for the proper localization of GLUT4 into insulin-responsive storage compartments [
19].
Because golgin-160 impacts efficient trafficking of β1AR and GLUT4 is mislocalized in its absence, we hypothesize that golgin-160 facilitates specialized sorting of cargo at the TGN. The complexity of post-Golgi sorting has become more apparent in recent years. In non-polarized cells, it was once believed that proteins moved from the TGN to the plasma membrane by a single route, but it is now apparent that there are many routes to the cell surface [
28]. In polarized cells, not only have distinct apical and basolateral sorting pathways been demonstrated, but proteins headed for the same cell surface also segregate into separate transport pathways at the TGN based on their mode of regulation [
29–
31]. Very little, however, is known about the composition of these various transport carriers or how this differential sorting occurs. We propose that golgin-160 plays a role in this sorting step, guiding cargo into vesicles so that specific cargo can be efficiently targeted. It is possible that golgin-160 accomplishes this directly by moving with cargo, or it could facilitate the interaction of its cargo with another protein, as proposed above. Both of these possibilities will be investigated by using the 3A mutant as a golgin-160-independent cargo protein. Since mutating only three basic residues dramatically interferes with this interaction, it may be that either a charged stretch or a three-dimensional conformation created by these three residues is critical for interaction with golgin-160. Our findings bring us closer to the identification of a binding motif, sequence-based or structural, among golgin-160 cargoes, which will then allow for a directed investigation into other potential cargoes including other GPCRs.
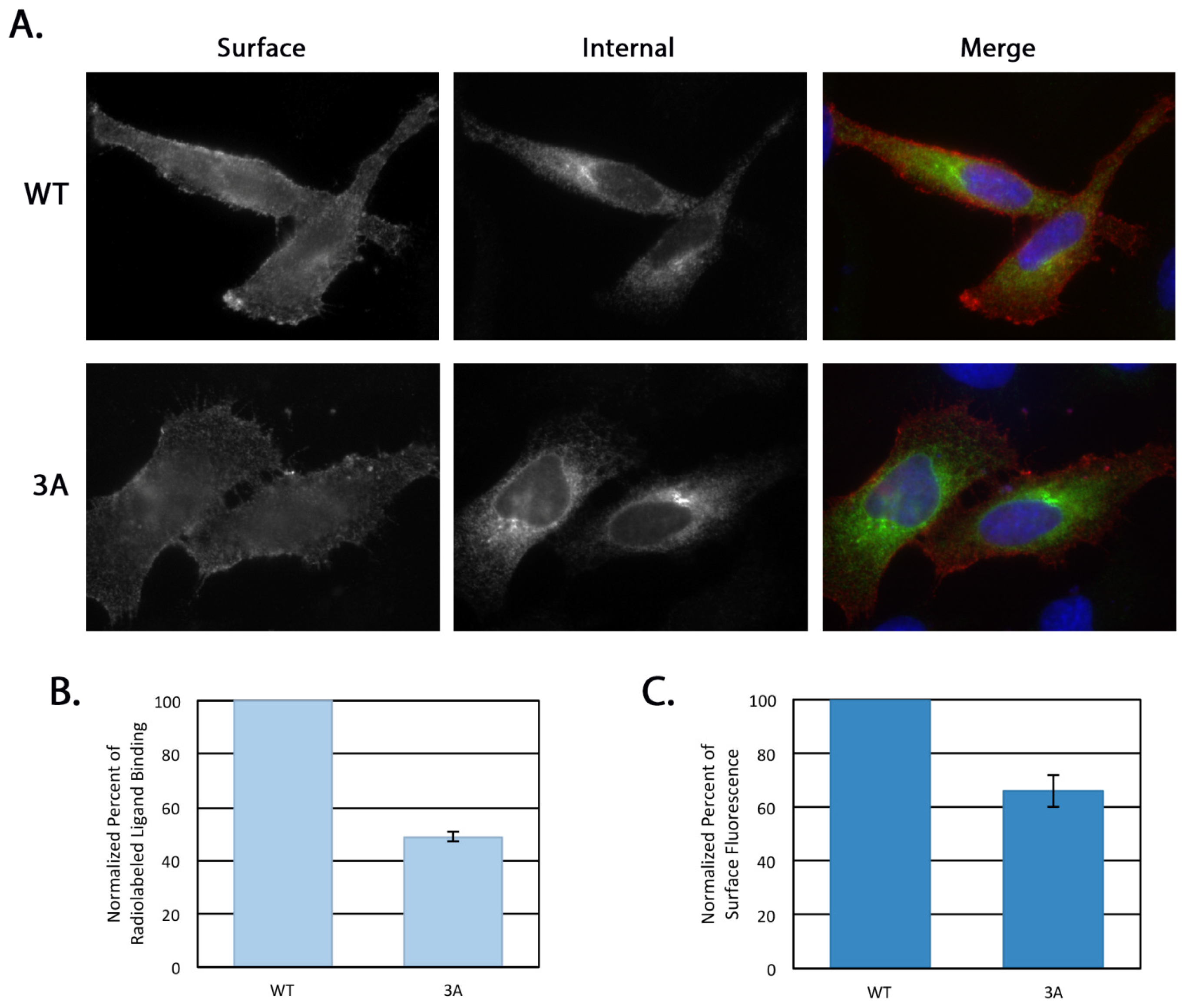
Neither depletion of golgin-160 nor disruption of its binding to β1AR completely disrupts β1AR localization—more than 50% of β1AR still makes it to the plasma membrane. This suggests that golgin-160 is playing a regulatory role, enhancing β1AR surface expression. This method of regulation is possible as golgin-160 can itself be subject to regulatory control. Golgin-160 is recruited to the Golgi through binding of GTP-bound ARF1, which is in turn rapidly cycling on and off the Golgi [
32,
33]. Its localization may also be affected by interactions with dynein [
32]. Additionally, golgin-160 can undergo cleavage by caspase-2, -3, and -7 as well as phosphorylation by MLK3 [
34,
35]. All of these interactions and modifications could alter the localization or conformation of golgin-160, which could mediate interaction with β1AR. In the future, it will prove useful to study the dynamics of endogenous golgin-160 and β1AR movement in the context of cardiomyocytes to gain a better understanding of this putative regulatory cycle.
4. Experimental Section
4.1. Cells and Transfection
HeLa cells were maintained in DMEM (Life technologies, Grand Island, NY, USA) with 10% Fetal Bovine Serum (Atlanta Biologicals, Flowery Branch, GA, USA) and 0.1 mg/mL Normocin (InvivoGen, San Diego, CA, USA) at 37 °C with 5% CO2. Cells were grown in 35 mm dishes, 6 cm dishes, or 12-well plates depending on the experiment, and transfected with 3 μL X-tremeGENE9 DNA transfection reagent (Roche, Indianapolis, IN, USA) per 1 μg DNA.
4.2. Expression Constructs
Glutathione S-transferase (GST) fusion proteins of golgin-160
(1–393) have been previously described [
36]. The untagged golgin-160
(1–393) protein was constructed by polymerase chain reaction (PCR) amplifying residues 1–393 from the previously described pBluescript SK+ golgin-160 construct [
34] and inserting a stop codon after residue 393, which was then inserted into New England Biolab’s pTBY12 vector for purification using the IMPACT system (New England Biolabs, Beverly, MD, USA). Human FLAG-β1AR cDNA in pcDNA3 was provided by Randy Hall (Emory University, Atlanta, GA, USA). The 3A mutation, converting K
308RR
310 to A
308AA
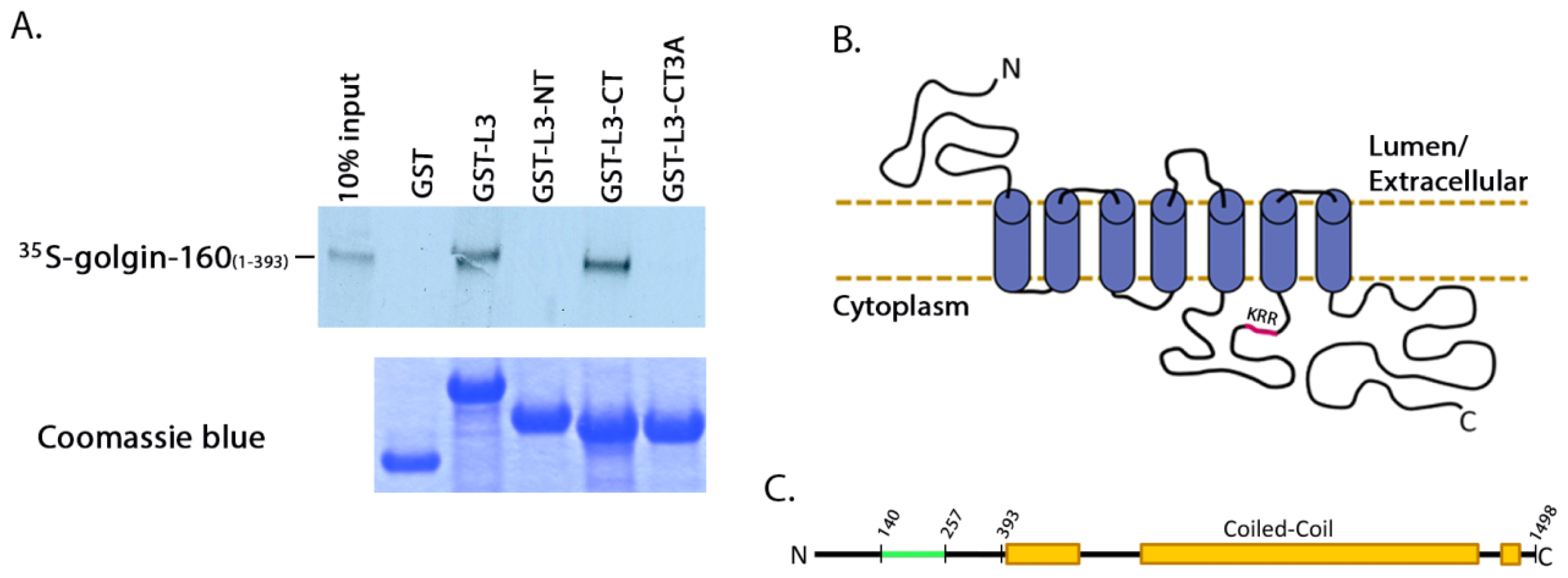
310, was introduced using Quikchange mutagenesis (Stratagene, La Jolla, CA, USA). GST-tagged β1AR loop 3 (L3) was constructed by PCR amplification of residues 249 to 325 of β1AR and inserting the fragment into the pGEX-2T expression plasmid (GE Healthcare, Little Chalfont, Buckinghamshire, UK). GST-β1AR L3
N-terminus (NT) was constructed by PCR amplifying residues 249 to 288 of β1AR, and the
C-terminus (CT) likewise by PCR amplifying residues 288 to 325, followed by insertion into the pGEX-2T expression plasmid. GST β1AR L3CT with the 3A mutation was created by amplifying residues 288 to 325 from the β1AR 3A plasmid (described above), followed by insertion into the pGET-2T expression plasmid. The cytosolic GFP vector pEGFP C1 was obtained from Clontech (Mountain View, CA, USA).
4.3. Antibodies
Mouse monoclonal anti-FLAG was from Sigma-Aldrich (St. Louis, MO, USA). Rabbit anti-β1AR was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit anti-golgin-160 was previously described [
34]. R-Phycoerythrin-conjugated IgG goat anti-mouse was from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA, USA). Alexa Fluor 568 anti-mouse IgG and Alexa Fluor 488 anti-rabbit IgG were from Life Technologies (Grand Island, NY, USA). Horseradish peroxidase (HRP)-conjugated secondary antibodies were from GE Healthcare (Little Chalfont, Buckinghamshire, UK).
4.4. In Vitro Binding
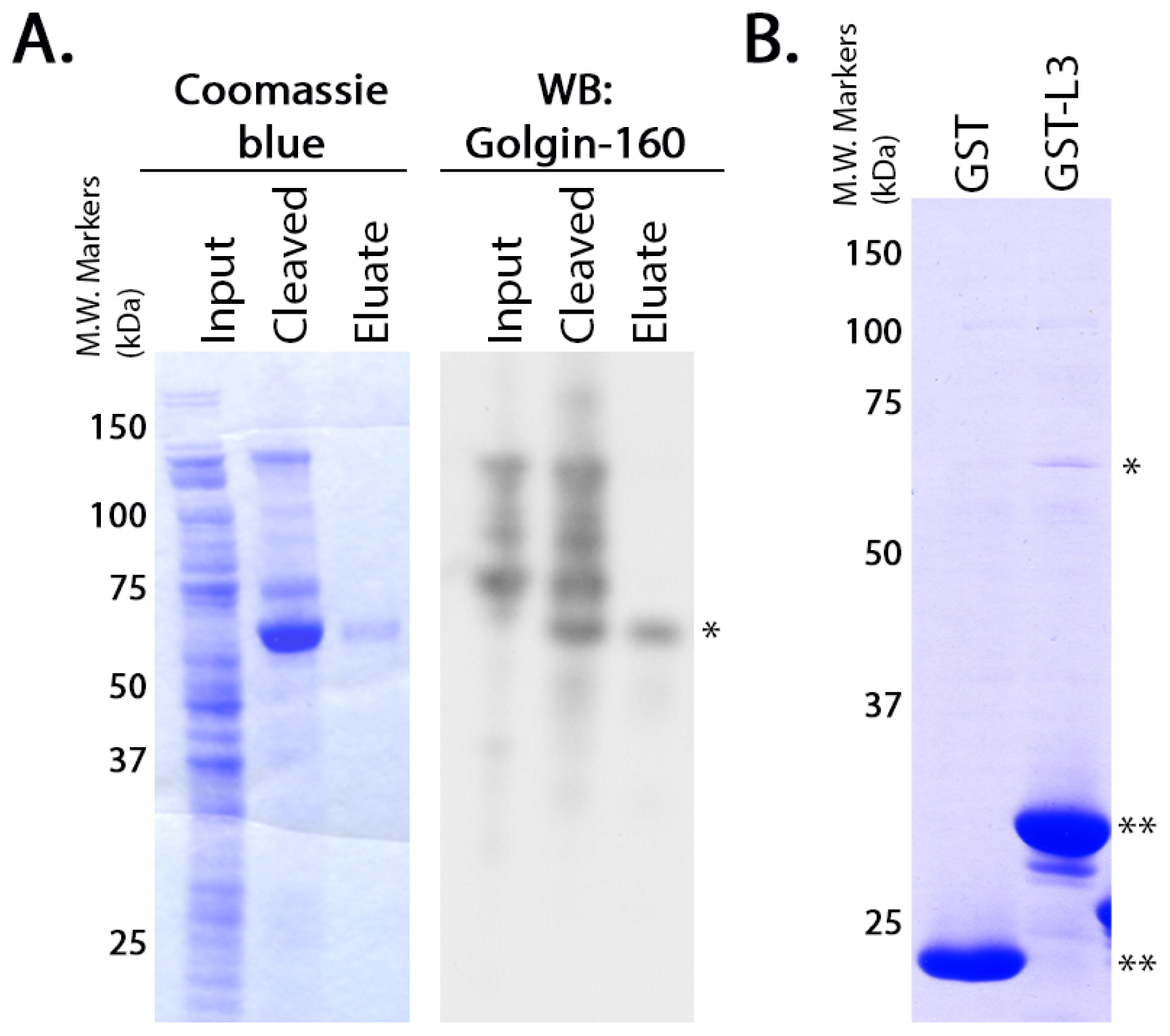
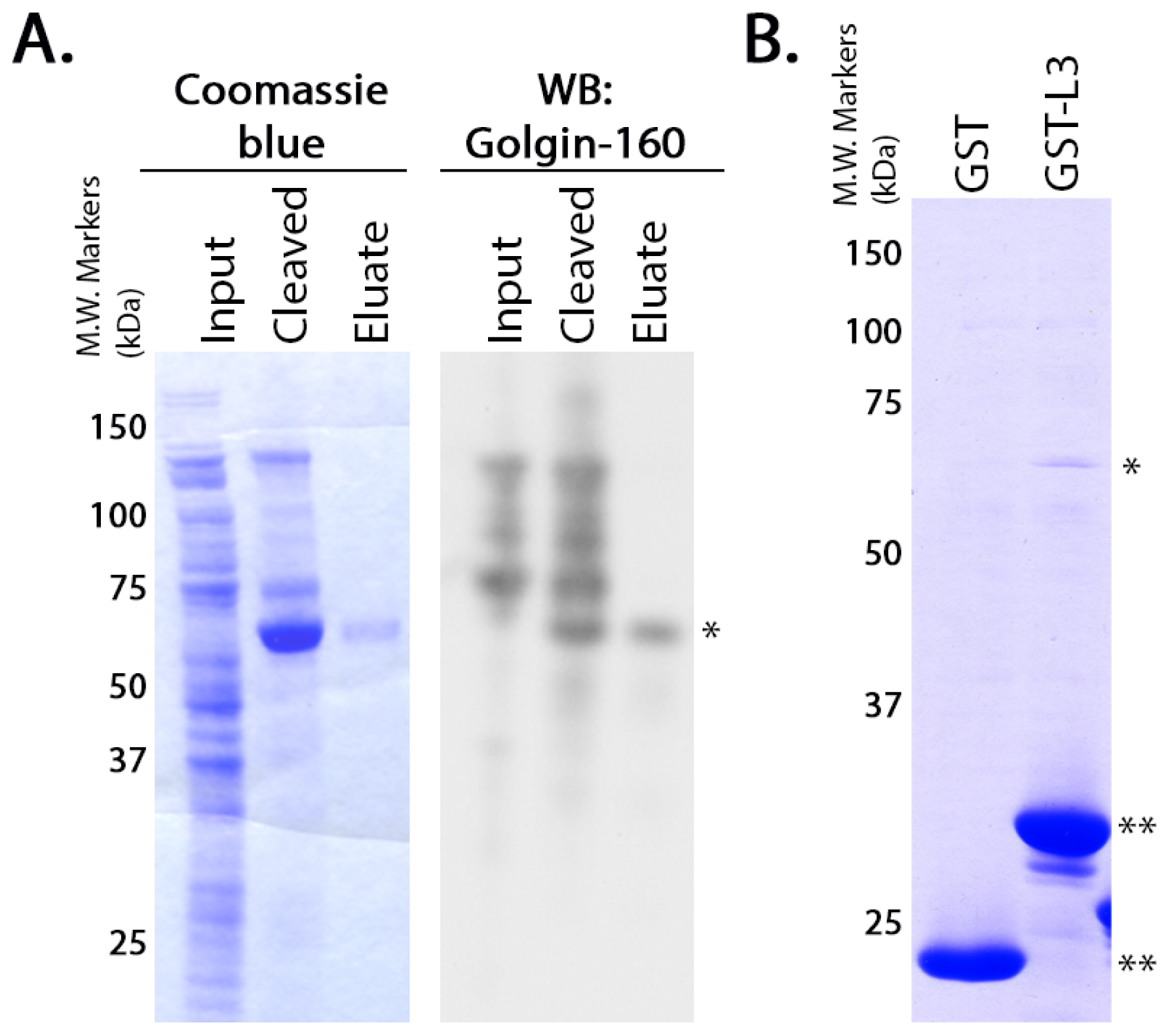
GST-fusion proteins were grown in
Escherichia coli BL21 cells (Stratagene) as previously described [
36]. GST or the GST-fusion proteins were purified from the soluble fraction of the cell lysates according to manufacturer instructions using glutathione-Sepharose 4B beads (GE Healthcare, Little Chalfont, Buckinghamshire, UK). The purified proteins (5 μg each) were diluted in PBS and rebound to glutathione-Sepharose 4B beads by incubating them overnight at 4 °C with rotation. Untagged golgin-160
(1–393) was purified using the NEB IMPACT system as described by the manufacturer (New England Biolabs, Beverly, MD, USA). To confirm the purity and identity of the resulting protein, the eluate was separated on NuPAGE 4%–12% Bis-Tris Gel (Invitrogen, Carlsbad, CA, USA) and was detected by Coomassie blue staining or immunoblotted with rabbit anti-golgin-160. Purified golgin-160
(1–393) in the elution buffer (20 mM HEPES pH 8.5, 500 mM NaCl, 1 mM EDTA, 50 mM DTT) was added to the conjugated GST-fusion proteins and incubated 4–6 h at 4 °C. After washing, protein remaining attached to the glutathione-Sepharose beads was eluted, separated by SDS-PAGE (10% gel) and detected by Coomassie blue staining.
For the binding assays using
in vitro transcribed and translated golgin-160, the GST-fusion proteins were purified and 3 μg of each was rebound to glutathione Sepharose beads as above. IVTT [
35S]-labeled golgin-160
(1–393) was prepared following the manufacturer’s instructions and as previously described (Promega, Madison, WI, USA, [
15]). Golgin-160 was then diluted in binding buffer (25 mM HEPES, 125 mM KOAc, 5 mM EDTA, 0.5% Nonidet P-40 (NP-40), 1 mM DTT) and incubated with the GST-fusion proteins for 2 h at 4 °C. After washing, bound golgin-160 was eluted and separated by SDS-PAGE followed by phosphorimaging on a PharosFX molecular imager (Bio-Rad, Hercules, CA, USA).
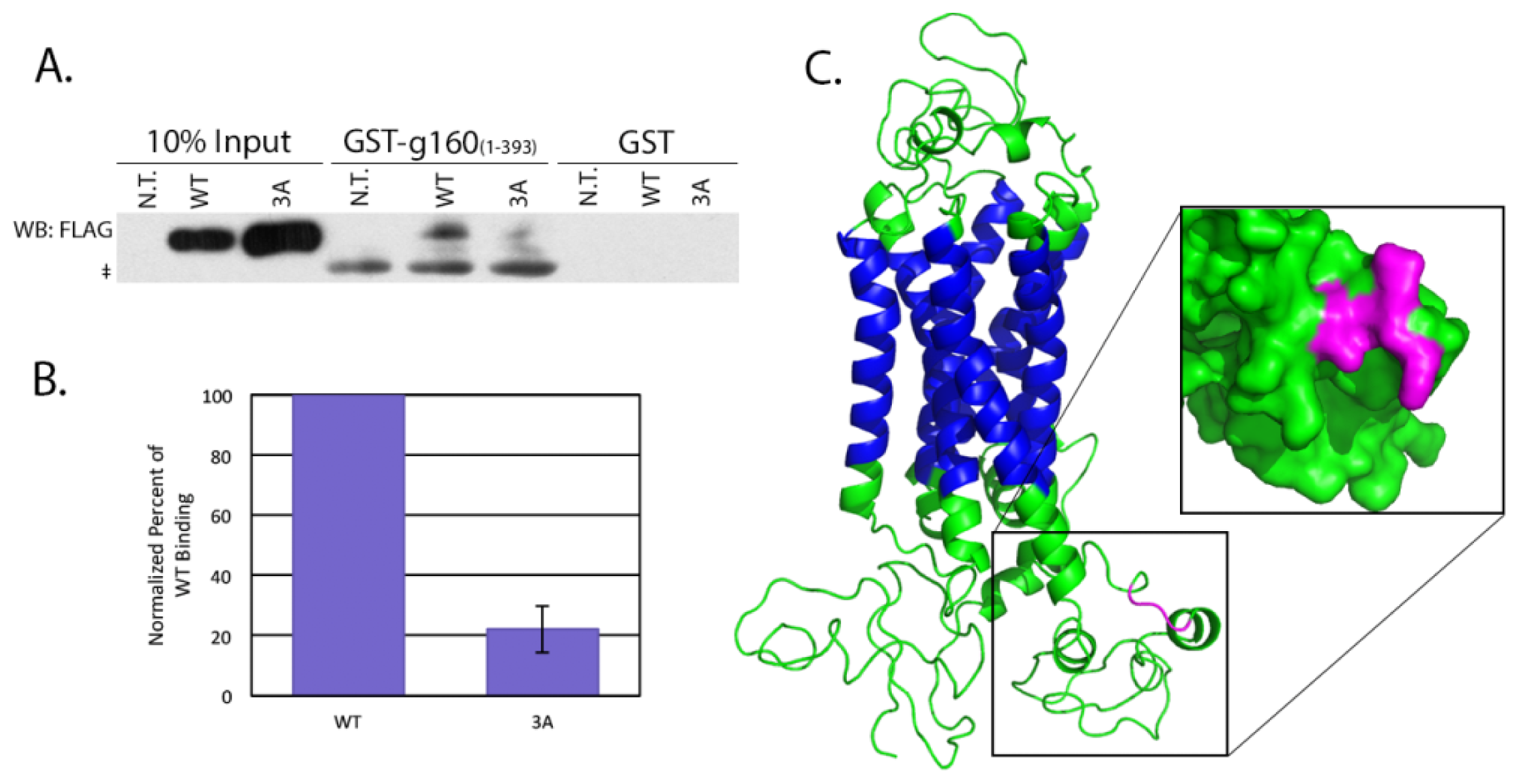
4.5. Pull Downs from Cell Lysates
HeLa cells grown to 70% confluency were left untransfected, or transfected with 0.5 μg of either pcDNA/WT or 3A β1AR. After incubating for an additional 16–18 h, cells were lysed in a potassium acetate lysis buffer (125 mM potassium acetate, 25 mM HEPES pH 7.1, 1 mM DTT, 1% NP-40, protease inhibitor cocktail). The soluble fraction of the lysed cells was incubated 2 h at 4 °C with 10 μg GST alone or GST-tagged golgin-160(1–393) that had been pre-conjugated to glutathione-Sepharose 4B beads. After washing the beads twice with lysis buffer, protein bound to the beads was separated by SDS-PAGE and β1AR was detected by immunoblotting with anti-FLAG antibody followed by ECL. The amount of protein in the input and pull down bands was measured after imaging on a VersaDoc Imaging System Model 5000 (Bio-Rad, Hercules, CA, USA) using Quantity One volume analysis tools (Bio-Rad, Hercules, CA, USA). Percent bound was calculated after first normalizing 3A and WT to their input and significance was calculated using a heteroscedastic two-tailed Student’s t-test.
4.6. Modeling of β1AR
The nucleotide sequence of the human β1AR mRNA was obtained from NCBI for the Homo sapiens adrenoceptor beta 1 (ADRB1) mRNA (Accession number NM_000684). The corresponding amino acid sequence was submitted for intensive modeling to the Protein Homology/analogY Recognition Engine (PHYRE) V 2.0 server (Structural Bioinformatics Group, London, UK). The resulting predicted protein structure was then visualized using PyMOL (Schrödinger, Portland, OR, USA).
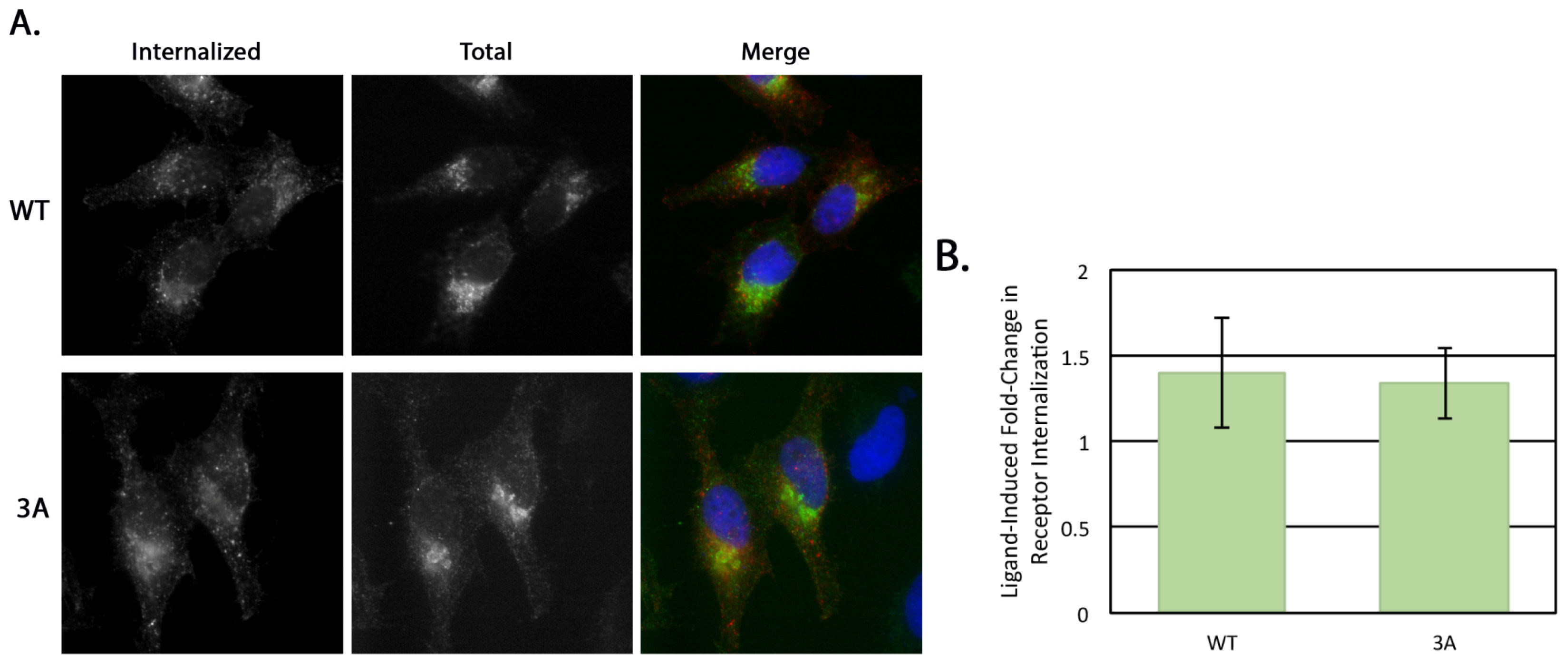
4.8. Receptor Internalization
HeLa cells at 70% confluency on coverslips in 35 mm dishes were transfected with 0.5 μg of either pcDNA/FLAG-β1AR WT or 3A. After 16–18 h the medium was switched to serum-free DMEM for 3 h. For the internalization assay, 1 μg/mL mouse anti-FLAG antibody was added to the serum-free medium with or without 10 uM isoproterenol (Iso, Sigma-Aldrich) for 30 min at 37 °C. Cells were then washed with PBS and left untreated or remaining surface antibody was removed with an acid wash (0.5 M NaCl, 0.5% HOAc, pH 1) for 1 min at room temperature. Cells were then fixed and permeabilized as previously described [
15] and the internal pool of β1AR detected using the rabbit anti-β1AR
C-terminal antibody described above. Alexa Fluor 568 anti-mouse IgG and Alexa Fluor 488 anti-rabbit IgG were used for secondary labeling and Hoescht 33258 was used as a DNA stain. Cells with similar rabbit anti-β1AR signal intensity were selected for comparison and all images were taken on the same day with the same exposure on an Axioskop microscope (Zeiss, Thornwood, NY, USA) equipped with epifluorescence using an ORCA-03G charge-coupled device camera (Hamamatsu, Japan) using iVision software (BioVision Technologies, Exton, PA, USA). The integrated pixel density of the anti-FLAG signal for each cell was determined using Image J (National Institutes of Health, Bethesda, MD, USA) and was normalized to background fluorescence. Statistical analysis was performed using a two-tailed heteroscedastic Student’s
t-test.
4.9. Surface Immunofluorescence
HeLa cells grown to 70% confluency on coverslips in 35 mm dishes were transfected with 0.5 μg of either pcDNA/FLAG-β1AR WT or 3A. After 5 h the cells were rinsed in ice cold PBS and incubated on ice with mouse anti-FLAG antibody for 15 min. Cells were then fixed and permeabilized as previously described [
15] and the internal pool of β1AR detected using the rabbit anti-β1AR
C-terminal antibody described above. Alexa Fluor 568 anti-mouse IgG and Alexa Fluor 488 anti-rabbit IgG were used for secondary labeling and Hoescht 33258 was used as a DNA stain. All images were taken on the same day with the same exposure on an Axioskop microscope (Zeiss, Thornwood, NY, USA) equipped with epifluorescence using an ORCA-03G charge-coupled device camera using iVision software (version 4.5.1, BioVision Technologies, Exton, PA, USA).
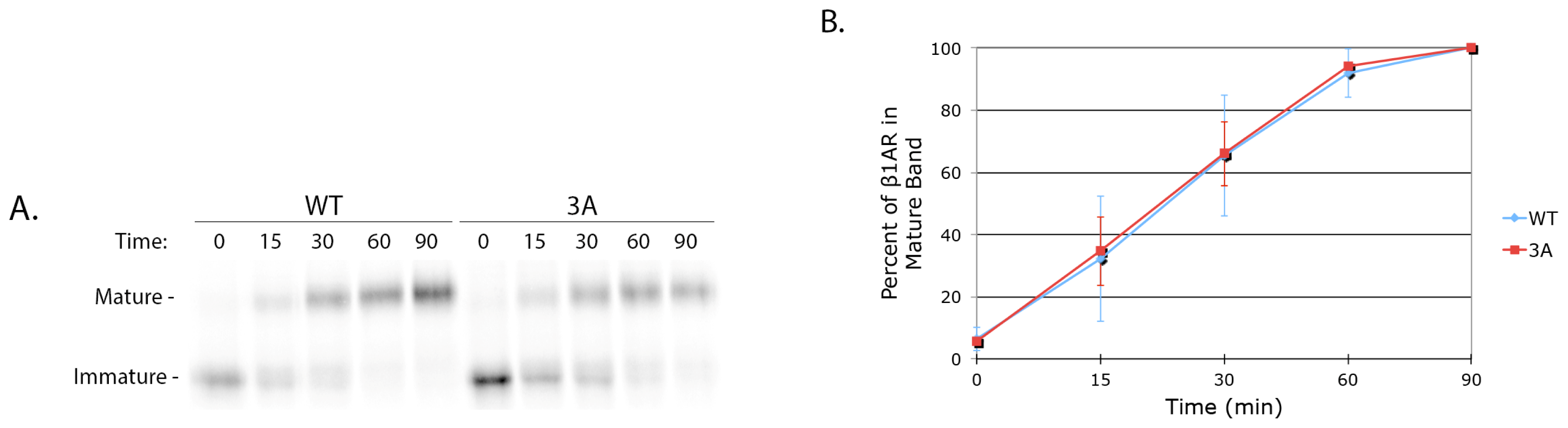
4.10. Cycloheximide Chase
HeLa cells were grown to 70% confluency in 35 mm dishes and were transfected with 0.5 μg of either pcDNA/FLAG-β1AR WT or 3A. After 16 h the cells were rinsed in PBS and incubated with DMEM containing 100 μg/mL cycloheximide (Calbiochem, La Jolla, CA, USA). After 0, 2, 4, 6, and 8 h, cells were lysed on ice for 5 min in 1% NP-40, 0.4% deoxycholic acid, 50 mM Tris pH 8, 62.5 mM EDTA pH 8, with protease inhibitor cocktail. Soluble protein was separated by SDS-PAGE and immunoblotted with rabbit anti-β1AR antibody.
4.11. Radiolabeled Ligand Binding
HeLa cells at 70% confluency on 12-well plates were transfected with 0.12 μg pcDNA/β1AR WT or 3A per well in triplicate. Nontransfected cells were used to determine background binding. After 14 h, cells were washed in cold PBS and incubated with 10 nM [3H]-labeled CGP-12177 (PerkinElmer Life Sciences, Waltham, MA, USA) diluted in KRH buffer (136 mM NaCl, 4.7 mM KCl, 1.25 mM MgSO4, 1.25 mM CaCl2, 20 mM HEPES, pH 7.4, and 2 mg/mL BSA) for 3 h at 4 °C. Cells were then washed in PBS and lysed as described above. Cell-associated radioactivity was determined using a Beckman Coulter LS6500 scintillation counter (Brea, CA, USA). Total expression level was determined by immunoblotting lysate prepared from a parallel transfected dish with anti-FLAG antibody followed by ECL. Three independent experiments (each done in triplicate) were averaged. Statistical analysis was performed using a two-tailed heteroscedastic Student’s t-test.
4.12. FACS Analysis
HeLa cells growing in 6 cm dishes to 70% confluency were co-transfected with 1.5 μg cytosolic GFP (pEGFP-C1) and 0.5 μg of either pcDNA/FLAG-β1AR WT or 3A. After 16–18 h, cells were harvested by trypsinization before being washed twice with DMEM and resuspended at (5–10) × 106 cells/mL in blocking solution (1% BSA in PBS). After blocking for 30 min on ice the cells were incubated 1 h on ice with monoclonal mouse anti-FLAG diluted in blocking solution. Cells were washed in PBS and then were incubated 1 h on ice in phycoerythrin-labeled goat anti-mouse IgG, also diluted in blocking solution. Cells were washed again in PBS before being resuspended in serum-free DMEM and analyzed using a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lake, NJ, USA). The mean phycoerythrin fluorescence intensity of cells expressing GFP was calculated for each sample using CellQuest Pro software (Becton Dickinson). Significance was determined using a two-tailed heteroscedastic Student’s t-test.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}