Shotgun Bisulfite Sequencing of the Betula platyphylla Genome Reveals the Tree’s DNA Methylation Patterning

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Generation of Methylation Data of B. platyphylla

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Raw Reads (M) | Raw Base Number (Gb) | Effective Reads (M) | Effective Base Number (Gb) | Genome Coverage (%) | Average Depth Per Base and Strand (X) |
|---|---|---|---|---|---|
| 178.02 | 15.65 | 148.78 | 13.08 | 83.58 | 15.02 |
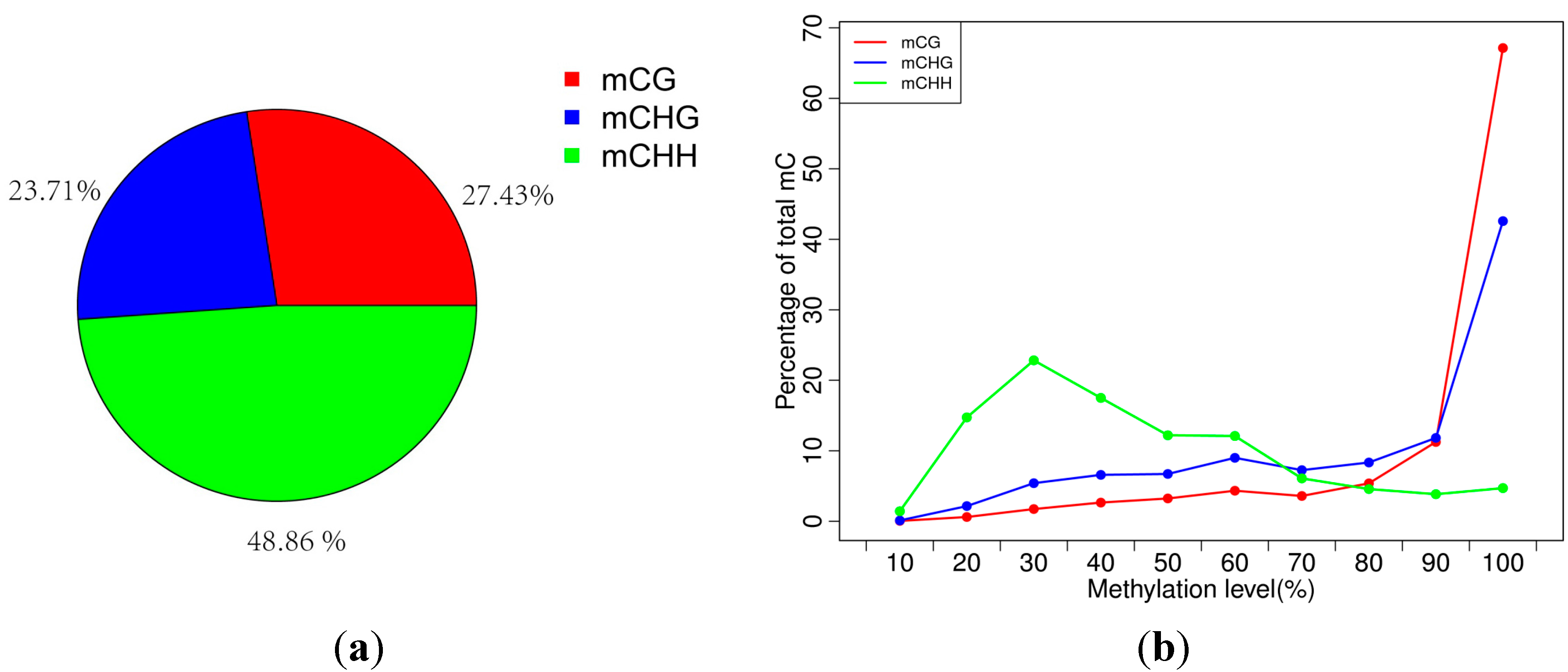
2.2. Analysis of Percentages of Methylated Cytosines (mCs) in CG, CHG and CHH
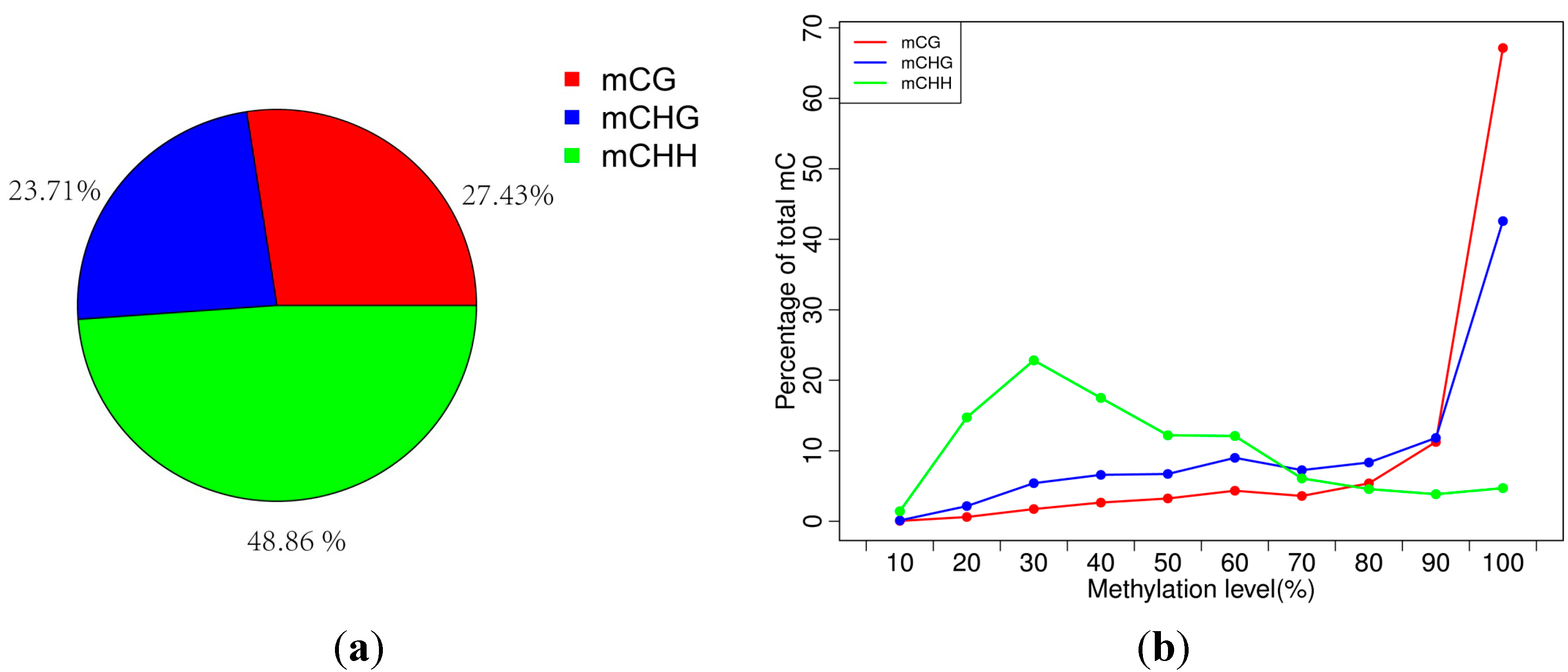
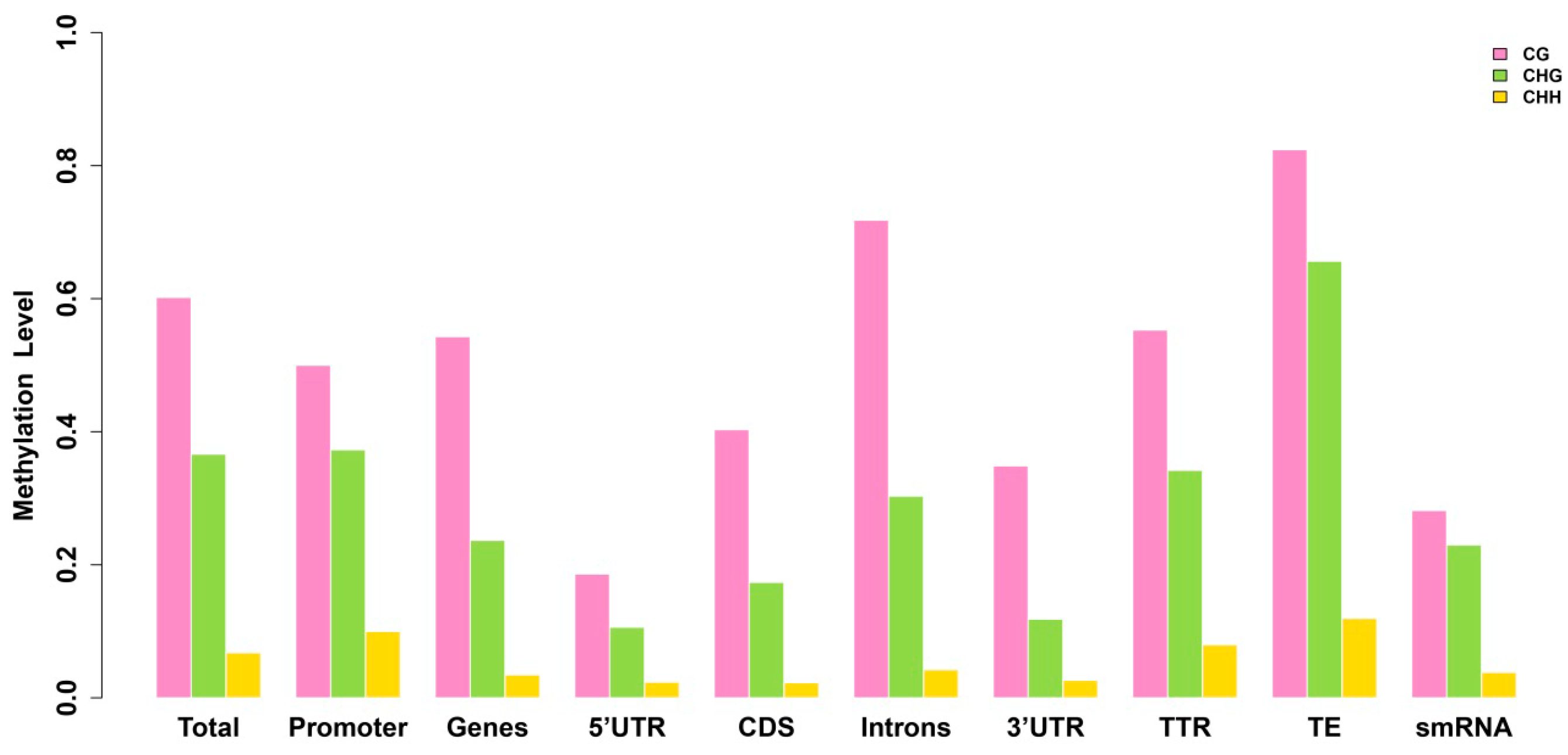
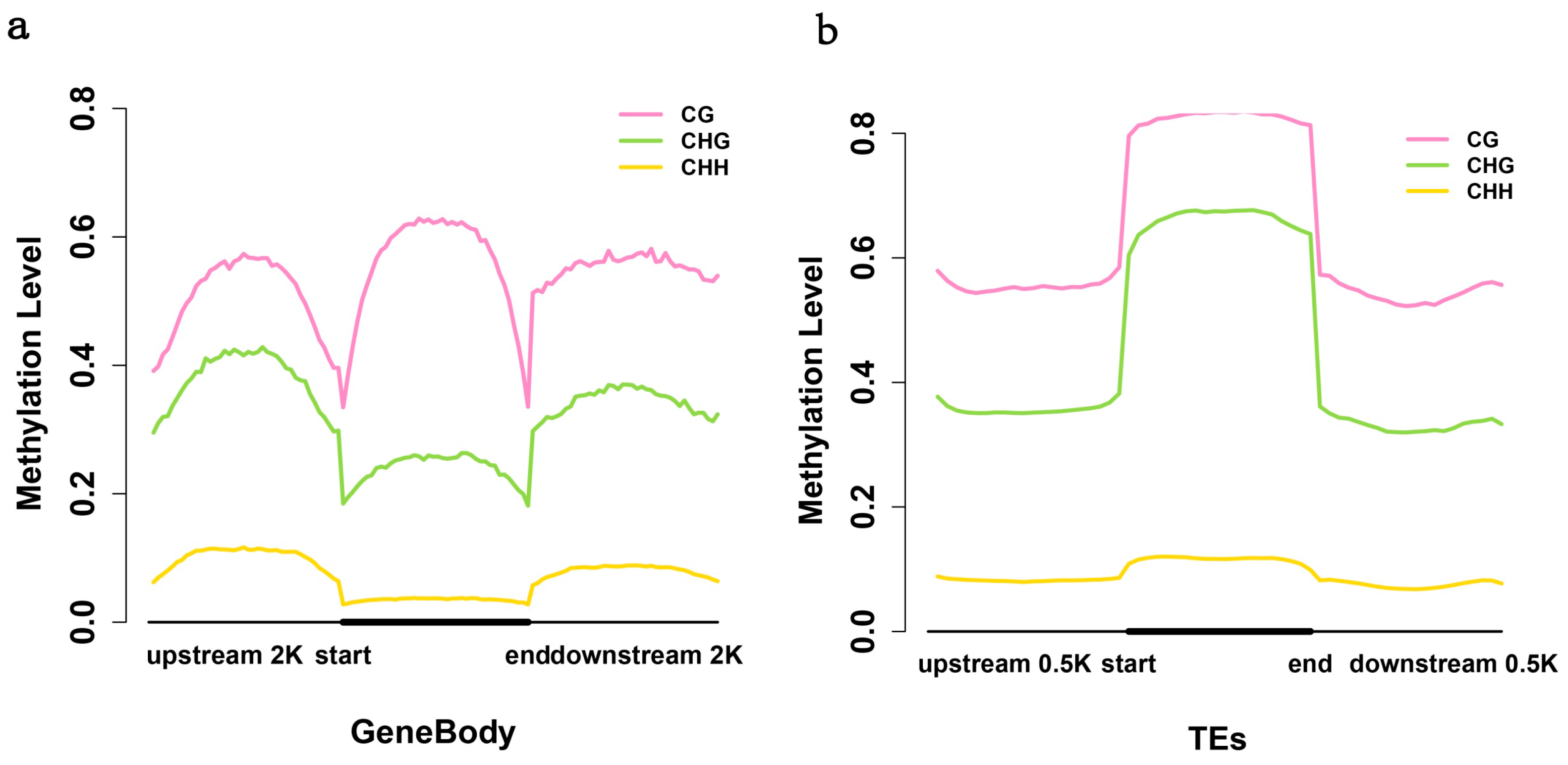
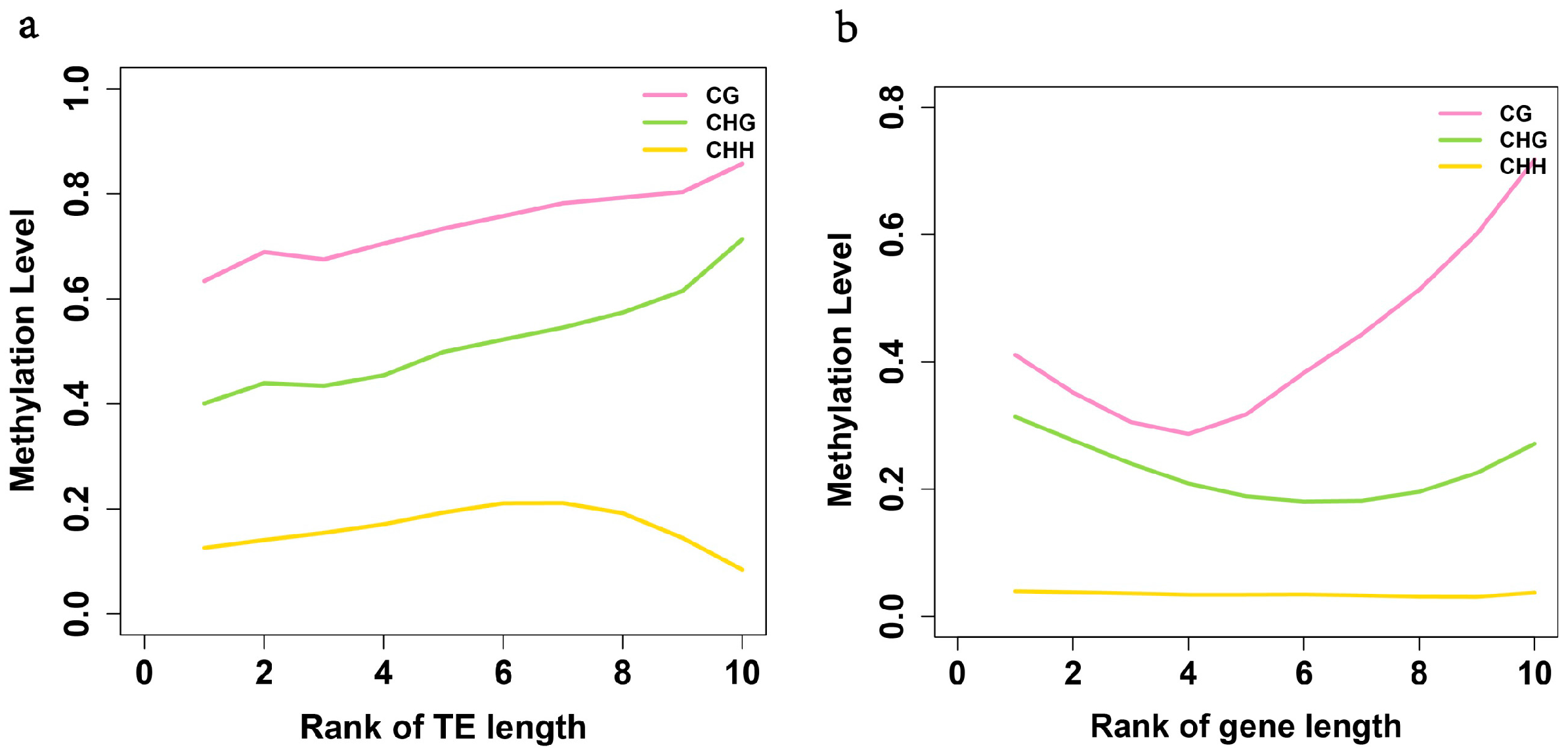
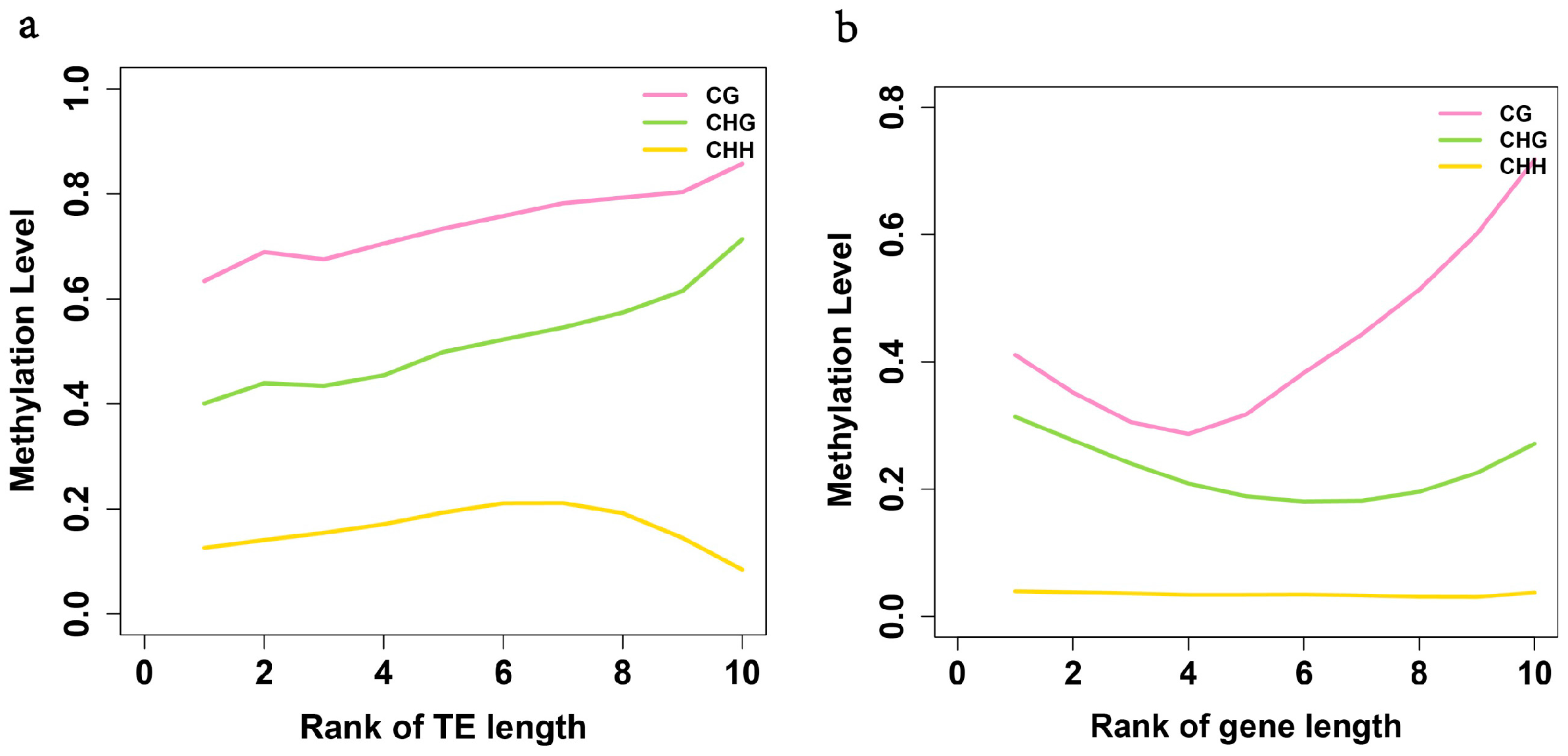
2.3. Analysis of the Methylation Profiles
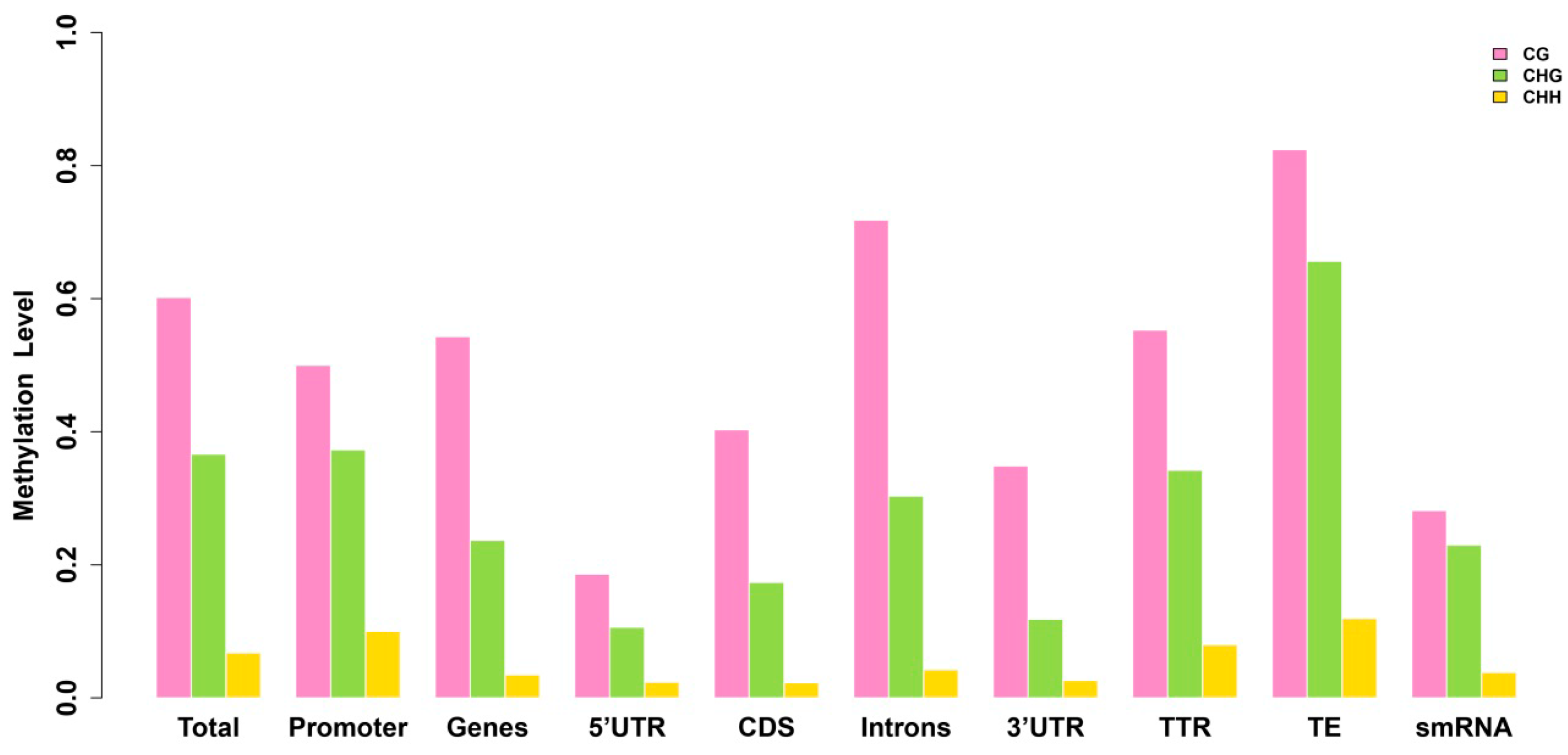
| Methylation Pattern | Total | Promoter | TTR | Genes | CDS | Introns | 5'-UTR | 3'-UTR | TE | smRNA |
|---|---|---|---|---|---|---|---|---|---|---|
| ML | 0.1728 | 0.1864 | 0.1674 | 0.1248 | 0.1051 | 0.1428 | 0.0607 | 0.0712 | 0.2759 | 0.1164 |
| MD | 0.4276 | 0.3829 | 0.4122 | 0.4800 | 0.5454 | 0.4465 | 0.4264 | 0.4777 | 0.4071 | 0.4218 |
| Pattern | mCG | mCHG | mCHH |
|---|---|---|---|
| Methylation level (depth ≥ 1×) | 42.64 | 28.8 | 5.16 |
| Methylation level (depth ≥ 5×) | 60.19 | 36.63 | 6.79 |
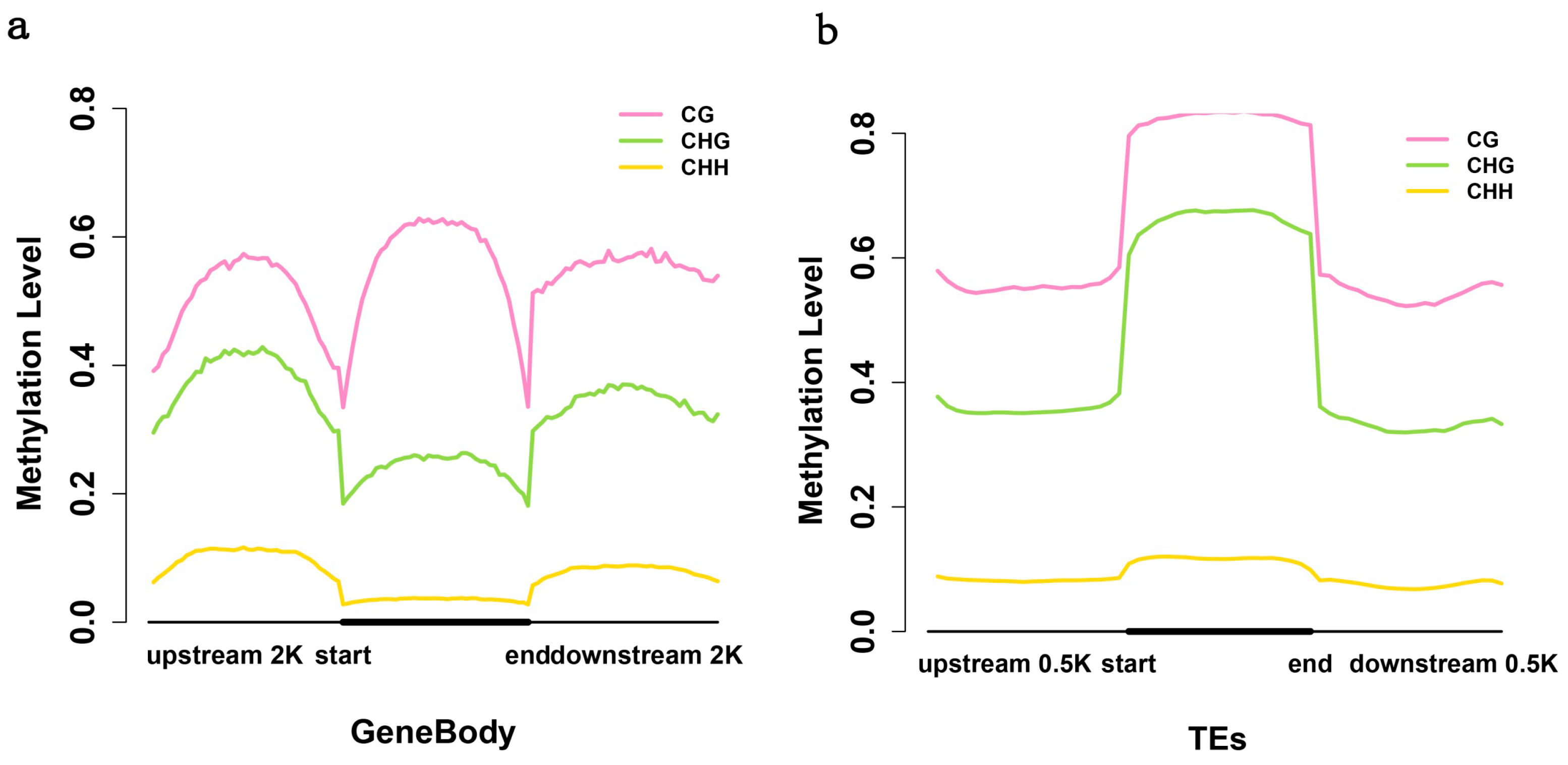
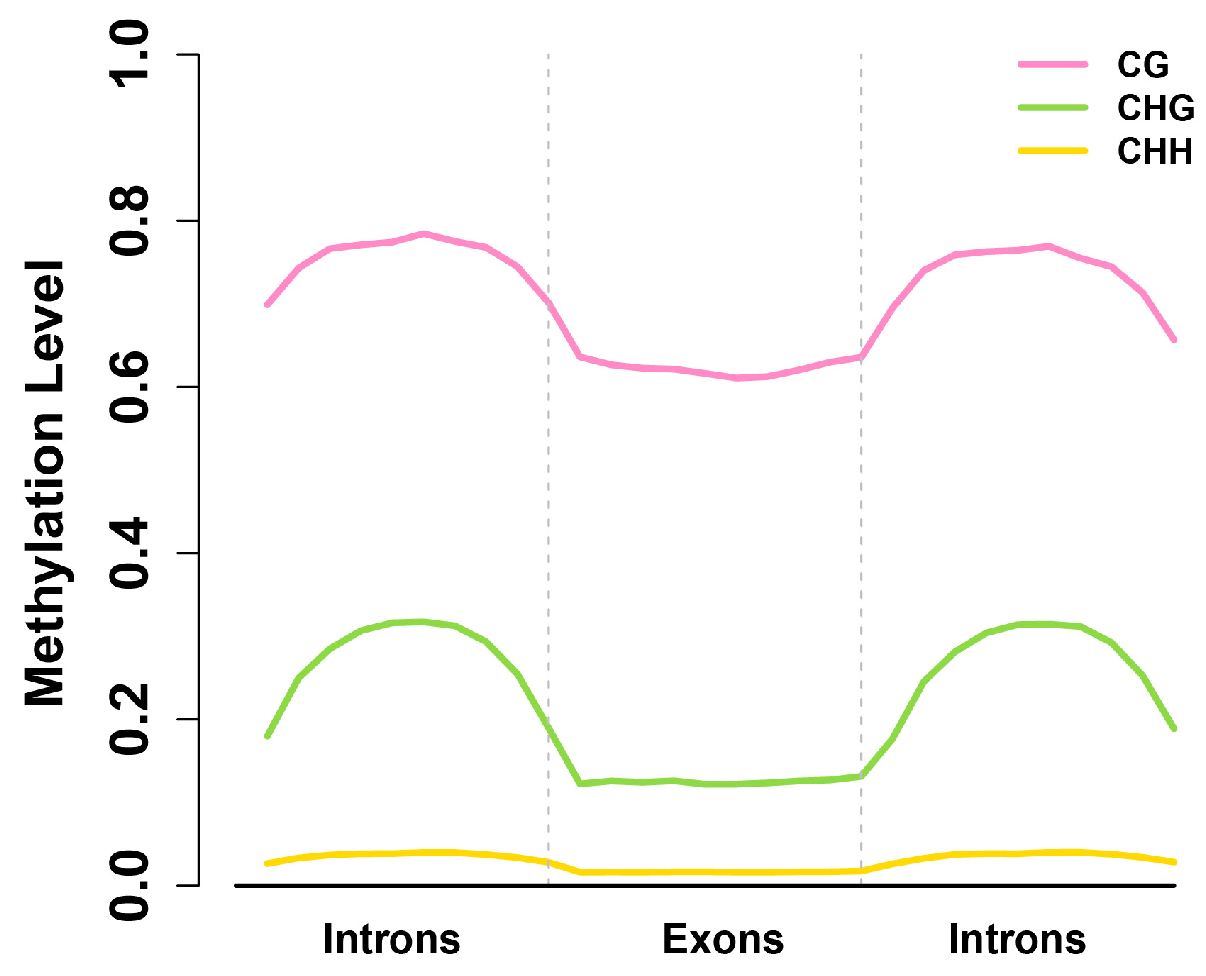
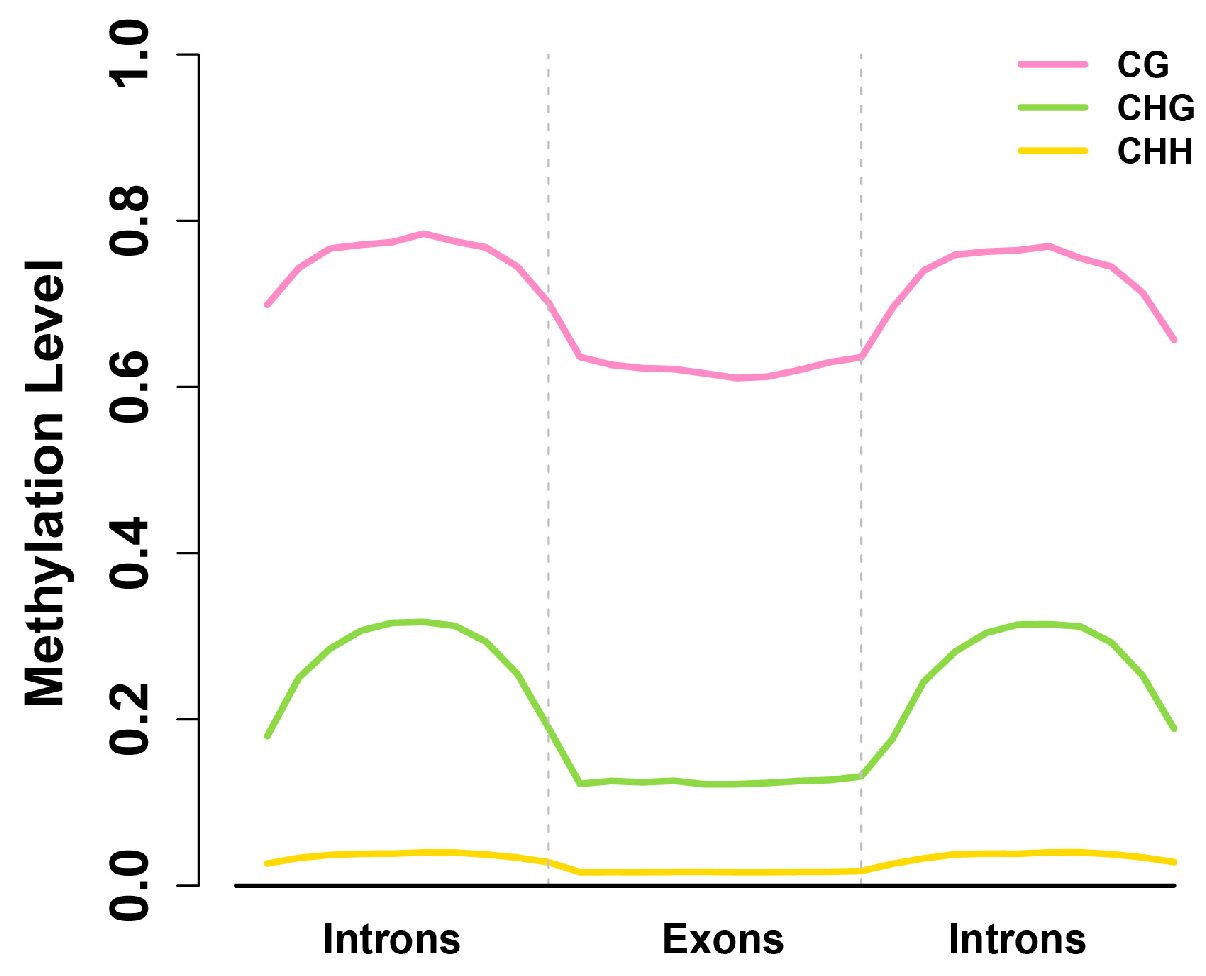
2.4. Exons Have Lower Methylation Levels than Introns in Birch Genes
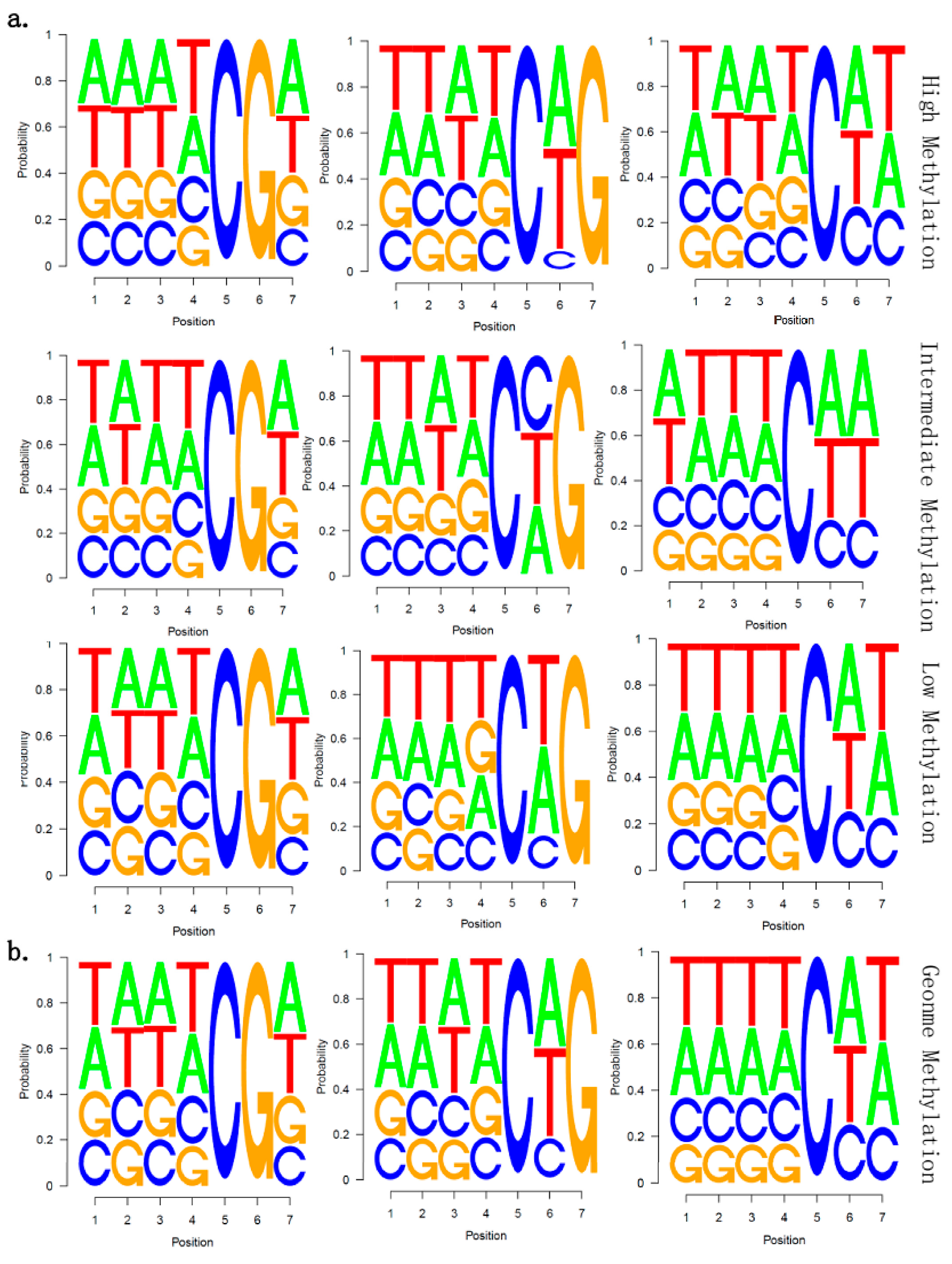
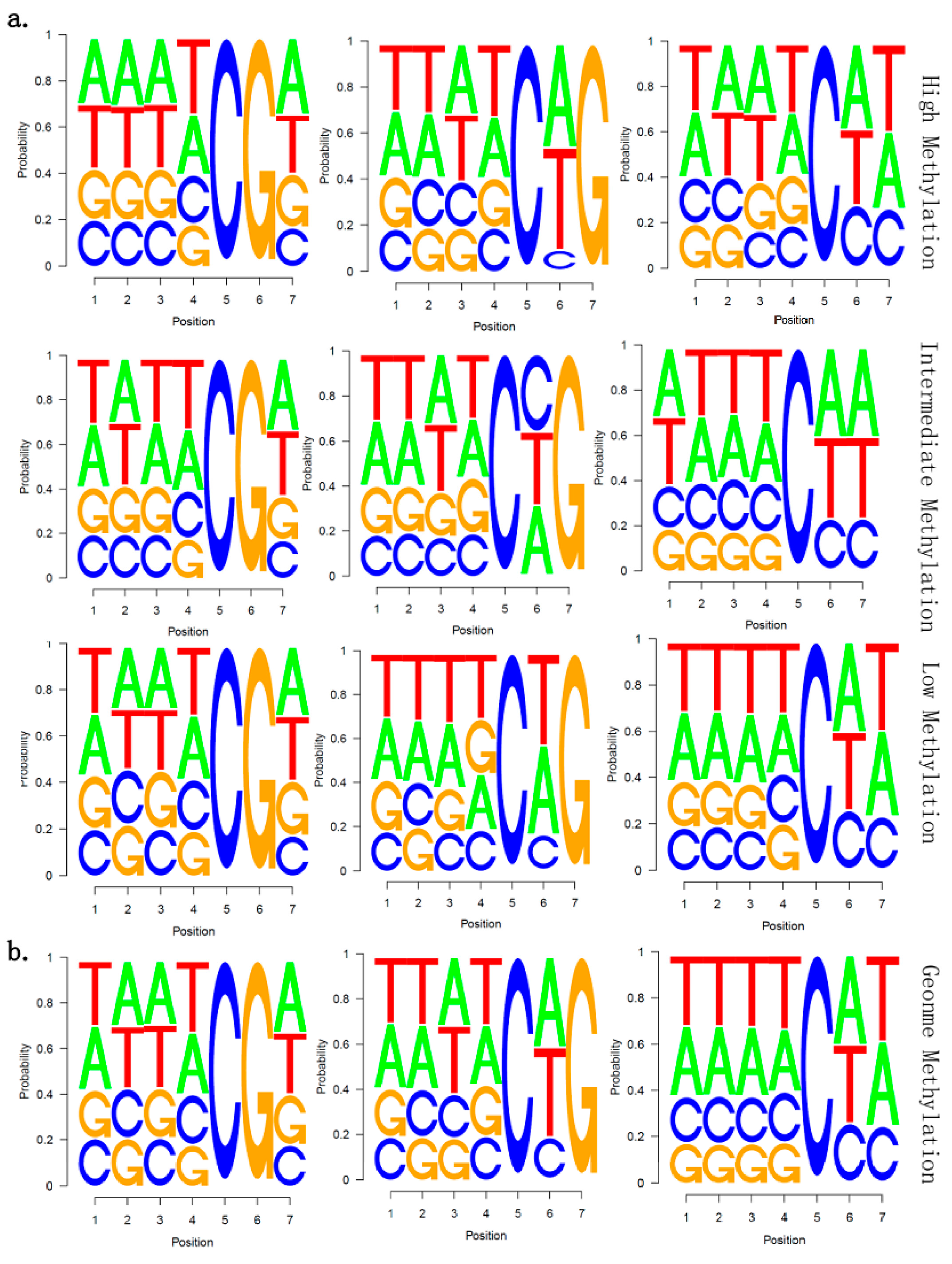
2.5. There Is no Correlation between Sequence Context and Methylation Preference in Birch
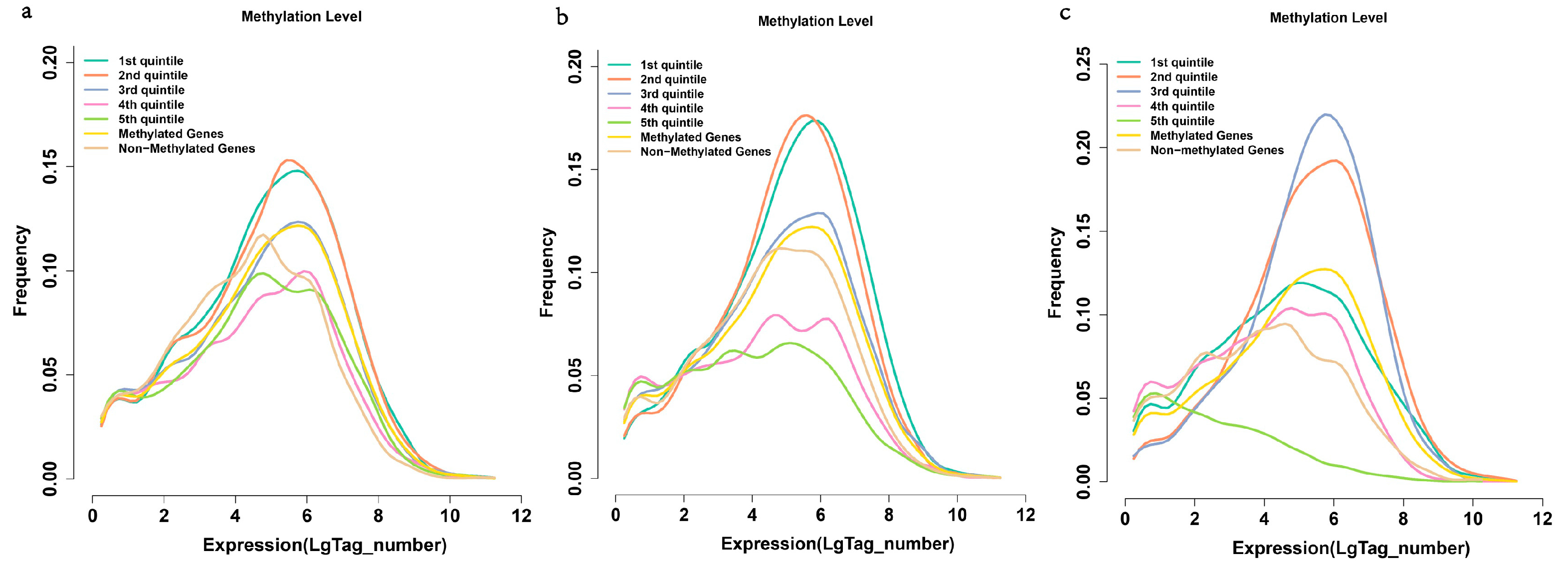
2.6. Methylation and Gene Expression
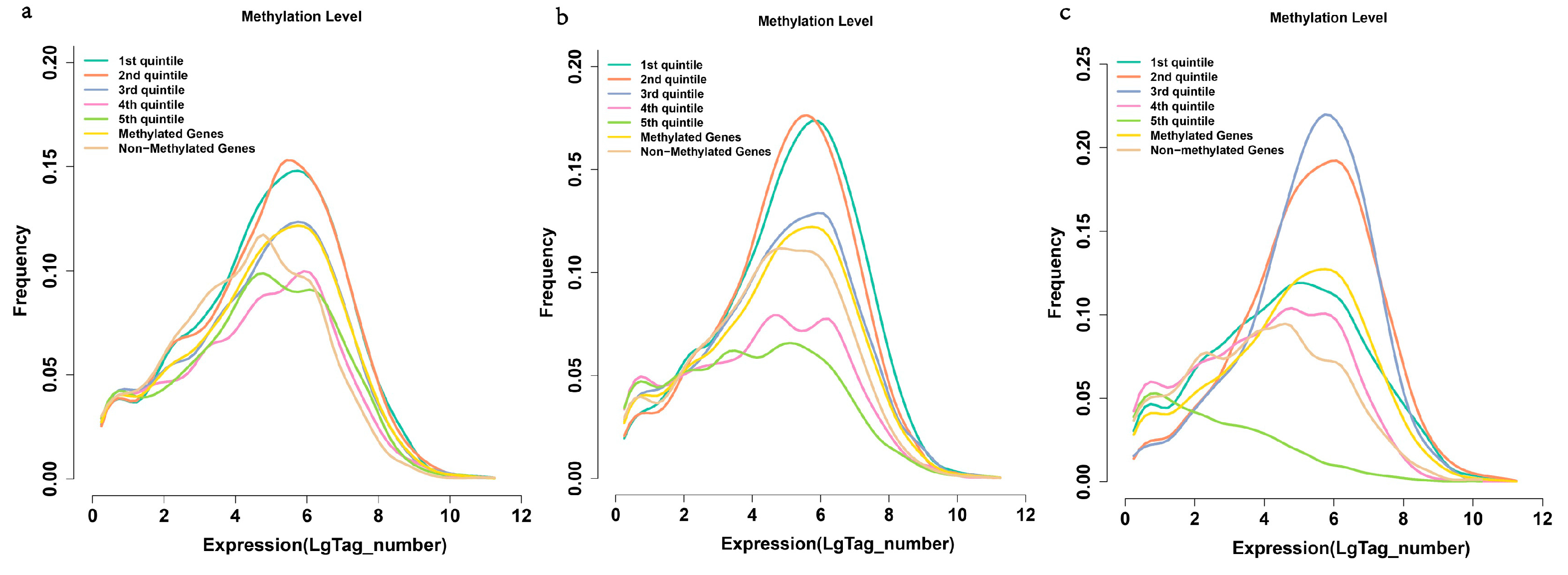
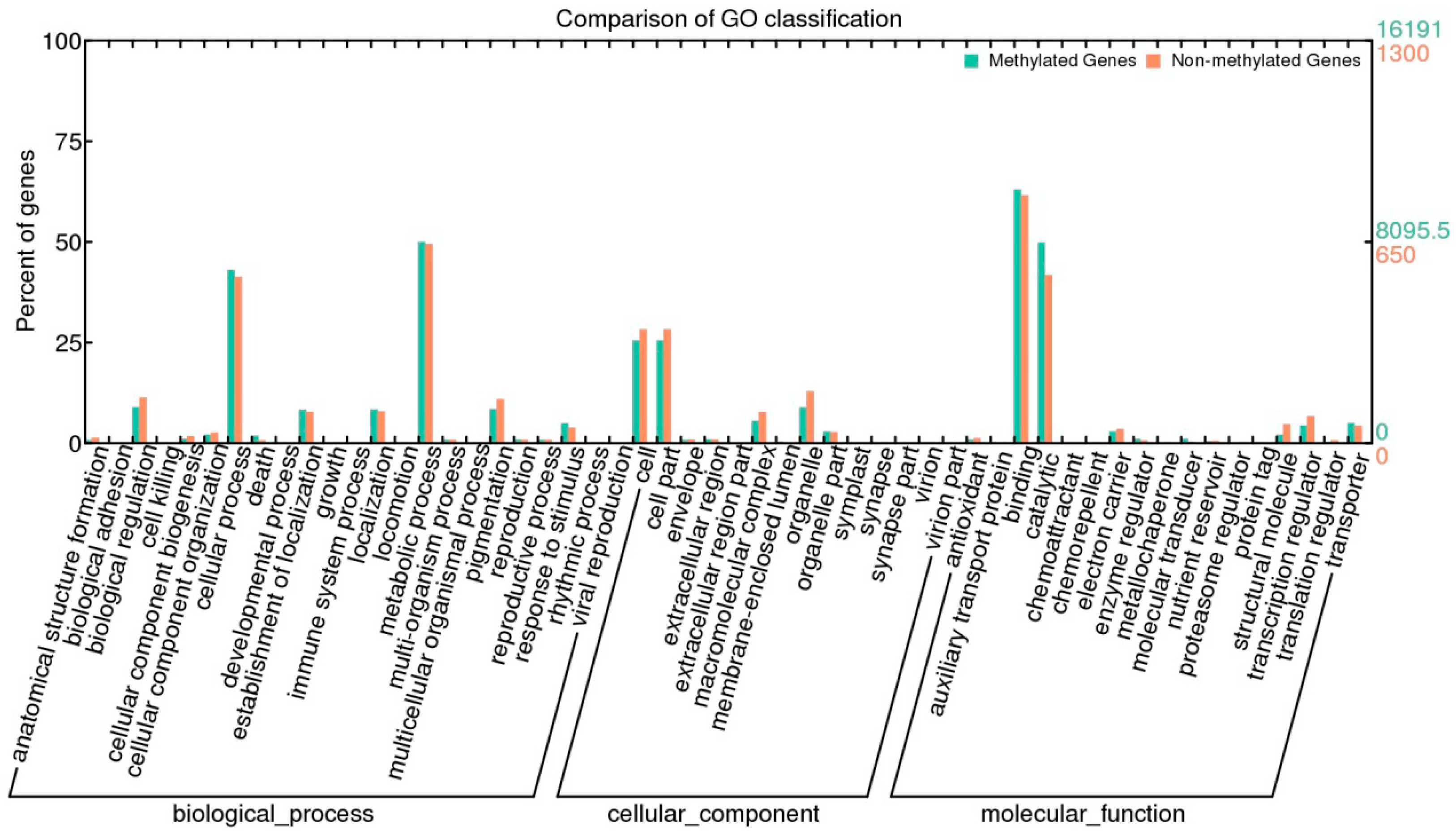
3. Experimental Section
3.1. Plant Materials
3.2. Mapping and Processing of Bisulphite Sequencing (BS-Seq) Reads
3.3. Construction of a cDNA Library and Illumina Sequencing
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Henderson, I.R.; Jacobsen, S.E. Epigenetic inheritance in plants. Nature 2007, 447, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.W.-L.; Henderson, I.R.; Jacobsen, S.E. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 2005, 6, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [PubMed]
- Zhang, X.Y.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.-L.; Chen, H.; Henderson, I.P.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.W.-L.; Zilberman, D.; Xie, Z.; Johansen, L.K.; Carrinqton, J.C.; Jacobsen, S.E. RNA silencing genes control de novo DNA methylation. Science 2004, 303, 1336–1336. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.F.; Ibarra, C.A.; Silva, P.; Zemach, A.; Eshed-Williams, L.; Fischer, R.L.; Zilberman, D. Genome-wide demethylation of Arabidopsis endosperm. Science 2009, 324, 1451–1454. [Google Scholar] [CrossRef] [PubMed]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef] [PubMed]
- Widma, N.; Feng, S.; Jacobsen, S.E.; Pellegrini, M. Epigenetic differences between shoots and roots in Arabidopsis reveals tissue-specific regulation. Epigenetics 2013, 9, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Fei, Z.; Chen, Y.-R.; Zheng, Y.; Huang, M.; Vrebalov, J.; McQuinn, R.; Gapper, N.; Liu, B.; Xiang, J.; et al. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 2013, 31, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Elling, A.A.; Li, X.; Peng, Z.; He, G.; Sun, H.; Qi, Y.; Liu, X.S.; Den, X.W. Ggenome-wide and organ-specific landscapes of epigenetic modifications and their relationships to mRNA and small RNA transcriptomes in maize. Plant Cell Online 2009, 21, 1053–1069. [Google Scholar] [CrossRef]
- Feng, S.H.; Cokus, S.J.; Zhang, X.; Chen, P.Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [PubMed]
- Tuskan, G.A.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Fojtova, M.; Kovarik, A.; Matyasek, R. Cytosine methylation of plastid genome in higher plants. Fact or artifact? Plant Sci. 2001, 160, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007, 39, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.F.; Aufsatz, W.; Zilberman, D.; Mette, M.F.; Huang, M.S.; Matzke, M.; Jacobsen, S.E. Role of the DRM and CMT3 Methyltransferases in RNA-directed DNA methylation. Curr. Biol. 2003, 13, 2212–2217. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, J.; Hu, F.; Ge, S.; Ye, M.; Xiang, H.; Zhang, G.; Zheng, X.; Zhang, H.; Zhang, S.; et al. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genomics 2012, 13, 300. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Tran, R.K.; Henikoff, J.G.; Zilberman, D.; Ditt, R.F.; Jacobsen, S.E.; Henikoff, S. DNA methylation profiling identifies CG methylation clusters Arabidopsis genes. Curr. Biol. 2005, 15, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Gruenbaum, Y.; Naveh-Many, T.; Cedar, H.; Razin, A. Sequence specificity of methylation in higher plant DNA. Nature 1981, 292, 860–862. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [PubMed]
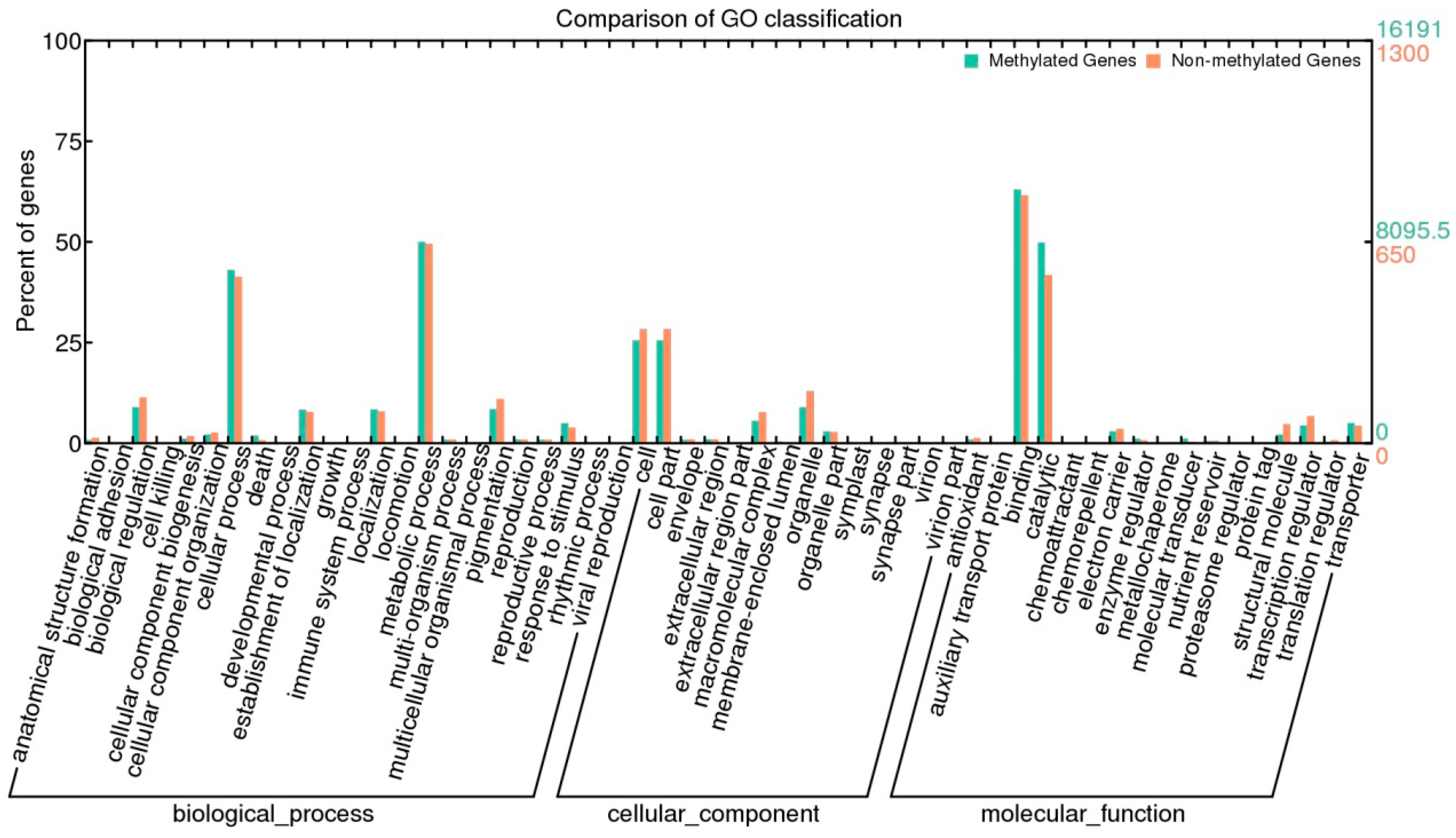
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; Wang, J. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Puryear, J.; Cairney, J. A simple and efficient method for isolating RNA from pine trees. Plant. Mol. Biol. Rep. 1993, 11, 113–116. [Google Scholar] [CrossRef]
- Xiang, H.; Zhu, J.; Chen, Q.; Dai, F.; Li, X.; Li, M.; Zhang, H.; Zhang, G.; Li, D.; Dong, Y.; et al. Single base-resolution methylome of the silkworm reveals a sparse epigenomic map. Nat. Biotechnol. 2010, 28, 756–756. [Google Scholar] [CrossRef]
- Information Site. Betula Platyphylla. Available online: http://birch.genomics.cn/ (accessed on 13 September 2014).
- Li, R.Q.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, C.; Wang, C.; He, L.; Yang, C.; Wang, Y. Shotgun Bisulfite Sequencing of the Betula platyphylla Genome Reveals the Tree’s DNA Methylation Patterning. Int. J. Mol. Sci. 2014, 15, 22874-22886. https://doi.org/10.3390/ijms151222874
Su C, Wang C, He L, Yang C, Wang Y. Shotgun Bisulfite Sequencing of the Betula platyphylla Genome Reveals the Tree’s DNA Methylation Patterning. International Journal of Molecular Sciences. 2014; 15(12):22874-22886. https://doi.org/10.3390/ijms151222874
Chicago/Turabian StyleSu, Chang, Chao Wang, Lin He, Chuanping Yang, and Yucheng Wang. 2014. "Shotgun Bisulfite Sequencing of the Betula platyphylla Genome Reveals the Tree’s DNA Methylation Patterning" International Journal of Molecular Sciences 15, no. 12: 22874-22886. https://doi.org/10.3390/ijms151222874