Survivin as a Preferential Target for Cancer Therapy

Abstract
:1. Apoptosis, a Developmental and Defense Mechanism
1.1. Intrinsic Apoptosis Pathway (Mitochondrial Pathway)
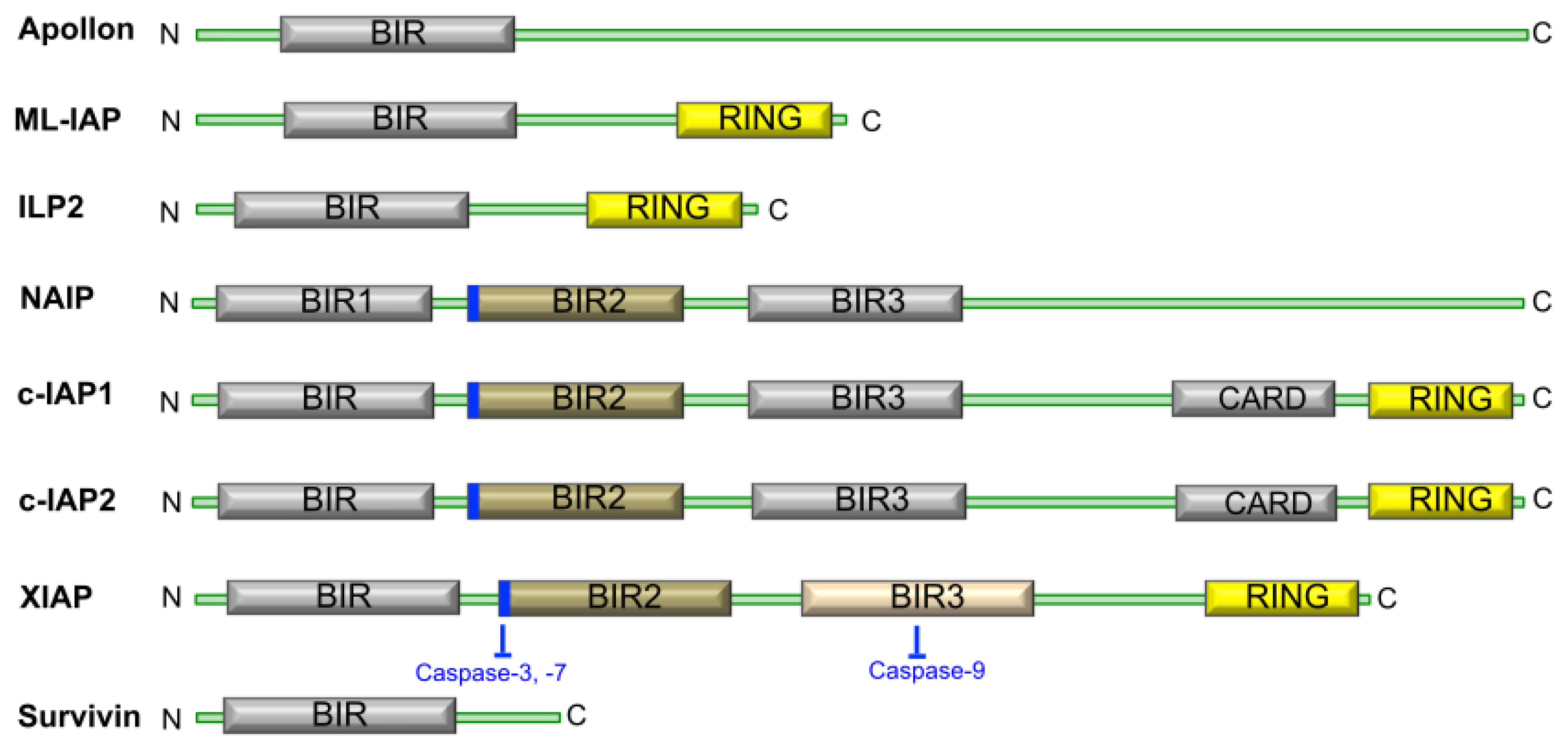
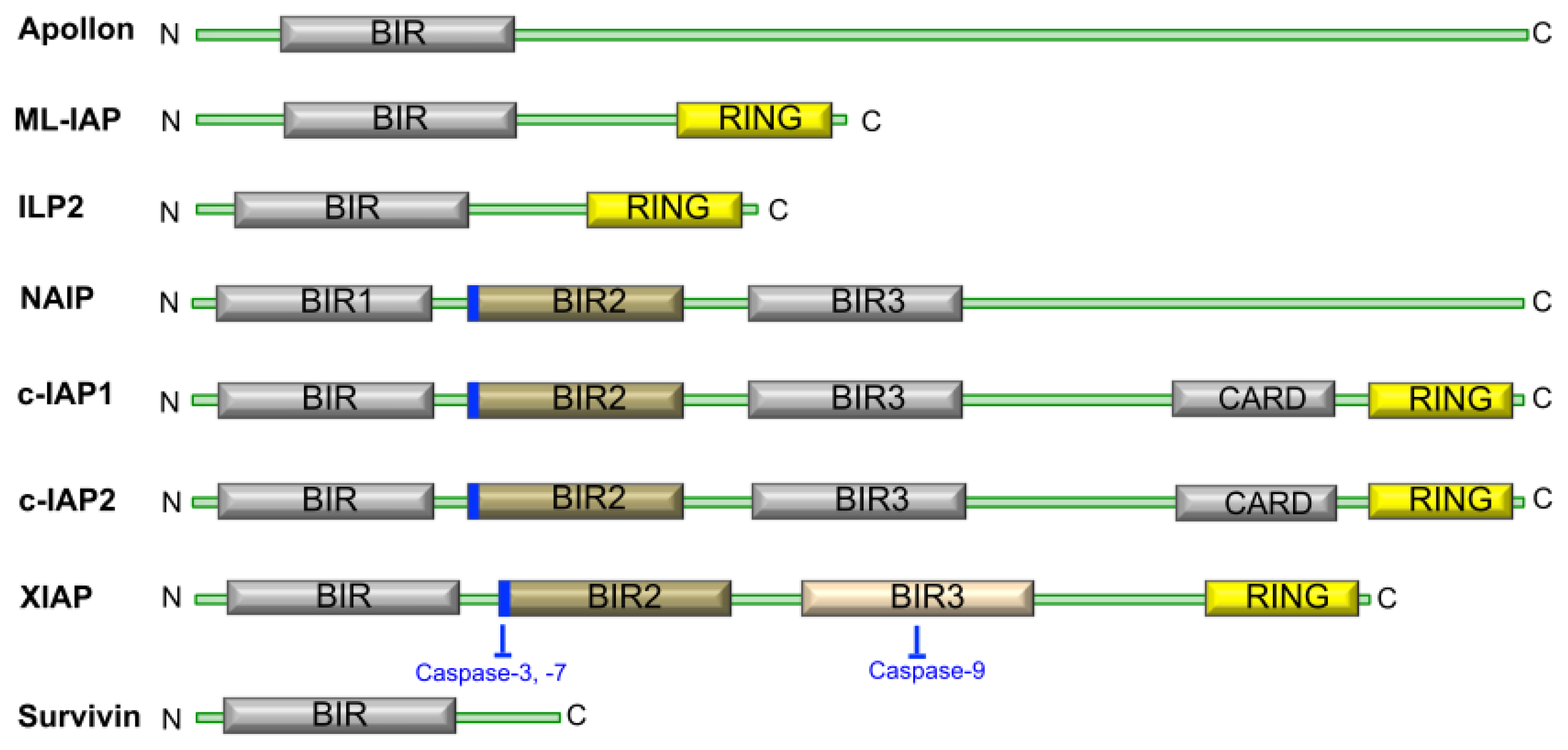
1.2. The Structure and Function of IAP Proteins
1.3. IAP Proteins and Cancer
1.4. Survivin, a Crucial IAP Target in Cancer Therapy
1.5. Function of Survivin
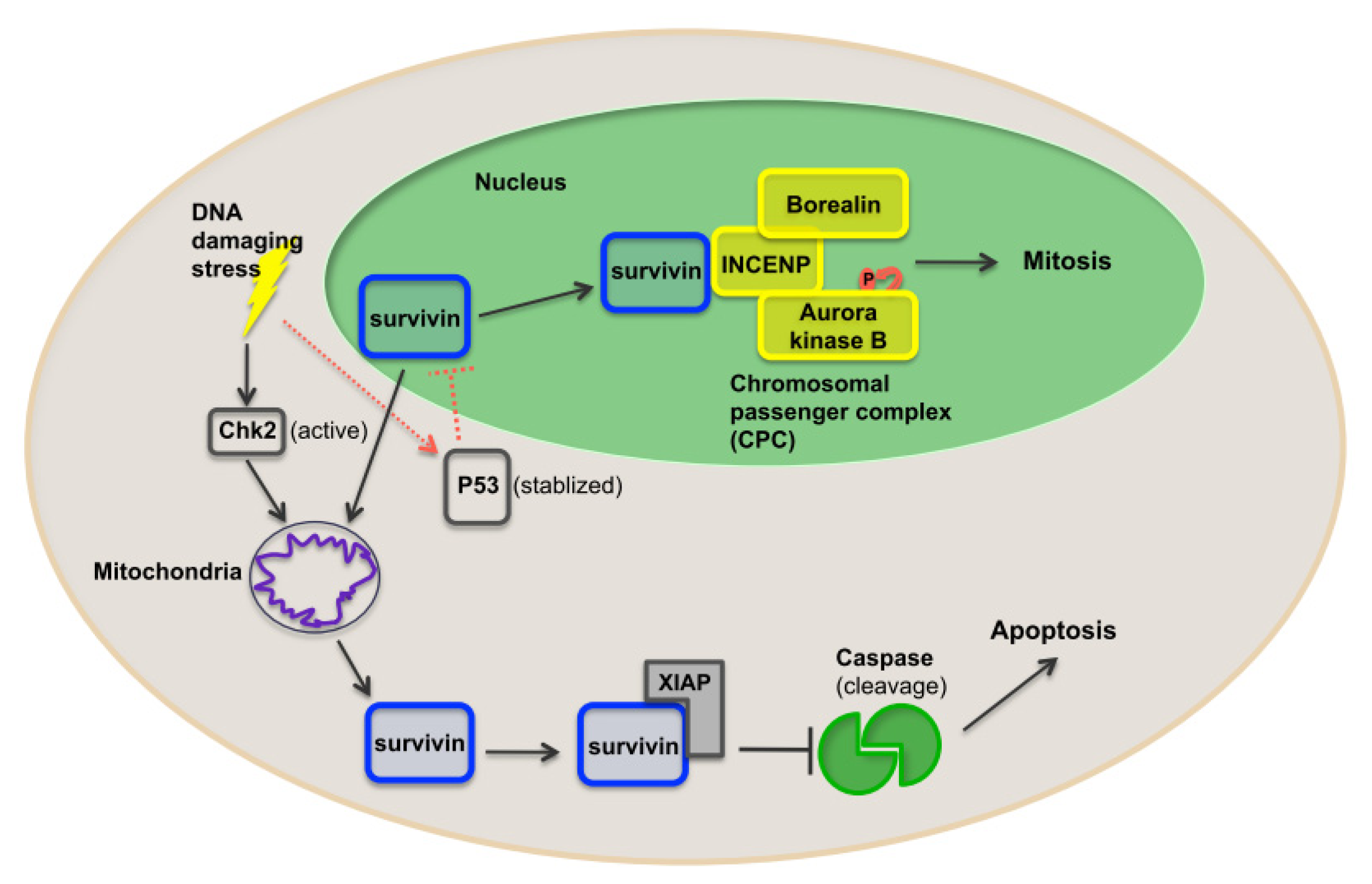
1.6. Survivin as a Nodal Protein
1.7. Survivin Induces Chemoresistence
1.8. Survivin Induces Radioresistence
1.9. Survivin as a Cancer Diagnostic Marker
1.10. Survivin as a Cancer Prognostic Marker
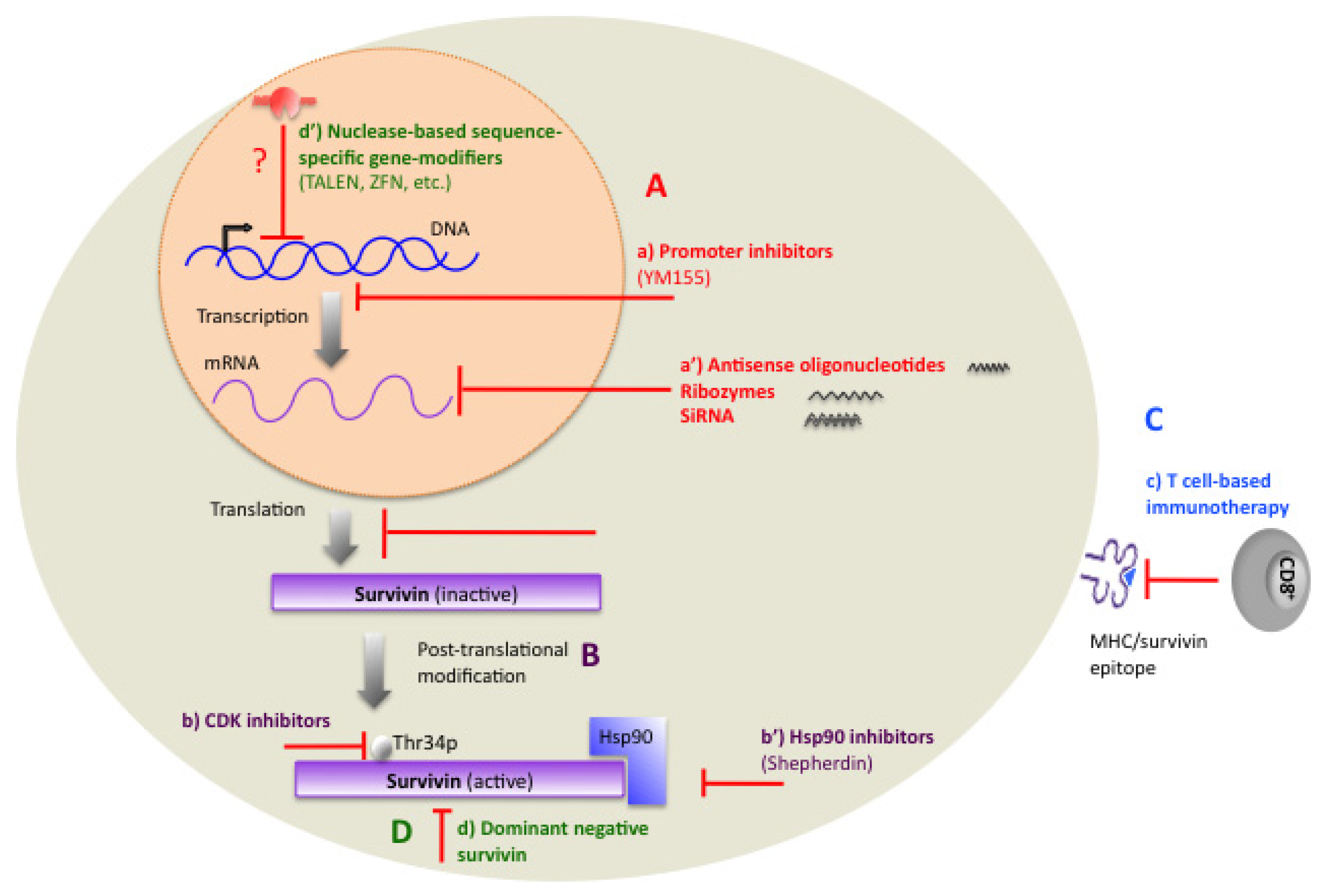
1.11. Therapeutic Targeting of Survivin
2. Transcriptional Inhibitors
3. Small-Molecule Antagonists
3.1. Hsp90 Inhibitors
3.2. Cyclin-Dependent Kinase (CDK) Inhibitors
3.3. Promoter Inhibitors
3.4. Other Low Molecular Weight Antagonists
4. Immunotherapy
5. Gene Therapy
5.1. Dominant-Negative Mutants
5.2. ZFN, TALEN, and CRISPR/Cas-Based Methods, as Potential Methods for Targeting the Survivin Gene in Tumor Cells
6. Potential Caveats and Alternative Approaches
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Reed, J.C. Bcl-2 family proteins: Regulators of apoptosis and chemoresistance in hematologic malignancies. Semin. Hematol 1997, 34, 9–19. [Google Scholar]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol 2008, 9, 47–59. [Google Scholar]
- Reed, J.C. Bcl-2 and the regulation of programmed cell death. J. Cell Biol 1994, 124, 1–6. [Google Scholar]
- Salvesen, G.S.; Abrams, J.M. Caspase activation—Stepping on the gas or releasing the brakes? Lessons from humans and flies. Oncogene 2004, 23, 2774–2784. [Google Scholar]
- Salvesen, G.S.; Duckett, C.S. IAP proteins: Blocking the road to death’s door. Nat. Rev. Mol. Cell Biol 2002, 3, 401–410. [Google Scholar]
- Hawkins, C.J.; Silke, J.; Verhagen, A.M.; Foster, R.; Ekert, P.G.; Ashley, D.M. Analysis of candidate antagonists of IAP-mediated caspase inhibition using yeast reconstituted with the mammalian Apaf-1-activated apoptosis mechanism. Apoptosis 2001, 6, 331–338. [Google Scholar]
- Hofmann, K.; Bucher, P.; Tschopp, J. The CARD domain: A new apoptotic signalling motif. Trends Biochem. Sci 1997, 22, 155–156. [Google Scholar]
- Eckelman, B.P.; Salvesen, G.S.; Scott, F.L. Human inhibitor of apoptosis proteins: Why XIAP is the black sheep of the family. EMBO Rep 2006, 7, 988–994. [Google Scholar]
- Suzuki, Y.; Nakabayashi, Y.; Takahashi, R. Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc. Natl. Acad. Sci. USA 2001, 98, 8662–8667. [Google Scholar]
- Chai, J.; Du, C.; Wu, J.W.; Kyin, S.; Wang, X.; Shi, Y. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature 2000, 406, 855–862. [Google Scholar]
- Liu, Z.; Sun, C.; Olejniczak, E.T.; Meadows, R.P.; Betz, S.F.; Oost, T.; Herrmann, J.; Wu, J.C.; Fesik, S.W. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature 2000, 408, 1004–1008. [Google Scholar]
- Srinivasula, S.M.; Datta, P.; Fan, X.J.; Fernandes-Alnemri, T.; Huang, Z.; Alnemri, E.S. Molecular determinants of the caspase-promoting activity of Smac/DIABLO and its role in the death receptor pathway. J. Biol. Chem 2000, 275, 36152–36157. [Google Scholar]
- Srinivasula, S.M.; Hegde, R.; Saleh, A.; Datta, P.; Shiozaki, E.; Chai, J.; Lee, R.A.; Robbins, P.D.; Fernandes-Alnemri, T.; Shi, Y.; Alnemri, E.S. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 2001, 410, 112–116. [Google Scholar]
- Wu, G.; Chai, J.; Suber, T.L.; Wu, J.W.; Du, C.; Wang, X.; Shi, Y. Structural basis of IAP recognition by Smac/DIABLO. Nature 2000, 408, 1008–1012. [Google Scholar]
- Fraser, A.G.; James, C.; Evan, G.I.; Hengartner, M.O. Caenorhabditis elegans inhibitor of apoptosis protein (IAP) homologue BIR-1 plays a conserved role in cytokinesis. Curr. Biol 1999, 9, 292–301. [Google Scholar]
- Uren, A.G.; Beilharz, T.; O’Connell, M.J.; Bugg, S.J.; van Driel, R.; Vaux, D.L.; Lithgow, T. Role for yeast inhibitor of apoptosis (IAP)-like proteins in cell division. Proc. Natl. Acad. Sci. USA 1999, 96, 10170–10175. [Google Scholar]
- Speliotes, E.K.; Uren, A.; Vaux, D.; Horvitz, H.R. The survivin-like C. elegans BIR-1 protein acts with the Aurora-like kinase AIR-2 to affect chromosomes and the spindle midzone. Mol. Cell 2000, 6, 211–223. [Google Scholar]
- Li, F.; Ambrosini, G.; Chu, E.Y.; Plescia, J.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 1998, 396, 580–584. [Google Scholar]
- Uren, A.G.; Wong, L.; Pakusch, M.; Fowler, K.J.; Burrows, F.J.; Vaux, D.L.; Choo, K.H. Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr. Biol 2000, 10, 1319–1328. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Tamm, I.; Richter, S.; Scholz, F.; Schmelz, K.; Oltersdorf, D.; Karawajew, L.; Schoch, C.; Haferlach, T.; Ludwig, W.D.; Wuchter, C. XIAP expression correlates with monocytic differentiation in adult de novo AML: Impact on prognosis. Hematol. J 2004, 5, 489–495. [Google Scholar]
- Yang, L.; Cao, Z.; Yan, H.; Wood, W.C. Coexistence of high levels of apoptotic signaling and inhibitor of apoptosis proteins in human tumor cells: Implication for cancer specific therapy. Cancer Res 2003, 63, 6815–6824. [Google Scholar]
- Tamm, I.; Kornblau, S.M.; Segall, H.; Krajewski, S.; Welsh, K.; Kitada, S.; Scudiero, D.A.; Tudor, G.; Qui, Y.H.; Monks, A.; et al. Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clin. Cancer Res 2000, 6, 1796–1803. [Google Scholar]
- Nachmias, B.; Ashhab, Y.; Ben-Yehuda, D. The inhibitor of apoptosis protein family (IAPs): An emerging therapeutic target in cancer. Semin. Cancer Biol 2004, 14, 231–243. [Google Scholar]
- Schimmer, A.D. Inhibitor of apoptosis proteins: Translating basic knowledge into clinical practice. Cancer Res 2004, 64, 7183–7190. [Google Scholar]
- Wright, C.W.; Duckett, C.S. Reawakening the cellular death program in neoplasia through the therapeutic blockade of IAP function. J. Clin. Invest 2005, 115, 2673–2678. [Google Scholar]
- Xu, R.; Zhang, P.; Huang, J.; Ge, S.; Lu, J.; Qian, G. Sp1 and Sp3 regulate basal transcription of the survivin gene. Biochem. Biophys. Res. Commun 2007, 356, 286–292. [Google Scholar]
- Jiang, Y.; Saavedra, H.I.; Holloway, M.P.; Leone, G.; Altura, R.A. Aberrant regulation of survivin by the RB/E2F family of proteins. J. Biol Chem 2004, 279, 40511–40520. [Google Scholar]
- Hoffman, W.H.; Biade, S.; Zilfou, J.T.; Chen, J.; Murphy, M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J. Biol. Chem 2002, 277, 3247–3257. [Google Scholar]
- Meng, R.D.; Shelton, C.C.; Li, Y.M.; Qin, L.X.; Notterman, D.; Paty, P.B.; Schwartz, G.K. gamma-Secretase inhibitors abrogate oxaliplatin-induced activation of the Notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res 2009, 69, 573–582. [Google Scholar]
- Chen, Y.; Li, D.; Liu, H.; Xu, H.; Zheng, H.; Qian, F.; Li, W.; Zhao, C.; Wang, Z.; Wang, X. Notch-1 signaling facilitates survivin expression in human non-small cell lung cancer cells. Cancer Biol. Ther 2011, 11, 14–21. [Google Scholar]
- Vaira, V.; Lee, C.W.; Goel, H.L.; Bosari, S.; Languino, L.R.; Altieri, D.C. Regulation of survivin expression by IGF-1/mTOR signaling. Oncogene 2007, 26, 2678–2684. [Google Scholar]
- Fortugno, P.; Beltrami, E.; Plescia, J.; Fontana, J.; Pradhan, D.; Marchisio, P.C.; Sessa, W.C.; Altieri, D.C. Regulation of survivin function by Hsp90. Proc. Natl. Acad. Sci. USA 2003, 100, 13791–13796. [Google Scholar]
- Wang, H.; Gambosova, K.; Cooper, Z.A.; Holloway, M.P.; Kassai, A.; Izquierdo, D.; Cleveland, K.; Boney, C.M.; Altura, R.A. EGF regulates survivin stability through the Raf-1/ERK pathway in insulin-secreting pancreatic beta-cells. BMC Mol. Biol 2010, 11, 66. [Google Scholar]
- Ju, J.H.; Yang, W.; Oh, S.; Nam, K.; Lee, K.M.; Noh, D.Y.; Shin, I. HER2 stabilizes survivin while concomitantly down-regulating survivin gene transcription by suppressing Notch cleavage. Biochem. J 2013, 451, 123–134. [Google Scholar]
- Siddiqa, A.; Long, L.M.; Li, L.; Marciniak, R.A.; Kazhdan, I. Expression of HER-2 in MCF-7 breast cancer cells modulates anti-apoptotic proteins Survivin and Bcl-2 via the extracellular signal-related kinase (ERK) and phosphoinositide-3 kinase (PI3K) signalling pathways. BMC Cancer 2008, 8, 129. [Google Scholar]
- Beierle, E.A.; Nagaram, A.; Dai, W.; Iyengar, M.; Chen, M.K. VEGF-mediated survivin expression in neuroblastoma cells. J. Surg. Res 2005, 127, 21–28. [Google Scholar]
- O’Connor, D.S.; Grossman, D.; Plescia, J.; Li, F.; Zhang, H.; Villa, A.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc. Natl. Acad. Sci. USA 2000, 97, 13103–13107. [Google Scholar]
- Zhou, S.; Li, L.; Jian, X.; Ou, X.; Jiang, H.; Yao, Z.; Xu, C.; Peng, J. The phosphorylation of survivin Thr34 by p34cdc2 in carcinogenesis of oral submucous fibrosis. Oncol. Rep 2008, 20, 1085–1091. [Google Scholar]
- Pannone, G.; Bufo, P.; Serpico, R.; Rubini, C.; Zamparese, R.; Corsi, F.; Pedicillo, M.C.; Staibano, S.; de Rosa, G.; Lo Muzio, L. Survivin phosphorylation and M-phase promoting factor in oral carcinogenesis. Histol. Histopathol 2007, 22, 1241–1249. [Google Scholar]
- Dohi, T.; Beltrami, E.; Wall, N.R.; Plescia, J.; Altieri, D.C. Mitochondrial survivin inhibits apoptosis and promotes tumorigenesis. J. Clin. Invest 2004, 114, 1117–1127. [Google Scholar]
- Dohi, T.; Xia, F.; Altieri, D.C. Compartmentalized phosphorylation of IAP by protein kinase A regulates cytoprotection. Mol. Cell 2007, 27, 17–28. [Google Scholar]
- Wang, H.; Holloway, M.P.; Ma, L.; Cooper, Z.A.; Riolo, M.; Samkari, A.; Elenitoba-Johnson, K.S.; Chin, Y.E.; Altura, R.A. Acetylation directs survivin nuclear localization to repress STAT3 oncogenic activity. J. Biol. Chem 2010, 285, 36129–36137. [Google Scholar]
- Fukuda, S.; Pelus, L.M. Survivin, a cancer target with an emerging role in normal adult tissues. Mol. Cancer Ther 2006, 5, 1087–1098. [Google Scholar]
- Xing, Z.; Conway, E.M.; Kang, C.; Winoto, A. Essential role of survivin, an inhibitor of apoptosis protein, in T cell development, maturation, and homeostasis. J. Exp. Med 2004, 199, 69–80. [Google Scholar]
- Fukuda, S.; Pelus, L.M. Regulation of the inhibitor-of-apoptosis family member survivin in normal cord blood and bone marrow CD34(+) cells by hematopoietic growth factors: Implication of survivin expression in normal hematopoiesis. Blood 2001, 98, 2091–2100. [Google Scholar]
- Mesri, M.; Morales-Ruiz, M.; Ackermann, E.J.; Bennett, C.F.; Pober, J.S.; Sessa, W.C.; Altieri, D.C. Suppression of vascular endothelial growth factor-mediated endothelial cell protection by survivin targeting. Am. J. Pathol 2001, 158, 1757–1765. [Google Scholar]
- Deguchi, M.; Shiraki, K.; Inoue, H.; Okano, H.; Ito, T.; Yamanaka, T.; Sugimoto, K.; Sakai, T.; Ohmori, S.; Murata, K.; et al. Expression of survivin during liver regeneration. Biochem. Biophys. Res. Commun 2002, 297, 59–64. [Google Scholar]
- Chiou, S.K.; Moon, W.S.; Jones, M.K.; Tarnawski, A.S. Survivin expression in the stomach: Implications for mucosal integrity and protection. Biochem. Biophys. Res. Commun 2003, 305, 374–379. [Google Scholar]
- Gurbuxani, S.; Xu, Y.; Keerthivasan, G.; Wickrema, A.; Crispino, J.D. Differential requirements for survivin in hematopoietic cell development. Proc. Natl. Acad. Sci. USA 2005, 102, 11480–11485. [Google Scholar]
- Altznauer, F.; Martinelli, S.; Yousefi, S.; Thurig, C.; Schmid, I.; Conway, E.M.; Schoni, M.H.; Vogt, P.; Mueller, C.; Fey, M.F.; et al. Inflammation-associated cell cycle-independent block of apoptosis by survivin in terminally differentiated neutrophils. J. Exp. Med 2004, 199, 1343–1354. [Google Scholar]
- Altieri, D.C. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene 2003, 22, 8581–8589. [Google Scholar]
- Adida, C.; Berrebi, D.; Peuchmaur, M.; Reyes-Mugica, M.; Altieri, D.C. Anti-apoptosis gene, survivin, and prognosis of neuroblastoma. Lancet 1998, 351, 882–883. [Google Scholar]
- Kawasaki, H.; Altieri, D.C.; Lu, C.D.; Toyoda, M.; Tenjo, T.; Tanigawa, N. Inhibition of apoptosis by survivin predicts shorter survival rates in colorectal cancer. Cancer Res 1998, 58, 5071–5074. [Google Scholar]
- Ambrosini, G.; Adida, C.; Altieri, D.C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med 1997, 3, 917–921. [Google Scholar]
- Lu, C.D.; Altieri, D.C.; Tanigawa, N. Expression of a novel antiapoptosis gene, survivin, correlated with tumor cell apoptosis and p53 accumulation in gastric carcinomas. Cancer Res 1998, 58, 1808–1812. [Google Scholar]
- Grossman, D.; Kim, P.J.; Blanc-Brude, O.P.; Brash, D.E.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Transgenic expression of survivin in keratinocytes counteracts UVB-induced apoptosis and cooperates with loss of p53. J. Clin Invest 2001, 108, 991–999. [Google Scholar]
- Yamamoto, T.; Manome, Y.; Nakamura, M.; Tanigawa, N. Downregulation of survivin expression by induction of the effector cell protease receptor-1 reduces tumor growth potential and results in an increased sensitivity to anticancer agents in human colon cancer. Eur J. Cancer 2002, 38, 2316–2324. [Google Scholar]
- Mirza, A.; McGuirk, M.; Hockenberry, T.N.; Wu, Q.; Ashar, H.; Black, S.; Wen, S.F.; Wang, L.; Kirschmeier, P.; Bishop, W.R.; et al. Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene 2002, 21, 2613–2622. [Google Scholar]
- Suzuki, A.; Ito, T.; Kawano, H.; Hayashida, M.; Hayasaki, Y.; Tsutomi, Y.; Akahane, K.; Nakano, T.; Miura, M.; Shiraki, K. Survivin initiates procaspase 3/p21 complex formation as a result of interaction with Cdk4 to resist Fas-mediated cell death. Oncogene 2000, 19, 1346–1353. [Google Scholar]
- Azuhata, T.; Scott, D.; Griffith, T.S.; Miller, M.; Sandler, A.D. Survivin inhibits apoptosis induced by TRAIL, and the ratio between survivin and TRAIL receptors is predictive of recurrent disease in neuroblastoma. J. Pediatr. Surg 2006, 41, 1431–1440. [Google Scholar]
- Tamm, I.; Wang, Y.; Sausville, E.; Scudiero, D.A.; Vigna, N.; Oltersdorf, T.; Reed, J.C. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res 1998, 58, 5315–5320. [Google Scholar]
- Kobayashi, K.; Hatano, M.; Otaki, M.; Ogasawara, T.; Tokuhisa, T. Expression of a murine homologue of the inhibitor of apoptosis protein is related to cell proliferation. Proc. Natl. Acad. Sci. USA 1999, 96, 1457–1462. [Google Scholar]
- Shin, S.; Sung, B.J.; Cho, Y.S.; Kim, H.J.; Ha, N.C.; Hwang, J.I.; Chung, C.W.; Jung, Y.K.; Oh, B.H. An anti-apoptotic protein human survivin is a direct inhibitor of caspase-3 and -7. Biochemistry 2001, 40, 1117–1123. [Google Scholar]
- Conway, E.M.; Pollefeyt, S.; Steiner-Mosonyi, M.; Luo, W.; Devriese, A.; Lupu, F.; Bono, F.; Leducq, N.; Dol, F.; Schaeffer, P.; et al. Deficiency of survivin in transgenic mice exacerbates Fas-induced apoptosis via mitochondrial pathways. Gastroenterology 2002, 123, 619–631. [Google Scholar]
- Banks, D.P.; Plescia, J.; Altieri, D.C.; Chen, J.; Rosenberg, S.H.; Zhang, H.; Ng, S.C. Survivin does not inhibit caspase-3 activity. Blood 2000, 96, 4002–4003. [Google Scholar]
- Vagnarelli, P.; Earnshaw, W.C. Chromosomal passengers: The four-dimensional regulation of mitotic events. Chromosoma 2004, 113, 211–222. [Google Scholar]
- Jeyaprakash, A.A.; Klein, U.R.; Lindner, D.; Ebert, J.; Nigg, E.A.; Conti, E. Structure of a Survivin-Borealin-INCENP core complex reveals how chromosomal passengers travel together. Cell 2007, 131, 271–285. [Google Scholar]
- Ghosh, J.C.; Dohi, T.; Raskett, C.M.; Kowalik, T.F.; Altieri, D.C. Activated checkpoint kinase 2 provides a survival signal for tumor cells. Cancer Res 2006, 66, 11576–11579. [Google Scholar]
- Altieri, D.C. The case for survivin as a regulator of microtubule dynamics and cell-death decisions. Curr. Opin. Cell Biol 2006, 18, 609–615. [Google Scholar]
- Giodini, A.; Kallio, M.J.; Wall, N.R.; Gorbsky, G.J.; Tognin, S.; Marchisio, P.C.; Symons, M.; Altieri, D.C. Regulation of microtubule stability and mitotic progression by survivin. Cancer Res 2002, 62, 2462–2467. [Google Scholar]
- Zaffaroni, N.; Pennati, M.; Colella, G.; Perego, P.; Supino, R.; Gatti, L.; Pilotti, S.; Zunino, F.; Daidone, M.G. Expression of the anti-apoptotic gene survivin correlates with taxol resistance in human ovarian cancer. Cell. Mol. Life Sci 2002, 59, 1406–1412. [Google Scholar]
- Zhang, M.; Mukherjee, N.; Bermudez, R.S.; Latham, D.E.; Delaney, M.A.; Zietman, A.L.; Shipley, W.U.; Chakravarti, A. Adenovirus-mediated inhibition of survivin expression sensitizes human prostate cancer cells to paclitaxel in vitro and in vivo. Prostate 2005, 64, 293–302. [Google Scholar]
- O’Connor, D.S.; Wall, N.R.; Porter, A.C.; Altieri, D.C. A p34(cdc2) survival checkpoint in cancer. Cancer Cell 2002, 2, 43–54. [Google Scholar]
- Cong, X.L.; Han, Z.C. Survivin and leukemia. Int. J. Hematol 2004, 80, 232–238. [Google Scholar]
- Van Geelen, C.M.; de Vries, E.G.; de Jong, S. Lessons from TRAIL-resistance mechanisms in colorectal cancer cells: Paving the road to patient-tailored therapy. Drug Resist. Updat 2004, 7, 345–358. [Google Scholar]
- Nomura, T.; Yamasaki, M.; Nomura, Y.; Mimata, H. Expression of the inhibitors of apoptosis proteins in cisplatin-resistant prostate cancer cells. Oncol. Rep 2005, 14, 993–997. [Google Scholar]
- Tirro, E.; Consoli, M.L.; Massimino, M.; Manzella, L.; Frasca, F.; Sciacca, L.; Vicari, L.; Stassi, G.; Messina, L.; Messina, A.; et al. Altered expression of c-IAP1, survivin, and Smac contributes to chemotherapy resistance in thyroid cancer cells. Cancer Res 2006, 66, 4263–4272. [Google Scholar]
- Zhang, M.; Latham, D.E.; Delaney, M.A.; Chakravarti, A. Survivin mediates resistance to antiandrogen therapy in prostate cancer. Oncogene 2005, 24, 2474–2482. [Google Scholar]
- Asanuma, K.; Moriai, R.; Yajima, T.; Yagihashi, A.; Yamada, M.; Kobayashi, D.; Watanabe, N. Survivin as a radioresistance factor in pancreatic cancer. Jpn J. Cancer Res 2000, 91, 1204–1209. [Google Scholar]
- Pennati, M.; Binda, M.; Colella, G.; Folini, M.; Citti, L.; Villa, R.; Daidone, M.G.; Zaffaroni, N. Radiosensitization of human melanoma cells by ribozyme-mediated inhibition of survivin expression. J. Invest. Dermatol 2003, 120, 648–654. [Google Scholar]
- Rodel, C.; Haas, J.; Groth, A.; Grabenbauer, G.G.; Sauer, R.; Rodel, F. Spontaneous and radiation-induced apoptosis in colorectal carcinoma cells with different intrinsic radiosensitivities: Survivin as a radioresistance factor. Int. J. Radiat. Oncol. Biol. Phys 2003, 55, 1341–1347. [Google Scholar]
- Lo Muzio, L.; Pannone, G.; Leonardi, R.; Staibano, S.; Mignogna, M.D.; de Rosa, G.; Kudo, Y.; Takata, T.; Altieri, D.C. Survivin, a potential early predictor of tumor progression in the oral mucosa. J. Dent. Res 2003, 82, 923–928. [Google Scholar]
- Ye, C.P.; Qiu, C.Z.; Huang, Z.X.; Su, Q.C.; Zhuang, W.; Wu, R.L.; Li, X.F. Relationship between survivin expression and recurrence, and prognosis in hepatocellular carcinoma. World J. Gastroenterol 2007, 13, 6264–6268. [Google Scholar]
- Rosato, A.; Pivetta, M.; Parenti, A.; Iaderosa, G.A.; Zoso, A.; Milan, G.; Mandruzzato, S.; Del Bianco, P.; Ruol, A.; Zaninotto, G.; et al. Survivin in esophageal cancer: An accurate prognostic marker for squamous cell carcinoma but not adenocarcinoma. Int. J. Cancer 2006, 119, 1717–1722. [Google Scholar]
- Shirai, K.; Suzuki, Y.; Oka, K.; Noda, S.E.; Katoh, H.; Itoh, J.; Itoh, H.; Ishiuchi, S.; Sakurai, H.; Hasegawa, M.; et al. Nuclear survivin expression predicts poorer prognosis in glioblastoma. J. Neurooncol 2009, 91, 353–358. [Google Scholar]
- Mohamed, S.; Yasufuku, K.; Nakajima, T.; Hiroshima, K.; Chiyo, M.; Yoshida, S.; Suzuki, M.; Sekine, Y.; Shibuya, K.; Agamy, G.; et al. Nuclear survivin in pN2 nonsmall cell lung cancer: Prognostic and clinical implications. Eur. Respir. J 2009, 33, 127–133. [Google Scholar]
- Brennan, D.J.; Rexhepaj, E.; O’Brien, S.L.; McSherry, E.; O’Connor, D.P.; Fagan, A.; Culhane, A.C.; Higgins, D.G.; Jirstrom, K.; Millikan, R.C.; et al. Altered cytoplasmic-to-nuclear ratio of survivin is a prognostic indicator in breast cancer. Clin. Cancer Res 2008, 14, 2681–2689. [Google Scholar]
- Yamashita, S.; Masuda, Y.; Kurizaki, T.; Haga, Y.; Murayama, T.; Ikei, S.; Kamei, M.; Takeno, S.; Kawahara, K. Survivin expression predicts early recurrence in early-stage breast cancer. Anticancer Res 2007, 27, 2803–2808. [Google Scholar]
- Fan, J.; Wang, L.; Jiang, G.N.; He, W.X.; Ding, J.A. The role of survivin on overall survival of non-small cell lung cancer, a meta-analysis of published literatures. Lung Cancer 2008, 61, 91–96. [Google Scholar]
- Grossman, D.; McNiff, J.M.; Li, F.; Altieri, D.C. Expression and targeting of the apoptosis inhibitor, survivin, in human melanoma. J. Invest. Dermatol 1999, 113, 1076–1081. [Google Scholar]
- Cao, C.; Mu, Y.; Hallahan, D.E.; Lu, B. XIAP and survivin as therapeutic targets for radiation sensitization in preclinical models of lung cancer. Oncogene 2004, 23, 7047–7052. [Google Scholar]
- Sharma, H.; Sen, S.; Lo Muzio, L.; Mariggio, A.; Singh, N. Antisense-mediated downregulation of anti-apoptotic proteins induces apoptosis and sensitizes head and neck squamous cell carcinoma cells to chemotherapy. Cancer Biol. Ther 2005, 4, 720–727. [Google Scholar]
- Du, Z.X.; Zhang, H.Y.; Gao da, X.; Wang, H.Q.; Li, Y.J.; Liu, G.L. Antisurvivin oligonucleotides inhibit growth and induce apoptosis in human medullary thyroid carcinoma cells. Exp. Mol. Med 2006, 38, 230–240. [Google Scholar]
- Fuessel, S.; Kueppers, B.; Ning, S.; Kotzsch, M.; Kraemer, K.; Schmidt, U.; Meye, A.; Wirth, M.P. Systematic in vitro evaluation of survivin directed antisense oligodeoxynucleotides in bladder cancer cells. J. Urol 2004, 171, 2471–2476. [Google Scholar]
- Ansell, S.M.; Arendt, B.K.; Grote, D.M.; Jelinek, D.F.; Novak, A.J.; Wellik, L.E.; Remstein, E.D.; Bennett, C.F.; Fielding, A. Inhibition of survivin expression suppresses the growth of aggressive non-Hodgkin’s lymphoma. Leukemia 2004, 18, 616–623. [Google Scholar]
- Fisker, N.; Westergaard, M.; Hansen, H.F.; Hansen, J.B. Survivin mRNA antagonists using locked nucleic acid, potential for molecular cancer therapy. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1427–1430. [Google Scholar]
- Carter, B.Z.; Mak, D.H.; Schober, W.D.; Cabreira-Hansen, M.; Beran, M.; McQueen, T.; Chen, W.; Andreeff, M. Regulation of survivin expression through Bcr-Abl/MAPK cascade: Targeting survivin overcomes imatinib resistance and increases imatinib sensitivity in imatinib-responsive CML cells. Blood 2006, 107, 1555–1563. [Google Scholar]
- Hayashi, N.; Asano, K.; Suzuki, H.; Yamamoto, T.; Tanigawa, N.; Egawa, S.; Manome, Y. Adenoviral infection of survivin antisense sensitizes prostate cancer cells to etoposide in vivo. Prostate 2005, 65, 10–19. [Google Scholar]
- Olie, R.A.; Simoes-Wust, A.P.; Baumann, B.; Leech, S.H.; Fabbro, D.; Stahel, R.A.; Zangemeister-Wittke, U. A novel antisense oligonucleotide targeting survivin expression induces apoptosis and sensitizes lung cancer cells to chemotherapy. Cancer Res 2000, 60, 2805–2809. [Google Scholar]
- Sah, N.K.; Munshi, A.; Hobbs, M.; Carter, B.Z.; Andreeff, M.; Meyn, R.E. Effect of downregulation of survivin expression on radiosensitivity of human epidermoid carcinoma cells. Int. J. Radiat. Oncol. Biol. Phys 2006, 66, 852–859. [Google Scholar]
- Kanwar, J.R.; Shen, W.P.; Kanwar, R.K.; Berg, R.W.; Krissansen, G.W. Effects of survivin antagonists on growth of established tumors and B7-1 immunogene therapy. J. Natl. Cancer Inst 2001, 93, 1541–1552. [Google Scholar]
- Pennati, M.; Colella, G.; Folini, M.; Citti, L.; Daidone, M.G.; Zaffaroni, N. Ribozyme-mediated attenuation of survivin expression sensitizes human melanoma cells to cisplatin-induced apoptosis. J. Clin. Invest 2002, 109, 285–286. [Google Scholar]
- Pennati, M.; Binda, M.; de Cesare, M.; Pratesi, G.; Folini, M.; Citti, L.; Daidone, M.G.; Zunino, F.; Zaffaroni, N. Ribozyme-mediated down-regulation of survivin expression sensitizes human melanoma cells to topotecan in vitro and in vivo. Carcinogenesis 2004, 25, 1129–1136. [Google Scholar]
- Carvalho, A.; Carmena, M.; Sambade, C.; Earnshaw, W.C.; Wheatley, S.P. Survivin is required for stable checkpoint activation in taxol-treated HeLa cells. J. Cell Sci 2003, 116, 2987–2998. [Google Scholar]
- Ling, X.; Li, F. Silencing of antiapoptotic survivin gene by multiple approaches of RNA interference technology. Biotechniques 2004, 36. [Google Scholar]
- Ai, Z.; Yin, L.; Zhou, X.; Zhu, Y.; Zhu, D.; Yu, Y.; Feng, Y. Inhibition of survivin reduces cell proliferation and induces apoptosis in human endometrial cancer. Cancer 2006, 107, 746–756. [Google Scholar]
- Paduano, F.; Villa, R.; Pennati, M.; Folini, M.; Binda, M.; Daidone, M.G.; Zaffaroni, N. Silencing of survivin gene by small interfering RNAs produces supra-additive growth suppression in combination with 17-allylamino-17-demethoxygeldanamycin in human prostate cancer cells. Mol. Cancer Ther 2006, 5, 179–186. [Google Scholar]
- Nakao, K.; Hamasaki, K.; Ichikawa, T.; Arima, K.; Eguchi, K.; Ishii, N. Survivin downregulation by siRNA sensitizes human hepatoma cells to TRAIL-induced apoptosis. Oncol. Rep 2006, 16, 389–392. [Google Scholar]
- Lens, S.M.; Wolthuis, R.M.; Klompmaker, R.; Kauw, J.; Agami, R.; Brummelkamp, T.; Kops, G.; Medema, R.H. Survivin is required for a sustained spindle checkpoint arrest in response to lack of tension. EMBO J 2003, 22, 2934–2947. [Google Scholar]
- Beltrami, E.; Plescia, J.; Wilkinson, J.C.; Duckett, C.S.; Altieri, D.C. Acute ablation of survivin uncovers p53-dependent mitotic checkpoint functions and control of mitochondrial apoptosis. J. Biol. Chem 2004, 279, 2077–2084. [Google Scholar]
- Song, H.; Xin, X.Y.; Xiao, F.; Wang, D.T.; Yue, Q.H.; Han, X. Survivin gene RNA interference inhibits proliferation, induces apoptosis, and enhances radiosensitivity in HeLa cells. Eur. J. Obstet. Gynecol. Reprod. Biol 2008, 136, 83–89. [Google Scholar]
- Huynh, T.; Walchli, S.; Sioud, M. Transcriptional targeting of small interfering RNAs into cancer cells. Biochem. Biophys. Res. Commun 2006, 350, 854–859. [Google Scholar]
- Kappler, M.; Taubert, H.; Bartel, F.; Blumke, K.; Panian, M.; Schmidt, H.; Dunst, J.; Bache, M. Radiosensitization, after a combined treatment of survivin siRNA and irradiation, is correlated with the activation of caspases 3 and 7 in a wt-p53 sarcoma cell line, but not in a mt-p53 sarcoma cell line. Oncol. Rep 2005, 13, 167–172. [Google Scholar]
- Yonesaka, K.; Tamura, K.; Kurata, T.; Satoh, T.; Ikeda, M.; Fukuoka, M.; Nakagawa, K. Small interfering RNA targeting survivin sensitizes lung cancer cell with mutant p53 to adriamycin. Int. J. Cancer 2006, 118, 812–820. [Google Scholar]
- Jiang, G.; Li, J.; Zeng, Z.; Xian, L. Lentivirus-mediated gene therapy by suppressing survivin in BALB/c nude mice bearing oral squamous cell carcinoma. Cancer Biol. Ther 2006, 5, 435–440. [Google Scholar]
- Plescia, J.; Salz, W.; Xia, F.; Pennati, M.; Zaffaroni, N.; Daidone, M.G.; Meli, M.; Dohi, T.; Fortugno, P.; Nefedova, Y.; et al. Rational design of shepherdin, a novel anticancer agent. Cancer Cell 2005, 7, 457–468. [Google Scholar]
- Pennati, M.; Campbell, A.J.; Curto, M.; Binda, M.; Cheng, Y.; Wang, L.Z.; Curtin, N.; Golding, B.T.; Griffin, R.J.; Hardcastle, I.R.; et al. Potentiation of paclitaxel-induced apoptosis by the novel cyclin-dependent kinase inhibitor NU6140: A possible role for survivin down-regulation. Mol. Cancer Ther 2005, 4, 1328–1337. [Google Scholar]
- Nakahara, T.; Takeuchi, M.; Kinoyama, I.; Minematsu, T.; Shirasuna, K.; Matsuhisa, A.; Kita, A.; Tominaga, F.; Yamanaka, K.; Kudoh, M.; et al. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res 2007, 67, 8014–8021. [Google Scholar]
- Chang, C.C.; Heller, J.D.; Kuo, J.; Huang, R.C. Tetra-O-methyl nordihydroguaiaretic acid induces growth arrest and cellular apoptosis by inhibiting Cdc2 and survivin expression. Proc. Natl. Acad. Sci. USA 2004, 101, 13239–13244. [Google Scholar]
- Smolewski, P. Terameprocol, a novel site-specific transcription inhibitor with anticancer activity. IDrugs 2008, 11, 204–214. [Google Scholar]
- Xiang, R.; Mizutani, N.; Luo, Y.; Chiodoni, C.; Zhou, H.; Mizutani, M.; Ba, Y.; Becker, J.C.; Reisfeld, R.A. A DNA vaccine targeting survivin combines apoptosis with suppression of angiogenesis in lung tumor eradication. Cancer Res 2005, 65, 553–561. [Google Scholar]
- Zhu, K.; Qin, H.; Cha, S.C.; Neelapu, S.S.; Overwijk, W.; Lizee, G.A.; Abbruzzese, J.L.; Hwu, P.; Radvanyi, L.; Kwak, L.W.; et al. Survivin DNA vaccine generated specific antitumor effects in pancreatic carcinoma and lymphoma mouse models. Vaccine 2007, 25, 7955–7961. [Google Scholar]
- Fuessel, S.; Meye, A.; Schmitz, M.; Zastrow, S.; Linne, C.; Richter, K.; Lobel, B.; Hakenberg, O.W.; Hoelig, K.; Rieber, E.P.; et al. Vaccination of hormone-refractory prostate cancer patients with peptide cocktail-loaded dendritic cells: Results of a phase I clinical trial. Prostate 2006, 66, 811–821. [Google Scholar]
- Tu, S.P.; Jiang, X.H.; Lin, M.C.; Cui, J.T.; Yang, Y.; Lum, C.T.; Zou, B.; Zhu, Y.B.; Jiang, S.H.; Wong, W.M.; et al. Suppression of survivin expression inhibits in vivo tumorigenicity and angiogenesis in gastric cancer. Cancer Res 2003, 63, 7724–7732. [Google Scholar]
- Mesri, M.; Wall, N.R.; Li, J.; Kim, R.W.; Altieri, D.C. Cancer gene therapy using a survivin mutant adenovirus. J. Clin. Invest 2001, 108, 981–990. [Google Scholar]
- Grossman, D.; Kim, P.J.; Schechner, J.S.; Altieri, D.C. Inhibition of melanoma tumor growth in vivo by survivin targeting. Proc. Natl. Acad. Sci. USA 2001, 98, 635–640. [Google Scholar]
- Blanc-Brude, O.P.; Mesri, M.; Wall, N.R.; Plescia, J.; Dohi, T.; Altieri, D.C. Therapeutic targeting of the survivin pathway in cancer: Initiation of mitochondrial apoptosis and suppression of tumor-associated angiogenesis. Clin. Cancer Res 2003, 9, 2683–2692. [Google Scholar]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 2013, 31, 397–405. [Google Scholar]
- Carroll, D. Genome engineering with zinc-finger nucleases. Genetics 2011, 188, 773–782. [Google Scholar]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010, 186, 757–761. [Google Scholar]
- Voll, R.E.; Herrmann, M.; Roth, E.A.; Stach, C.; Kalden, J.R.; Girkontaite, I. Immunosuppressive effects of apoptotic cells. Nature 1997, 390, 350–351. [Google Scholar]
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest 1998, 101, 890–898. [Google Scholar]
- McDonald, P.P.; Fadok, V.A.; Bratton, D.; Henson, P.M. Transcriptional and translational regulation of inflammatory mediator production by endogenous TGF-beta in macrophages that have ingested apoptotic cells. J. Immunol 1999, 163, 6164–6172. [Google Scholar]
- Fadok, V.A.; Bratton, D.L.; Guthrie, L.; Henson, P.M. Differential effects of apoptotic versus lysed cells on macrophage production of cytokines: Role of proteases. J. Immunol 2001, 166, 6847–6854. [Google Scholar]
- Morimoto, K.; Amano, H.; Sonoda, F.; Baba, M.; Senba, M.; Yoshimine, H.; Yamamoto, H.; Ii, T.; Oishi, K.; Nagatake, T. Alveolar macrophages that phagocytose apoptotic neutrophils produce hepatocyte growth factor during bacterial pneumonia in mice. Am. J. Respir Cell Mol. Biol 2001, 24, 608–615. [Google Scholar]
- Hristov, M.; Erl, W.; Linder, S.; Weber, P.C. Apoptotic bodies from endothelial cells enhance the number and initiate the differentiation of human endothelial progenitor cells in vitro. Blood 2004, 104, 2761–2766. [Google Scholar]
- Golpon, H.A.; Fadok, V.A.; Taraseviciene-Stewart, L.; Scerbavicius, R.; Sauer, C.; Welte, T.; Henson, P.M.; Voelkel, N.F. Life after corpse engulfment: Phagocytosis of apoptotic cells leads to VEGF secretion and cell growth. FASEB J 2004, 18, 1716–1718. [Google Scholar]
- White, G.E.; Tan, T.C.; John, A.E.; Whatling, C.; McPheat, W.L.; Greaves, D.R. Fractalkine has anti-apoptotic and proliferative effects on human vascular smooth muscle cells via epidermal growth factor receptor signalling. Cardiovasc. Res 2010, 85, 825–835. [Google Scholar]
- Hermens, A.F.; Barendsen, G.W. Changes of cell proliferation characteristics in a rat rhabdomyosarcoma before and after x-irradiation. Eur. J. Cancer 1969, 5, 173–189. [Google Scholar]
- Stephens, T.C.; Currie, G.A.; Peacock, J.H. Repopulation of gamma-irradiated Lewis lung carcinoma by malignant cells and host macrophage progenitors. Br. J. Cancer 1978, 38, 573–582. [Google Scholar]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.C.; et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med 2011, 17, 860–866. [Google Scholar]
- Garg, H.; Salcedo, R.; Trinchieri, G.; Blumenthal, R. Improved nonviral cancer suicide gene therapy using survivin promoter-driven mutant Bax. Cancer Gene Ther 2010, 17, 155–163. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Type of regulation | Reported interaction | Cell line(s) | References |
|---|---|---|---|
| Transcriptional regulation | |||
| Sp1/Sp3 | As transcription factors, positively regulate survivin promoter activity | Human HeLa cervical adenocarcinoma cells | [27] |
| RB/E2F | E2F activators (such as E2F1, E2F2 and E2F3) positively and RB negatively regulates survivin promoter activity | Rat embryo fibroblasts, human WI-38 fibroblasts, normal human melanocytes | [28,29] |
| p53 | Competes with the binding of E2F activators to survivin promoter, hence inhibits survivin transcription | Normal human melanocytes | [29] |
| NICD (Notch-intracellular domain) | Translocation of NICD to the nucleus activates survivin promoter | Colon cancer cells and human non-small cell lung cancer cells | [30,31] |
| IGF-1 | Enhances translation of survivin | Human DU145 prostate cancer cell line | [32] |
| Regulation of survivin protein stability | |||
| HSP90 | Stabilizes survivin through physical association | HeLa cervical carcinoma cells | [33] |
| EGF and EGFR pathways such ERK and AKT signaling pathways | Inhibits poly-ubiquitination and thus degradation of survivin | Mouse MIN6 and rat INS-1 pancreatic β cells, MCF-7 breast cancer cells | [34–36] |
| VEGF (vascular endothelial growth factor) | Increased VEGF enhances survivin protein levels through activating PI3K/AKT pathway | Neuroblastoma cells | [37] |
| Post-translational regulation of survivin | |||
| Phosphorylation on threonine 34 | Thr34-phosphorylated survivin binds caspase-9 and inhibits apoptosis | Human oral squamous adenocarcinoma cells, oral fibrosis, HeLa cells | [38–40] |
| Dephosphorylation on serine20 | Ser20-dephosphorylation translocates survivin from mitochondria to cytoplasm to inhibit caspase cleavage | Insulinoma INS-1 cells | [41,42] |
| Acetylation on lysine 129 | CREB-binding protein acetylates survivin on lysine 129 to increase survivin nuclear accumulation, decreasing cell survival | HEK293T, HeLa cells and MCF-7 breast cancer cells | [43] |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mobahat, M.; Narendran, A.; Riabowol, K. Survivin as a Preferential Target for Cancer Therapy. Int. J. Mol. Sci. 2014, 15, 2494-2516. https://doi.org/10.3390/ijms15022494
Mobahat M, Narendran A, Riabowol K. Survivin as a Preferential Target for Cancer Therapy. International Journal of Molecular Sciences. 2014; 15(2):2494-2516. https://doi.org/10.3390/ijms15022494
Chicago/Turabian StyleMobahat, Mahsa, Aru Narendran, and Karl Riabowol. 2014. "Survivin as a Preferential Target for Cancer Therapy" International Journal of Molecular Sciences 15, no. 2: 2494-2516. https://doi.org/10.3390/ijms15022494